DOXERCALCIFEROL injection
Doxercalciferol by
Drug Labeling and Warnings
Doxercalciferol by is a Prescription medication manufactured, distributed, or labeled by West-Ward Pharmaceutical Corp., Hikma Farmaceutica. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
Doxercalciferol, the active ingredient in Doxercalciferol Injection, is a synthetic vitamin D2 analog that undergoes metabolic activation in vivo to form 1a,25-dihydroxyvitamin D2 (1a,25-(OH)2D2), a naturally occurring, biologically active form of vitamin D2. Doxercalciferol injection is available as a sterile, clear, colorless aqueous solution for intravenous injection.
Doxercalciferol single-use injection is supplied in a 2 mL amber glass ampul containing 4 mcg/2 mL or 2 mcg/mL. Each milliliter (mL) of solution contains doxercalciferol, 2 mcg; Polysorbate 20, 4 mg; sodium chloride, 1.5 mg; sodium ascorbate, 10 mg; sodium phosphate, dibasic, anhydrous 7.6 mg; sodium dihydrogen phosphate, dihydrate, 2.34 mg; and disodium edetate, 1.1 mg.
Doxercalciferol is a colorless crystalline compound with a calculated molecular weight of 412.66 and a molecular formula of C28H44O2. It is soluble in oils and organic solvents, but is relatively insoluble in water. Chemically, doxercalciferol is (1α,3β,5Z,7E,22E)-9,10-secoergosta-5,7,10(19),22-tetraene-1,3-diol and has the structural formula presented in Figure 1.
Figure 1: Chemical Structure of Doxercalciferol
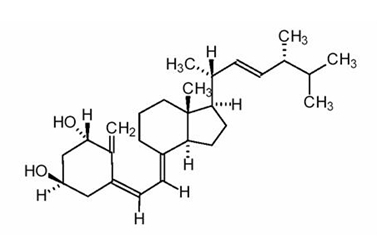
Other names frequently used for doxercalciferol are 1α-hydroxyvitamin D2, 1α-OH-D2, and 1α-hydroxyergocalciferol.
-
CLINICAL PHARMACOLOGY
Vitamin D levels in humans depend on two sources: (1) exposure to the ultraviolet rays of the sun for conversion of 7-dehydrocholesterol in the skin to vitamin D3 (cholecalciferol) and (2) dietary intake of either vitamin D2 (ergocalciferol) or vitamin D3. Vitamin D2 and vitamin D3 must be metabolically activated in the liver and kidney before becoming fully active on target tissues. The initial step in the activation process is the introduction of a hydroxyl group in the side chain at C-25 by the hepatic enzyme, CYP 27 (a vitamin D-25-hydroxylase). The products of this reaction are 25-(OH)D2 and 25-(OH)D3, respectively. Further hydroxylation of these metabolites occurs in the mitochondria of kidney tissue, catalyzed by renal 25-hydroxyvitamin D-1-α-hydroxylase to produce 1α,25-(OH)2D2, the primary biologically active form of vitamin D2, and 1α,25-(OH)2D3 (calcitriol), the biologically active form of vitamin D3.
Mechanism of Action
Calcitriol (1α,25-(OH)2D3) and 1α,25-(OH)2D2 regulate blood calcium at levels required for essential body functions. Specifically, the biologically active vitamin D metabolites control the intestinal absorption of dietary calcium, the tubular reabsorption of calcium by the kidney and, in conjunction with parathyroid hormone (PTH), the mobilization of calcium from the skeleton. They act directly on bone cells (osteoblasts) to stimulate skeletal growth, and on the parathyroid glands to suppress PTH synthesis and secretion. These functions are mediated by the interaction of these biologically active metabolites with specific receptor proteins in the various target tissues. In uremic patients, deficient production of biologically active vitamin D metabolites (due to lack of or insufficient 25-hydroxyvitamin D-1-alpha-hydroxylase activity) leads to secondary hyperparathyroidism, which contributes to the development of metabolic bone disease in patients with renal failure.
