EVRYSDI- risdiplam powder, for solution EVRYSDI- risdiplam tablet
EVRYSDI by
Drug Labeling and Warnings
EVRYSDI by is a Prescription medication manufactured, distributed, or labeled by Genentech Inc., F. Hoffmann-La Roche AG, F. Hoffmann-La Roche Ltd.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use EVRYSDI safely and effectively. See full prescribing information for EVRYSDI.
EVRYSDI® (risdiplam) for oral solution
EVRYSDI® (risdiplam) tablets, for oral use
Initial U.S. Approval: 2020INDICATIONS AND USAGE
EVRYSDI is a survival of motor neuron 2 (SMN2) splicing modifier indicated for the treatment of spinal muscular atrophy (SMA) in pediatric and adult patients. (1)
DOSAGE AND ADMINISTRATION
- Administer once daily with or without food per the table below (2.1):
Age and Body Weight Recommended Daily Dosage Dosage Form Less than 2 months of age 0.15 mg/kg EVRYSDI for Oral Solution 2 months to less than
2 years of age0.2 mg/kg 2 years of age and older weighing less than 20 kg 0.25 mg/kg 2 years of age and older weighing 20 kg or more 5 mg EVRYSDI for Oral Solution
or
EVRYSDI Tablet- Swallow EVRYSDI tablet whole with water or dispersed in non-chlorinated drinking water (e.g., filtered water). (2.2)
- Administer EVRYSDI for oral solution with the provided oral syringe. (2.2)
- EVRYSDI for oral solution must be constituted by a healthcare provider prior to dispensing. (2.4)
- See Full Prescribing Information for important preparation and administration instructions. (2.2, 2.4)
DOSAGE FORMS AND STRENGTHS
- For Oral Solution: 60 mg of risdiplam as a powder for constitution to provide 0.75 mg/mL solution. (3)
- Tablets: 5 mg
CONTRAINDICATIONS
None. (4)
ADVERSE REACTIONS
The most common adverse reactions in later-onset SMA (incidence at least 10% of patients treated with EVRYSDI and more frequent than control) were fever, diarrhea, and rash. (6.1)
The most common adverse reactions in infantile-onset SMA were similar to those observed in later-onset SMA patients. Additionally, adverse reactions with an incidence of at least 10% were upper respiratory tract infection, lower respiratory tract infection, constipation, vomiting, and cough. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Genentech at 1-888-835-2555 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Avoid coadministration with drugs that are substrates of multidrug and toxin extrusion (MATE) transporters. (7.1)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 2/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
2.2 Important Administration Instructions
2.3 Missed Dose
2.4 Preparation of Powder for Oral Solution by Healthcare Provider
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of EVRYSDI on Substrates of Multidrug and Toxin Extrusion (MATE) Protein Transporters
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Infantile-Onset SMA
14.2 Later-Onset SMA
14.3 Pre-Symptomatic SMA
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 EVRYSDI for Oral Solution
16.2 EVRYSDI Tablets
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
EVRYSDI is administered orally once daily with or without food at approximately the same time each day. The recommended dosage is determined by age and body weight (see Table 1). EVRYSDI tablets are available for patients prescribed the 5 mg dose.
Table 1 Adult and Pediatric Dosing Regimen by Age and Body Weight Age and Body Weight Recommended Daily Dosage Dosage Form Less than 2 months of age 0.15 mg/kg EVRYSDI for Oral Solution 2 months to less than 2 years of age 0.2 mg/kg 2 years of age and older weighing less than 20 kg 0.25 mg/kg 2 years of age and older weighing 20 kg or more 5 mg EVRYSDI for Oral Solution
or
EVRYSDI Tablet2.2 Important Administration Instructions
It is recommended that a healthcare provider discuss with the patient or caregiver how to prepare the prescribed daily dose prior to administration of the first dose [see Instructions for Use for EVRYSDI for Oral Solution and EVRYSDI Tablets and Patient Information].
EVRYSDI for Oral Solution
In infants who are breastfed, EVRYSDI for oral solution can be administered before or after breastfeeding. EVRYSDI cannot be mixed with formula or milk.
Instruct patients or caregivers to administer the dose using the reusable oral syringe provided.
EVRYSDI for oral solution must be taken immediately after it is drawn up into the oral syringe. If EVRYSDI is not taken within 5 minutes, EVRYSDI should be discarded from the oral syringe, and a new dose should be prepared.
Instruct patients to drink water after taking EVRYSDI for oral solution to ensure the drug has been completely swallowed.
EVRYSDI for oral solution can be administered via a nasogastric or gastrostomy tube. The tube should be flushed with water after delivering EVRYSDI for oral solution [see Instructions for Use].
EVRYSDI Tablets
Swallow EVRYSDI tablets whole with water. Do not chew, cut, or crush the tablets.
Alternatively, the EVRYSDI tablet can also be dispersed in one teaspoon (5 mL) of room temperature non-chlorinated drinking water (e.g., filtered water). EVRYSDI tablets must not be dispersed in any liquid other than non-chlorinated drinking water. Do not expose the prepared dispersion to sunlight. Swirl the small cup gently for up to 3 minutes until fully mixed (though some particles will remain). Administer the dispersed tablet immediately. To ensure no particles are left in the small cup, refill it with at least one tablespoon (15 mL) of non-chlorinated drinking water, swirl, and administer immediately again.
EVRYSDI must be taken immediately after it is dispersed in non-chlorinated drinking water. Discard the prepared dispersion if it is not used within 10 minutes of adding non-chlorinated drinking water.
The dispersed EVRYSDI tablet can be administered via a nasogastric or gastrostomy tube that is 8 French or higher. Flush the tube with the non-chlorinated drinking water [at least one tablespoon (15 mL)] used to rinse the dispersion cup [see Instructions for Use].
2.3 Missed Dose
If a dose of EVRYSDI is missed, EVRYSDI should be administered as soon as possible if still within 6 hours of the missed dose, and the usual dosing schedule can be resumed on the next day. Otherwise, the missed dose should be skipped, and the next dose should be taken at the regularly scheduled time on the next day.
If a dose is not fully swallowed or vomiting occurs after taking a dose of EVRYSDI, another dose should not be administered to make up for the lost dose. The patient should wait until the next day to take the next dose at the regularly scheduled time.
2.4 Preparation of Powder for Oral Solution by Healthcare Provider
EVRYSDI powder must be constituted to the oral solution by a pharmacist or other healthcare provider prior to dispensing to the patient.
Preparation of the EVRYSDI Oral Solution 0.75 mg/mL
The EVRYSDI "Instructions for Constitution" booklet contains more detailed instructions on the preparation of the oral solution [see Instructions for Constitution].
Caution should be exercised when handling EVRYSDI powder for oral solution. Avoid inhalation and direct contact with skin or mucous membranes with the dry powder and the constituted solution. If such contact occurs, wash thoroughly with soap and water; rinse eyes with water. Wear disposable gloves during the preparation and cleanup procedure.
- Gently tap the bottom of the closed glass bottle to loosen the powder.
- Remove the cap. Do not throw away the cap.
- Carefully pour 79 mL of purified water into the EVRYSDI bottle to yield the 0.75 mg/mL oral solution. Do not mix EVRYSDI with formula or milk.
- Insert the press-in bottle adapter into the bottle opening by pushing it down against the bottle lip. Ensure it is completely pressed against the bottle lip.
- Re-cap the bottle tightly and shake well for 15 seconds. Wait for 10 minutes. You should have obtained a clear solution. If not, shake well again for another 15 seconds or until you have obtained a clear solution.
- Write the date of expiration of the constituted oral solution (calculated as 64 days after constitution) and the lot number on the bottle label. Peel off the part of the bottle label that has the expiration date of the powder.
- Put the bottle back in its original carton.
- Select the appropriate oral syringes (1 mL, 6 mL, or 12 mL) based on the patient's dosage and remove the other oral syringes from the carton.
- Dispense with the "Instructions for Use" and FDA-approved patient labeling. Alert patients to read the important handling information described in the Instructions for Use.
Storage
Keep the constituted oral solution of EVRYSDI in the original amber bottle to protect from light. Store in a refrigerator at 2°C to 8°C (36°F to 46°F). Do not freeze. Discard any unused portion 64 days after constitution. Keep the bottle in an upright position with the cap tightly closed. If refrigeration is not available, EVRYSDI can be kept at room temperature up to 40°C (up to 104°F) for a combined total of 5 days. EVRYSDI can be removed from, and returned to, a refrigerator. The total combined time out of refrigeration should not exceed 5 days.
-
3 DOSAGE FORMS AND STRENGTHS
EVRYSDI for oral solution: 60 mg as a light yellow, pale yellow, yellow, greyish yellow, greenish yellow, or light green powder for constitution. Following constitution, the volume of the greenish yellow to yellow solution is 80 mL, providing 60 mg/80 mL (0.75 mg/mL) risdiplam.
EVRYSDI tablet: 5 mg as a pale yellow film-coated tablet, round and curved, with EVR debossed on one side.
- 4 CONTRAINDICATIONS
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in clinical trials of another drug and may not reflect the rates observed in practice.
In clinical trials including patients with infantile-onset SMA, later-onset SMA, and pre-symptomatic SMA, a total of 491 patients (51% female, 74% Caucasian) were exposed to EVRYSDI for up to a median duration of 48.1 months (range: 0.6 to 63.4 months), with 231 patients receiving treatment for more than 24 months. At the time of first EVRYSDI dose, 90 (18%) patients were 18 years and older, 119 (24%) were 12 years to less than 18 years, 189 (39%) were 2 years to less than 12 years, 67 (14%) 2 months to less than 2 years, and 26 (5%) were less than 2 months.
Clinical Trial in Later-Onset SMA
The safety of EVRYSDI for later-onset SMA is based on data from a randomized, double-blinded, placebo-controlled study (Study 2 Part 2) in patients with SMA Type 2 or 3 (n = 180) [see Clinical Studies (14.2)]. The patient population in Study 2 Part 2 ranged in age from 2 to 25 years at the time of the first dose.
The most common adverse reactions (reported in at least 10% of patients treated with EVRYSDI and at an incidence greater than on placebo) in Study 2 Part 2 were fever, diarrhea, and rash. Table 2 lists the adverse reactions that occurred in at least 5% of patients treated with EVRYSDI and at an incidence ≥ 5% greater than on placebo in Study 2 Part 2.
Table 2 Adverse Reactions Reported in ≥ 5% of Patients Treated with EVRYSDI and with an Incidence ≥ 5% Greater Than on Placebo in Study 2 Part 2 Adverse Reaction EVRYSDI
(N = 120)
%Placebo
(N = 60)
%- * Includes pyrexia and hyperpyrexia.
