PANTOPRAZOLE tablet, delayed release
Pantoprazole by
Drug Labeling and Warnings
Pantoprazole by is a Prescription medication manufactured, distributed, or labeled by ST. MARY'S MEDICAL PARK PHARMACY. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use PANTOPRAZOLE SODIUM DELAYED-RELEASE TABLETS safely and effectively. See full prescribing information for PANTOPRAZOLE SODIUM DELAYED-RELEASE TABLETS .
PANTOPRAZOLE SODIUM delayed-release tablets, for oral use
Initial U.S. approval: 2000RECENT MAJOR CHANGES
Warnings and Precautions, Acute Tubulointerstitial Nephritis (5.2) 11/2020
INDICATIONS AND USAGE
Pantoprazole sodium is a proton pump inhibitor (PPI) indicated for the following:
- Short-Term Treatment of Erosive Esophagitis Associated with Gastroesophageal Reflux Disease (GERD) ( 1.1)
- Maintenance of Healing of Erosive Esophagitis ( 1.2)
- Pathological Hypersecretory Conditions Including Zollinger-Ellison (ZE) Syndrome ( 1.3)
DOSAGE AND ADMINISTRATION
Indication Dose Frequency Short-Term Treatment of Erosive Esophagitis Associated With GERD ( 2.1) Adults 40 mg Once Daily for up to 8 wks Children (5 years and older) ≥ 15 kg to < 40 kg 20 mg Once Daily for up to 8 wks ≥ 40 kg 40 mg Maintenance of Healing of Erosive Esophagitis ( 2.1) Adults 40 mg Once Daily* Pathological Hypersecretory Conditions Including Zollinger-Ellison Syndrome ( 2.1) Adults 40 mg Twice Daily * Controlled studies did not extend beyond 12 months
See full prescribing information for administration instructions
DOSAGE FORMS AND STRENGTHS
- Delayed-Release Tablets: 20 mg and 40 mg pantoprazole ( 3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
Gastric Malignancy: In adults, symptomatic response does not preclude presence of gastric malignancy. Consider additional follow-up and diagnostic testing. ( 5.1)
Acute Tubulointerstitial Nephritis: Discontinue treatment and evaluate patients. ( 5.2)
Clostridium difficile-Associated Diarrhea : PPI therapy may be associated with increased risk of Clostridium difficile-associated diarrhea. ( 5.3)
Bone Fracture: Long-term and multiple daily dose PPI therapy may be associated with an increased risk for osteoporosis-related fractures of the hip, wrist or spine. ( 5.4)
Cutaneous and Systemic Lupus Erythematosus: Mostly cutaneous; new onset or exacerbation of existing disease; discontinue pantoprazole sodium and refer to specialist for evaluation. ( 5.5)
Cyanocobalamin (Vitamin B-12) Deficiency: Daily long-term use (e.g., longer than 3 years) may lead to malabsorption or a deficiency of cyanocobalamin. ( 5.6)
Hypomagnesemia: Reported rarely with prolonged treatment with PPIs. ( 5.7)
Fundic Gland Polyps: Risk increases with long-term use, especially beyond one year. Use the shortest duration of therapy. ( 5.9)
ADVERSE REACTIONS
Most common adverse reactions are:
- For adult use (>2%): headache, diarrhea, nausea, abdominal pain, vomiting, flatulence, dizziness, and arthralgia. ( 6.1)
- For pediatric use (>4%): URI, headache, fever, diarrhea, vomiting, rash, and abdominal pain. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Dr. Reddy’s Laboratories, Inc. at 1-888-375-3784 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See full prescribing information for a list of clinically important drug interactions ( 7)
USE IN SPECIFIC POPULATIONS
Pregnancy: Based on animal data, may cause fetal harm. ( 8.1)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 10/2022
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Short-Term Treatment of Erosive Esophagitis Associated With Gastroesophageal Reflux Disease (GERD)
1.2 Maintenance of Healing of Erosive Esophagitis
1.3 Pathological Hypersecretory Conditions Including Zollinger-Ellison Syndrome
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing Schedule
2.2 Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Presence of Gastric Malignancy
5.2 Acute Tubulointerstitial Nephritis
5.3 Clostridium difficile-Associated Diarrhea
5.4 Bone Fracture
5.5 Cutaneous and Systemic Lupus Erythematosus
5.6 Cyanocobalamin (Vitamin B-12) Deficiency
5.7 Hypomagnesemia
5.8 Tumorigenicity
5.9 Fundic Gland Polyps
5.10 Interference with Investigations for Neuroendocrine Tumors
5.11 Interference with Urine Screen for THC
5.12 Concomitant Use of Pantoprazole Sodium with Methotrexate
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.5 Pharmacogenomics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Erosive Esophagitis (EE) Associated with Gastroesophageal Reflux Disease (GERD)
14.2 Long-Term Maintenance of Healing of Erosive Esophagitis
14.3 Pathological Hypersecretory Conditions Including Zollinger-Ellison Syndrome
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
Pantoprazole sodium delayed-release tablets are indicated for:
1.1 Short-Term Treatment of Erosive Esophagitis Associated With Gastroesophageal Reflux Disease (GERD)
Pantoprazole sodium delayed-release tablets are indicated in adults and pediatric patients five years of age and older for the short-term treatment (up to 8 weeks) in the healing and symptomatic relief of erosive esophagitis (EE). For those adult patients who have not healed after 8 weeks of treatment, an additional 8-week course of pantoprazole sodium delayed-release tablets may be considered. Safety of treatment beyond 8 weeks in pediatric patients has not been established.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing Schedule
Pantoprazole sodium is supplied as delayed-release tablets. The recommended dosages are outlined in Table 1.
Table 1: Recommended Dosing Schedule for Pantoprazole Sodium Delayed-Release Tablets
Indication Dose Frequency Short-Term Treatment of Erosive Esophagitis Associated With GERD Adults 40 mg Once daily for up to 8 weeks* Children (5 years and older) ≥ 15 kg to < 40 kg 20 mg Once daily for up to 8 weeks ≥ 40 kg 40 mg Maintenance of Healing of Erosive Esophagitis Adults 40 mg Once daily*** Pathological Hypersecretory Conditions Including Zollinger-Ellison Syndrome Adults 40 mg Twice daily ** * For adult patients who have not healed after 8 weeks of treatment, an additional 8-week course of pantoprazole sodium delayed-release tablets may be considered.
** Dosage regimens should be adjusted to individual patient needs and should continue for as long as clinically indicated. Doses up to 240 mg daily have been administered.
*** Controlled studies did not extend beyond 12 months
2.2 Administration Instructions
Directions for method of administration for each dosage form are presented in Table 2.
Table 2: Administration Instructions
Formulation Route Instructions* Delayed-Release Tablets Oral Swallowed whole, with or without food * Do not split, chew, or crush pantoprazole sodium delayed-release tablets.
Take a missed dose as soon as possible. If it is almost time for the next dose, skip the missed dose and take the next dose at the regular scheduled time. Do not take 2 doses at the same time.
Pantoprazole Sodium Delayed-Release Tablets
Swallow pantoprazole sodium delayed-release tablets whole, with or without food in the stomach. For patients unable to swallow a 40 mg tablet, two 20 mg tablets may be taken. Concomitant administration of antacids does not affect the absorption of pantoprazole sodium delayed-release tablets.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Pantoprazole sodium delayed-release tablets are contraindicated in patients with known hypersensitivity to any component of the formulation or any substituted benzimidazole. Hypersensitivity reactions may include anaphylaxis, anaphylactic shock, angioedema, bronchospasm, acute tubulointerstitial nephritis, and urticaria [see Warnings and Precautions (5.2), [see Adverse Reactions (6) ].
Proton pump inhibitors (PPIs), including pantoprazole sodium delayed-release tablets, are contraindicated in patients receiving rilpivirine-containing products [see Drug Interactions ( 7) ].
-
5 WARNINGS AND PRECAUTIONS
5.1 Presence of Gastric Malignancy
In adults, symptomatic response to therapy with pantoprazole sodium does not preclude the presence of gastric malignancy. Consider additional follow-up and diagnostic testing in adult patients who have a suboptimal response or an early symptomatic relapse after completing treatment with a PPI. In older patients, also consider an endoscopy.
5.2 Acute Tubulointerstitial Nephritis
Acute tubulointerstitial nephritis (TIN) has been observed in patients taking PPIs and may occur at any point during PPI therapy. Patients may present with varying signs and symptoms from symptomatic hypersensitivity reactions to non-specific symptoms of decreased renal function (e.g., malaise, nausea, anorexia). In reported case series, some patients were diagnosed on biopsy and in the absence of extra-renal manifestations (e.g., fever, rash or arthralgia). Discontinue pantoprazole sodium and evaluate patients with suspected acute TIN [see Contraindications ( 4) ].
5.3 Clostridium difficile-Associated Diarrhea
Published observational studies suggest that PPI therapy like pantoprazole sodium may be associated with an increased risk of Clostridium difficile associated diarrhea, especially in hospitalized patients. This diagnosis should be considered for diarrhea that does not improve [see Adverse Reactions ( 6.2)].
Patients should use the lowest dose and shortest duration of PPI therapy appropriate to the condition being treated.
5.4 Bone Fracture
Several published observational studies suggest that PPI therapy may be associated with an increased risk for osteoporosis-related fractures of the hip, wrist, or spine. The risk of fracture was increased in patients who received high-dose, defined as multiple daily doses, and long-term PPI therapy (a year or longer). Patients should use the lowest dose and shortest duration of PPI therapy appropriate to the condition being treated. Patients at risk for osteoporosis-related fractures should be managed according to established treatment guidelines [see Dosage and Administration ( 2), Adverse Reactions ( 6.2)].
