VARUBI- rolapitant tablet VARUBI- rolapitant injection, emulsion
Varubi by
Drug Labeling and Warnings
Varubi by is a Prescription medication manufactured, distributed, or labeled by Tesaro, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VARUBI safely and effectively. See full prescribing information for VARUBI.
VARUBI® (rolapitant) tablets, for oral use
VARUBI® (rolapitant) injectable emulsion, for intravenous use
Initial U.S. Approval: 2015RECENT MAJOR CHANGES
INDICATIONS AND USAGE
VARUBI is a substance P/neurokinin 1 (NK1) receptor antagonist indicated in combination with other antiemetic agents in adults for the prevention of delayed nausea and vomiting associated with initial and repeat courses of emetogenic cancer chemotherapy, including, but not limited to, highly emetogenic chemotherapy. (1)
DOSAGE AND ADMINISTRATION
- Administer in combination with dexamethasone and a 5-HT3 receptor antagonist; see full prescribing information for dosing information. (2)
- No dosage adjustment for dexamethasone is required. (2)
- Administer VARUBI within 2 hours prior to the initiation of chemotherapy on Day 1. (2)
- Tablets: The recommended dosage is 180 mg as a single dose. (2)
- Injectable emulsion: The recommended dosage is 166.5 mg administered as an intravenous infusion over 30 minutes. (2)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Anaphylaxis, Anaphylactic Shock and Other Serious Hypersensitivity Reactions: May occur during or soon after infusion. Most occur immediately following initiation of infusion. If symptoms occur, stop VARUBI injectable emulsion, initiate appropriate medical management including epinephrine and/or an antihistamine. Permanently discontinue VARUBI injectable emulsion. (5.1)
-
CYP2D6 Substrates: Rolapitant is a moderate inhibitor of CYP2D6 and significantly increases the plasma concentrations of CYP2D6 substrates for at least 28 days following single dose administration of VARUBI. Before starting VARUBI, consider if patients require:
- thioridazine or pimozide; if so, use an alternative antiemetic to VARUBI or an alternative to thioridazine or pimozide that is not metabolized by CYP2D6.
- other CYP2D6 substrates; if so, consult the prescribing information for the CYP2D6 substrate for additional information about interactions with CYP2D6 inhibitors. (4, 5.2, 7.1)
ADVERSE REACTIONS
Most common adverse reactions (≥5%) are:
- Cisplatin Based Highly Emetogenic Chemotherapy: neutropenia and hiccups (6.1)
- Moderately Emetogenic Chemotherapy and Combinations of Anthracycline and Cyclophosphamide: decreased appetite, neutropenia and dizziness (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact TESARO at 1-844-4-TESARO or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Strong CYP3A4 Inducers (e.g., rifampin): significantly reduced plasma concentrations of rolapitant can decrease the efficacy of VARUBI; avoid use of VARUBI in patients who require chronic administration of such drugs. (7.2)
- BCRP and P-gp Substrates with a Narrow Therapeutic Index: Oral VARUBI is an inhibitor of BCRP and P-gp and can increase plasma concentrations of the concomitant drug and potential for adverse reactions. See full prescribing information for specific examples. (7.3, 7.4)
- Warfarin: Monitor for increased INR or prothrombin time; adjust the dose of warfarin as needed. (7.5)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 3/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosage
2.2 Instructions for Administration of VARUBI Injectable Emulsion
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Anaphylaxis, Anaphylactic Shock and Other Serious Hypersensitivity Reactions
5.2 Interaction with CYP2D6 Substrates
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 CYP2D6 Substrates
7.2 Strong CYP3A4 Inducers
7.3 BCRP Substrates with a Narrow Therapeutic Index
7.4 P-gp Substrates with a Narrow Therapeutic Index
7.5 Warfarin
7.6 CYP3A4 Substrates
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosage
The recommended dosage of VARUBI tablets and VARUBI injectable emulsion in adults in combination with a 5-HT3 receptor antagonist and dexamethasone for the prevention of nausea and vomiting with emetogenic cancer chemotherapy is shown in Table 1. There is no drug interaction between rolapitant and dexamethasone, so no dosage adjustment for dexamethasone is required. Administer a dexamethasone dose of 20 mg on Day 1 [see Clinical Pharmacology (12.3)].
Administer VARUBI prior to the initiation of each chemotherapy cycle, but at no less than 2 week intervals.
Administer VARUBI tablets without regards to meals.