- † Includes rash, erythema, rash maculo-papular, rash erythematous, rash papular, dermatitis allergic, and folliculitis.
- ‡ Includes urinary tract infection and cystitis.
Fever* 22 17 Diarrhea 17 8 Rash† 17 2 Mouth and aphthous ulcers 7 0 Arthralgia 5 0 Urinary tract infection‡ 5 0 Clinical Trial in Infantile-Onset SMA
The safety of EVRYSDI therapy for infantile-onset SMA is based on data from an open-label study in 62 patients (Study 1) [see Clinical Studies (14.1)]. The patient population ranged in age from 2 to 7 months at the time of the first EVRYSDI dose (weight range: 4.1 to 10.6 kg).
The most frequent adverse reactions reported in infantile-onset SMA patients treated with EVRYSDI in Study 1 were similar to those observed in later-onset SMA patients in Study 2. Additionally, the following adverse reactions reported in ≥ 10% of patients were: upper respiratory tract infection (including nasopharyngitis, rhinitis), lower respiratory tract infection (including pneumonia, bronchitis), constipation, vomiting, and cough.
Clinical Trial in Pre-Symptomatic SMA
The safety of EVRYSDI therapy for pre-symptomatic SMA is based on data from an open-label, single-arm study in 26 patients (Study 3) [see Clinical Studies (14.3)]. The patient population ranged in age from 16 to 41 days at the time of the first dose (weight range: 3.1 to 5.7 kg). The safety profile of EVRYSDI in pre-symptomatic patients in Study 3 is consistent with the safety profile for symptomatic SMA patients treated with EVRYSDI in clinical trials.
-
7 DRUG INTERACTIONS
7.1 Effect of EVRYSDI on Substrates of Multidrug and Toxin Extrusion (MATE) Protein Transporters
Based on in vitro data, EVRYSDI may increase plasma concentrations of drugs eliminated via MATE1 or MATE2-K [see Clinical Pharmacology (12.3)], such as metformin. Avoid coadministration of EVRYSDI with MATE substrates. If coadministration cannot be avoided, monitor for drug-related toxicities and consider dosage reduction of the coadministered drug (based on the labeling of that drug) if needed.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure RegistryThere is a pregnancy exposure registry that monitors pregnancy and fetal/neonatal/infant outcomes in women exposed to EVRYSDI during pregnancy. Physicians are encouraged to register patients and pregnant women are encouraged to register themselves by calling 1-833-760-1098 or visiting https://www.evrysdipregnancyregistry.com.
Risk Summary
There are no adequate data on the developmental risk associated with the use of EVRYSDI in pregnant women. In animal studies, administration of risdiplam during pregnancy or throughout pregnancy and lactation resulted in adverse effects on development (embryofetal mortality, malformations, decreased fetal body weights, and reproductive impairment in offspring) at or above clinically relevant drug exposures [see Data].
The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. In the U.S. general population, the estimated background risk of major birth defect and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. Based on animal data, advise pregnant women of the potential risk to the fetus.
Data
Animal Data
Oral administration of risdiplam (0, 1, 3, or 7.5 mg/kg/day) to pregnant rats throughout organogenesis resulted in decreased fetal body weights and increased incidences of fetal structural variations at the highest dose tested, which was not associated with maternal toxicity. The no-effect level for adverse effects on embryofetal development (3 mg/kg/day) was associated with maternal plasma exposure (AUC) approximately 2 times that in humans at the maximum recommended human dose (MRHD) of 5 mg.
Oral administration of risdiplam (0, 1, 4, or 12 mg/kg/day) to pregnant rabbits throughout organogenesis resulted in embryofetal mortality, fetal malformations (hydrocephaly), and structural variations at the highest dose tested, which was associated with maternal toxicity. The no-effect dose for adverse effects on embryofetal development (4 mg/kg/day) was associated with maternal plasma exposure (AUC) approximately 4 times that in humans at the MRHD.
When risdiplam (0, 0.75, 1.5, or 3 mg/kg/day) was orally administered to rats throughout pregnancy and lactation, gestation was prolonged in the dams, and delayed sexual maturation (vaginal opening) and impaired reproductive function (decreased numbers of corpora lutea, implantation sites, and live embryos) were observed in female offspring at the highest dose. The no-effect dose for adverse effects on pre- and postnatal development in rats (1.5 mg/kg/day) was associated with maternal plasma exposure (AUC) similar to that in humans at the MRHD.
8.2 Lactation
Risk Summary
There are no data on the presence of risdiplam in human milk, the effects on the breastfed infant, or the effects on milk production. Risdiplam was excreted in the milk of lactating rats orally administered risdiplam.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for EVRYSDI and any potential adverse effects on the breastfed infant from EVRYSDI or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Studies of risdiplam in juvenile and adult rats and in monkeys demonstrated adverse effects on the reproductive organs, including germ cells, in males at clinically relevant plasma exposures [see Use in Specific Populations (8.4) and Nonclinical Toxicology (13.1)].
Pregnancy Testing
Pregnancy testing is recommended for females of reproductive potential prior to initiating EVRYSDI [see Use in Specific Populations (8.1)].
Contraception
EVRYSDI may cause embryofetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Infertility
Male Patients
Male fertility may be compromised by treatment with EVRYSDI [see Nonclinical Toxicology (13.1)].
Counsel male patients of reproductive potential receiving EVRYSDI about the potential effects on fertility. Male patients may consider sperm preservation prior to treatment.
8.4 Pediatric Use
The safety and effectiveness of EVRYSDI in pediatric patients (neonates and older) have been established. Use of EVRYSDI for SMA is supported by evidence from adequate and well-controlled studies of EVRYSDI in patients 2 months of age and older with SMA. Use of EVRYSDI for SMA in patients 2 months of age and younger is supported by pharmacokinetic and safety data from pediatric patients 16 days and older, and pharmacokinetic modeling and simulation to identify the dosing regimen [see Clinical Pharmacology (12.3) and Clinical Studies (14)].
Juvenile Animal Toxicity Data
Oral administration of risdiplam (0, 0.75, 1.5, 2.5 mg/kg/day) to young rats from postnatal day (PND) 4 through PND 31 resulted in decreased growth (body weight, tibia length) and delayed sexual maturation in males at the mid and high dose. The skeletal and body weight deficits persisted after cessation of dosing. Ophthalmic changes consisting of vacuoles in the anterior vitreous were seen at the high dose. Decreases in absolute B lymphocyte counts were observed at all doses after cessation of dosing. Decreases in testis and epididymis weights, which correlated with degeneration of the seminiferous epithelium in the testis, occurred at the mid and high doses; the histopathology findings were reversible, but organ weight persisted after cessation of dosing. Impaired female reproductive performance (decreased mating index, fertility index, and conception rate) was observed at the high dose. A no-effect dose for adverse developmental effects on preweaning rats was not identified. The lowest dose tested (0.75 mg/kg/day) was associated with plasma exposures (AUC) lower than that in humans at the maximum recommended human dose (MRHD) of 5 mg/day.
Oral administration of risdiplam (0, 1, 3, or 7.5 mg/kg/day) to young rats from PND 22 through PND 112 produced a marked increase in micronuclei in the bone marrow, male reproductive organ histopathology (degeneration/necrosis of the seminiferous tubule epithelium, oligo/aspermia in the epididymis, spermatic granulomas), and adverse effects on sperm parameters (decreased sperm concentration and motility, increased sperm morphology abnormalities) at the highest dose tested. Increases in T lymphocytes (total, helper, and cytotoxic) were observed at the mid and high doses. The reproductive and immune effects persisted after cessation of dosing. The no-effect dose (1 mg/kg/day) for adverse effects on postweaning juvenile rats was associated with plasma exposures (AUC) lower than that in humans at the MRHD.
-
11 DESCRIPTION
EVRYSDI for oral solution and EVRYSDI tablets for oral use contain risdiplam, which is a survival of motor neuron 2 (SMN2)-directed RNA splicing modifier.
The chemical name of risdiplam is 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8 dimethylimidazo[1,2-b]pyridazin-6-yl)pyrido-4H-[1,2-a]pyrimidin-4-one. Risdiplam has a molecular weight of 401.46 g/mol. Risdiplam demonstrates pH-dependent aqueous solubility; the greatest solubility is at low pH, and solubility decreases with increasing pH. Risdiplam has a pKa1 of 3.78 (base) and pKa2 of 6.62 (base).
The molecular formula of risdiplam is C22H23N7O and the chemical structure is shown below.
EVRYSDI for oral solution is supplied as a powder in an amber glass bottle. Each bottle contains 60 mg of risdiplam. The inactive ingredients of EVRYSDI are: ascorbic acid, disodium edetate dihydrate, isomalt, mannitol, polyethylene glycol 6000, sodium benzoate, strawberry flavor, sucralose, and tartaric acid.
The powder is constituted with purified water to yield 60 mg/80 mL (0.75 mg/mL) of risdiplam after constitution [see Dosage and Administration (2.4)].
Each EVRYSDI tablet contains 5 mg of risdiplam. The inactive ingredients of EVRYSDI tablet are colloidal silicon dioxide, crospovidone, mannitol, microcrystalline cellulose, polyethylene glycol 3350, polyvinyl alcohol, sodium stearyl fumarate, strawberry flavor, talc, tartaric acid, titanium dioxide, and yellow iron oxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Risdiplam is a survival of motor neuron 2 (SMN2) splicing modifier designed to treat patients with spinal muscular atrophy (SMA) caused by mutations in chromosome 5q that lead to SMN protein deficiency. Using in vitro assays and studies in transgenic animal models of SMA, risdiplam was shown to increase exon 7 inclusion in SMN2 messenger ribonucleic acid (mRNA) transcripts and production of full-length SMN protein in the brain.
In vitro and in vivo data indicate that risdiplam may cause alternative splicing of additional genes, including FOXM1 and MADD. FOXM1 and MADD are thought to be involved in cell cycle regulation and apoptosis, respectively, and have been identified as possible contributors to adverse effects seen in animals.
12.2 Pharmacodynamics
In clinical trials for infantile-onset SMA and later-onset SMA patients, EVRYSDI led to an increase in SMN protein with a greater than 2-fold median change from baseline within 4 weeks of treatment initiation across all SMA types studied. The increase was sustained throughout the treatment period (of at least 24 months).
12.3 Pharmacokinetics
Pharmacokinetics of EVRYSDI have been characterized in healthy adult subjects and in patients with SMA.