5.5 Cutaneous and Systemic Lupus Erythematosus
Cutaneous lupus erythematosus (CLE) and systemic lupus erythematosus (SLE) have been reported in patients taking PPIs, including pantoprazole sodium. These events have occurred as both new onset and an exacerbation of existing autoimmune disease. The majority of PPI-induced lupus erythematous cases were CLE.
The most common form of CLE reported in patients treated with PPIs was subacute CLE (SCLE) and occurred within weeks to years after continuous drug therapy in patients ranging from infants to the elderly. Generally, histological findings were observed without organ involvement.
Systemic lupus erythematosus (SLE) is less commonly reported than CLE in patients receiving PPIs. PPI associated SLE is usually milder than non-drug induced SLE. Onset of SLE typically occurred within days to years after initiating treatment primarily in patients ranging from young adults to the elderly. The majority of patients presented with rash; however, arthralgia and cytopenia were also reported.
Avoid administration of PPIs for longer than medically indicated. If signs or symptoms consistent with CLE or SLE are noted in patients receiving pantoprazole sodium, discontinue the drug and refer the patient to the appropriate specialist for evaluation. Most patients improve with discontinuation of the PPI alone in 4 to 12 weeks. Serological testing (e.g. ANA) may be positive and elevated serological test results may take longer to resolve than clinical manifestations.
5.6 Cyanocobalamin (Vitamin B-12) Deficiency
Generally, daily treatment with any acid-suppressing medications over a long period of time (e.g., longer than 3 years) may lead to malabsorption of cyanocobalamin (Vitamin B-12) caused by hypo- or achlorhydria. Rare reports of cyanocobalamin deficiency occurring with acid-suppressing therapy have been reported in the literature. This diagnosis should be considered if clinical symptoms consistent with cyanocobalamin deficiency are observed.
5.7 Hypomagnesemia
Hypomagnesemia, symptomatic and asymptomatic, has been reported rarely in patients treated with PPIs for at least three months, and in most cases after a year of therapy. Serious adverse events include tetany, arrhythmias, and seizures. In most patients, treatment of hypomagnesemia required magnesium replacement and discontinuation of the PPI.
For patients expected to be on prolonged treatment or who take PPIs with medications such as digoxin or drugs that may cause hypomagnesemia (e.g., diuretics), health care professionals may consider monitoring magnesium levels prior to initiation of PPI treatment and periodically [see Adverse Reactions(6.2)].
5.8 Tumorigenicity
Due to the chronic nature of GERD, there may be a potential for prolonged administration of pantoprazole sodium. In long-term rodent studies, pantoprazole was carcinogenic and caused rare types of gastrointestinal tumors. The relevance of these findings to tumor development in humans is unknown [see Nonclinical Toxicology( 13.1)].
5.9 Fundic Gland Polyps
PPI use is associated with an increased risk of fundic gland polyps that increases with long-term use, especially beyond one year. Most PPI users who developed fundic gland polyps were asymptomatic and fundic gland polyps were identified incidentally on endoscopy. Use the shortest duration of PPI therapy appropriate to the condition being treated.
5.10 Interference with Investigations for Neuroendocrine Tumors
Serum chromogranin A (CgA) levels increase secondary to drug-induced decreases in gastric acidity. The increased CgA level may cause false positive results in diagnostic investigations for neuroendocrine tumors. Healthcare providers should temporarily stop pantoprazole sodium treatment at least 14 days before assessing CgA levels and consider repeating the test if initial CgA levels are high. If serial tests are performed (e.g. for monitoring), the same commercial laboratory should be used for testing, as reference ranges between tests may vary [see Clinical Pharmacology ( 12.2) ] .
5.11 Interference with Urine Screen for THC
There have been reports of false-positive urine screening tests for tetrahydrocannabinol (THC) in patients receiving PPIs, including pantoprazole sodium [see Drug Interactions ( 7)] .
5.12 Concomitant Use of Pantoprazole Sodium with Methotrexate
Literature suggests that concomitant use of PPIs with methotrexate (primarily at high dose; see methotrexate prescribing information) may elevate and prolong serum levels of methotrexate and/or its metabolite, possibly leading to methotrexate toxicities. In high-dose methotrexate administration, a temporary withdrawal of the PPI may be considered in some patients [see Drug Interactions(7)].
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described below and elsewhere in labeling:
Acute Tubulointerstitial Nephritis [see Warnings and Precautions(5.2)]
Clostridium difficile-Associated Diarrhea [see Warnings and Precautions(5.3)]
Bone Fracture [see Warnings and Precautions(5.4)]
Cutaneous and Systemic Lupus Erythematosus [see Warnings and Precautions(5.5)]
Cyanocobalamin (Vitamin B-12) Deficiency [see Warnings and Precautions(5.6)]
Hypomagnesemia [see Warnings and Precautions(5.7)]
Fundic Gland Polyps [see Warnings and Precautions ( 5.9)]
6.1 Clinical Trials Experience
The adverse reaction profiles for pantoprazole sodium delayed-release oral suspension and pantoprazole sodium delayed-release tablets are similar.
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Adults
Safety in nine randomized comparative US clinical trials in patients with GERD included 1,473 patients on oral pantoprazole sodium (20 mg or 40 mg), 299 patients on an H 2-receptor antagonist, 46 patients on another PPI, and 82 patients on placebo. The most frequently occurring adverse reactions are listed in Table 3.
Table 3: Adverse Reactions Reported in Clinical Trials of Adult Patients with GERD at a Frequency of > 2%
Pantoprazole Sodium
(n=1473)
%Comparators
(n=345)
%Placebo
(n=82)
%Headache 12.2 12.8 8.5 Diarrhea 8.8 9.6 4.9 Nausea 7 5.2 9.8 Abdominal pain 6.2 4.1 6.1 Vomiting 4.3 3.5 2.4 Flatulence 3.9 2.9 3.7 Dizziness 3 2.9 1.2 Arthralgia 2.8 1.4 1.2 Additional adverse reactions that were reported for pantoprazole sodium in clinical trials with a frequency of ≤ 2% are listed below by body system:
Body as a Whole: allergic reaction, pyrexia, photosensitivity reaction, facial edema
Gastrointestinal: constipation, dry mouth, hepatitis
Hematologic: leukopenia, thrombocytopenia
Metabolic/Nutritional: elevated CK (creatine kinase), generalized edema, elevated triglycerides, liver enzymes elevated
Musculoskeletal: myalgia
Nervous: depression, vertigo
Skin and Appendages: urticaria, rash, pruritus
Special Senses: blurred vision
Pediatric Patients
Safety of pantoprazole sodium in the treatment of EE associated with GERD was evaluated in pediatric patients ages 1 year through 16 years in three clinical trials. Safety trials involved pediatric patients with EE; however, as EE is uncommon in the pediatric population, 249 pediatric patients with endoscopically-proven or symptomatic GERD were also evaluated. All adult adverse reactions to pantoprazole sodium are considered relevant to pediatric patients. In patients ages 1 year through 16 years, the most commonly reported (> 4%) adverse reactions include: URI, headache, fever, diarrhea, vomiting, rash, and abdominal pain .
For safety information in patients less than 1 year of age see Use in Specific Populations ( 8.4).
Additional adverse reactions that were reported for pantoprazole sodium in pediatric patients in clinical trials with a frequency of ≤ 4% are listed below by body system:
Body as a Whole: allergic reaction, facial edema Gastrointestinal: constipation, flatulence, nausea
Metabolic/Nutritional: elevated triglycerides, elevated liver enzymes, elevated CK (creatine kinase)
Musculoskeletal: arthralgia, myalgia
Nervous: dizziness, vertigo
Skin and Appendages: urticaria
The following adverse reactions seen in adults in clinical trials were not reported in pediatric patients in clinical trials, but are considered relevant to pediatric patients: photosensitivity reaction, dry mouth, hepatitis, thrombocytopenia, generalized edema, depression, pruritus, leukopenia, and blurred vision.
Zollinger-Ellison (ZE) Syndrome
In clinical studies of ZE Syndrome, adverse reactions reported in 35 patients taking pantoprazole sodium 80 mg/day to 240 mg/day for up to 2 years were similar to those reported in adult patients with GERD.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of pantoprazole sodium. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
These adverse reactions are listed below by body system:
Gastrointestinal Disorders: fundic gland polyps
General Disorders and Administration Conditions: asthenia, fatigue, malaise
Hematologic: pancytopenia, agranulocytosis
Hepatobiliary Disorders: hepatocellular damage leading to jaundice and hepatic failure
Immune System Disorders: anaphylaxis (including anaphylactic shock), systemic lupus erythematosus
Infections and Infestations: Clostridium difficile associated diarrhea
Investigations: weight changes
Metabolism and Nutritional Disorders: hyponatremia, hypomagnesemia
Musculoskeletal Disorders: rhabdomyolysis, bone fracture
Nervous: ageusia, dysgeusia
Psychiatric Disorders: hallucination, confusion, insomnia, somnolence
Renal and Urinary Disorders: acute tubulointerstitial nephritis
Skin and Subcutaneous Tissue Disorders: severe dermatologic reactions (some fatal), including erythema multiforme, Stevens-Johnson syndrome, toxic epidermal necrolysis (TEN, some fatal), angioedema (Quincke's edema) and cutaneous lupus erythematosus
-
7 DRUG INTERACTIONS
Table 4 includes drugs with clinically important drug interactions and interaction with diagnostics when administered concomitantly with pantoprazole sodium and instructions for preventing or managing them.