Table 1: Recommended Dosing Regimen of VARUBI Tablets or VARUBI Injectable Emulsion Day 1 Day 2 Day 3 Day 4 Prevention of Nausea and Vomiting Associated with Cisplatin-Based Highly Emetogenic Cancer Chemotherapy VARUBI Administer orally or intravenously within 2 hours prior to initiation of chemotherapy
Oral: 180 mg as a single dose
Injectable emulsion: Infuse 166.5 mg over 30 minutes.None Dexamethasone 20 mg; 30 min prior to initiation of chemotherapy 8 mg twice daily 8 mg twice daily 8 mg twice daily 5-HT3 receptor antagonist See the prescribing information for the co-administered 5-HT3 receptor antagonist for appropriate dosing information. None Prevention of Nausea and Vomiting Associated with Moderately Emetogenic Cancer Chemotherapy and Combinations of Anthracycline and Cyclophosphamide VARUBI Administer orally or intravenously within 2 hours prior to initiation of chemotherapy
Oral: 180 mg as a single dose
Injectable emulsion: Infuse 166.5 mg over 30 minutes.None Dexamethasone 20 mg; 30 min prior to initiation of chemotherapy None 5-HT3 receptor antagonist See the prescribing information for the co-administered 5-HT3 receptor antagonist for appropriate dosing information. See the prescribing information for the co-administered 5-HT3 receptor antagonist for appropriate dosing information. 2.2 Instructions for Administration of VARUBI Injectable Emulsion
- VARUBI injectable emulsion is supplied as a single-dose vial for intravenous administration. The emulsion is homogeneous (no shaking required) and translucent white in appearance.
- Inspect VARUBI injectable emulsion for particulate matter and discoloration prior to administration; discard if present. Do not use if contamination is suspected.
- Use and maintain aseptic technique while handling VARUBI injectable emulsion.
- Do not add any diluents or other medications directly to the vial.
- Administer the dose by inserting a vented intravenous set through the septum of the vial. Once the stopper is punctured, use immediately. Do not dilute VARUBI injectable emulsion.
- Infuse 92.5 mL from the vial over 30 minutes.
-
VARUBI injectable emulsion is compatible with the following other intravenous fluids through a Y-site connection:
- Sodium Chloride 0.9% Injection, USP
- Dextrose 5% Injection, USP
- Dextrose 5% in Lactated Ringer's Injection, USP
- Lactated Ringer's Injection, USP
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
VARUBI is contraindicated in patients:
- who are hypersensitive to any component of the product (including soybean oil). Serious hypersensitivity reactions, including anaphylaxis and anaphylactic shock, have been reported with VARUBI injectable emulsion [see Warnings and Precautions (5.1), Adverse Reactions (6.2), Description (11)].
- taking CYP2D6 substrates with a narrow therapeutic index, such as thioridazine and pimozide. VARUBI can significantly increase the plasma concentrations of thioridazine and pimozide, which may result in QT prolongation and Torsades de Pointes [see Warnings and Precautions (5.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Anaphylaxis, Anaphylactic Shock and Other Serious Hypersensitivity Reactions
Anaphylaxis, anaphylactic shock and other serious hypersensitivity reactions have been reported in the postmarketing setting, some requiring hospitalization [see Adverse Reactions (6.2)]. These reactions have occurred during or soon after the infusion of VARUBI injectable emulsion. Most reactions have occurred within the first few minutes of administration. Symptoms of anaphylaxis can include wheezing or difficulty breathing; swelling of the face or throat; hives or flushing; itching; abdominal cramping, abdominal pain or vomiting; back pain or chest pain; hypotension or shock.
Consult with patients to determine if the patient is hypersensitive to any component of the product (including soybean oil). Furthermore, as cross reactions to other allergens is possible, patients with known allergies to legumes or other related allergens should be monitored closely. Patients with a potential hypersensitivity should not be administered VARUBI injectable emulsion [see Contraindications (4)].
Appropriate treatment should be available for immediate use in the event of an anaphylactic reaction during treatment with VARUBI injectable emulsion.
If anaphylaxis or any other serious hypersensitivity/infusion reaction occurs:
- administration of VARUBI injectable emulsion should be stopped immediately.
- appropriate medical management (including epinephrine and or antihistamines) should be initiated, and
- VARUBI injectable emulsion should be permanently discontinued.
5.2 Interaction with CYP2D6 Substrates
Rolapitant is a moderate inhibitor of CYP2D6. Exposure to dextromethorphan, a CYP2D6 substrate, following a single dose of rolapitant increased about 3-fold on Days 8 and Day 22. The inhibition of CYP2D6 persisted on Day 28 with a 2.3-fold increase in dextromethorphan concentrations, the last time point measured. The inhibitory effect of rolapitant on CYP2D6 is expected to persist beyond 28 days for an unknown duration following administration of VARUBI [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Narrow Therapeutic Index Drugs (Thioridazine and Pimozide)
VARUBI is contraindicated in patients taking CYP2D6 substrates with a narrow therapeutic index such as thioridazine and pimozide. Increased plasma concentrations of thioridazine and pimozide are associated with serious and/or life-threatening events of QT prolongation and Torsades de Pointes [see Contraindications (4)].