Pharmacokinetics and Metabolism
After intravenous administration, doxercalciferol is activated by CYP 27 in the liver to form 1α,25-(OH)2D2 (major metabolite) and 1α,24-dihydroxyvitamin D2 (minor metabolite). Activation of doxercalciferol does not require the involvement of the kidneys.
Peak blood levels of 1α,25-(OH)2D2 are reached at 8 +/- 5.9 hours (mean +/- SD) after a single intravenous dose of 5 mcg of doxercalciferol. The mean elimination half-life of 1α,25-(OH)2D2 after an oral dose is approximately 32 to 37 hours with a range of up to 96 hours. The mean elimination half-life in patients with end stage renal disease (ESRD) and in healthy volunteers appears to be similar following an oral dose. Hemodialysis causes a temporary increase in 1α,25-(OH)2D2 mean concentrations presumably due to volume contraction. 1α,25-(OH)2D2 is not removed from blood during hemodialysis.
Clinical Studies
The safety and effectiveness of doxercalciferol injection were evaluated in two open-label, single-arm, multi-centered clinical studies (Study C and Study D) in a total of 70 patients with chronic kidney disease on hemodialysis (Stage 5 CKD). Patients in Study C were an average age of 54 years (range: 23 to 73), were 50% male, and were 61% African-American, 25% Caucasian, and 14% Hispanic, and had been on hemodialysis for an average of 65 months. Patients in Study D were an average age of 51 years (range: 28 to 76), were 48% male, and 100% African-American and had been on hemodialysis for an average of 61 months. This group of 70 of the 138 patients who had been treated with doxercalciferol capsules in prior clinical studies (Study A and Study B) received doxercalciferol injection in an open-label fashion for 12 weeks following an 8-week washout (control) period. Dosing of doxercalciferol injection was initiated at the rate of 4 mcg administered at the end of each dialysis session (3 times weekly) for a total of 12 mcg per week. The dosage of doxercalciferol was adjusted in an attempt to achieve iPTH levels within a targeted range of 150 to 300 pg/mL. The dosage was increased by 2 mcg per dialysis session after 8 weeks of treatment if the iPTH levels remained above 300 pg/mL and were greater than 50% of baseline levels. The maximum dosage was limited to 18 mcg per week. If at any time during the trial iPTH fell below 150 pg/mL, doxercalciferol injection was immediately suspended and restarted at a lower dosage the following week.
Results
Fifty-two of the 70 patients who were treated with doxercalciferol injection achieved iPTH levels ≤ 300 pg/mL. Forty-one of these patients exhibited plasma iPTH levels ≤ 300 pg/mL on at least three occasions. Thirty-six patients had plasma iPTH levels < 150 pg/mL on at least one occasion during study participation.
Mean weekly doses in Study C ranged from 8.9 mcg to 12.5 mcg. In Study D, the mean weekly doses ranged from 9.1 mcg to 11.6 mcg.
Decreases in plasma iPTH from baseline values were calculated using as baseline the average of the last three values obtained during the 8-week washout period and are displayed in the table below. Plasma iPTH levels were measured weekly during the 12-week study.
Table 1: iPTH Summary Data for Patients Receiving Doxercalciferol Injection - * Values were carried forward for the two patients on study for 10 weeks
- †
Treatment iPTH minus baseline iPTH
- ‡ Wilcoxon one-sample test
iPTH Level Study C
(n = 28)Study D
(n = 42)Combined Protocols
(n = 70)Baseline (Mean of Weeks -2, -1 and 0) Mean (SE) 698 (60) 762 (65) 736 (46) Median 562 648 634 On-treatment (Week 12*) Mean (SE) 406 (63) 426 (60) 418 (43) Median 311 292 292 Change from Baseline† Mean (SE) -292 (55) -336 (41) -318 (33) Median -274 -315 -304 P-value‡ 0.004 0.001 < 0.001 In both studies, iPTH levels increased progressively and significantly in 62.9% of patients during the 8-week washout (control) period during which no vitamin D derivatives were administered. In contrast, doxercalciferol injection treatment resulted in a clinically significant reduction (at least 30%) from baseline in mean iPTH levels during the 12-week open-label treatment period in more than 92% of the 70 treated patients.