After administration of EVRYSDI as an oral solution, pharmacokinetics of risdiplam were approximately linear between 0.6 and 18 mg in a single-ascending-dose study in healthy adult subjects, and between 0.02 and 0.25 mg/kg once daily in a multiple-ascending-dose study in patients with SMA. Following once-daily oral administration of risdiplam in healthy subjects, approximately 3-fold accumulation of peak plasma concentrations (Cmax) and area under the plasma concentration-time curve (AUC0-24h) was observed. Risdiplam exposures reach steady state 7 to 14 days after once daily administration. EVRYSDI tablet (swallowed whole or dispersed in water) demonstrated comparable bioavailability to EVRYSDI for oral solution in adult healthy volunteers under fasted and fed states.
Absorption
Following oral administration of risdiplam in fasted state, the median time to reach maximum plasma concentration (Tmax) was 3.26 to 4 hours. The Tmax was delayed by up to1 hour in fed state compared to that under fasted state.
Distribution
The apparent volume of distribution at steady state is 190.4 L for a 31.3 kg patient.
Risdiplam is predominantly bound to serum albumin, without any binding to alpha-1 acid glycoprotein, with a free fraction of 11%.
Elimination
The apparent clearance (CL/F) of risdiplam is 2.45 L/h for a 31.3 kg patient.
The terminal elimination half-life of risdiplam was approximately 50 hours in healthy adults.
Metabolism
Risdiplam is primarily metabolized by flavin monooxygenase 1 and 3 (FMO1 and FMO3) and also by CYPs 1A1, 2J2, 3A4, and 3A7.
Parent drug was the major component found in plasma, accounting for 83% of drug-related material in circulation. The pharmacologically-inactive metabolite M1 was identified as the major circulating metabolite.
Specific Populations
There were no clinically significant differences in the pharmacokinetics of EVRYSDI based on race or gender. Renal impairment is not expected to alter the exposures to risdiplam.
The impact of geriatric age on the pharmacokinetics of EVRYSDI has not been studied.
Hepatic Impairment
The pharmacokinetics and safety of risdiplam have been studied in subjects with mild or moderate hepatic impairment (as defined by Child-Pugh class A and B, respectively, n = 8 each) compared to subjects with normal hepatic function (n = 10). Following the administration of 5 mg EVRYSDI, the AUCinf and Cmax of risdiplam were approximately 20% and 5% lower, respectively, in subjects with mild hepatic impairment and were approximately 8% and 20% higher, respectively, in subjects with moderate hepatic impairment, versus matched healthy control subjects. The magnitude of these changes is not considered to be clinically meaningful. The pharmacokinetics and safety in patients with severe hepatic impairment (Child-Pugh class C) have not been studied.
Pediatric Patients
Body weight and age were found to have significant effect on the pharmacokinetics of risdiplam. The estimated exposure (mean AUC0-24h) in pre-symptomatic infants at the age of 1 to 2 months was 2090 ng.h/mL at the recommended dose of 0.15 mg/kg once daily. The estimated exposure for infantile-onset SMA patients (age 2 to 7 months at enrollment) at the recommended dose of 0.2 mg/kg once daily was 1930 ng.h/mL. The estimated exposure for later-onset SMA patients (2 to 25 years old at enrollment) at the recommended dose was 2070 ng.h/mL (0.25 mg/kg once daily for patients with a body weight < 20 kg and 5 mg once daily for patients with a body weight ≥ 20 kg).
No data on risdiplam pharmacokinetics are available in patients less than 16 days of age [see Use in Specific Populations (8.4)].
Drug Interaction Studies
Effect of Other Drugs on EVRYSDI
Coadministration of 200 mg itraconazole (a strong CYP3A inhibitor) twice daily with a single 6 mg oral dose of risdiplam did not have a clinically relevant effect on the pharmacokinetics of risdiplam (11% increase in AUC and 9% decrease in Cmax).
Risdiplam is a weak substrate of human MDR-1 and breast cancer resistant protein (BCRP) transporters in vitro. Human MDR-1 or BCRP inhibitors are not expected to result in a clinically significant increase of risdiplam concentrations.
The effect of omeprazole (an acid-reducing agent/proton pump inhibitor) on risdiplam pharmacokinetics was investigated in healthy subjects. No clinically significant difference was observed in the pharmacokinetics of risdiplam administered as a tablet when used concomitantly with omeprazole. Based on these results, no clinically significant differences are expected with other acid-reducing agents, including H2-receptor antagonists and antacids.
Effect of EVRYSDI on Other Drugs
Risdiplam and its major circulating metabolite M1 did not induce CYP1A2, 2B6, 2C8, 2C9, 2C19, or 3A4 in vitro. Risdiplam and M1 did not inhibit (reversible or time-dependent inhibition) any of the CYP enzymes tested (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6), with the exception of CYP3A in vitro.
EVRYSDI is a weak inhibitor of CYP3A. In healthy adult subjects, administration of EVRYSDI once daily for 2 weeks slightly increased the exposure of midazolam, a sensitive CYP3A substrate (AUC 11%; Cmax 16%); this increase is not considered clinically relevant. Based on physiologically-based pharmacokinetic (PBPK) modeling, a similar increase is expected in children and infants as young as 2 months of age.
In vitro studies have shown that risdiplam and its major metabolite are not significant inhibitors of human MDR1, organic anion-transporting polypeptide (OATP) 1B1, OATP1B3, organic anion transporter 1 and 3 (OAT 1 and 3) transporters, and human organic cation transporter 2 (OCT2), at clinically relevant concentrations. Risdiplam and its metabolite are, however, in vitro inhibitors of the multidrug and toxin extrusion (MATE) 1 and MATE2-K transporters [see Drug Interactions (7.1)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Risdiplam was not carcinogenic in Tg.rasH2 mice when administered at oral doses of up to 9 mg/kg/day for 26 weeks.
In a 2-year carcinogenicity study in rats, oral administration of risdiplam (0, 0.3, 1, or 3 mg/kg/day) resulted in increased incidences of preputial gland squamous cell carcinomas in males and combined thyroid follicular cell adenomas and carcinomas in females at the highest dose tested. The higher dose not associated with an increase in tumors (1 mg/kg/day) was associated with plasma drug exposures (AUC) similar to that in humans at the maximum recommended human dose (MRHD) of 5 mg/day.
Mutagenesis
Risdiplam was negative in an in vitro Ames assay. In an in vivo combined bone marrow micronucleus and comet assay in rat, risdiplam was clastogenic, as evidenced by an increase in micronuclei in bone marrow, but was negative in the comet assay. A pronounced increase in bone marrow micronuclei was also observed in toxicity studies in adult and juvenile rats [see Use in Specific Populations (8.4)].
Impairment of Fertility
Oral administration of risdiplam to rats for 4 (0, 1, 3, or 9 mg/kg/day) or 26 (0, 1, 3, or 7.5 mg/kg/day) weeks resulted in histopathological effects in the testis (degenerated spermatocytes, degeneration/atrophy of the seminiferous tubules) and epididymis (degeneration/necrosis of ductular epithelium) at the mid and/or high doses. At the high dose in the 26-week study, the testicular lesions persisted to the end of the recovery period, which corresponds, in rat, to approximately one spermatogenic cycle. The no-effect dose for adverse reproductive system effects in adult male rats (1 mg/kg/day) was associated with plasma drug exposures (AUC) similar to that in humans at the maximum recommended human dose (MRHD) of 5 mg/day.
Adverse effects of risdiplam on the testis could not be fully evaluated in the monkey because the majority of monkeys tested were sexually immature. However, oral administration of risdiplam (0, 2, 4, or 6 mg/kg/day) for 2 weeks resulted in histopathological changes in the testis (increases in multinucleate cells, germ cell degeneration) at the highest dose. At the no-effect dose for testicular toxicity in monkeys, plasma exposures were approximately 3 times that in humans at the MRHD.
Oral administration of risdiplam to postweaning juvenile rats resulted in male reproductive toxicity (degeneration/necrosis of the testis seminiferous epithelium with associated oligo/aspermia in the epididymis and abnormal sperm parameters). The no-effect dose for adverse reproductive effects in postweaning male juvenile rats was associated with plasma exposures approximately 4 times that in humans at the MRHD [see Use in Specific Populations (8.4)].
13.2 Animal Toxicology and/or Pharmacology
Retinal toxicity
Risdiplam-induced functional and structural retinal abnormalities were seen in animal studies. In a 39-week toxicity study in monkeys, oral administration of risdiplam (0, 1.5, 3, or 7.5/5 mg/kg/day; high dose lowered after 4 weeks) produced functional abnormalities on the electroretinogram (ERG) in all mid- and high-dose animals at the earliest examination time (Week 20). These findings were associated with retinal degeneration, detected by optical coherence tomography (OCT), on Week 22, the first examination time. The retinal degeneration, with peripheral photoreceptor loss, was irreversible. A no-effect dose for the retinal findings (1.5 mg/kg/day) was associated with plasma exposures (AUC) similar to that in humans at the maximum recommended human dose (MRHD) of 5 mg.
Effect on Epithelial Tissues
Oral administration of risdiplam to rats and monkeys resulted in histopathological changes in epithelium of the gastrointestinal (GI) tract (apoptosis/single cell necrosis), lamina propria (vacuolation), the exocrine pancreas (single cell necrosis), the skin, tongue, and larynx (parakeratosis/hyperplasia/degeneration) with associated inflammation. The skin and GI epithelial effects were reversible. The no-effect doses for effects on epithelial tissues in rats and monkeys were associated with plasma exposures (AUC) similar to that in humans at the MRHD.
-
14 CLINICAL STUDIES
The efficacy of EVRYSDI for the treatment of patients with infantile-onset, later-onset, and pre-symptomatic SMA was evaluated in three clinical studies, Study 1 (NCT02913482) and Study 2 (NCT02908685), and Study 3 (NCT03779334), respectively.
The overall findings of these studies support the effectiveness of EVRYSDI in SMA pediatric and adult patients and appear to support the early initiation of treatment with EVRYSDI.
14.1 Infantile-Onset SMA
Study 1 was an open-label, 2-part study to investigate the efficacy, safety, pharmacokinetics, and pharmacodynamics of EVRYSDI for oral solution in patients with Type 1 SMA (symptom onset between 28 days and 3 months of age). All patients had genetic confirmation of homozygous deletion or compound heterozygosity predictive of loss of function of the SMN1 gene, and two SMN2 gene copies.
Part 1 of Study 1 was designed as a dose-finding study. Part 2 of Study 1 assessed the safety and efficacy of EVRYSDI at 0.20 mg/kg, the recommended dose determined in Part 1 [see Dosage and Administration (2.4)]. Patients from Part 1 did not take part in Part 2.
A total of 62 patients with symptomatic Type 1 SMA were enrolled in FIREFISH Part 1 (n = 21) and Part 2 (n = 41), of which 58 patients received the recommended dosage [see Dosage and Administration (2.1)]. The median age of onset of clinical signs and symptoms was 1.5 months (range: 0.9 to 3.0 months). The median age at enrollment was 5.6 months (range: 2.2 to 6.9 months), and the median time between onset of symptoms and the first dose was 3.7 months (range 1.0 to 6.0 months). Of these patients, 60% were female, 57% were Caucasian, and 29% were Asian. The demographics and baseline disease characteristics were comparable between Part 1 and Part 2 of the study.