Before starting treatment with VARUBI, consider whether patients require treatment with thioridazine or pimozide. If patients require these drugs, use an alternative antiemetic to VARUBI or an alternative to thioridazine or pimozide that is not metabolized by CYP2D6.
Other Drugs
VARUBI can also increase plasma concentrations of other CYP2D6 substrates for at least 28 days following administration of VARUBI and may result in adverse reactions.
Before starting treatment with VARUBI, consult the prescribing information for CYP2D6 substrates to obtain additional information about interactions with CYP2D6 inhibitors.
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Anaphylaxis, Anaphylactic Shock and Other Serious Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
- Interaction with CYP2D6 Substrates [see Contraindications (4) and Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
VARUBI Tablets
In 4 controlled clinical trials in patients receiving emetogenic cancer chemotherapy, VARUBI tablets were given in combination with a 5-HT3 receptor antagonist and dexamethasone. On Day 1 of Cycle 1 of chemotherapy, 1567 patients were treated with VARUBI tablets and 1198 of these patients continued into the optional multiple cycle extension for up to 6 cycles of chemotherapy. The median number of cycles administered 180 mg of VARUBI tablets was four. VARUBI tablets 180 mg were administered to 1294 patients.
In Cycle 1 adverse reactions were reported in approximately 7% of patients treated with VARUBI tablets compared with approximately 6% of patients treated with control therapy. The most common adverse reactions reported with an incidence of ≥3% and greater than control are listed in Table 2 and Table 3.
Table 2: Most Common Adverse Reactions in Patients Receiving Cisplatin-Based Highly Emetogenic Chemotherapy (Cycle 1)* VARUBI Regimen
(VARUBI tablets, Dexamethasone, and 5-HT3 Receptor Antagonist)
N = 624Control
(Placebo, Dexamethasone, and 5-HT3 Receptor Antagonist)
N = 627- * all reactions occurring at ≥3% in the VARUBI group and for which the rate for VARUBI exceeds the rate for control
Neutropenia 9% 8% Hiccups 5% 4% Abdominal Pain 3% 2% Table 3: Most Common Adverse Reactions in Patients Receiving Moderately Emetogenic Chemotherapy and Combinations of Anthracycline and Cyclophosphamide (Cycle 1)* VARUBI Regimen
(VARUBI tablets, Dexamethasone, and 5-HT3 Receptor Antagonist)
N = 670Control
(Placebo, Dexamethasone, and 5-HT3 Receptor Antagonist)
N = 674- * all reactions occurring at ≥3% in the VARUBI group and for which the rate for VARUBI exceeds the rate for control.
Decreased appetite 9% 7% Neutropenia 7% 6% Dizziness 6% 4% Dyspepsia 4% 2% Urinary tract infection 4% 3% Stomatitis 4% 2% Anemia 3% 2% Adverse reactions in the multiple-cycle extensions of highly and moderately emetogenic chemotherapy studies for up to 6 cycles of chemotherapy were generally similar to that observed in Cycle 1.
VARUBI Injectable Emulsion
In healthy subjects receiving the recommended therapeutic dose of VARUBI injectable emulsion, infusion-related adverse reactions were reported in 2.6% (2/78) of subjects during the infusion and included sensation of warmth, abdominal pain, dizziness and paresthesia.
Since VARUBI injectable emulsion is the same active substance (rolapitant) as VARUBI tablets, adverse reactions associated with VARUBI tablets might also be expected to occur with VARUBI injectable emulsion.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of VARUBI injectable emulsion. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
In postmarketing experience, severe systemic reactions have occurred within minutes of the start of infusion with VARUBI injectable emulsion.
Immune system disorders: hypersensitivity reactions including anaphylaxis, anaphylactoid reactions and anaphylactic shock, including back pain and chest pain
Gastrointestinal disorders: abdominal pain
Skin and subcutaneous disorders: erythema
General disorders and administration site conditions: feeling hot
Vascular disorders: generalized flushing, dizziness
-
7 DRUG INTERACTIONS
7.1 CYP2D6 Substrates
Rolapitant is a moderate CYP2D6 inhibitor. Exposure to dextromethorphan, CYP2D6 substrate, following a single dose of rolapitant increased about 3-fold on Days 8 and Day 22. The inhibition of CYP2D6 persisted on Day 28 with a 2.3-fold increase in dextromethorphan concentrations, the last time point measured. The inhibitory effect of rolapitant on CYP2D6 is expected to persist beyond 28 days for an unknown duration following administration of VARUBI [see Clinical Pharmacology (12.3)].