Table 2 shows the numbers of patients who achieved iPTH levels below 300 pg/mL on one, two, or three or more non-consecutive occasions during the 12-week treatment period. Thirty-seven of 70 patients (53%) had plasma iPTH levels within the targeted range (150 to 300 pg/mL) during Weeks 10 to 12.
Table 2: Number of Times iPTH ≤ 300 pg/mL 1 2 ≥3 Study C 3/28 0/28 16/28 Study D 4/42 4/42 25/42 - INDICATIONS AND USAGE
-
CONTRAINDICATIONS
Doxercalciferol injection should not be given to patients with a tendency towards hypercalcemia or current evidence of vitamin D toxicity.
Doxercalciferol injection is contraindicated in patients with previous hypersensitivity to doxercalciferol or any of its ingredients (see WARNINGS and ADVERSE REACTIONS).
-
WARNINGS
Overdosage of any form of vitamin D, including doxercalciferol injection is dangerous (see OVERDOSAGE). Progressive hypercalcemia due to overdosage of vitamin D and its metabolites may be so severe as to require emergency attention. Acute hypercalcemia may exacerbate tendencies for cardiac arrhythmias and seizures and may potentiate the action of digitalis drugs. Chronic hypercalcemia can lead to generalized vascular calcification and other soft-tissue calcification. The serum calcium times serum phosphorus (Ca X P) product should be maintained at < 55 mg2/dL2 in patients with chronic kidney disease. Radiographic evaluation of suspect anatomical regions may be useful in the early detection of this condition.
Since doxercalciferol is a precursor for 1α,25-(OH)2D2, a potent metabolite of vitamin D2, pharmacologic doses of vitamin D and its derivatives should be withheld during doxercalciferol injection treatment to avoid possible additive effects and hypercalcemia.
Oral calcium-based or other non-aluminum-containing phosphate binders and a low phosphate diet should be used to control serum phosphorus levels in patients undergoing dialysis. Uncontrolled serum phosphorus exacerbates secondary hyperparathyroidism and can lessen the effectiveness of doxercalciferol injection in reducing blood PTH levels. If hypercalcemia occurs after initiating doxercalciferol injection therapy, the dose of doxercalciferol injection and/or calcium-containing phosphate binders should be decreased. If hyperphosphatemia occurs after initiating doxercalciferol injection, the dose of doxercalciferol injection should be decreased and/or the dose of phosphate binders increased. (See dosing recommendations for doxercalciferol injection under DOSAGE AND ADMINISTRATION.)
Magnesium-containing antacids and doxercalciferol injection should not be used concomitantly in patients on chronic renal dialysis because such use may lead to the development of hypermagnesemia.
Serious hypersensitivity reactions, including fatal outcome, have been reported post marketing in patients on hemodialysis following administration of doxercalciferol injection. Hypersensitivity reactions include anaphylaxis with symptoms of angioedema (involving face, lips, tongue and airways), hypotension, unresponsiveness, chest discomfort, shortness of breath, and cardiopulmonary arrest. These reactions may occur separately or together.
Monitor patients receiving doxercalciferol injection upon initiation of treatment for hypersensitivity reactions. Should a hypersensitivity reaction occur, discontinue doxercalciferol, monitor and treat if indicated (see CONTRAINDICATIONS).
-
PRECAUTIONS
General
The principal adverse effects of treatment with doxercalciferol injection are hypercalcemia, hyperphosphatemia, and oversuppression of PTH (iPTH less than 150 pg/mL). Prolonged hypercalcemia can lead to calcification of soft tissues, including the heart and arteries, and hyperphosphatemia can exacerbate hyperparathyroidism. Oversuppression of PTH may lead to adynamic bone syndrome. All of these potential adverse effects should be managed by regular patient monitoring and appropriate dosage adjustments. During treatment with doxercalciferol injection, patients usually require dose titration, as well as adjustment in co-therapy (i.e., dietary phosphate binders) in order to maximize PTH suppression while maintaining serum calcium and phosphorus levels within prescribed ranges.