Consult the labeling of concomitantly used drugs to obtain further information about interactions with PPIs.
Table 4: Clinically Relevant Interactions Affecting Drugs Co-Administered with Pantoprazole sodium and Interactions with Diagnostics
Antiretrovirals Clinical Impact: The effect of PPIs on antiretroviral drugs is variable. The clinical importance and the mechanisms behind these interactions are not always known. - Decreased exposure of some antiretroviral drugs (e.g., rilpivirine atazanavir, and nelfinavir) when used concomitantly with pantoprazole may reduce antiviral effect and promote the development of drug resistance.
- Increased exposure of other antiretroviral drugs (e.g., saquinavir) when used concomitantly with pantoprazole may increase toxicity of the antiretroviral drugs.
- There are other antiretroviral drugs which do not result in clinically relevant interactions with pantoprazole.
Intervention: Rilpivirine-containing products: Concomitant use with pantoprazole sodium is contraindicated [see Contraindications ( 4) ]. See prescribing information.
Atazanavir: See prescribing information for atazanavir for dosing information. Nelfinavir: Avoid concomitant use with pantoprazole sodium. See prescribing information for nelfinavir.
Saquinavir: See the prescribing information for saquinavir and monitor for potential saquinavir toxicities.
Other antiretrovirals: See prescribing information.Warfarin Clinical Impact: Increased INR and prothrombin time in patients receiving PPIs, including pantoprazole, and warfarin concomitantly. Increases in INR and prothrombin time may lead to abnormal bleeding and even death. Intervention: Monitor INR and prothrombin time. Dose adjustment of warfarin may be needed to maintain target INR range. See prescribing information for warfarin. Clopidogrel Clinical Impact: Concomitant administration of pantoprazole and clopidogrel in healthy subjects had no clinically important effect on exposure to the active metabolite of clopidogrel or clopidogrel-induced platelet inhibition [see Clinical Pharmacology ( 12.3) ]. Intervention: No dose adjustment of clopidogrel is necessary when administered with an approved dose of pantoprazole sodium. Methotrexate Clinical Impact: Concomitant use of PPIs with methotrexate (primarily at high dose) may elevate and prolong serum concentrations of methotrexate and/or its metabolite hydroxymethotrexate, possibly leading to methotrexate toxicities. No formal drug interaction studies of high-dose methotrexate with PPIs have been conducted [see Warnings and Precautions ( 5.12) ]. Intervention: A temporary withdrawal of pantoprazole sodium may be considered in some patients receiving high-dose methotrexate. Drugs Dependent on Gastric pH for Absorption (e.g., iron salts, erlotinib, dasatinib,nilotinib, mycophenolate mofetil, ketoconazole/ itraconazole) Clinical Impact: Pantoprazole can reduce the absorption of other drugs due to its effect on reducing intragastric acidity. Intervention: Mycophenolate mofetil (MMF): Co-administration of pantoprazole sodium in healthy subjects and in transplant patients receiving MMF has been reported to reduce the exposure to the active metabolite, mycophenolic acid (MPA), possibly due to a decrease in MMF solubility at an increased gastric pH [see Clinical Pharmacology ( 12.3) ]. The clinical relevance of reduced MPA exposure on organ rejection has not been established in transplant patients receiving pantoprazole sodium and MMF. Use pantoprazole sodium with caution in transplant patients receiving MMF. See the prescribing information for other drugs dependent on gastric pH for absorption. Interactions with Investigations of Neuroendocrine Tumors Clinical Impact: CgA levels increase secondary to PPI-induced decreases in gastric acidity. The increased CgA level may cause false positive results in diagnostic investigations for neuroendocrine tumors [see Warnings and Precautions ( 5.10) , Clinical Pharmacology ( 12.2) ]. Intervention: Temporarily stop pantoprazole sodium treatment at least 14 days before assessing CgA levels and consider repeating the test if initial CgA levels are high. If serial tests are performed (e.g. for monitoring), the same commercial laboratory should be used for testing, as reference ranges between tests may vary. False Positive Urine Tests for THC Clinical Impact: There have been reports of false positive urine screening tests for tetrahydrocannabinol (THC) in patients receiving PPIs [see Warnings and Precautions ( 5.11) ] . Intervention: An alternative confirmatory method should be considered to verify positive results. -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from published observational studies did not demonstrate an association of major malformations or other adverse pregnancy outcomes with pantoprazole.
In animal reproduction studies, no evidence of adverse development outcomes was observed with pantoprazole. Reproduction studies have been performed in rats at oral doses up to 450 mg/kg/day (about 88 times the recommended human dose) and rabbits at oral doses up to 40 mg/kg/day (about 16 times the recommended human dose) with administration of pantoprazole during organogenesis in pregnant animals and have revealed no evidence of harm to the fetus due to pantoprazole in this study (see Data).
A pre-and postnatal development toxicity study in rats with additional endpoints to evaluate the effect on bone development was performed with pantoprazole sodium. Oral pantoprazole doses of 5, 15, and 30 mg/kg/day (approximately 1, 3, and 6 times the human dose of 40 mg/day) were administered to pregnant females from gestation day (GD) 6 through lactation day (LD) 21. Changes in bone morphology were observed in pups exposed to pantoprazole in utero and through milk during the period of lactation as well as by oral dosing from postnatal day (PND) 4 through PND 21 [see Use in Specific Populations ( 8.4)]. There were no drug-related findings in maternal animals. Advise pregnant women of the potential risk of fetal harm.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in the clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Human Data
Available data from published observational studies failed to demonstrate an association of adverse pregnancy-related outcomes and pantoprazole use. Methodological limitations of these observational studies cannot definitely establish or exclude any drug-associated risk during pregnancy. In a prospective study by the European Network of Teratology Information Services, outcomes from a group of 53 pregnant women administered median daily doses of 40 mg pantoprazole were compared to a control group of 868 pregnant women who did not take any proton pump inhibitors (PPIs). There was no difference in the rate of major malformations between women exposed to PPIs and the control group, corresponding to a Relative Risk (RR) = 0.55, [95% Confidence Interval (CI) 0.08-3.95]. In a population-based retrospective cohort study covering all live births in Denmark from 1996 to 2008, there was no significant increase in major birth defects during analysis of first trimester exposure to pantoprazole in 549 live births. A meta-analysis that compared 1,530 pregnant women exposed to PPIs in at least the first trimester with 133,410 unexposed pregnant women showed no significant increases in risk for congenital malformations or spontaneous abortion with exposure to PPIs (for major malformations OR=1.12 ([95% CI 0.86-1.45] and for spontaneous abortions OR=1.29 [95% CI 0.84-1.97]).
Animal Data
Reproduction studies have been performed in rats at oral pantoprazole doses up to 450 mg/kg/day (about 88 times the recommended human dose based on body surface area) and in rabbits at oral doses up to 40 mg/kg/day (about 16 times the recommended human dose based on body surface area) with administration of pantoprazole sodium during organogenesis in pregnant animals. The studies have revealed no evidence of impaired fertility or harm to the fetus due to pantoprazole.
A pre- and postnatal development toxicity study in rats with additional endpoints to evaluate the effect on bone development was performed with pantoprazole sodium. Oral pantoprazole doses of 5, 15, and 30 mg/kg/day (approximately 1, 3, and 6 times the human dose of 40 mg/day on a body surface area basis) were administered to pregnant females from gestation day (GD) 6 through lactation day (LD) 21. On postnatal day (PND 4) through PND 21, the pups were administered oral doses at 5, 15, and 30 mg/kg/day (approximately 1, 2.3, and 3.2 times the exposure (AUC) in humans at a dose of 40 mg). There were no drug-related findings in maternal animals. During the preweaning dosing phase (PND 4 to 21) of the pups, there were increased mortality and/or moribundity and decreased body weight and body weight gain at 5 mg/kg/day (approximately equal exposures (AUC) in humans receiving the 40 mg dose) and higher doses. On PND 21, decreased mean femur length and weight and changes in femur bone mass and geometry were observed in the offspring at 5 mg/kg/day (approximately equal exposures (AUC) in humans at the 40 mg dose) and higher doses. The femur findings included lower total area, bone mineral content and density, periosteal and endosteal circumference, and cross-sectional moment of inertia. There were no microscopic changes in the distal femur, proximal tibia, or stifle joints. Changes in bone parameters were partially reversible following a recovery period, with findings on PND 70 limited to lower femur metaphysis cortical/subcortical bone mineral density in female pups at 5 mg/kg/day (approximately equal exposures (AUC) in humans at the 40 mg dose) and higher doses.
8.2 Lactation
Risk Summary
Pantoprazole has been detected in breast milk of a nursing mother after a single 40 mg oral dose of pantoprazole. There were no effects on the breastfed infant (see Data). There are no data on pantoprazole effects on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for pantoprazole and any potential adverse effects on the breastfed child from pantoprazole or from the underlying maternal condition.
Data
The breast milk of a 42-year-old woman receiving 40 mg of oral pantoprazole, at 10 months postpartum, was studied for 24 hours, to demonstrate low levels of pantoprazole present in the breast milk. Pantoprazole was detectable in milk only 2 and 4 hours after the dose with milk levels of approximately 36 mcg/L and 24 mcg/L, respectively. A milk-to-plasma ratio of 0.022 was observed at 2 hours after drug administration. Pantoprazole was not detectable (<10 mcg/L) in milk at 6, 8 and 24 hours after the dose. The relative dose to the infant was estimated to be 7.3 mcg of pantoprazole, which is equivalent to 0.14% of the weight-adjusted maternal dose. No adverse events in the infant were reported by the mother.