Effectiveness was established based on the ability to sit without support for at least 5 seconds (as measured by Item 22 of the Bayley Scales of Infant and Toddler Development – Third Edition (BSID-III) gross motor scale) and on the basis of survival without permanent ventilation. Permanent ventilation was defined as requiring a tracheostomy or more than 21 consecutive days of either non-invasive ventilation (≥ 16 hours per day) or intubation, in the absence of an acute reversible event.
The primary endpoint was the proportion of patients with the ability to sit without support for at least 5 seconds (BSID-III gross motor scale, Item 22) after 12 months of treatment in Part 2; 29% of patients (n = 12/41) achieved this milestone.
Other efficacy endpoints of EVRYSDI-treated patients in Study 1 (pooled Part 1 and Part 2) are shown in Table 3.
Table 3 Key Efficacy Results at Month 12 and Month 24 (Study 1, Parts 1 and Part 2) Efficacy Endpoints Proportion of Patients Parts 1 & 2 at Month 12 Proportion of Patients Parts 1 & 2 at Month 24 - * Results were pooled from all patients who received the recommended dose of risdiplam (all patients in Part 2 and those in the high-dose cohort of Part 1; n = 58).
- † Results were pooled from all patients who received any dose of risdiplam in Part 1 and Part 2 (n = 62).
Motor Function and Development Milestones N = 58 * BSID-III, Item 22: sitting without support for at least 5 seconds 32.8% 60.3% Survival and Event-Free Survival N = 62 † Alive without Permanent Ventilation 87.1% 83.8% At Month 24, 40% (23/58) of patients who received the recommended dose achieved sitting without support for 30 seconds (BSID-III, Item 26). In addition at Month 24, patients continued to achieve additional motor milestones; 28% (16/58) of patients achieved a standing measure (16% [9/58] supporting weight and 12% [7/58] standing with support), as measured by Section 2 of the Hammersmith Infant Neurological Examination (HINE-2) which assesses motor milestones.
The proportion of patients alive without permanent ventilation (event-free survival) was 84% for all patients at Month 24 (Table 3). Out of 62 patients, 6 infants died (4 within the first 3 months following study enrollment) and one additional patient withdrew from treatment and died 3.5 months later. Four patients required permanent ventilation by Month 24. These results indicate a clinically meaningful deviation from the natural history of untreated infantile-onset SMA. As described in the natural history of untreated infantile-onset SMA, patients would not be expected to attain the ability to sit independently, and no more than 25% of these patients would be expected to survive without permanent ventilation beyond 14 months of age.
14.2 Later-Onset SMA
Study 2 was a 2-part, multicenter trial to investigate the efficacy, safety, pharmacokinetics, and pharmacodynamics of EVRYSDI for oral solution in patients diagnosed with SMA Type 2 or Type 3. Part 1 of Study 2 was dose-finding and exploratory in 51 patients (14% ambulatory). Part 2 was randomized, double-blind, placebo-controlled, and is described below.
The primary endpoint in Study 2 Part 2 was the change from baseline to Month 12 in the Motor Function Measure 32 (MFM32) score. A key secondary endpoint was the proportion of patients with a 3-point or greater change from baseline to Month 12 in the MFM32 total score. The MFM32 measures motor function abilities that relate to daily functions. The total MFM32 score is expressed as a percentage (range: 0 to 100) of the maximum possible score, with higher scores indicating greater motor function. Another key secondary endpoint was the Revised Upper Limb Module (RULM). The RULM is a tool used to assess motor performance of the upper limb in SMA patients. It tests proximal and distal motor functions of the arm. The total score ranges from 0 (all the items cannot be performed) to 37 (all the activities are achieved fully without any compensatory maneuvers).
Study 2 Part 2 enrolled 180 non-ambulatory patients with Type 2 (71%) or Type 3 (29%) SMA. Patients were randomized 2:1 to receive EVRYSDI at the recommended dosage [see Dosage and Administration (2.1)] or placebo. Randomization was stratified by age group (2 to 5, 6 to 11, 12 to 17, or 18 to 25 years of age).
The median age of patients at the start of treatment was 9.0 years (range: 2 to 25), and the median time between onset of initial SMA symptoms and first treatment was 102.6 months (range: 1 to 275). Of the 180 patients included in the trial, 51% were female, 67% were Caucasian, and 19% were Asian. At baseline, 67% of patients had scoliosis (32% of them with severe scoliosis). Patients had a mean baseline MFM32 score of 46.1, and RULM score of 20.1. Overall baseline demographic characteristics were reasonably balanced between the treatment groups (EVRYSDI and placebo), with the exception of scoliosis (63% in the EVRYSDI arm vs. 73% in the placebo group).
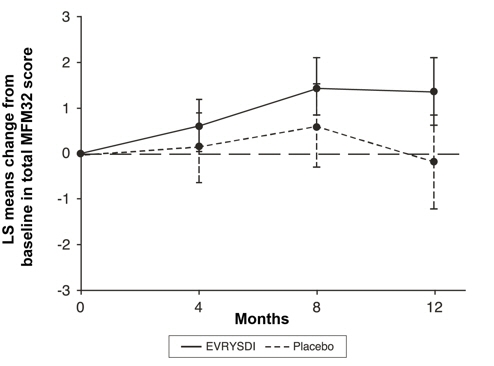
The primary analysis on the change from baseline in MFM32 total score at Month 12 showed a clinically meaningful and statistically significant difference between patients treated with EVRYSDI and placebo. The results of the primary analysis and key secondary endpoints are shown in Table 4 and Figure 1.
Table 4 Summary of Efficacy in Patients with Later-Onset SMA at Month 12 of Treatment (Study 2 Part 2) Endpoint EVRYSDI
(N = 120)Placebo
(N = 60)- * The Mixed Model Repeated Measure (MMRM) analysis included the change from baseline total score as the dependent variable and as independent variables the baseline total score, treatment group, time, treatment-by-time interaction, and the randomization stratification variable of age group (2 to 5, 6 to 11, 12 to 17, 18 to 25).
- † The MFM total score was calculated according to the user manual, expressed as a percentage of the maximum score possible for the scale (i.e., sum of the 32 item scores divided by 96 and multiplied by 100).
- ‡ Based on the missing data rule for MFM32, 6 patients were excluded from the analysis (EVRYSDI n = 115; placebo control n = 59).
- § The adjusted p-value was obtained for the endpoints included in the hierarchical testing and was derived based on all the p-values from endpoints in order of the hierarchy up to the current endpoint.
- ¶ The logistic regression analysis included the baseline total score, treatment and age group as independent variables.
- # Based on the missing data rule for RULM, 3 patients were excluded from the analysis (EVRYSDI n = 119; placebo control n = 58).
Primary Endpoint: Change from baseline in total MFM32 score at Month 12, LS means (95% CI) *,†,‡ 1.36 (0.61, 2.11) -0.19 (-1.22, 0.84) Difference from Placebo, Estimate (95% CI)*
p-value1.55 (0.30, 2.81)
0.0156Secondary Endpoints: Proportion of patients with a change from baseline MFM32 total score of 3 or more at Month 12 (95% CI)†,‡ 38.3% (28.9, 47.6) 23.7% (12.0, 35.4) Odds ratio for overall response (95% CI)
adjusted§ (unadjusted) p-value¶2.35 (1.01, 5.44)
0.0469 (0.0469)Change from baseline in total score of RULM at Month 12, LS means (95% CI)*, # 1.61 (1.00, 2.22) 0.02 (-0.83, 0.87) Difference from Placebo, Estimate (95% CI)
adjusted§ (unadjusted) p-value*1.59 (0.55, 2.62)
0.0469 (0.0028)- * Error bars denote the 95% confidence interval.
- † The MFM total score was calculated according to the user manual, expressed as a percentage of the maximum score possible for the scale (i.e., sum of the 32 item scores divided by 96 and multiplied by 100).
Figure 1 Mean Change from Baseline in Total MFM32 Score Over 12 Months (Study 2 Part 2)*,†
14.3 Pre-Symptomatic SMA
Study 3 was an open-label, single-arm, multicenter clinical study to investigate the efficacy, safety, pharmacokinetics, and pharmacodynamics of EVRYSDI in infants up to 6 weeks of age (at first dose) who have been genetically diagnosed with SMA but do not yet present with symptoms.
The efficacy in pre-symptomatic SMA patients was evaluated at Month 12 in 26 patients treated with EVRYSDI in Study 3: 8 patients had 2 copies of the SMN2 gene, 13 patients had 3 copies, and 5 patients had 4 or more copies. The median age of these patients at first dose was 25 days (range: 16 to 41), 62% were female, and 85% were Caucasian. The primary efficacy population (N = 5) included patients with 2 SMN2 copies and a baseline CMAP amplitude ≥1.5 mV.
The primary efficacy endpoint was the proportion of patients with the ability to sit without support for at least 5 seconds (BSID-III gross motor scale, Item 22) at Month 12. This milestone was achieved by 80% (4/5) of patients in the primary efficacy population. This milestone was also achieved by 87.5% (7/8) of all patients with 2 copies of SMN2 and 96.2% (25/26) of patients in the full treated population.
At Month 12, 80.8% (21/26) of patients in the full treated population achieved sitting without support for 30 seconds (BSID-III, Item 26). Of the 26 patients treated with EVRYSDI, 25 patients had motor milestones measured by the HINE-2 at Month 12. Of these, 24 (96%) achieved sitting (23 patients could pivot/rotate and 1 achieved stable sit); 21 (84%) could stand (13 patients could stand unaided and 8 could stand with support); and 12 (48%) could walk independently. Seven patients were not tested for walking at Month 12. All 26 patients were alive at 12 months without permanent ventilation.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 EVRYSDI for Oral Solution
How Supplied
Each amber glass bottle of EVRYSDI for oral solution is packaged with a bottle adapter, two 1 mL reusable oral syringes, two 6 mL reusable oral syringes, and one 12 mL reusable oral syringe. EVRYSDI for oral solution is a light yellow, pale yellow, yellow, greyish yellow, greenish yellow, or light green powder. Each bottle contains 60 mg of risdiplam (NDC: 50242-175-07).
Storage and Handling
Store the dry powder at 20°C to 25°C (68°F to 77°F), excursions permitted between 15°C to 30°C (59°F to 86°F) [see USP controlled room temperature]. Keep in the original carton.
Keep the constituted oral solution of EVRYSDI in the original amber bottle to protect from light. Store in a refrigerator at 2°C to 8°C (36°F to 46°F) [see Dosage and Administration (2.4)].