In two open-label, single-arm, multi-centered studies, the incidence of hypercalcemia and hyperphosphatemia increased during therapy with doxercalciferol injection (see ADVERSE REACTIONS). The observed increases during doxercalciferol injection treatment underscore the importance of regular safety monitoring of serum calcium and phosphorus levels throughout treatment. Patients with higher pre-treatment serum levels of calcium (> 10.5 mg/dL) or phosphorus (> 6.9 mg/dL) were more likely to experience hypercalcemia or hyperphosphatemia. Therefore, doxercalciferol injection should not be given to patients with a recent history of hypercalcemia or hyperphosphatemia, or evidence of vitamin D toxicity.
Table 3: Incidence Rates of Hypercalcemia and Hyperphosphatemia in Two Phase 3 Studies with Doxercalciferol Injection Study Hypercalcemia
(per 100 patient weeks)Hyperphosphatemia
(per 100 patient weeks)Washout
(Off Treatment)Open-Label
(Treatment)Washout
(Off Treatment)Open-Label
(Treatment)Study C 0.9 0.9 0.9 2.4 Study D 0.3 1 1.2 3.7 Information for the Patient
The patient, spouse, or guardian should be informed about adherence to instructions about diet, calcium supplementation, and avoidance of the use of nonprescription drugs without prior approval from the patient’s physician. Patients should also be carefully informed about the symptoms of hypercalcemia (see ADVERSE REACTIONS).
Laboratory Tests
Serum levels of iPTH, calcium, and phosphorus should be determined prior to initiation of doxercalciferol injection treatment. During the early phase of treatment (i.e., first 12 weeks), serum iPTH, calcium, and phosphorus levels should be determined weekly. For dialysis patients in general, serum or plasma iPTH and serum calcium, phosphorus, and alkaline phosphatase should be determined periodically.
Drug Interactions
Specific drug interaction studies have not been conducted. Magnesium-containing antacids and doxercalciferol should not be used concomitantly because such use may lead to the development of hypermagnesemia (see WARNINGS). Although not examined specifically, enzyme inducers (such as glutethimide and phenobarbital) may affect the 25-hydroxylation of doxercalciferol and may necessitate dosage adjustments. Cytochrome P450 inhibitors (such as ketoconazole and erythromycin) may inhibit the 25-hydroxylation of doxercalciferol. Hence, formation of the active doxercalciferol moiety may be hindered.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies in animals to evaluate the carcinogenic potential of doxercalciferol have not been conducted. No evidence of genetic toxicity was observed in an in vitro bacterial mutagenicity assay (Ames test) or a mouse lymphoma gene mutation assay. Doxercalciferol caused structural chromatid and chromosome aberrations in an in vitro human lymphocyte clastogenicity assay with metabolic activation. However, doxercalciferol was negative in an in vivo mouse micronucleus clastogenicity assay. Doxercalciferol had no effect on male or female fertility in rats at oral doses up to 2.5 mcg/kg/day (approximately 3 times the maximum recommended human oral dose of 60 mcg/wk based on mcg/m2 body surface area).
Use in Pregnancy
Pregnancy Category B
Reproduction studies in rats and rabbits, at doses up to 20 mcg/kg/day and 0.1 mcg/kg/day (approximately 25 times and less than the maximum recommended human oral dose of 60 mcg/week based on mcg/m2 body surface area, respectively) have revealed no teratogenic or fetotoxic effects due to doxercalciferol. There are, however, no adequate and well controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Nursing Mothers
It is not known whether doxercalciferol is excreted in human milk. Because other vitamin D derivatives are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from doxercalciferol, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
Safety and efficacy of doxercalciferol injection in pediatric patients have not been established.