8.4 Pediatric Use
The safety and effectiveness of pantoprazole sodium for short-term treatment (up to eight weeks) of EE associated with GERD have been established in pediatric patients 1 year through 16 years of age. Effectiveness for EE has not been demonstrated in patients less than 1 year of age. In addition, for patients less than 5 years of age, there is no appropriate dosage strength in an age-appropriate formulation available. Therefore, pantoprazole sodium is indicated for the short-term treatment of EE associated with GERD for patients 5 years and older. The safety and effectiveness of pantoprazole sodium for pediatric uses other than EE have not been established.
1 year through 16 years of age
Use of pantoprazole sodium in pediatric patients 1 year through 16 years of age for short-term treatment (up to eight weeks) of EE associated with GERD is supported by: a) extrapolation of results from adequate and well-controlled studies that supported the approval of pantoprazole sodium for treatment of EE associated with GERD in adults, and b) safety, effectiveness, and pharmacokinetic studies performed in pediatric patients [see Clinical Studies ( 14.1) , Clinical Pharmacology ( 12.3) ].
Safety of pantoprazole sodium in the treatment of EE associated with GERD in pediatric patients 1 through 16 years of age was evaluated in three multicenter, randomized, double-blind, parallel-treatment studies, involving 249 pediatric patients, including 8 with EE (4 patients ages 1 year to 5 years and 4 patients 5 years to 11 years). The children ages 1 year to 5 years with endoscopically diagnosed EE (defined as an endoscopic Hetzel-Dent score ≥ 2) were treated once daily for 8 weeks with one of two dose levels of pantoprazole sodium (approximating 0.6 mg/kg or 1.2 mg/kg). All 4 of these patients with EE were healed (Hetzel-Dent score of 0 or 1) at 8 weeks. Because EE is uncommon in the pediatric population, predominantly pediatric patients with endoscopically-proven or symptomatic GERD were also included in these studies. Patients were treated with a range of doses of pantoprazole sodium once daily for 8 weeks. For safety findings see Adverse Reactions ( 6.1). Because these pediatric trials had no placebo, active comparator, or evidence of a dose response, the trials were inconclusive regarding the clinical benefit of pantoprazole sodium for symptomatic GERD in the pediatric population. The effectiveness of pantoprazole sodium for treating symptomatic GERD in pediatric patients has not been established.
Although the data from the clinical trials support use of pantoprazole sodium for the short-term treatment of EE associated with GERD in pediatric patients 1 year through 5 years, there is no commercially available dosage formulation appropriate for patients less than 5 years of age [see Dosage and Administration ( 2) ].
In a population pharmacokinetic analysis, clearance values in the children 1 to 5 years old with endoscopically proven GERD had a median value of 2.4 L/h. Following a 1.2 mg/kg equivalent dose (15 mg for ≤ 12.5 kg and 20 mg for > 12.5 to < 25 kg), the plasma concentrations of pantoprazole were highly variable and the median time to peak plasma concentration was 3 to 6 hours. The estimated AUC for patients 1 to 5 years old was 37% higher than for adults receiving a single 40 mg tablet, with a geometric mean AUC value of 6.8 mcghr/mL.
Neonates to less than one year of age
Pantoprazole sodium was not found to be effective in a multicenter, randomized, double-blind, placebo-controlled, treatment-withdrawal study of 129 pediatric patients 1 through 11 months of age. Patients were enrolled if they had symptomatic GERD based on medical history and had not responded to non-pharmacologic interventions for GERD for two weeks. Patients received pantoprazole sodium daily for four weeks in an open-label phase, then patients were randomized in equal proportion to receive pantoprazole sodium treatment or placebo for the subsequent four weeks in a double-blind manner. Efficacy was assessed by observing the time from randomization to study discontinuation due to symptom worsening during the four-week treatment-withdrawal phase. There was no statistically significant difference between pantoprazole sodium and placebo in the rate of discontinuation.
In this trial, the adverse reactions that were reported more commonly (difference of ≥ 4%) in the treated population compared to the placebo population were elevated CK, otitis media, rhinitis, and laryngitis.
In a population pharmacokinetic analysis, the systemic exposure was higher in patients less than 1 year of age with GERD compared to adults who received a single 40 mg dose (geometric mean AUC was 103% higher in preterm infants and neonates receiving single dose of 2.5 mg of pantoprazole sodium, and 23% higher in infants 1 through 11 months of age receiving a single dose of approximately 1.2 mg/kg). In these patients, the apparent clearance (CL/F) increased with age (median clearance: 0.6 L/hr, range: 0.03 to 3.2 L/hr).
These doses resulted in pharmacodynamic effects on gastric but not esophageal pH. Following once daily dosing of 2.5 mg of pantoprazole sodium in preterm infants and neonates, there was an increase in the mean gastric pH (from 4.3 at baseline to 5.2 at steady-state) and in the mean % time that gastric pH was > 4 (from 60% at baseline to 80% at steady-state). Following once daily dosing of approximately 1.2 mg/kg of pantoprazole sodium in infants 1 through 11 months of age, there was an increase in the mean gastric pH (from 3.1 at baseline to 4.2 at steady-state) and in the mean % time that gastric pH was > 4 (from 32% at baseline to 60% at steady-state). However, no significant changes were observed in mean intraesophageal pH or % time that esophageal pH was < 4 in either age group.
Because pantoprazole sodium was not shown to be effective in the randomized, placebo-controlled study in this age group, the use of pantoprazole sodium for treatment of symptomatic GERD in infants less than 1 year of age is not indicated.
Animal Toxicity Data
In a pre- and post-natal development study in rats, the pups were administered oral doses of pantoprazole at 5, 15, and 30 mg/kg/day (approximately 1, 2.3, and 3.2 times the exposure (AUC) in children aged 6 to 11 years at a dose of 40 mg) on postnatal day (PND 4) through PND 21, in addition to lactational exposure through milk. On PND 21, decreased mean femur length and weight and changes in femur bone mass and geometry were observed in the offspring at 5 mg/kg/day (approximately equal exposures (AUC) in children aged 6 to 11 years at the 40 mg dose) and higher doses. Changes in bone parameters were partially reversible following a recovery period.
In neonatal/juvenile animals (rats and dogs) toxicities were similar to those observed in adult animals, including gastric alterations, decreases in red cell mass, increases in lipids, enzyme induction and hepatocellular hypertrophy. An increased incidence of eosinophilic chief cells in adult and neonatal/juvenile rats, and atrophy of chief cells in adult rats and in neonatal/juvenile dogs, was observed in the fundic mucosa of stomachs in repeated-dose studies. Full to partial recovery of these effects were noted in animals of both age groups following a recovery period.
8.5 Geriatric Use
In short-term U.S. clinical trials, EE healing rates in the 107 elderly patients (≥ 65 years old) treated with pantoprazole sodium were similar to those found in patients under the age of 65. The incidence rates of adverse reactions and laboratory abnormalities in patients aged 65 years and older were similar to those associated with patients younger than 65 years of age.
-
10 OVERDOSAGE
Experience in patients taking very high doses of pantoprazole sodium (greater than 240 mg) is limited. Spontaneous post-marketing reports of overdose are generally within the known safety profile of pantoprazole sodium.
Pantoprazole is not removed by hemodialysis. In case of overdosage, treatment should be symptomatic and supportive.
Single oral doses of pantoprazole at 709 mg/kg, 798 mg/kg, and 887 mg/kg were lethal to mice, rats, and dogs, respectively. The symptoms of acute toxicity were hypoactivity, ataxia, hunched sitting, limb-splay, lateral position, segregation, absence of ear reflex, and tremor.
If overexposure to pantoprazole sodium occurs, call your Poison Control Center at 1-800-222-1222 for current information on the management of poisoning or overdosage.
-
11 DESCRIPTION
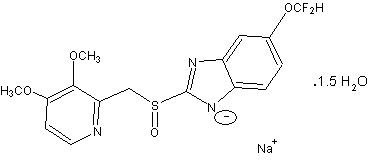
The active ingredient in pantoprazole sodium delayed-release tablets USP, a PPI, is a substituted benzimidazole, sodium 5-(difluoromethoxy)-2-[[(3,4-dimethoxy-2-pyridinyl)methyl] sulfinyl]-1H- benzimidazole sesquihydrate, a compound that inhibits gastric acid secretion. Its molecular formula is C 16H 14F 2N 3NaO 4S x 1.5 H 2O, with a molecular weight of 432.4. The structural formula is:
Pantoprazole sodium sesquihydrate is a white to cream color powder and is racemic. Pantoprazole has weakly basic and acidic properties. Pantoprazole sodium sesquihydrate is freely soluble in water and ethanol.
The stability of the compound in aqueous solution is pH-dependent. The rate of degradation increases with decreasing pH. At ambient temperature, the degradation half-life is approximately 2.8 hours at pH 5 and approximately 220 hours at pH 7.8.
Pantoprazole sodium USP is supplied as a delayed-release tablet, available in two strengths 20 mg pantoprazole (equivalent to 22.6 mg of pantoprazole sodium sesquihydrate) and 40 mg pantoprazole (equivalent to 45.1 mg of pantoprazole sodium sesquihydrate).