Narrow Therapeutic Index Drugs (Thioridazine and Pimozide)
VARUBI is contraindicated in patients taking CYP2D6 substrates with a narrow therapeutic index such as thioridazine and pimozide. Increased plasma concentrations of thioridazine and pimozide are associated with serious and/or life-threatening events of QT prolongation and Torsades de Pointes [see Contraindications (4), Warnings and Precautions (5.2)].
Before starting treatment with VARUBI, consider whether patients require treatment with thioridazine or pimozide. If patients require these drugs, use an alternative antiemetic to VARUBI or an alternative to thioridazine or pimozide that is not metabolized by CYP2D6.
Other Drugs
VARUBI can also increase plasma concentrations of other CYP2D6 substrates for at least 28 days following administration of VARUBI and may result in adverse reactions.
Before starting treatment with VARUBI consult the prescribing information of CYP2D6 substrates to obtain further information about interactions with CYP2D6 inhibitors.
7.2 Strong CYP3A4 Inducers
Co-administration of VARUBI with strong CYP3A4 inducers (e.g., rifampin) can significantly reduce the plasma concentrations of rolapitant and decrease the efficacy of VARUBI. Avoid the use of VARUBI in patients who require chronic administration of strong CYP3A4 inducers [see Clinical Pharmacology (12.3)].
7.3 BCRP Substrates with a Narrow Therapeutic Index
Oral rolapitant is an inhibitor of Breast-Cancer-Resistance Protein (BCRP). Increased plasma concentrations of BCRP substrates (e.g., methotrexate, topotecan, or irinotecan) may result in potential adverse reactions. Monitor for adverse reactions related to the concomitant drug if use of VARUBI tablets cannot be avoided. Use the lowest effective dose of rosuvastatin (see prescribing information for additional information on recommended dosing) [see Clinical Pharmacology (12.3)].
7.4 P-gp Substrates with a Narrow Therapeutic Index
Oral rolapitant is an inhibitor of p-glycoprotein (P-gp). Increased plasma concentrations of P-gp substrates (e.g., digoxin) may result in potential adverse reactions. Monitor digoxin concentrations with concomitant use of VARUBI and adjust the dosage as needed to maintain therapeutic concentrations. Monitor for adverse reactions if concomitant use of VARUBI tablets with other P-gp substrates with a narrow therapeutic index cannot be avoided.
7.5 Warfarin
Although co-administration of intravenous VARUBI with warfarin did not substantially increase the systemic exposure to S-warfarin, the active enantiomer, the effects on INR and prothrombin time were not studied. Monitor INR and prothrombin time and adjust the dosage of warfarin, as needed with concomitant use of VARUBI, to maintain the target INR range.
7.6 CYP3A4 Substrates
Rolapitant, given as a single intravenous or oral dose, is not an inhibitor or inducer of CYP3A4 [see Clinical Pharmacology (12.3)]. Therefore, no dosage adjustment for dexamethasone (CYP3A4 substrate) is needed when co-administered with VARUBI [see Dosage and Administration (2)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The limited data with VARUBI use in pregnant women are insufficient to inform a drug associated risk of adverse developmental outcomes. In animal reproduction studies, there were no adverse developmental effects observed with oral administration of rolapitant in rats and rabbits during the period of organogenesis at doses up to 1.3 times and 2.9-times, respectively, the maximum recommended human dose (MRHD) [see Data].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
The potential embryo-fetal toxicity of rolapitant was assessed in pregnant rats administered oral doses up to 22.5 mg/kg per day throughout the period of organogenesis. Rats administered doses of 13.5 or 22.5 mg/kg per day rolapitant exhibited evidence of maternal toxicity including decreased body weight gain and/or body weight loss and a concomitant decrease in food consumption during the first week of dosing. No adverse embryo-fetal developmental effects were observed at doses up to 22.5 mg/kg per day rolapitant (approximately 1.3 times the recommended intravenous human dose on a body surface area basis). In rabbits administered rolapitant throughout the period of organogenesis, oral doses up to 27 mg/kg per day (approximately 3 times the recommended intravenous human dose on a body surface area basis) were without effects on the developing fetus.