Geriatric Use
Of the 70 patients treated with doxercalciferol injection in the two Phase 3 clinical studies, 12 patients were 65 years or over. In these studies, no overall differences in efficacy or safety were observed between patients 65 years or older and younger patients.
Hepatic Insufficiency
Studies examining the influence of hepatic insufficiency on the metabolism of doxercalciferol were inconclusive. Since patients with hepatic insufficiency may not metabolize doxercalciferol appropriately, the drug should be used with caution in patients with impaired hepatic function. More frequent monitoring of iPTH, calcium, and phosphorus levels should be done in such individuals.
-
ADVERSE REACTIONS
Doxercalciferol injection has been evaluated for safety in 70 patients with chronic renal disease on hemodialysis (who had been previously treated with oral doxercalciferol) from two 12-week, open-label, single-arm, multi-centered studies. (Dosage titrated to achieve target plasma iPTH levels, see CLINICAL PHARMACOLOGY, Clinical Studies.)
Because there was no placebo group included in the studies of doxercalciferol injection, Table 4 provides the adverse event incidence rates from placebo-controlled studies of oral doxercalciferol.
Table 4: Adverse Events Reported by ≥ 2% of Doxercalciferol Injection Treated Patients and More Frequently than Placebo During the Double-Blind Phase of Two Clinical Studies Adverse Event Doxercalciferol Injection (n = 61)
%Placebo (n = 61)
%Body as a Whole Abscess 3.3 0.0 Headache 27.9 18.0 Malaise 27.9 19.7 Cardiovascular System Bradycardia 6.6 4.9 Digestive System Anorexia 4.9 3.3 Constipation 3.3 3.3 Dyspepsia 4.9 1.6 Nausea/Vomiting 21.3 19.7 Musculoskeletal System Arthralgia 4.9 0.0 Metabolic and Nutritional Edema 34.4 21.3 Weight increase 4.9 0.0 Nervous System Dizziness 11.5 9.8 Sleep Disorder 3.3 0.0 Respiratory System Dyspnea 11.5 6.6 Skin Pruritus 8.2 6.6 A patient who reported the same medical term more than once was counted only once for that medical term. Potential adverse effects of doxercalciferol injection are, in general, similar to those encountered with excessive vitamin D intake. The early and late signs and symptoms of vitamin D intoxication associated with hypercalcemia include:
Early
Weakness, headache, somnolence, nausea, vomiting, dry mouth, constipation, muscle pain, bone pain, metallic taste, and anorexia.Late
Polyuria, polydipsia, anorexia, weight loss, nocturia, conjunctivitis (calcific), pancreatitis, photophobia, rhinorrhea, pruritus, hyperthermia, decreased libido, elevated blood urea nitrogen (BUN), albuminuria, hypercholesterolemia, elevated serum aspartate transaminase (AST) and alanine transaminase (ALT), ectopic calcification, hypertension, cardiac arrhythmias, sensory disturbances, dehydration, apathy, arrested growth, urinary tract infections, and, rarely, overt psychosis.Postmarketing Experience
Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to estimate their frequency or to establish a causal relationship to drug exposure.
Hypersensitivity reactions, including fatal outcome, have been reported in patients on hemodialysis following administration of doxercalciferol injection. Hypersensitivity reactions include anaphylaxis with symptoms of angioedema (involving face, lips, tongue and airways), hypotension, unresponsiveness, chest discomfort, shortness of breath, cardiopulmonary arrest, pruritus and skin burning sensation (see WARNINGS). These reactions may occur separately or together.
-
OVERDOSAGE
Administration of doxercalciferol injection to patients in excess doses can cause hypercalcemia, hypercalciuria, hyperphosphatemia, and over-suppression of PTH secretion leading in certain cases to adynamic bone disease. High intake of calcium and phosphate concomitant with doxercalciferol injection may lead to similar abnormalities. High levels of calcium in the dialysate bath may contribute to hypercalcemia.