Pantoprazole sodium delayed-release tablets USP contains the following inactive ingredients: ammonium hydroxide, calcium stearate, crospovidone, hydroxypropyl cellulose, hypromellose, iron oxide black, isopropyl alcohol, mannitol, methacrylic acid copolymer, methylated spirit, N-butyl alcohol, polyethylene glycol, propylene glycol, shellac, sodium carbonate anhydrous, synthetic yellow iron oxide, talc, titanium dioxide, triethyl citrate, zein.
Pantoprazole sodium delayed-release tablets meets USP dissolution test 2.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Pantoprazole is a PPI that suppresses the final step in gastric acid production by covalently binding to the (H +, K +)-ATPase enzyme system at the secretory surface of the gastric parietal cell. This effect leads to inhibition of both basal and stimulated gastric acid secretion, irrespective of the stimulus. The binding to the (H +, K +)-ATPase results in a duration of antisecretory effect that persists longer than 24 hours for all doses tested (20 mg to 120 mg).
12.2 Pharmacodynamics
Antisecretory Activity
Under maximal acid stimulatory conditions using pentagastrin, a dose-dependent decrease in gastric acid output occurs after a single dose of oral (20 to 80 mg) or a single dose of intravenous (20 to 120 mg) pantoprazole in healthy subjects. Pantoprazole given once daily results in increasing inhibition of gastric acid secretion. Following the initial oral dose of 40 mg pantoprazole, a 51% mean inhibition was achieved by 2.5 hours. With once-a-day dosing for 7 days, the mean inhibition was increased to 85%. Pantoprazole suppressed acid secretion in excess of 95% in half of the subjects. Acid secretion had returned to normal within a week after the last dose of pantoprazole; there was no evidence of rebound hypersecretion.
In a series of dose-response studies, pantoprazole, at oral doses ranging from 20 to 120 mg, caused dose-related increases in median basal gastric pH and in the percent of time gastric pH was > 3 and > 4. Treatment with 40 mg of pantoprazole produced significantly greater increases in gastric pH than the 20 mg dose. Doses higher than 40 mg (60, 80, 120 mg) did not result in further significant increases in median gastric pH. The effects of pantoprazole on median pH from one double-blind crossover study are shown in Table 5.
Table 5: Effect of Single Daily Doses of Oral Pantoprazole on Intragastric pH
–––––––––––—––––––––Median pH on day 7–––––––––––––––––– Time Placebo 20 mg 40 mg 80 mg 8 a.m. - 8 a.m.(24 hours) 1.3 2.9* 3.8*# 3.9*# 8 a.m. - 10 p.m.(Daytime) 1.6 3.2* 4.4*# 4.8*# 10 p.m. - 8 a.m.(Nighttime) 1.2 2.1* 3* 2.6* * Significantly different from placebo
# Significantly different from 20 mg
Serum Gastrin Effects
Fasting serum gastrin levels were assessed in two double-blind studies of the acute healing of EE in which 682 patients with gastroesophageal reflux disease (GERD) received 10, 20, or 40 mg of pantoprazole sodium for up to 8 weeks. At 4 weeks of treatment there was an increase in mean gastrin levels of 7%, 35%, and 72% over pretreatment values in the 10, 20, and 40 mg treatment groups, respectively. A similar increase in serum gastrin levels was noted at the 8-week visit with mean increases of 3%, 26%, and 84% for the three pantoprazole dose groups. Median serum gastrin levels remained within normal limits during maintenance therapy with pantoprazole sodium delayed-release tablets.
In long-term international studies involving over 800 patients, a 2 to 3 fold mean increase from the pretreatment fasting serum gastrin level was observed in the initial months of treatment with pantoprazole at doses of 40 mg per day during GERD maintenance studies and 40 mg or higher per day in patients with refractory GERD. Fasting serum gastrin levels generally remained at approximately 2 to 3 times baseline for up to 4 years of periodic follow-up in clinical trials.
Following short-term treatment with pantoprazole sodium, elevated gastrin levels return to normal by at least 3 months.
Enterochromaffin-Like (ECL) Cell Effects
In 39 patients treated with oral pantoprazole 40 mg to 240 mg daily (majority receiving 40 mg to 80 mg) for up to 5 years, there was a moderate increase in ECL-cell density, starting after the first year of use, which appeared to plateau after 4 years.
In a nonclinical study in Sprague-Dawley rats, lifetime exposure (24 months) to pantoprazole at doses of 0.5 to 200 mg/kg/day resulted in dose-related increases in gastric ECL-cell proliferation and gastric neuroendocrine (NE)-cell tumors. Gastric NE-cell tumors in rats may result from chronic elevation of serum gastrin concentrations. The high density of ECL cells in the rat stomach makes this species highly susceptible to the proliferative effects of elevated gastrin concentrations produced by PPIs. However, there were no observed elevations in serum gastrin following the administration of pantoprazole at a dose of 0.5 mg/kg/day. In a separate study, a gastric NE-cell tumor without concomitant ECL-cell proliferative changes was observed in 1 female rat following 12 months of dosing with pantoprazole at 5 mg/kg/day and a 9 month off-dose recovery [see Nonclinical Toxicology ( 13.1)].
Endocrine Effects
In a clinical pharmacology study, pantoprazole sodium delayed-release tablets 40 mg given once daily for 2 weeks had no effect on the levels of the following hormones: cortisol, testosterone, triiodothyronine (T 3), thyroxine (T 4), thyroid-stimulating hormone (TSH), thyronine-binding protein, parathyroid hormone, insulin, glucagon, renin, aldosterone, follicle-stimulating hormone, luteinizing hormone, prolactin, and growth hormone.
In a 1-year study of GERD patients treated with pantoprazole sodium delayed-release tablets 40 mg or 20 mg, there were no changes from baseline in overall levels of T 3, T 4, and TSH.
12.3 Pharmacokinetics
Pantoprazole sodium delayed-release tablets are prepared as enteric-coated tablets so that absorption of pantoprazole begins only after the tablet leaves the stomach. Peak serum concentration (C max) and area under the serum concentration time curve (AUC) increase in a manner proportional to oral and intravenous doses from 10 mg to 80 mg. Pantoprazole does not accumulate, and its pharmacokinetics are unaltered with multiple daily dosing. Following oral or intravenous administration, the serum concentration of pantoprazole declines biexponentially, with a terminal elimination half-life of approximately one hour.
In extensive metabolizers with normal liver function receiving an oral dose of the enteric-coated 40 mg pantoprazole tablet, the peak concentration (C max) is 2.5 mcg/mL; the time to reach the peak concentration (t max) is 2.5 h, and the mean total area under the plasma concentration versus time curve (AUC) is 4.8 mcgh/mL (range 1.4 to 13.3 mcgh/mL). Following intravenous administration of pantoprazole to extensive metabolizers, its total clearance is 7.6 to 14 L/h, and its apparent volume of distribution is 11 to 23.6 L.
Absorption
After administration of a single or multiple oral 40 mg doses of pantoprazole sodium delayed-release tablets, the peak plasma concentration of pantoprazole was achieved in approximately 2.5 hours, and Cmax was 2.5 mcg/mL. Pantoprazole undergoes little first-pass metabolism, resulting in an absolute bioavailability of approximately 77%. Pantoprazole absorption is not affected by concomitant administration of antacids.
Administration of pantoprazole sodium delayed-release tablets with food may delay its absorption up to 2 hours or longer; however, the C max and the extent of pantoprazole absorption (AUC) are not altered. Thus, pantoprazole sodium delayed-release tablets may be taken without regard to timing of meals.
Distribution
The apparent volume of distribution of pantoprazole is approximately 11 to 23.6 L, distributing mainly in extracellular fluid. The serum protein binding of pantoprazole is about 98%, primarily to albumin.
Elimination
Metabolism
Pantoprazole is extensively metabolized in the liver through the cytochrome P450 (CYP) system. Pantoprazole metabolism is independent of the route of administration (intravenous or oral). The main metabolic pathway is demethylation, by CYP2C19, with subsequent sulfation; other metabolic pathways include oxidation by CYP3A4. There is no evidence that any of the pantoprazole metabolites have significant pharmacologic activity.
Excretion
After a single oral or intravenous dose of 14C-labeled pantoprazole to healthy, normal metabolizer subjects, approximately 71% of the dose was excreted in the urine, with 18% excreted in the feces through biliary excretion. There was no renal excretion of unchanged pantoprazole.
Specific Populations
Geriatric Patients
Only slight to moderate increases in the AUC (43%) and Cmax (26%) of pantoprazole were found in elderly subjects (64 to 76 years of age) after repeated oral administration, compared with younger subjects [see Use in Specific Populations ( 8.5) ]
Pediatric Patients
The pharmacokinetics of pantoprazole were studied in children less than 16 years of age in four randomized, open-label clinical trials in pediatric patients with presumed/proven GERD. A pediatric granule formulation was studied in children through 5 years of age, and pantoprazole sodium delayed-release tablets were studied in children older than 5 years.
In a population PK analysis, total clearance increased with increasing bodyweight in a non-linear fashion. The total clearance also increased with increasing age only in children under 3 years of age.
Neonate through 5 Years of Age [See Use in Specific Populations (8.4)].
Children and Adolescents 6 through 16 Years of Age
The pharmacokinetics of pantoprazole sodium delayed-release tablets were evaluated in children ages 6 through 16 years with a clinical diagnosis of GERD. The PK parameters following a single oral dose of 20 mg or 40 mg of pantoprazole sodium delayed-release tablets in children ages 6 through 16 years were highly variable (%CV ranges 40 to 80%). The geometric mean AUC estimated from population PK analysis after a 40 mg pantoprazole sodium delayed-release tablet in pediatric patients was about 39% and 10% higher respectively in 6 to 11 and 12 to 16 year-old children, compared to that of adults (Table 7).