The pre- and postnatal developmental effects of rolapitant were assessed in rats administered oral doses of 2.25, 9 or 22.5 mg/kg per day during the periods of organogenesis and lactation. Maternal toxicity was evident based on mortality/moribund condition, decreased body weight and food consumption, total litter loss, prolonged parturition, decreased length of gestation, and increased number of unaccounted for implantation sites at a dose of 22.5 mg/kg per day (approximately 1.3 times the recommended intravenous human dose on a body surface area basis). Effects on offspring at this dose included decreased postnatal survival, and decreased body weights and body weight gain, and may be related to the maternal toxicity observed. At a maternal dose of 9 mg/kg per day rolapitant (approximately 0.5 times the recommended intravenous human dose on a body surface area basis), there was a decrease in memory in female pups in a maze test and a decrease in pup body weight.
8.2 Lactation
Risk Summary
There are no data on the presence of rolapitant in human milk, the effects of rolapitant in the breastfed infant, or the effects of rolapitant on milk production. Rolapitant administered orally to lactating female rats was present in milk [see Data]. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for VARUBI and any potential adverse effects on the breastfed infant from VARUBI or from the underlying maternal condition or the use of concomitant chemotherapy.
Data
Radioactivity from labeled [14C] rolapitant was transferred into milk of lactating rats following a single oral dose of 22.5 mg/kg, and the maximum radioactivity in milk was observed at 12 hours post-dose. The mean milk/plasma radioactivity concentration ratios in dams at 1 to 48 hours post-dose ranged from 1.24 to 3.25. Based on average daily consumption of milk (2 mL/day) and the maximum milk radioactivity determined, pup exposure is expected to be 0.32% of the orally administered dose.
8.3 Females and Males of Reproductive Potential
Infertility
Females
In animal fertility studies, rolapitant impaired the fertility in females in a reversible fashion [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and efficacy of VARUBI have not been established in pediatric patients.
In non-clinical studies, sexual development and fertility of juvenile rats (human age equivalent of birth to 16 years) were affected following oral administration of VARUBI, as described below in Juvenile Animal Toxicity Data.
Juvenile Animal Toxicity Data
In an oral juvenile toxicity study in rats, at rolapitant doses of 11.3, 22.5 and 45 mg/kg/day from postnatal (PND) Day 7 through PND 70 (equivalent human age of newborn to 16 years), there was a delay in the attainment of balanopreputial separation in males and an acceleration of the attainment of vaginal patency in females with rolapitant doses of 22.5 and 45 mg/kg/day (approximately 1.3 and 2.6 times, respectively, the recommended intravenous human dose on a body surface area basis). Treated males and females were mated following a 2-week wash-out period after the last dose. There were lower mean numbers of implantation sites, corpora lutea, and mean number of viable embryos at 22.5 and 45 mg/kg/day (approximately 1.3 and 2.6 times, respectively, the recommended intravenous human dose on a body surface area basis) when compared to control.
8.5 Geriatric Use
Of the 1294 subjects treated with VARUBI, 25% were 65 years and over, while 5% were 75 and over. No overall differences in safety or efficacy were reported between the elderly subjects and younger subjects, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Hepatic Impairment
No dosage adjustment is needed in patients with mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment. There are no clinical or pharmacokinetic data in patients with severe hepatic impairment (Child-Pugh Class C). Avoid use of VARUBI in patients with severe hepatic impairment. If use cannot be avoided, monitor patients for adverse reactions related to rolapitant [see Adverse Reactions (6.1)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
VARUBI contains rolapitant, a substance P/neurokinin 1 (NK1) receptor antagonist. Rolapitant hydrochloride is chemically described as (5S,8S)-8- { [(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethoxy]methyl]}-8-phenyl-1,7-diazaspiro[4.5]decan-2-one hydrochloride. Its empirical formula is C25H26F6N2O2. HCl.H2O, and its structural formula is:
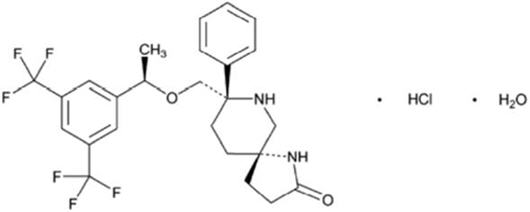
rolapitant hydrochloride
Rolapitant hydrochloride is a white to off-white powder, with a molecular weight of 554.95. Solubility of rolapitant hydrochloride in aqueous solution is pH-dependent and is more soluble at lower pH. Rolapitant has good solubility in common pharmaceutical solvents such as ethanol, propylene glycol and 40% hydroxypropyl beta-cyclodextrin.