16.2 EVRYSDI Tablets
How Supplied
Pale yellow film-coated tablet, round and curved, with EVR debossed on one side; available in HDPE bottles of 30 tablets with a child-resistant cap (NDC: 50242-202-01).
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Pregnancy and Fetal Risk
Inform pregnant women and women of reproductive potential that, based on animal studies, EVRYSDI may cause fetal harm [see Use in Specific Populations (8.1)].
Discuss with women of childbearing age whether they are pregnant, might be pregnant, or are trying to become pregnant.
Advise women of childbearing potential to use effective contraception during treatment with EVRYSDI and for at least 1 month after stopping EVRYSDI.
Advise a female patient to immediately inform the prescriber if she is pregnant or planning to become pregnant [see Use in Specific Populations (8.3)].
Pregnancy Registry
Encourage patients to enroll in the EVRYSDI Pregnancy Registry if they become pregnant while taking EVRYSDI [see Use in Specific Populations (8.1)].
Potential Effects on Male Fertility
Advise male patients that their fertility may be compromised while on treatment with EVRYSDI [see Use in Specific Populations (8.3)].
Instructions for Preparation of Oral Solution
Advise patients/caregivers to ensure that EVRYSDI oral solution is in liquid form when received from the pharmacy.
Instruct patients/caregivers to take EVRYSDI oral solution with or without food or before or after breastfeeding at approximately the same time each day. However, instruct caregivers not to mix EVRYSDI with formula or milk.
Instruct patients/caregivers to take EVRYSDI oral solution immediately after it is drawn up into the reusable oral syringe [see Dosage and Administration (2.2)].
Instructions for EVRYSDI Tablets
Advise patients/caregivers to swallow EVRYSDI tablets whole with water. Do not chew, cut, or crush the tablets.
Alternatively, the tablet can be dispersed in one teaspoon (5 mL) of room temperature non-chlorinated drinking water (e.g., filtered water) and taken immediately. EVRYSDI tablets must not be dispersed in any liquid other than non-chlorinated drinking water. Instruct the patient/caregivers that the dispersion must be administered within 10 minutes of adding non-chlorinated drinking water, or it must be discarded [see Dosage and Administration (2.2)].
Advise patients/caregivers to wash their hands before and after preparing or taking EVRYSDI tablets.
Advise patients/caregivers to avoid getting the dispersed tablet on their skin or in their eyes. Advise patients/caregivers to wash the area with soap and water if the dispersed tablet gets on the skin. Advise patients/caregivers to rinse their eyes with water if the dispersed tablet gets in the eyes.
Advise patients/caregivers to use a dry paper towel to dry the area if the dispersion is spilled and then clean with soap and water. Advise patients/caregivers to throw the paper towel away in the trash and wash their hands with soap and water.
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Issued: 2/2026 Patient Information EVRYSDI® [ev-RIZ-dee]
(risdiplam)
for oral solutionEVRYSDI® [ev-RIZ-dee]
(risdiplam)
tablets, for oral useWhat is EVRYSDI? - EVRYSDI is a prescription medicine used to treat spinal muscular atrophy (SMA) in children and adults.
Before taking EVRYSDI, tell your healthcare provider about all of your medical conditions, including if you: - are pregnant or plan to become pregnant. If you are pregnant, or are planning to become pregnant, ask your healthcare provider for advice before taking this medicine. EVRYSDI may harm your unborn baby.
- are a woman who can become pregnant:
- Before you start your treatment with EVRYSDI, your healthcare provider may test you for pregnancy. Because EVRYSDI may harm your unborn baby, you and your healthcare provider will decide if taking EVRYSDI is right for you during this time.
- Talk to your healthcare provider about birth control methods that may be right for you. Use birth control while on treatment and for at least 1 month after stopping EVRYSDI.
- Pregnancy Registry. There is a pregnancy registry for women who take EVRYSDI during pregnancy. The purpose of this registry is to collect information about the health of the pregnant woman and her baby. If you are pregnant or become pregnant while receiving EVRYSDI, tell your healthcare provider right away. Talk to your healthcare provider about registering with the EVRYSDI Pregnancy Registry. Your healthcare provider can enroll you in this registry or you can enroll by calling 1-833-760-1098 or visiting https://www.evrysdipregnancyregistry.com.
- are an adult male planning to have children: EVRYSDI may affect a man's ability to have children (fertility). If this is of concern to you, make sure to ask a healthcare provider for advice.
- are breastfeeding or plan to breastfeed. It is not known if EVRYSDI passes into breast milk and may harm your baby. If you plan to breastfeed, discuss with your healthcare provider about the best way to feed your baby while on treatment with EVRYSDI.
How should I take EVRYSDI? - For infants and children, your healthcare provider will determine the daily dose of EVRYSDI needed based on your child's age and weight. For adults, take 5 mg of EVRYSDI daily.
- Your healthcare provider will either prescribe:
EVRYSDI for oral solution
Or
EVRYSDI tablet - Your healthcare provider will tell you how long you or your child needs to take EVRYSDI. Do not stop treatment with EVRYSDI unless your healthcare provider tells you to.
- Take EVRYSDI exactly as your healthcare provider tells you to take it. Do not change the dose without talking to your healthcare provider.
- Avoid getting EVRYSDI on your skin or in your eyes. If it gets on your skin, wash the area with soap and water. If it gets in your eyes, rinse your eyes with water.
If you are taking EVRYSDI for oral solution: - See the detailed Instructions for Use that comes with it for information on how to take or give EVRYSDI for oral solution.
- You should receive EVRYSDI from the pharmacy as a liquid that can be given by mouth or through a feeding tube. The liquid solution is prepared by your pharmacist or other healthcare provider. If the medicine in the bottle is a powder, do not use it. Contact your pharmacist for a replacement.
- Take EVRYSDI one time daily with or without a meal at about the same time each day. Drink water afterwards to make sure EVRYSDI has been completely swallowed.
- In infants who are breastfed, EVRYSDI can be given before or after breastfeeding.
- Do not mix EVRYSDI with formula or milk.
- If you are unable to swallow and have a nasogastric (NG-tube) or gastrostomy tube (G-tube), EVRYSDI for oral solution can be given through the tube.
Reusable Oral Syringes for EVRYSDI for Oral Solution - Your pharmacist will provide you with the reusable oral syringes that are needed for taking your medicine and explain how to use them. You should receive 1 or 2 identical oral syringes depending on your prescribed daily dose.
- From the bottle, draw up (measure) the dose of EVRYSDI with these syringes, as they are made to protect the medicine from light.
- Take EVRYSDI right away after it has been drawn into the syringe. Do not store the EVRYSDI solution in the syringe. If EVRYSDI is not taken within 5 minutes of when it is drawn up, throw away the solution by pressing the plunger and prepare a new dose with the same syringe.
- Do not throw the syringes away because they are reusable.
- Wash the syringes per instructions after use.
- Contact your healthcare provider or pharmacist if your oral syringes are lost or damaged.
If you are taking EVRYSDI Tablets: - See the detailed Instructions for Use that comes with it for information on how to take or give EVRYSDI tablets.
- Wash your hands before and after preparing or taking EVRYSDI tablets.
- Take EVRYSDI one time daily with or without a meal at about the same time each day.
- Do not chew, cut, or crush the tablet.
- Swallow the EVRYSDI tablet whole with some water.
- If you cannot swallow tablets whole or if you have a nasogastric (NG-tube) or gastrostomy (G-tube), see the detailed Instructions for Use that comes with it for information on how to take or give EVRYSDI tablets.
If you miss a dose of EVRYSDI: - If you remember the missed dose within 6 hours of when you normally take EVRYSDI, then take the dose. Continue taking EVRYSDI at your usual time the next day.
- If you remember the missed dose more than 6 hours after you normally take EVRYSDI, skip the missed dose. Take your next dose at your usual time the next day.
- If you do not fully swallow the dose, or you vomit after taking a dose, do not take another dose of EVRYSDI to make up for that dose. Wait until the next day to take the next dose at your usual time.
What are the possible side effects of EVRYSDI?
The most common side effects of EVRYSDI include:- For later-onset SMA:
- fever
- diarrhea
- rash
- For infantile-onset SMA:
- fever
- diarrhea
- rash
- runny nose, sneezing, and sore throat (upper respiratory infection)
- lung infection (lower respiratory infection)
- constipation
- vomiting
- cough
These are not all of the possible side effects of EVRYSDI. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store EVRYSDI?
EVRYSDI for Oral Solution:- Store EVRYSDI in the refrigerator between 36°F to 46°F (2°C to 8°C). Do not freeze.
- If necessary, EVRYSDI can be kept at room temperature up to 104°F (up to 40°C) for a combined total of 5 days. EVRYSDI can be removed from, and returned to, a refrigerator. The total combined time out of refrigeration should not be more than 5 days.
- Keep EVRYSDI in an upright position in the original amber bottle to protect from light.
- Throw away (discard) any unused portion of EVRYSDI 64 days after it is mixed by the pharmacist (constitution) or if EVRYSDI has been kept at room temperature (below 104°F [40°C]) for more than a total combined time of 5 days. Discard EVRYSDI if it has been kept above 104°F (40°C). Please see the Discard After date written on the bottle label. (See the Instructions for Use that comes with EVRYSDI for oral solution).
- Store at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep the tablets in their original bottle with the cap tightly closed to protect from moisture.
General information about the safe and effective use of EVRYSDI.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use EVRYSDI for a condition for which it was not prescribed. Do not give EVRYSDI to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about EVRYSDI that is written for health professionals.What are the ingredients in EVRYSDI?
Active ingredient: risdiplam
Inactive ingredients:
EVRYSDI for Oral Solution: ascorbic acid, disodium edetate dihydrate, isomalt, mannitol, polyethylene glycol 6000, sodium benzoate, strawberry flavor, sucralose, and tartaric acid.
EVRYSDI Tablets: colloidal silicon dioxide, crospovidone, mannitol, microcrystalline cellulose, polyethylene glycol 3350, polyvinyl alcohol, sodium stearyl fumarate, strawberry flavor, talc, tartaric acid, titanium dioxide, and yellow iron oxide.
Distributed by: Genentech USA, Inc., A Member of the Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990
EVRYSDI is a registered trademark of Genentech, Inc.
©2026 Genentech, Inc. All rights reserved.
For more information, go to www.EVRYSDI.com or call 1-833-387-9734. -
INSTRUCTIONS FOR USE
INSTRUCTIONS FOR USE
EVRYSDI® [ev-RIZ-dee]
(risdiplam)
for oral solutionPlease read and understand this Instructions for Use and the Patient Information leaflet before you start taking EVRYSDI for information about EVRYSDI and how to prepare and give EVRYSDI through an oral syringe, gastrostomy tube (G-tube), or nasogastric tube (NG-tube).