Treatment of Hypercalcemia and Overdosage
General treatment of hypercalcemia (greater than 1 mg/dL above the upper limit of the normal range) consists of immediate suspension of doxercalciferol injection therapy, institution of a low calcium diet, and withdrawal of calcium supplements. Serum calcium levels should be determined at least weekly until normocalcemia ensues. Hypercalcemia usually resolves in 2 to 7 days. When serum calcium levels have returned to within normal limits, doxercalciferol injection therapy may be reinstituted at a dose that is at least 1 mcg lower than prior therapy. Serum calcium levels should be obtained weekly after all dosage changes and during subsequent dosage titration. Persistent or markedly elevated serum calcium levels may be corrected by dialysis against a reduced calcium or calcium-free dialysate.
Treatment of Accidental Overdosage of Doxercalciferol Injection
The treatment of acute accidental overdosage of doxercalciferol injection should consist of general supportive measures. Serial serum electrolyte determinations (especially calcium), rate of urinary calcium excretion, and assessment of electrocardiographic abnormalities due to hypercalcemia should be obtained. Such monitoring is critical in patients receiving digitalis. Discontinuation of supplemental calcium and institution of a low calcium diet are also indicated in accidental overdosage. If persistent and markedly elevated serum calcium levels occur, treatment with standard medical care should be followed, as needed. Based on similarities between doxercalciferol and its active metabolite, 1α,25-(OH)2D2, it is expected that doxercalciferol is not removed from the blood by dialysis.
-
DOSAGE AND ADMINISTRATION
Adult Administration:
For intravenous use only. The optimal dose of doxercalciferol injection must be carefully determined for each patient.
The recommended initial dose of doxercalciferol injection is 4 mcg administered intravenously as a bolus dose 3 times weekly at the end of dialysis (approximately every other day). The initial dose should be adjusted, as needed, in order to lower blood iPTH into the range of 150 to 300 pg/mL. The dose may be increased at 8-week intervals by 1 mcg to 2 mcg if iPTH is not lowered by 50% and fails to reach the target range. Dosages higher than 18 mcg weekly have not been studied. Drug administration should be suspended if iPTH falls below 100 pg/mL and restarted one week later at a dose that is at least 1 mcg lower than the last administered dose. During titration, iPTH, serum calcium, and serum phosphorus levels should be obtained weekly. If hypercalcemia, hyperphosphatemia, or a serum calcium times phosphorus product greater than 55 mg2/dL2 is noted, the dose of doxercalciferol injection should be decreased or suspended and/or the dose of phosphate binders should be appropriately adjusted. If suspended, the drug should be restarted at a dose that is 1 mcg lower.
Dosing must be individualized and based on iPTH levels with monitoring of serum calcium and serum phosphorus levels. Table 5 presents a suggested approach in dose titration:
Table 5: Initial Dosing iPTH Level Doxercalciferol Injection Dose > 400 pg/mL 4 mcg 3 times per week at the end of dialysis, or approximately every other day Dose Titration iPTH Level Doxercalciferol Injection Dose Decreased by < 50% and above 300 pg/mL Increase by 1 mcg to 2 mcg at 8-week intervals as necessary Decreased by > 50% and above 300 pg/mL Maintain 150 to 300 pg/mL Maintain < 100 pg/mL Suspend for one week, then resume at a dose that is at least 1 mcg lower -
HOW SUPPLIED
Doxercalciferol Injection is available in the following package:
4 mcg/2 mL (2 mcg/mL), 2 mL pre-scored amber ampuls packaged in 25s (NDC: 0641-6154-25)
Store at 25°C (77°F), excursions permitted to 15 to 30°C (59 to 86°F) [See USP Controlled Room Temperature].
Protect from light.
To report SUSPECTED ADVERSE REACTIONS, contact West-Ward Pharmaceuticals Corp. at 1-877-845-0689, or the FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
For Product Inquiry call 1-877-845-0689.
Manufactured by:
HIKMA FARMACÊUTICA (PORTUGAL), S.A.