VARUBI (rolapitant) Tablets
Each tablet contains 90 mg rolapitant (equivalent to 100 mg rolapitant hydrochloride) and the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone, and pregelatinized starch. The tablets are coated in non-functional blue and clear coats. The tablet coating comprises the following inactive ingredients: FD&C Blue No. 2-Indigo Carmine Lake, polyethylene glycol, polysorbate 80, polyvinyl alcohol, talc, and titanium dioxide.
VARUBI (rolapitant) Injectable Emulsion
Each single-dose vial of 92.5 mL sterile emulsion for intravenous use contains 166.5 mg rolapitant (equivalent to 185 mg of rolapitant hydrochloride) and the following inactive ingredients: dibasic sodium phosphate, anhydrous (2.8 mg/mL), medium chain triglycerides (11 mg/mL), polyoxyl 15 hydroxystearate (44 mg/mL), sodium chloride (6.2 mg/mL), soybean oil (6.6 mg/mL), water for injection and may contain hydrochloric acid and/or sodium hydroxide to adjust pH to 7.0 to 8.0. VARUBI injectable emulsion is a sterile, translucent white liquid and has some opalescence.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Rolapitant is a selective and competitive antagonist of human substance P/NK1 receptors. Rolapitant does not have significant affinity for the NK2 or NK3 receptors or for a battery of other receptors, transporters, enzymes and ion channels. Rolapitant is also active in animal models of chemotherapy-induced emesis.
12.2 Pharmacodynamics
NK1 Receptor Occupancy
A human Positron Emission Tomography (PET) study with rolapitant demonstrated that rolapitant crosses the blood brain barrier and occupies brain NK1 receptors. A dose-dependent increase in mean NK1 receptor occupancy was observed in the oral dose range from 4.5 mg to 180 mg of rolapitant. At the 180 mg oral dose of rolapitant, the mean NK1 receptor occupancy was 73% in the striatum at 120 hours after a single dose administration in healthy subjects. The relationship between NK1 receptor occupancy and the clinical efficacy of rolapitant has not been established.
Cardiac Electrophysiology
At an oral dose of 720 mg (4 times the recommended oral dose), VARUBI does not prolong the QT interval to any clinically relevant extent. The maximum rolapitant concentration after a single 720 mg dose of oral rolapitant was 30% to 50% higher than that achieved with the recommended dose of 166.5 mg of intravenous rolapitant.
12.3 Pharmacokinetics
Absorption
Oral tablet
Following a single oral dose administration of 180 mg VARUBI tablets under fasting conditions to healthy subjects, rolapitant was measurable in plasma between 30 minutes and the peak plasma concentration (Cmax) for rolapitant which was reached in about 4 hours and mean Cmax was 968 ng/mL (%CV:28%).
Following multiple oral doses of 9 to 45 mg once daily of rolapitant (5% to 25% of the recommended dose) for 10 days, accumulation of rolapitant (ratio of AUC0-24hr) ranged from 5.0 to 5.3 fold.
The systemic exposures (Cmax and AUC) to rolapitant increased in a dose-proportional manner when single oral doses of rolapitant increased from 4.5 mg to 180 mg. With 4 times the recommended clinical oral dose of 180 mg, the Cmax and AUC of rolapitant increased 3.1 fold and 3.7 fold, respectively.
Concomitant administration of a high-fat meal did not significantly affect the pharmacokinetics of rolapitant after administration of 180 mg VARUBI tablets [see Dosage and Administration (2)].
Injectable emulsion
Following a 30-minute infusion of a single intravenous dose of 166.5 mg VARUBI injectable emulsion to healthy subjects, the mean Cmax at the end of the infusion (30 min) was 1986 ng/mL (%CV:39%).
Following multiple intravenous doses of 18 to 54 mg once daily of rolapitant (approximately 10% to 33% of the recommended dose) for 10 days, accumulation of rolapitant (ratio of AUC0-24hr) ranged from 4.3 fold to 5.4 fold.
The systemic exposures (Cmax and AUC) to rolapitant increased in a dose-proportional manner when single intravenous doses of rolapitant increased from 18 mg to 180 mg. The Cmax and AUC of rolapitant increased 11.6 fold and 10.1 fold, respectively.
At the recommended dose, the absolute bioavailability of rolapitant is 91%.
Distribution
Rolapitant was highly protein bound to human plasma (99.8%). The volume of distribution (Vd) was 392 L in healthy subjects following a single intravenous dose of 166.5 mg rolapitant. The apparent volume of distribution (Vd/F) following a single oral dose of 180 mg rolapitant was 460 L in healthy subjects. The large Vd indicated an extensive tissue distribution of rolapitant. In a population pharmacokinetic analysis of oral rolapitant, the Vd/F was 387 L in cancer patients.