If you have any questions about how to take EVRYSDI, contact your healthcare provider.
EVRYSDI should come as a liquid in a bottle when you receive it from the pharmacy. Do not take EVRYSDI and contact your pharmacist if the medicine in the bottle is a powder.
Each EVRYSDI carton contains (see Figure A):

1 Cap  Figure A
Figure A
1 Bottle adapter 
1 EVRYSDI bottle 
1 or 2 Reusable oral syringe(s) 
1 Instructions for Use (not shown) 
1 Prescribing Information and Patient Information (not shown) Reusable Oral Syringe Overview (see Figure B)  Figure B
Figure BImportant information about EVRYSDI
- Ask your healthcare provider to show you the correct oral syringe you should use and how to measure your prescribed daily dose.
- Always use the reusable oral syringes that come with EVRYSDI to measure your prescribed daily dose. If your carton does not contain two identical syringes, contact your pharmacist.
- Always take EVRYSDI exactly as your healthcare provider tells you to take it.
- Take EVRYSDI 1 time daily with or without a meal at approximately the same time each day.
- Do not take EVRYSDI if the bottle adapter is not in the bottle. If the bottle adapter is not in the bottle, contact your pharmacist.
- Do not mix EVRYSDI into food or liquids. Do not mix EVRYSDI with formula or milk.
- Do not take EVRYSDI if the bottle or oral syringes are damaged.
- Avoid getting EVRYSDI on your skin or in your eyes. If EVRYSDI gets on your skin, wash the area with soap and water. If EVRYSDI gets in your eyes, rinse your eyes with water.
- If you spill EVRYSDI, dry the area with a dry paper towel and then clean with water. Throw away the paper towel in the trash and wash your hands well with soap and water.
- If there is not enough EVRYSDI left in the bottle for your prescribed dose, throw away (discard) the bottle with remaining EVRYSDI and used oral syringes according to your local requirements.
- Use a new bottle of EVRYSDI to get your prescribed dose.
Do not mix EVRYSDI from the new bottle with the bottle you are currently using.
How to store EVRYSDI
- Store EVRYSDI in the refrigerator between 36°F to 46°F (2°C to 8°C). Do not freeze.
- If necessary, EVRYSDI can be kept at room temperature up to 104°F (up to 40°C) for a combined total of 5 days. EVRYSDI can be removed from, and returned to, a refrigerator. The total combined time out of refrigeration should not be more than 5 days. Throw away EVRYSDI if it has been kept at room temperature for more than 5 days.
- Store EVRYSDI in the original amber bottle in an upright position with the cap tightly closed.
- Throw away (discard) any unused portion of EVRYSDI 64 days after mixed by the pharmacist (constitution) when stored in the refrigerator at 36°F to 46°F (2°C to 8°C). Please see the Discard After date written on the bottle label (see Figure C).
- Ask your pharmacist for the Discard After date if it is not written on the bottle label.
- Throw away any unused portion of EVRYSDI that has been kept above 104°F (40°C).
- Keep EVRYSDI, all medicines and syringes out of the reach of children.
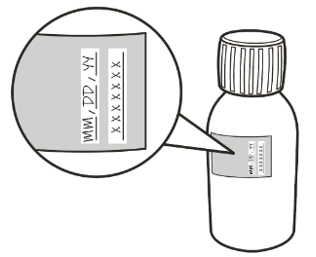 Figure C
Figure CA) Preparing and withdrawing your dose
How to prepare your dose of EVRYSDI
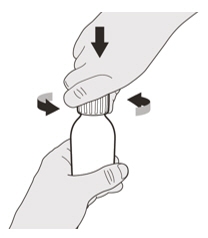 Figure D
Figure DStep A1
Remove the cap by pushing it down and then twisting the cap to the left (counterclockwise) (See Figure D).
Do not throw away the cap.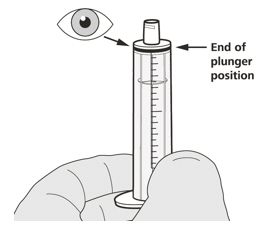 Figure E
Figure EStep A2
Push the plunger of the oral syringe all the way down to remove any air in the oral syringe (See Figure E).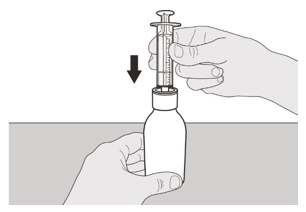 Figure F
Figure FStep A3
Place the EVRYSDI bottle on a flat surface. While keeping the bottle in an upright position, insert the syringe tip into the bottle adapter (See Figure F).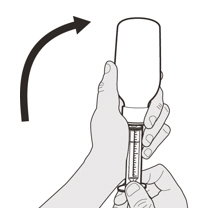 Figure G
Figure GStep A4
Carefully turn the bottle upside down with the syringe tip firmly inserted into the bottle adapter (See Figure G).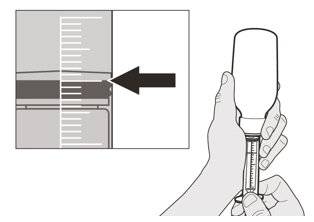 Figure H
Figure HStep A5
Slowly pull back on the plunger to withdraw your prescribed dose of EVRYSDI.
The top of the black plunger stopper must line up with the mL marking on the oral syringe for your prescribed daily dose (See Figure H).
After the correct dose is withdrawn, hold the plunger in place to keep the plunger from moving.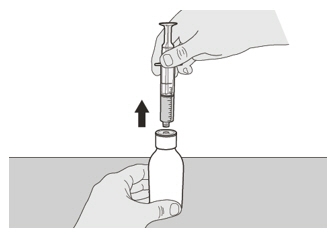 Figure I
Figure IStep A6
Continue to hold the plunger in place to keep the plunger from moving. Leave the oral syringe in the bottle adapter and turn the bottle to an upright position. Place the bottle onto a flat surface. Remove the oral syringe from the bottle adapter by gently pulling straight up on the oral syringe while holding the plunger in place (See Figure I).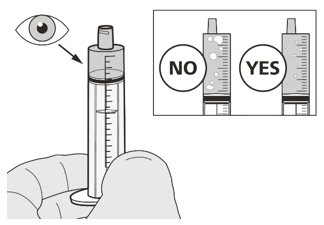 Figure J
Figure JStep A7
Hold the oral syringe with the syringe tip pointing up. Check the EVRYSDI in the oral syringe. If there are large air bubbles in the oral syringe (See Figure J) or if you have drawn up the wrong dose of EVRYSDI, insert the syringe tip firmly into the bottle adapter while the bottle is in an upright position. Push the plunger all the way down so that EVRYSDI flows back into the bottle and repeat Steps A4 through A7.
Take or give EVRYSDI right away after it is drawn up into the oral syringe. If it is not taken within 5 minutes, throw away EVRYSDI liquid from your oral syringe into the household trash. Do this by pushing the plunger all the way down to remove EVRYSDI from the oral syringe. Prepare a new dose starting with Step A2.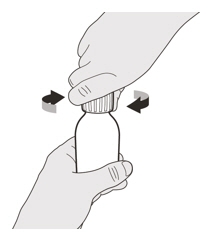 Figure K
Figure KStep A8
Put the cap back on the bottle. Turn the cap to the right (clockwise) to tightly close the bottle (See Figure K). Do not remove the bottle adapter from the bottle.If you are taking your dose of EVRYSDI by mouth, follow the instructions in "B) How to take a dose of EVRYSDI by mouth".
If you are taking your dose of EVRYSDI through a gastrostomy tube, follow the instructions in "C) How to give a dose of EVRYSDI through a gastrostomy tube".
If you are taking your dose of EVRYSDI through a nasogastric tube, follow the instructions in "D) How to give a dose of EVRYSDI through a nasogastric tube".
B) How to take a dose of EVRYSDI by mouth
Sit upright when taking a dose of EVRYSDI by mouth.
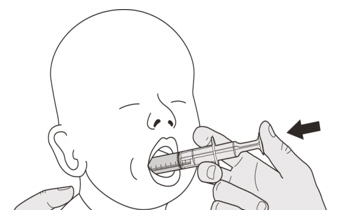 Figure L
Figure LStep B1
Place the oral syringe into the mouth with the tip along either cheek.
Slowly push the plunger all the way down to give the full dose of EVRYSDI (See Figure L).
Giving EVRYSDI into the throat or too fast may cause choking.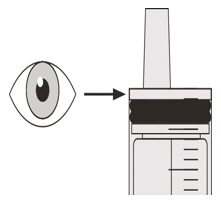 Figure M
Figure MStep B2
Check that there is no EVRYSDI left in the oral syringe (See Figure M).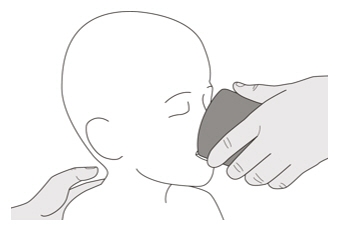 Figure N
Figure NStep B3
Drink about a tablespoon (15 mL) of water right after taking the prescribed dose of EVRYSDI to make sure the drug has been completely swallowed (See Figure N).
Go to Step E for cleaning of the syringe.C) How to give a dose of EVRYSDI through a gastrostomy tube
If you are giving EVRYSDI through a gastrostomy tube, ask your healthcare provider to show you how to inspect the gastrostomy tube before giving EVRYSDI.
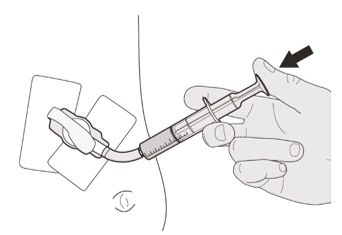 Figure O
Figure OStep C1
Place the oral syringe tip into the gastrostomy tube. Slowly push the plunger all the way down to give the full dose of EVRYSDI (See Figure O).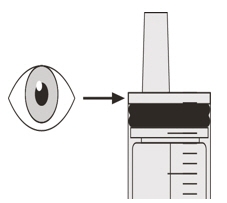 Figure P
Figure PStep C2
Check that there is no EVRYSDI left in the oral syringe (See Figure P).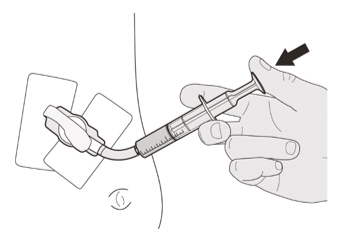 Figure Q
Figure QStep C3
Flush the gastrostomy tube with 10 mL to 20 mL of water right after giving the prescribed dose of EVRYSDI (See Figure Q).