Table 7: PK Parameters in Children and Adolescents 6 through 16 years with GERD receiving 40 mg Pantoprazole Sodium Delayed-Release Tablets
6 to 11 years (n=12) 12 to 16 years (n=11) C max (mcg/mL) a 1.8 1.8 t max (h) b 2 2 AUC (mcgh/mL) a 6.9 5.5 CL/F (L/h) b 6.6 6.8 a Geometric mean values
b Median values
Male and Female Patients
There is a modest increase in pantoprazole AUC and C max in women compared to men. However, weight-normalized clearance values are similar in women and men.
In pediatric patients ages 1 through 16 years there were no clinically relevant effects of gender on clearance of pantoprazole, as shown by population pharmacokinetic analysis.
Patients with Renal Impairment
In patients with severe renal impairment, pharmacokinetic parameters for pantoprazole were similar to those of healthy subjects.
Patients with Hepatic Impairment
In patients with mild to severe hepatic impairment (Child-Pugh A to C cirrhosis), maximum pantoprazole concentrations increased only slightly (1.5-fold) relative to healthy subjects. Although serum half-life values increased to 7 to 9 hours and AUC values increased by 5 to 7 fold in hepatic-impaired patients, these increases were no greater than those observed in CYP2C19 poor metabolizers, where no dosage adjustment is warranted. These pharmacokinetic changes in hepatic-impaired patients result in minimal drug accumulation following once-daily, multiple-dose administration. Doses higher than 40 mg/day have not been studied in hepatically impaired patients.
Drug Interaction Studies
Effect of Other Drugs on Pantoprazole
Pantoprazole is metabolized mainly by CYP2C19 and to minor extents by CYPs 3A4, 2D6, and 2C9. In in vivo drug-drug interaction studies with CYP2C19 substrates (diazepam [also a CYP3A4 substrate] and phenytoin [also a CYP3A4 inducer] and clopidogrel), nifedipine, midazolam, and clarithromycin (CYP3A4 substrates), metoprolol (a CYP2D6 substrate), diclofenac, naproxen and piroxicam (CYP2C9 substrates), and theophylline (a CYP1A2 substrate) in healthy subjects, the pharmacokinetics of pantoprazole were not significantly altered.
Effect of Pantoprazole on Other Drugs
Clopidogrel
Clopidogrel is metabolized to its active metabolite in part by CYP2C19. In a crossover clinical study, 66 healthy subjects were administered clopidogrel (300 mg loading dose followed by 75 mg per day) alone and with pantoprazole (80 mg at the same time as clopidogrel) for 5 days. On Day 5, the mean AUC of the active metabolite of clopidogrel was reduced by approximately 14% (geometric mean ratio was 86%, with 90% CI of 79 to 93%) when pantoprazole was coadministered with clopidogrel as compared to clopidogrel administered alone. Pharmacodynamic parameters were also measured and demonstrated that the change in inhibition of platelet aggregation (induced by 5 μM ADP) was correlated with the change in the exposure to clopidogrel active metabolite. The clinical significance of this finding is not clear.
Mycophenolate Mofetil (MMF)
Administration of pantoprazole 40 mg twice daily for 4 days and a single 1,000 mg dose of MMF approximately one hour after the last dose of pantoprazole to 12 healthy subjects in a cross-over study resulted in a 57% reduction in the Cmax and 27% reduction in the AUC of MPA.
Transplant patients receiving approximately 2,000 mg per day of MMF (n=12) were compared to transplant patients receiving approximately the same dose of MMF and pantoprazole 40 mg per day (n=21). There was a 78% reduction in the C max and a 45% reduction in the AUC of MPA in patients receiving both pantoprazole and MMF [see Drug Interactions ( 7)] .
Other Drugs
In vivo studies also suggest that pantoprazole does not significantly affect the kinetics of the following drugs (cisapride, theophylline, diazepam [and its active metabolite, desmethyldiazepam], phenytoin, metoprolol, nifedipine, carbamazepine, midazolam, clarithromycin, diclofenac, naproxen, piroxicam, and oral contraceptives [levonorgestrel/ethinyl estradiol]). In other in vivo studies, digoxin, ethanol, glyburide, antipyrine, caffeine, metronidazole, and amoxicillin had no clinically relevant interactions with pantoprazole.
Although no significant drug-drug interactions have been observed in clinical studies, the potential for significant drug-drug interactions with more than once-daily dosing with high doses of pantoprazole has not been studied in poor metabolizers or individuals who are hepatically impaired.
Antacids
There was also no interaction with concomitantly administered antacids.
12.5 Pharmacogenomics
CYP2C19 displays a known genetic polymorphism due to its deficiency in some subpopulations (e.g., approximately 3% of Caucasians and African-Americans and 17% to 23% of Asians are poor metabolizers). Although these subpopulations of pantoprazole poor metabolizers have elimination half-life values of 3.5 to 10 hours in adults, they still have minimal accumulation (23% or less) with once-daily dosing. For adult patients who are CYP2C19 poor metabolizers, no dosage adjustment is needed.
Similar to adults, pediatric patients who have the poor metabolizer genotype of CYP2C19 (CYP2C19 *2/*2) exhibited greater than a 6-fold increase in AUC compared to pediatric extensive (CYP2C19 *1/*1) and intermediate (CYP2C19 *1/*x) metabolizers. Poor metabolizers exhibited approximately 10-fold lower apparent oral clearance compared to extensive metabolizers.
For known pediatric poor metabolizers, a dose reduction should be considered.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 24-month carcinogenicity study, Sprague-Dawley rats were treated orally with pantoprazole doses of 0.5 to 200 mg/kg/day, about 0.1 to 40 times the exposure on a body surface area basis of a 50 kg person dosed with 40 mg/day. In the gastric fundus, treatment with 0.5 to 200 mg/kg/day produced enterochromaffin-like (ECL) cell hyperplasia and benign and malignant neuroendocrine cell tumors in a dose-related manner. In the forestomach, treatment with 50 and 200 mg/kg/day (about 10 and 40 times the recommended human dose on a body surface area basis) produced benign squamous cell papillomas and malignant squamous cell carcinomas. Rare gastrointestinal tumors associated with pantoprazole treatment included an adenocarcinoma of the duodenum with 50 mg/kg/day and benign polyps and adenocarcinomas of the gastric fundus with 200 mg/kg/day. In the liver, treatment with 0.5 to 200 mg/kg/day produced dose-related increases in the incidences of hepatocellular adenomas and carcinomas. In the thyroid gland, treatment with 200 mg/kg/day produced increased incidences of follicular cell adenomas and carcinomas for both male and female rats.
In a 24-month carcinogenicity study, Fischer 344 rats were treated orally with doses of 5 to 50 mg/kg/day of pantoprazole, approximately 1 to 10 times the recommended human dose based on body surface area. In the gastric fundus, treatment with 5 to 50 mg/kg/day produced enterochromaffin-like (ECL) cell hyperplasia and benign and malignant neuroendocrine cell tumors. Dose selection for this study may not have been adequate to comprehensively evaluate the carcinogenic potential of pantoprazole.
In a 24-month carcinogenicity study, B6C3F1 mice were treated orally with doses of 5 to 150 mg/kg/day of pantoprazole, 0.5 to 15 times the recommended human dose based on body surface area. In the liver, treatment with 150 mg/kg/day produced increased incidences of hepatocellular adenomas and carcinomas in female mice. Treatment with 5 to 150 mg/kg/day also produced gastric-fundic ECL cell hyperplasia.
A 26-week p53 +/- transgenic mouse carcinogenicity study was not positive.
Pantoprazole was positive in the in vitro human lymphocyte chromosomal aberration assays, in one of two mouse micronucleus tests for clastogenic effects, and in the in vitro Chinese hamster ovarian cell/HGPRT forward mutation assay for mutagenic effects. Equivocal results were observed in the in vivo rat liver DNA covalent binding assay. Pantoprazole was negative in the in vitro Ames mutation assay, the in vitro unscheduled DNA synthesis (UDS) assay with rat hepatocytes, the in vitro AS52/GPT mammalian cell-forward gene mutation assay, the in vitro thymidine kinase mutation test with mouse lymphoma L5178Y cells, and the in vivo rat bone marrow cell chromosomal aberration assay.
There were no effects on fertility or reproductive performance when pantoprazole was given at oral doses up to 500 mg/kg/day in male rats (98 times the recommended human dose based on body surface area) and 450 mg/kg/day in female rats (88 times the recommended human dose based on body surface area).
-
14 CLINICAL STUDIES
Pantoprazole sodium delayed-release tablets were used in the following clinical trials.
14.1 Erosive Esophagitis (EE) Associated with Gastroesophageal Reflux Disease (GERD)
Adult Patients
A US multicenter, double-blind, placebo-controlled study of pantoprazole sodium 10 mg, 20 mg, or 40 mg once daily was conducted in 603 patients with reflux symptoms and endoscopically diagnosed EE of grade 2 or above (Hetzel-Dent scale). In this study, approximately 25% of enrolled patients had severe EE of grade 3, and 10% had grade 4. The percentages of patients healed (per protocol, n = 541) in this study are shown in Table 8.