Estrada do Rio da Mό, 8, 8A e 8B – Fervença – 2705-906 Terrugem SNT, PORTUGALDistributed by:
WEST-WARD
A Hikma Company
Eatontown, NJ 07724 USAPIN349-WES/2
Revised July 2016
-
PRINCIPAL DISPLAY PANEL - Container
NDC: 0641-6154-01
2 mL Ampul
Doxercalciferol
Injection
Rx only
4 mcg/2 mL (2 mcg/mL)
For Intravenous Use Only
Protect From Light
Discard Unused Portion
-
PRINCIPAL DISPLAY PANEL - Carton
NDC: 0641-6154-25 Rx only
Doxercalciferol
Injection
4 mcg/2 mL
(2 mg/mL)
For Intravenous Use Only
Protect From Light
Discard Unused Portion
25 x 2 mL AmpulsCONTENTS
Active Ingredient: Doxercalciferol, 0.0002%
Inactive ingredients: Polysorbate 20, 0.4%;
Sodium Chloride, 0.15%; Sodium Ascorbate, 1.0%;
Sodium Phosphate, Dibasic, Anhydrous, 0.76%;
Sodium Dihydrogen Phosphate, Dihydrate, 0.234%;
Edetate, Disodium, Dihydrate, 0.11%;
Water for Injection, 97.35%Usual Dosage: See accompanying prescribing
information.
Store at 20° to 25°C (68° to 77°F)
[See USP Controlled Room Temperature].
PROTECT FROM LIGHT: Keep covered in carton
until time of use.![NDC: <a href=/NDC/0641-6154-25>0641-6154-25</a> Rx only Doxercalciferol Injection 4 mcg/2 mL (2 mg/mL) For Intravenous Use Only Protect From Light Discard Unused Portion 25 x 2 mL Ampuls CONTENTS Active Ingredient: Doxercalciferol, 0.0002% Inactive ingredients: Polysorbate 20, 0.4%; Sodium Chloride, 0.15%; Sodium Ascorbate, 1.0%; Sodium Phosphate, Dibasic, Anhydrous, 0.76%; Sodium Dihydrogen Phosphate, Dihydrate, 0.234%; Edetate, Disodium, Dihydrate, 0.11%; Water for Injection, 97.35% Usual Dosage: See accompanying prescribing information. Store at 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature]. PROTECT FROM LIGHT: Keep covered in carton until time of use.](https://fda.report/DailyMed/e99a1d29-3989-4437-999e-4e9cab8b9b1e/doxercalciferol-injection-3.jpg)
-
INGREDIENTS AND APPEARANCE
DOXERCALCIFEROL
doxercalciferol injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0641-6154 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DOXERCALCIFEROL (UNII: 3DIZ9LF5Y9) (DOXERCALCIFEROL - UNII:3DIZ9LF5Y9) DOXERCALCIFEROL 2 ug in 1 mL Inactive Ingredients Ingredient Name Strength POLYSORBATE 20 (UNII: 7T1F30V5YH) 4 mg in 1 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) 1.5 mg in 1 mL SODIUM ASCORBATE (UNII: S033EH8359) 10 mg in 1 mL SODIUM PHOSPHATE, DIBASIC, ANHYDROUS (UNII: 22ADO53M6F) 7.6 mg in 1 mL SODIUM PHOSPHATE, MONOBASIC (UNII: 3980JIH2SW) 2.3 mg in 1 mL EDETATE DISODIUM (UNII: 7FLD91C86K) 1.1 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0641-6154-25 25 in 1 BOX 08/30/2013 1 NDC: 0641-6154-01 2 mL in 1 AMPULE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA091101 08/30/2013 Labeler - Hikma Pharmaceuticals USA Inc. (946499746) Establishment Name Address ID/FEI Business Operations HIKMA FARMACEUTICA (PORTUGAL), S.A 452742943 ANALYSIS(0641-6154) , LABEL(0641-6154) , MANUFACTURE(0641-6154) , PACK(0641-6154)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.