Go to Step E for cleaning of the syringe.D) How to give a dose of EVRYSDI through a nasogastric tube
If you are giving EVRYSDI through a nasogastric tube, ask your healthcare provider to show you how to inspect the nasogastric tube before giving EVRYSDI.
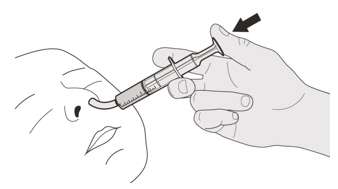 Figure R
Figure RStep D1
Place the oral syringe tip into the nasogastric tube. Slowly press the plunger all the way down to give the full dose of EVRYSDI (See Figure R). Figure S
Figure SStep D2
Check that there is no EVRYSDI left in the oral syringe (See Figure S).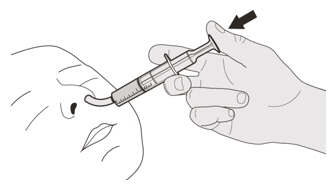 Figure T
Figure TStep D3
Flush the nasogastric tube with 10 mL to 20 mL of water right after giving the prescribed dose of EVRYSDI (See Figure T).
Go to Step E for cleaning of the syringe.E) How to clean the oral syringe after use
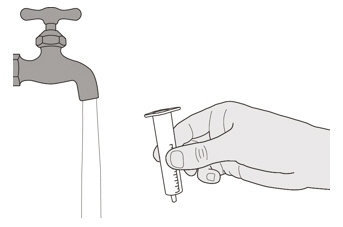 Figure U
Figure UStep E1
Remove the plunger from the oral syringe by pulling the plunger away from the syringe until the plunger comes out of the syringe.
Rinse the oral syringe barrel well under clean water (See Figure U).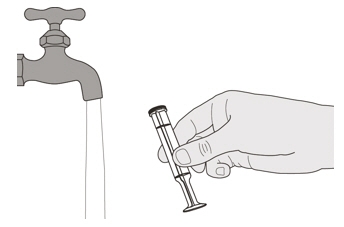 Figure V
Figure VStep E2
Rinse the plunger well under clean water (See Figure V). Figure W
Figure WStep E3
Check that the oral syringe barrel and plunger are clean.
Place the oral syringe barrel and plunger on a clean surface in a safe place to dry (See Figure W).
Wash your hands with soap and water.
After the oral syringe barrel and plunger are dry, put the plunger back into the oral syringe barrel and store the syringe with your medicine.EVRYSDI is a registered trademark of Genentech, Inc.
Distributed by:
Genentech, Inc.
1 DNA Way
South San Francisco, CA 94080-4990
Approved: 2/2025
This Instructions for Use has been Approved by the U.S. Food and Drug Administration.©2025 Genentech, Inc. All Rights Reserved
-
SPL UNCLASSIFIED SECTION
EVRYSDI®
(risdiplam) for oral solutionInstructions for Constitution
(FOR HEALTHCARE PROVIDERS ONLY)Each EVRYSDI carton contains (see Figure A):
1 Cap 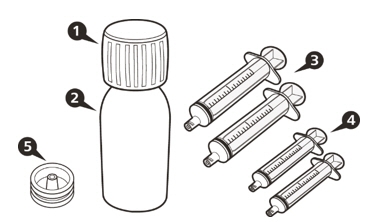 Figure A
Figure A1 EVRYSDI bottle 1 Reusable oral syringe 12 mL 2 Reusable oral syringes 6 mL 2 Reusable oral syringes 1 mL 1 Press-in bottle adapter 
1 Prescribing Information (not shown) 
1 Instructions for Constitution (not shown) 
1 Instructions for Use (not shown) Important information about EVRYSDI
- Do not use if the powder expiration date has passed. The powder expiration date is printed on the bottle label.
- Do not use the medicine if any of the supplies are damaged or missing.
- Use purified water to constitute the medicine.
- Select the appropriate oral syringes (1 mL, 6 mL, or 12 mL) based on the patient's dose and provide instruction to the patient/caregiver on how to administer their dose. The oral syringes provided in the carton are intended to be reusable.
- Only use the amber oral syringe provided for use with EVRYSDI. Other syringes may not be compatible with the press-in-bottle adapter and may not protect the product from light.
How to store EVRYSDI
- Store the dry powder at 20°C to 25°C (68°F to 77°F), excursions permitted between 15°C to 30°C (59°F to 86°F) [see USP controlled room temperature]. Keep in the original carton.
- Store the constituted oral solution of EVRYSDI upright in the original amber bottle in a refrigerator at 2°C to 8°C (36°F to 46°F). Do not freeze.
Important precautions for preparation of EVRYSDI
- Avoid inhalation and direct contact with skin or mucous membranes with the dry powder and the constituted solution. If such contact occurs, wash thoroughly with soap and water; rinse eyes with water.
- Wear disposable gloves during the preparation and clean up procedure.
Constitution
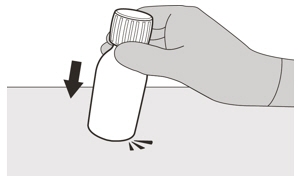 Figure B
Figure BStep 1
Gently tap the bottom of the bottle to loosen the powder (See Figure B).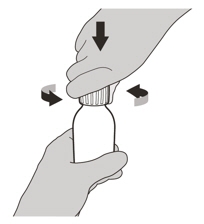 Figure C
Figure CStep 2
Remove the cap by pushing it down and then twisting to the left (counter-clockwise) (See Figure C). Do not throw away the cap.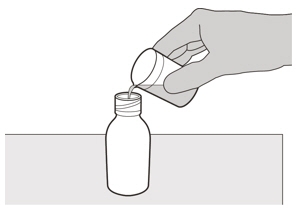 Figure D
Figure DStep 3
Carefully pour 79 mL of purified water into the medicine bottle (See Figure D).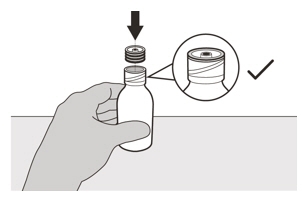 Figure E
Figure EStep 4
Hold the medicine bottle on a table with one hand.
Insert the press-in bottle adapter into the opening by pushing it down with the other hand. Ensure it is completely pressed against the bottle lip (See Figure E).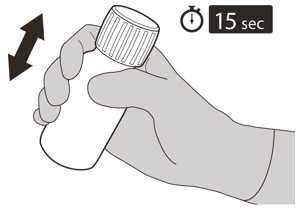 Figure F
Figure FStep 5
Put the cap back on the bottle. Turn the cap to the right (clockwise) to close the bottle.
Ensure it is completely closed and then shake well for 15 seconds (See Figure F).
Wait for 10 minutes. You should have obtained a clear solution.
If not, shake well again for another 15 seconds or until you have obtained a clear solution.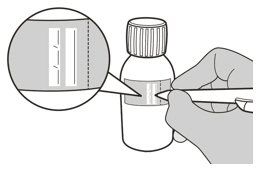 Figure G
Figure GStep 6
Calculate the Discard After date of the oral solution as 64 days after constitution (Note: The day of constitution is counted as day 0. For example, if constitution is on the 1st of April, the Discard After date will be the 4th of June).
Write the Discard After date of the solution and the Lot number on the bottle label (See Figure G).
Do not dispense the constituted solution if the solution's Discard After date exceeds the original powder expiration date.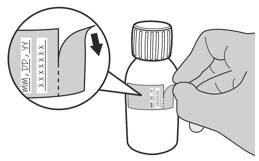 Figure H
Figure HStep 7
Remove and discard the peel-off part of the bottle label with the expiration date of the powder (See Figure H).Selecting the oral syringe for the prescribed daily dose of EVRYSDI
For the calculation of dosing volume, the syringe increments need to be considered. Round the dose volume to the closest increment marked on the selected oral syringe.
Select the correct oral syringe(s) (1 mL, 6 mL, or 12 mL) for the calculated dosing volume according to the table below and remove the other oral syringes.
Dose Strength Syringe Size Dosing Volume Syringe Increments 0.75 mg/mL 1 mL 0.3 mL to 1 mL 0.01 mL 6 mL 1 mL to 6 mL 0.1 mL 12 mL 6.2 mL to 6.6 mL 0.2 mL Put the bottle back in its original carton with the correct oral syringes, Prescribing Information, and Instructions for Use.
Store the constituted oral solution of EVRYSDI upright in the original amber bottle in a refrigerator at 2°C to 8°C (36°F to 46°F). Do not freeze. Discard any unused portion 64 days after constitution.
EVRYSDI is a registered trademark of Genentech, Inc.
Distributed by:
Genentech, Inc.
1 DNA Way
South San Francisco, CA 94080-4990
Revision Date: 2/2025
©2025 Genentech, Inc. All Rights Reserved. -
SPL UNCLASSIFIED SECTION
Representative sample of labeling (see the HOW SUPPLIED section for complete listing):
-
INSTRUCTIONS FOR USE
EVRYSDI® [ev-RIZ-dee]
(risdiplam)
Tablets, for oral useBefore you start
This Instructions for Use contains information on how to prepare and take EVRYSDI tablets.
Read this Instructions for Use and Patient Information leaflet before you start taking or giving EVRYSDI tablets for the first time and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or treatment.
If you have any questions about how to take EVRYSDI, contact your healthcare provider.
Important Information
- If you are taking EVRYSDI tablets, the daily dose is one tablet.
- EVRYSDI tablets can be swallowed whole with water. Do not chew, cut, or crush the tablet.
- If you are unable to swallow EVRYSDI tablets whole or have a nasogastric (NG) tube or gastrostomy tube (G-tube), your healthcare provider will show you how to prepare and take EVRYSDI tablets. Always take EVRYSDI tablets exactly as your healthcare provider tells you.
- Do not take or give EVRYSDI tablets until you have been shown the right way to prepare and take or give EVRYSDI.
- Wash your hands before and after preparing, taking or giving EVRYSDI.
- Check the expiration date and check the product for damage before use. Do not use if expired or damaged.
- Avoid getting the EVRYSDI tablet mixture on your skin or in your eyes. If the EVRYSDI tablet mixture gets on your skin, wash the area with soap and water. If the tablet mixture gets in your eyes, rinse your eyes with water.
- Keep the EVRYSDI tablet mixture out of sunlight.
- If you spill the EVRYSDI tablet mixture, dry the area with a dry paper towel and then clean with soap and water. Throw away the paper towel in the trash and wash your hands with soap and water.
- Do not take an extra dose if you vomit at any time after taking EVRYSDI.
How to store EVRYSDI tablets - Store EVRYSDI tablets at room temperature between 68°F to 77°F (20°C to 25°C)
- Do not take or give EVRYSDI if it has been exposed to temperatures above 86°F (30°C).