Table 8: Erosive Esophagitis Healing Rates (Per Protocol)
–––––––––––––––Pantoprazole Sodium––––––––––––––– Placebo 10 mg daily 20 mg daily 40 mg daily Week (n = 153) (n = 158) (n = 162) (n = 68) 4 45.6% + 58.4% +# 75% +* 14.3% 8 66% + 83.5 % +# 92.6% +* 39.7% + (p < 0.001) pantoprazole sodium versus placebo
* (p < 0.05) versus 10 mg or 20 mg pantoprazole sodium
# (p < 0.05) versus 10 mg pantoprazole sodium
In this study, all pantoprazole sodium treatment groups had significantly greater healing rates than the placebo group. This was true regardless of H. pylori status for the 40 mg and 20 mg pantoprazole sodium treatment groups. The 40 mg dose of pantoprazole sodium resulted in healing rates significantly greater than those found with either the 20 mg or 10 mg dose.
A significantly greater proportion of patients taking pantoprazole sodium 40 mg experienced complete relief of daytime and nighttime heartburn and the absence of regurgitation, starting from the first day of treatment, compared with placebo. Patients taking pantoprazole sodium consumed significantly fewer antacid tablets per day than those taking placebo.
Pantoprazole sodium 40 mg and 20 mg once daily were also compared with nizatidine 150 mg twice daily in a US multicenter, double-blind study of 243 patients with reflux symptoms and endoscopically diagnosed EE of grade 2 or above. The percentages of patients healed (per protocol, n = 212) are shown in Table 9.
Table 9: Erosive Esophagitis Healing Rates (Per Protocol)
–––––––––––– Pantoprazole Sodium–––––––––––– Nizatidine 20 mg daily 40 mg daily 150 mg twice daily Week (n = 72) (n = 70) (n = 70) 4 61.4% + 64.0% + 22.2% 8 79.2% + 82.9% + 41.4% + (p < 0.001) pantoprazole sodium versus nizatidine
Once-daily treatment with pantoprazole sodium 40 mg or 20 mg resulted in significantly superior rates of healing at both 4 and 8 weeks compared with twice-daily treatment with 150 mg of nizatidine. For the 40 mg treatment group, significantly greater healing rates compared to nizatidine were achieved regardless of the H. pylori status.
A significantly greater proportion of the patients in the pantoprazole sodium treatment groups experienced complete relief of nighttime heartburn and regurgitation, starting on the first day and of daytime heartburn on the second day, compared with those taking nizatidine 150 mg twice daily. Patients taking pantoprazole sodium consumed significantly fewer antacid tablets per day than those taking nizatidine.
Pediatric Patients Ages 5 Years through 16 Years
The efficacy of pantoprazole sodium in the treatment of EE associated with GERD in pediatric patients ages 5 years through 16 years is extrapolated from adequate and well-conducted trials in adults, as the pathophysiology is thought to be the same. Four pediatric patients with endoscopically diagnosed EE were studied in multicenter, randomized, double-blind, parallel-treatment trials. Children with endoscopically diagnosed EE (defined as an endoscopic Hetzel-Dent score ≥ 2) were treated once daily for 8 weeks with one of two dose levels of pantoprazole sodium (20 mg or 40 mg). All 4 patients with EE were healed (Hetzel-Dent score of 0 or 1) at 8 weeks.
14.2 Long-Term Maintenance of Healing of Erosive Esophagitis
Two independent, multicenter, randomized, double-blind, comparator-controlled trials of identical design were conducted in adult GERD patients with endoscopically confirmed healed EE to demonstrate efficacy of pantoprazole sodium in long-term maintenance of healing. The two US studies enrolled 386 and 404 patients, respectively, to receive either 10 mg, 20 mg, or 40 mg of pantoprazole sodium delayed-release tablets once daily or 150 mg of ranitidine twice daily. As demonstrated in Table 10, pantoprazole sodium 40 mg and 20 mg were significantly superior to ranitidine at every timepoint with respect to the maintenance of healing. In addition, pantoprazole sodium 40 mg was superior to all other treatments studied.
Table 10: Long-Term Maintenance of Healing of Erosive Gastroesophageal Reflux Disease (GERD Maintenance): Percentage of Patients Who Remained Healed
Pantoprazole Sodium
20 mg dailyPantoprazole Sodium
40 mg dailyRanitidine150 mg
twice dailyStudy 1 n = 75 n = 74 n = 75 Month 1 91* 99* 68 Month 3 82* 93* # 54 Month 6 76* 90* # 44 Month 12 70* 86* # 35 Study 2 n = 74 n = 88 n = 84 Month 1 89* 92* # 62 Month 3 78* 91* # 47 Month 6 72* 88* # 39 Month 12 72* 83* 37 * (p < 0.05 vs. ranitidine)
# (p < 0.05 vs. pantoprazole sodium 20 mg)
Note: Pantoprazole sodium 10 mg was superior (p < 0.05) to ranitidine in Study 2, but not Study 1.
Pantoprazole sodium 40 mg was superior to ranitidine in reducing the number of daytime and nighttime heartburn episodes from the first through the twelfth month of treatment. Pantoprazole sodium 20 mg, administered once daily, was also effective in reducing episodes of daytime and nighttime heartburn in one trial, as presented in Table 11.
Table 11: Number of Episodes of Heartburn (mean ± SD)
Pantoprazole Sodium
40 mg dailyRanitidine150 mg
twice dailyMonth 1 Daytime 5.1 ± 1.6* 18.3 ± 1.6 Nighttime 3.9 ± 1.1* 11.9 ± 1.1 Month 12 Daytime 2.9 ± 1.5* 17.5 ± 1.5 Nighttime 2.5 ± 1.2* 13.8 ± 1.3 * (p < 0.001 vs. ranitidine, combined data from the two US studies)
14.3 Pathological Hypersecretory Conditions Including Zollinger-Ellison Syndrome
In a multicenter, open-label trial of 35 patients with pathological hypersecretory conditions, such as Zollinger-Ellison Syndrome, with or without multiple endocrine neoplasia-type I, pantoprazole sodium successfully controlled gastric acid secretion. Doses ranging from 80 mg daily to 240 mg daily maintained gastric acid output below 10 mEq/h in patients without prior acid-reducing surgery and below 5 mEq/h in patients with prior acid-reducing surgery.
Doses were initially titrated to the individual patient needs, and adjusted in some patients based on the clinical response with time [see Dosage and Administration(2)]. Pantoprazole sodium was well tolerated at these dose levels for prolonged periods (greater than 2 years in some patients).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Pantoprazole sodium delayed-release tablets USP 20 mg are yellow, round, biconvex coated tablets printed with “R332” on one side with black ink and plain on the other side.
60760-574-30 BOTTLES OF 30
60760-574-60 BOTTLES OF 60
Storage
Store pantoprazole sodium delayed-release tablets, USP at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Gastric Malignancy
Advise patients to return to their healthcare provider if they have a suboptimal response or an early symptomatic relapse [see Warnings and Precautions ( 5.1)].
Acute Tubulointerstitial Nephritis
Advise patients to call their healthcare provider immediately if they experience signs and/or symptoms associated with acute tubulointerstitial nephritis [see Contraindications (4), Warnings and Precautions ( 5.2) ].
Clostridium difficile-Associated Diarrhea
Advise patients to immediately call their healthcare provider if they experience diarrhea that does not improve [see Warnings and Precautions ( 5.3) ].
Bone Fracture
Advise patients to report any fractures, especially of the hip, wrist or spine, to their healthcare provider [see Warnings and Precautions ( 5.4) ].
Cutaneous and Systemic Lupus Erythematosus
Advise patients to immediately call their healthcare provider for any new or worsening of symptoms associated with cutaneous or systemic lupus erythematosus [see Warnings and Precautions ( 5.5) ].
Cyanocobalamin (Vitamin B-12)
Deficiency Advise patients to report any clinical symptoms that may be associated with cyancobalamin deficiency to their healthcare provider if they have been receiving pantoprazole sodium for longer than 3 years [see Warnings and Precautions ( 5.6) ].
Hypomagnesemia
Advise patients to report any clinical symptoms that may be associated with hypomagnesemia to their healthcare provider, if they have been receiving pantoprazole sodium for at least 3 months [see Warnings and Precautions ( 5.7) ].
Drug Interactions
Instruct patients to inform their healthcare provider of any other medications they are currently taking, including rilpivirine-containing products [see Contraindications (4)] digoxin [see Warnings and Precautions ( 5.7) ] and high dose methotrexate [see Warnings and Precautions ( 5.12) ].
Pregnancy
Advise a pregnant woman of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Use in Specific Populations( 8.1) ].
Administration
- Do not split, crush, or chew pantoprazole sodium delayed-release tablets.
- Swallow pantoprazole sodium delayed-release tablets whole, with or without food in the stomach.
- Concomitant administration of antacids does not affect the absorption of pantoprazole sodium delayed-release tablets.
- Take a missed dose as soon as possible. If it is almost time for the next dose, skip the missed dose and take the next dose at the regular scheduled time. Do not take 2 doses at the same time.
-
MEDICATION GUIDE
MEDICATION GUIDE
Pantoprazole Sodium Delayed-Release Tablets, USP
(pan toe' pra zole soe' dee um)
What is the most important information I should know about pantoprazole sodium delayed-release tablets?
You should take pantoprazole sodium delayed-release tablets exactly as prescribed, at the lowest dose possible and for the shortest time needed.
Pantoprazole sodium delayed-release tablets may help your acid-related symptoms, but you could still have serious stomach problems. Talk with your doctor.