Elimination
Following single intravenous doses (18 to 180 mg) and single oral doses (4.5 to 180 mg) of rolapitant, the mean terminal half-life (t1/2) of rolapitant ranged from 138 to 205 hours and 169 to 183 hours (approximately 7 days), respectively, and was independent of dose. In a population pharmacokinetic analysis, the apparent total clearance (CL/F) of oral rolapitant was 0.96 L/hour in cancer patients. The total clearance following intravenous administration of 166.5 mg rolapitant in healthy subjects was 1.65 L/hour.
Metabolism
Rolapitant is metabolized primarily by CYP3A4 to form a major active metabolite, M19 (C4-pyrrolidine-hydroxylated rolapitant). In a mass balance study, the metabolite M19 was the major circulating metabolite. The formation of M19 was significantly delayed with the median Tmax of 120 hours (range: 24-168 hours) with Cmax of 183 ng/mL. The mean half-life of M19 was 158 hours.
The exposure ratio of M19 to rolapitant was approximately 50% in plasma.
The Cmax and AUC of M19 following single intravenous administration of 166.5 mg of rolapitant were 149 ng/mL and 68,600 ng∙h/mL, respectively. The median Tmax was 168 hours. The mean half-life was 182 hours.
Excretion
Rolapitant is eliminated primarily through the hepatic/biliary route. Following administration of a single oral 180-mg dose of [14C]-rolapitant, on average 14.2% (range 9% to 20%) and 73% (range 52% to 89%) of the dose was recovered in the urine and feces, respectively over 6 weeks. In pooled samples collected over 2 weeks, 8.3% of the dose was recovered in the urine primarily as metabolites and 37.8% of the dose was recovered in the feces primarily as unchanged rolapitant. Unchanged rolapitant or M19 were not found in pooled urine sample.
Specific Populations
Age, Male and Female Patients and Racial or Ethnic Groups
Population pharmacokinetic analyses indicated that age, sex and race had no significant impact on the pharmacokinetics of rolapitant.
Patients with Hepatic Impairment
Following administration of a single oral dose of 180 mg rolapitant to patients with mild hepatic impairment (Child-Pugh Class A), the pharmacokinetics of rolapitant were comparable with those of healthy subjects. In patients with moderate hepatic impairment (Child-Pugh Class B), the mean Cmax was 25% lower while mean AUC of rolapitant was similar compared to those of healthy subjects. The median Tmax for M19 was delayed to 204 hours in patients with mild or moderate hepatic impairment compared to 168 hours in healthy subjects. The pharmacokinetics of rolapitant were not studied in patients with severe hepatic impairment (Child-Pugh Class C) [see Use in Specific Populations (8.6)].
Patients with Renal Impairment
In population pharmacokinetic analyses, creatinine clearance (CLcr) at baseline did not show a significant effect on rolapitant pharmacokinetics in cancer patients with mild (CLcr: 60 to 90 mL/min) or moderate (CLcr: 30 to 60 mL/min) renal impairment compared to cancer patients with normal kidney function. Information is insufficient for the effect of severe renal impairment. The pharmacokinetics of rolapitant was not studied in patients with end-stage renal disease requiring hemodialysis.
Drug Interaction Studies
Effect of Other Drugs on Rolapitant
Rolapitant is a substrate for CYP3A4.
CYP3A4 inducers
When 600 mg rifampin was administered once daily for 7 days before and 7 days after administration of a single oral dose of 180 mg rolapitant, the mean Cmax of rolapitant was reduced by 30% and the mean AUC was reduced by 85% compared to administration of rolapitant alone. The mean half-life of rolapitant decreased from 176 hours without rifampin to 41 hours with concurrent rifampin [see Drug Interactions (7.2)].
CYP3A4 inhibitors
Concurrent administration of 400 mg ketoconazole, a strong CYP3A4 inhibitor, once daily for 21 days following a single 90 mg oral dose of rolapitant, did not affect the Cmax of rolapitant while the AUC increased by 21%. These pharmacokinetic differences are not clinically significant.
Effect of Rolapitant on Other Drugs
The effect of VARUBI on CYP450 enzymes and transporters is summarized below.
CYP3A4 substrates
Rolapitant is neither an inhibitor nor an inducer of CYP3A4.
Midazolam: A single oral dose of 180 mg rolapitant had no significant effects on the pharmacokinetics of midazolam when oral midazolam 3 mg was co-administered on Day 1 and administered alone on Days 6 and 9. A single intravenous dose of 166.5 mg rolapitant had no significant effects on the pharmacokinetics of midazolam when oral midazolam 3 mg was co-administered on Day 1 and administered alone on Day 8.