- Keep tablets in the original bottle. Keep the bottle tightly closed to protect from moisture.
- Keep EVRYSDI and all medicines out of the sight and reach of children.
If able to swallow the EVRYSDI tablet whole: Swallow the tablet whole with water. Do not chew, cut, or crush the tablet. Do not swallow with any liquids other than water. Wash your hands with soap and water If unable to swallow the EVRYSDI tablet whole:
Prepare an EVRYSDI tablet mixtureGather supplies
(Figure A):- 1 EVRYSDI tablet
- a small, clean empty cup
- 1 teaspoon (5 mL) of room temperature non-chlorinated drinking water (such as filtered water) for mixing
- at least 1 tablespoon (15 mL) of non-chlorinated drinking water (such as filtered water) for rinsing
- If giving EVRYSDI through a feeding tube: 1 dosing syringe that connects to the feeding tube

Figure AThe cup and the dosing syringe are not included in the package. Obtain a dosing syringe from your healthcare provider or pharmacist. Your healthcare provider or pharmacist can help you choose the right dosing syringe. Step 1. Wash your hands with soap and water (Figure B). 
Figure BStep 2. Remove the cap by pushing down and then twisting the child resistant cap to the left (counterclockwise) (Figure C). 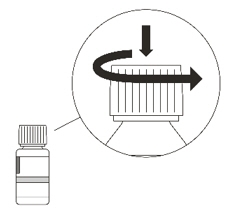
Figure CStep 3. Take 1 tablet out of the bottle (Figure D). 
Figure DStep 4. Replace the child resistant cap on the bottle, turn the cap to the right (clockwise) until tightly closed. (Figure E).
Do not store EVRYSDI outside the original container due to moisture sensitivity.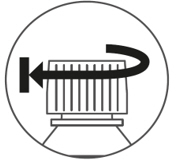
Figure EStep 5
Put 1 teaspoon (5 mL) of room temperature non-chlorinated drinking water (such as filtered water) in a small cup and add 1 tablet.- Do not use any liquids other than non-chlorinated drinking water (such as filtered water).
- Do not expose the mixture to sunlight.
Figure FStep 6
Gently swirl the cup until it is fully mixed. Some particles may remain after swirling. This may take up to 3 minutes. (Figure F).Option A: To take or give the tablet mixture by mouth, see Step A1
Option B: To take or give the tablet mixture by feeding tube, see Step B1Option A: Take or give EVRYSDI tablet mixture by mouth Step A1
Drink or give the tablet mixture right away. Throw away the mixture if not used within 10 minutes of adding the tablet to the cup with non-chlorinated drinking water (such as filtered water) (Figure G).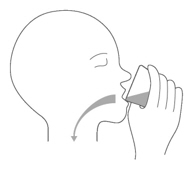
Figure GStep A2
Refill the cup with at least 1 tablespoon (15 mL) of non-chlorinated drinking water (such as filtered water) and swirl to get any medicine left in the cup (Figure H).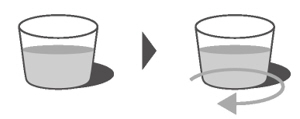
Figure HStep A3
Drink the 2nd mixture right away. (Figure I).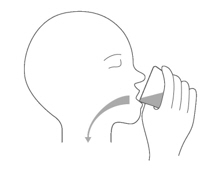
Figure IStep A4
Wash your hands and all the items used to give EVRYSDI (Figure J).
Figure JOption B: Take or give EVRYSDI tablet mixture through a feeding tube (gastrostomy or nasogastric tube) You can take or give the EVRYSDI tablet mixture through a feeding tube (gastrostomy or nasogastric tube) placed by a healthcare provider.
Check the manufacturer's instructions for the size and dimensions of the gastrostomy or nasogastric tube.
Make sure that the gastrostomy or nasogastric tube size is at least 8 French or higher to prevent clogging of the feeding tube. Ask your healthcare provider to show you how to check the feeding tube before giving EVRYSDI.Step B1
Take or give EVRYSDI tablet mixture right away. Throw away the mixture if not used within 10 minutes of adding the tablet to the cup with non-chlorinated drinking water (such as filtered water).
Place the dosing syringe tip into the cup and slowly pull up the plunger to draw up all the mixture (Figure K).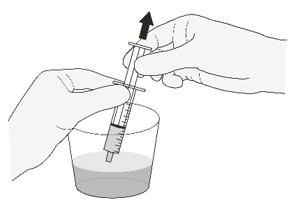
Figure KStep B2
Place the dosing syringe tip into the gastrostomy or nasogastric tube. Slowly push the plunger all the way down to give the full dose of EVRYSDI (Figure L1 or L2).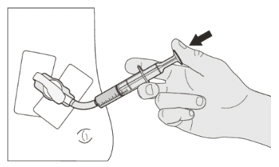
Figure L1
(gastrostomy tube)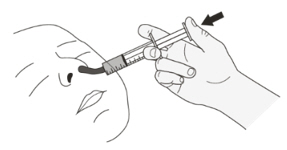 Figure L2
Figure L2
(nasogastric tube)Step B3
Check that there is no EVRYSDI left in the dosing syringe (Figure M).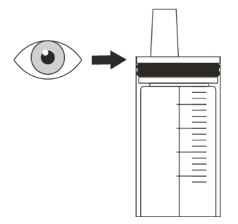
Figure MStep B4
Refill the cup with at least 1 tablespoon (15 mL) of non-chlorinated drinking water (such as filtered water) and swirl to get any medicine left in the cup (Figure N).
Place the dosing syringe into the cup and pull up the plunger to draw up all the mixture.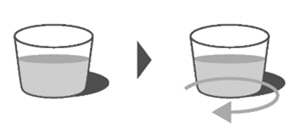
Figure NStep B5
Flush the gastrostomy or nasogastric tube with the mixture (Figure O1 or O2).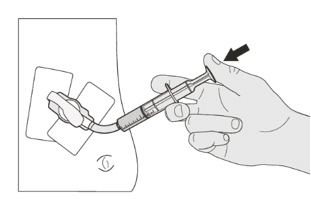
Figure O1
(gastrostomy tube)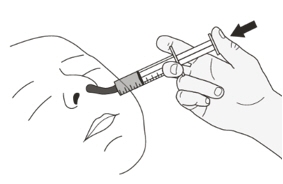
Figure O2
(nasogastric tube)Step B6
Wash your hands and all the items used to give EVRYSDI (Figure P).
Check with your pharmacist if the dosing syringe provided is for single use only or can be re-used multiple times. Follow the manufacturer's instructions to throw away or immediately clean your dosing syringe.
Figure P
Distributed by:
Genentech, USA Inc., A Member of the Roche Group
1 DNA Way, South San Francisco, CA 94080-4990Evrysdi® is a registered trademark of Genentech, Inc.
©2026 Genentech, Inc.This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Approved: 2/2026
-
PRINCIPAL DISPLAY PANEL - 60 mg/80 mL Bottle Carton
NDC: 50242-175-07
Evrysdi®
(risdiplam)
for oral solution60 mg/80 mL
(0.75 mg/mL)Attention pharmacist: Evrysdi must be
constituted with water prior to dispensing.80 mL (2.71 fl oz) total volume after constitution
Rx only
Genentech
11044976

-
PRINCIPAL DISPLAY PANEL - 5 mg Tablet Bottle Carton
NDC: 50242-202-01
Evrysdi®
(risdiplam)
Tablets5 mg
Do not chew, cut,
or crush tablet.30 Tablets
Rx only
Genentech
11044980

-
INGREDIENTS AND APPEARANCE
EVRYSDI
risdiplam powder, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 50242-175 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RISDIPLAM (UNII: 76RS4S2ET1) (RISDIPLAM - UNII:76RS4S2ET1) RISDIPLAM 0.75 mg in 1 mL Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) 16.81 mg in 1 mL ISOMALT (UNII: S870P55O2W) 2.97 mg in 1 mL MALTODEXTRIN (UNII: 7CVR7L4A2D) 1.88 mg in 1 mL TARTARIC ACID (UNII: W4888I119H) 1.51 mg in 1 mL SODIUM BENZOATE (UNII: OJ245FE5EU) 0.38 mg in 1 mL POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) 0.25 mg in 1 mL SUCRALOSE (UNII: 96K6UQ3ZD4) 0.2 mg in 1 mL ASCORBIC ACID (UNII: PQ6CK8PD0R) 0.18 mg in 1 mL EDETATE DISODIUM (UNII: 7FLD91C86K) 0.09 mg in 1 mL Product Characteristics Color Score Shape Size Flavor STRAWBERRY (Strawberry) Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 50242-175-07 1 in 1 CARTON 08/07/2020 1 80 mL in 1 BOTTLE, GLASS; Type 1: Convenience Kit of Co-Package Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA213535 08/07/2020 EVRYSDI
risdiplam tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 50242-202 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RISDIPLAM (UNII: 76RS4S2ET1) (RISDIPLAM - UNII:76RS4S2ET1) RISDIPLAM 5 mg Inactive Ingredients Ingredient Name Strength TARTARIC ACID (UNII: W4888I119H) 3 mg MANNITOL (UNII: 3OWL53L36A) 39.6 mg MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) 63.5 mg SILICON DIOXIDE (UNII: ETJ7Z6XBU4) 2.5 mg CROSPOVIDONE, UNSPECIFIED (UNII: 2S7830E561) 7.5 mg SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) 3.7 mg POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) 1 mg TITANIUM DIOXIDE (UNII: 15FIX9V2JP) 0.6 mg POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) 0.5 mg TALC (UNII: 7SEV7J4R1U) 0.3 mg Product Characteristics Color yellow (pale yellow) Score no score Shape ROUND Size 7mm Flavor STRAWBERRY Imprint Code EVR Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 50242-202-01 1 in 1 CARTON 02/11/2025 1 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA219285 02/11/2025 Labeler - Genentech, Inc. (080129000) Registrant - Genentech, Inc. (080129000) Establishment Name Address ID/FEI Business Operations F. Hoffmann-La Roche AG 482242971 ANALYSIS(50242-175, 50242-202) , API MANUFACTURE(50242-175, 50242-202) Establishment Name Address ID/FEI Business Operations F. Hoffmann-La Roche AG 485244961 ANALYSIS(50242-175, 50242-202) , MANUFACTURE(50242-175, 50242-202) , PACK(50242-175, 50242-202) , LABEL(50242-175, 50242-202)
Trademark Results [EVRYSDI]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 EVRYSDI 90622036 not registered Live/Pending |
Genentech, Inc. 2021-04-02 |
 EVRYSDI 88769281 not registered Live/Pending |
Genentech, Inc. 2020-01-22 |
 EVRYSDI 88350381 not registered Live/Pending |
Genentech, Inc. 2019-03-21 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.