Pantoprazole sodium delayed-release tablets can cause serious side effects, including:
A type of kidney problem (acute tubulointerstitial nephritis). Some people who take proton pump inhibitor (PPI) medicines, including pantoprazole sodium delayed-release tablets, may develop a kidney problem called acute tubulointerstitial nephritis that can happen at any time during treatment with pantoprazole sodium delayed-release tablets. Call your doctor right away if you have a decrease in the amount that you urinate or if you have blood in your urine.
Diarrhea caused by an infection ( Clostridium difficile) in your intestines. Call your doctor right away if you have watery stools or stomach pain that does not go away. You may or may not have a fever.
Bone fractures (hip, wrist, or spine). Bone fractures in the hip, wrist, or spine may happen in people who take multiple daily doses of PPI medicines and for a long period of time (a year or longer). Tell your doctor if you have a bone fracture, especially in the hip, wrist, or spine.
Certain types of lupus erythematosus. Lupus erythematosus is an autoimmune disorder (the body’s immune cells attack other cells or organs in the body). Some people who take PPI medicines, including pantoprazole sodium delayed-release tablets, may develop certain types of lupus erythematosus or have worsening of the lupus they already have. Call your doctor right away if you have new or worsening joint pain or a rash on your cheeks or arms that gets worse in the sun.
Talk to your doctor about your risk of these serious side effects.
Pantoprazole sodium delayed-release tablets can have other serious side effects. See “ What are the possible side effects of pantoprazole sodium delayed-release tablets?”
What are pantoprazole sodium delayed-release tablets?
A prescription medicine called a proton pump inhibitor (PPI) used to reduce the amount of acid in your stomach.
In adults, pantoprazole sodium delayed-release tablets are used for:
up to 8 weeks for the healing and symptom relief of acid-related damage to the lining of the esophagus (called erosive esophagitis or EE). Your doctor may prescribe another 8 weeks of pantoprazole sodium delayed-release tablets in patients whose EE does not heal.
maintaining healing of EE and to help prevent the return of heartburn symptoms caused by GERD. It is not known if pantoprazole sodium delayed-release tablets are safe and effective when used for longer than 12 months for this purpose.
the long-term treatment of conditions where your stomach makes too much acid. This includes a rare condition called Zollinger-Ellison Syndrome.
In children 5 years of age and older, pantoprazole sodium delayed-release tablets are used for:
up to 8 weeks for the healing and symptom relief of EE.
It is not known if pantoprazole sodium delayed-release tablets are safe if used longer than 8 weeks in children.
Pantoprazole sodium delayed-release tablets are not for use in children under 5 years of age.
It is not known if pantoprazole sodium delayed-release tablets are safe and effective in children for treatment other than EE.
Do not take pantoprazole sodium delayed-release tablets if you are:
allergic to pantoprazole sodium, any other PPI medicine, or any of the ingredients in pantoprazole sodium delayed-release tablets. See the end of this Medication Guide for a complete list of ingredients.
taking a medicine that contains rilpivirine (EDURANT, COMPLERA, ODEFSEY, JULUCA) used to treat HIV-1 (Human Immunodeficiency Virus).
Before taking pantoprazole sodium delayed-release tablets, tell your doctor about all of your medical conditions, including if you:
have low magnesium levels in your blood.
are pregnant or plan to become pregnant. Pantoprazole sodium delayed-release tablets may harm your unborn baby. Tell your doctor if you become pregnant or think you may be pregnant during treatment with pantoprazole sodium delayed-release tablets.
are breastfeeding or plan to breastfeed. Pantoprazole sodium can pass into your breast milk. Talk with your doctor about the best way to feed your baby if you take pantoprazole sodium delayed-release tablets.
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins and herbal supplements. Especially tell your doctor if you take methotrexate (Otrexup, Rasuvo, Trexall, XATMEP), digoxin (LANOXIN), or a water pill (diuretic).
How should I take pantoprazole sodium delayed-release tablets?
Take pantoprazole sodium delayed-release tablets exactly as prescribed by your doctor.
Pantoprazole sodium delayed-release tablets:
o Do not split, chew, or crush pantoprazole sodium delayed-release tablets.
o Swallow pantoprazole sodium delayed-release tablets whole, with or without food.
o Tell your doctor if you are not able to swallow your pantoprazole sodium delayed-release tablet.
o You may use antacids while taking pantoprazole sodium delayed-release tablets.
If you miss a dose of pantoprazole sodium delayed-release tablet, take it as soon as possible. If it is almost time for your next dose, do not take the missed dose. Take the next dose at your regular time. Do not take 2 doses at the same time.
If you take too much pantoprazole sodium, call your doctor or your poison control center at 1-800-222-1222 right away or go to the nearest emergency room.
What are the possible side effects of pantoprazole sodium delayed-release tablets?
Pantoprazole sodium delayed-release tablets can cause serious side effects, including:
See “What is the most important information I should know about pantoprazole sodium delayed-release tablets?”
Low vitamin B-12 levels in your body can happen in people who have taken pantoprazole sodium delayed-release tablets for a long time (more than 3 years). Tell your doctor if you have symptoms of low vitamin B-12 levels, including shortness of breath, lightheadedness, irregular heartbeat, muscle weakness, pale skin, feeling tired, mood changes, and tingling or numbness in the arms and legs.
Low magnesium levels in your body can happen in people who have taken pantoprazole sodium delayed-release tablets for at least 3 months. Tell your doctor if you have symptoms of low magnesium levels, including seizures, dizziness, irregular heartbeat, jitteriness, muscle aches or weakness, and spasms of hands, feet or voice.
Stomach growths (fundic gland polyps). People who take PPI medicines for a long time have an increased risk of developing a certain type of stomach growths called fundic gland polyps, especially after taking PPI medicines for more than 1 year.
The most common side effects of pantoprazole sodium delayed-release tablets in adults include: headache, diarrhea, nausea, stomach-area (abdominal) pain, vomiting, gas, dizziness, and joint pain.
The most common side effects of pantoprazole sodium delayed-release tablets in children include: upper respiratory infection, headache, fever, diarrhea, vomiting, rash, and stomach-area (abdominal) pain.
These are not all the possible side effects of pantoprazole sodium delayed-release tablets. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store pantoprazole sodium delayed-release tablets?
Store pantoprazole sodium delayed-release tablets at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F).
Keep pantoprazole sodium delayed-release tablets and all medicines out of the reach of children.
General information about the safe and effective use of pantoprazole sodium delayed-release tablets.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use pantoprazole sodium delayed-release tablets for a condition for which it was not prescribed. Do not give pantoprazole sodium delayed-release tablets to other people, even if they have the same symptoms that you have. It may harm them. You can ask your doctor or pharmacist for information about pantoprazole sodium delayed-release tablets that is written for health professionals.
What are the ingredients in pantoprazole sodium delayed-release tablets?
Active ingredient: pantoprazole sodium sesquihydrate
Inactive ingredients in pantoprazole sodium delayed-release tablets: ammonium hydroxide, calcium stearate, crospovidone, hydroxypropyl cellulose, hypromellose, iron oxide black, isopropyl alcohol, mannitol, methacrylic acid copolymer, methylated spirit, N-butyl alcohol, polyethylene glycol, propylene glycol, shellac, sodium carbonate anhydrous, synthetic yellow iron oxide, talc, titanium dioxide, triethyl citrate, zein.
For more information, call toll-free 1-888-375-3784.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
The brands listed are trademarks of their respective owners and are not trademarks of Dr. Reddy's Laboratories Limited.
Rx Only
Manufactured by:
Dr. Reddy’s Laboratories Limited
Bachupally –500 090 INDIA
Revised: 1220
Dispense with Medication Guide available at : www.drreddys.com/medguide/pantoprazoledrtabs.pdf
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
PANTOPRAZOLE
pantoprazole tablet, delayed releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 60760-574(NDC:55111-332) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PANTOPRAZOLE SODIUM (UNII: 6871619Q5X) (PANTOPRAZOLE - UNII:D8TST4O562) PANTOPRAZOLE 20 mg Inactive Ingredients Ingredient Name Strength AMMONIA (UNII: 5138Q19F1X) CALCIUM STEARATE (UNII: 776XM7047L) CROSPOVIDONE (UNII: 2S7830E561) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) HYPROMELLOSES (UNII: 3NXW29V3WO) FERROSOFERRIC OXIDE (UNII: XM0M87F357) ISOPROPYL ALCOHOL (UNII: ND2M416302) MANNITOL (UNII: 3OWL53L36A) METHACRYLIC ACID - ETHYL ACRYLATE COPOLYMER (1:1) TYPE A (UNII: NX76LV5T8J) BUTYL ALCOHOL (UNII: 8PJ61P6TS3) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SHELLAC (UNII: 46N107B71O) SODIUM CARBONATE (UNII: 45P3261C7T) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TRIETHYL CITRATE (UNII: 8Z96QXD6UM) ZEIN (UNII: 80N308T1NN) ALCOHOL (UNII: 3K9958V90M) Product Characteristics Color yellow Score no score Shape ROUND Size 6mm Flavor Imprint Code R332 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 60760-574-60 60 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 10/31/2022 2 NDC: 60760-574-30 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 08/29/2023 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077619 10/31/2022 Labeler - ST. MARY'S MEDICAL PARK PHARMACY (063050751) Establishment Name Address ID/FEI Business Operations ST. MARY'S MEDICAL PARK PHARMACY 063050751 relabel(60760-574) , repack(60760-574)

© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.