Ondansetron: Rolapitant had no significant effects on the pharmacokinetics of intravenous ondansetron when concomitantly administered with a single 180 mg oral dose of rolapitant on the same day.
Dexamethasone: Rolapitant had no significant effects on the pharmacokinetics of dexamethasone when oral dexamethasone was administered on Days 1 to 3 after a single 180 mg oral dose of rolapitant was co-administered on Day 1 [see Dosage and Administration (2)].
CYP2D6 substrates
Rolapitant is a moderate inhibitor of CYP2D6 [see Contraindications (4), Warnings and Precautions (5.2), and Drug Interactions (7.1)]. See Table 4 and Table 5 for a summary of the effects of VARUBI on the pharmacokinetics of co-administered dextromethorphan, a CYP2D6 substrate.
Table 4: Effect of Oral Rolapitant on the Systemic Exposure of Co-administered CYP2D6 Substrate (Dextromethorphan)* Rolapitant Dose % Change for Dextromethorphan Day 1 with rolapitant Day 8 without rolapitant Change in Cmax Change in AUC Change in Cmax Change in AUC ↑ Denotes a mean increase in exposure by the percentage indicated. - * A single oral dose of 180 mg rolapitant was administered on Day 1; the interacting drug (dextromethorphan 30 mg) was administered orally on Day 1 with rolapitant and alone on Day 8.
180 mg 120% ↑ 160% ↑ 180% ↑ 230% ↑ Table 5: Effect of Intravenous Rolapitant on the Systemic Exposure of Co-administered CYP2D6 Substrate (Dextromethorphan)* Rolapitant Dose % Change for Dextromethorphan Day 1 with rolapitant Day 8 without rolapitant Day 15 without rolapitant Day 22 without rolapitant Day 29 without rolapitant Change in Cmax Change in AUC Change in Cmax Change in AUC Change in Cmax Change in AUC Change in Cmax Change in AUC Change in Cmax Change in AUC ↑ Denotes a mean increase in exposure by the percentage indicated. - * A single intravenous dose of 166.5 mg rolapitant was administered on Day 1; the interacting drug (dextromethorphan 30 mg) was administered orally on Day 1 with rolapitant and alone on Days 8, 15, 22, and 29.
166.5 mg 75% ↑ 110% ↑ 140% ↑ 220% ↑ 170% ↑ 220% ↑ 120% ↑ 180% ↑ 96% ↑ 130% ↑ BCRP transporter
In vitro, rolapitant is a BCRP transporter inhibitor.
When sulfasalazine (BCRP substrate) was administered with a single intravenous dose of 166.5 mg rolapitant on Day 1 and without rolapitant on Day 11, no effect on Cmax and AUC of sulfasalazine 500 mg was observed on Day 1, an 18% decrease in Cmax and an 11% decrease in AUC were observed on Day 11. These differences in systemic exposures are not clinically significant.
When sulfasalazine was administered with a single oral dose of 180 mg rolapitant on Day 1 and without rolapitant on Day 8, a 140% increase in Cmax and a 130% increase in AUC of sulfasalazine 500 mg was observed on Day 1, a 17% increase in Cmax and a 32% increase in AUC was observed on Day 8 [see Drug Interactions (7.3)].
P-glycoprotein substrates
In vitro, rolapitant is a P-gp inhibitor.
When digoxin (P-gp substrate) was administered with a single oral dose of 180 mg rolapitant, a 70% increase in Cmax and a 30% increase in AUC of digoxin 0.5 mg were observed [see Drug Interactions (7.4)].
When digoxin was administered with a single intravenous dose of 166.5 mg rolapitant, no effect on AUC and a 21% increase in the Cmax of digoxin 0.5 mg was observed [see Drug Interactions (7.4)].
Warfarin
When warfarin was administered with a single intravenous dose of 166.5 mg rolapitant, 3% and 18% increases in Cmax and AUC of S-warfarin were observed on Day 1, respectively. On Day 8, the increases were 3% for Cmax and 21% for AUC. The effect on INR or prothrombin time was not measured [see Drug Interactions (7.5)].
Substrates for other CYP enzymes
In vitro studies suggest that rolapitant is not an inhibitor of CYP1A2 and CYP2E1. In vitro studies suggest that rolapitant inhibits CYP2A6; however, a clinically meaningful drug interaction via an inhibition of CYP2A6 appears unlikely.
No clinically significant interaction was seen on the systemic exposures of the following drugs when administered with a single intravenous dose of 166.5 mg rolapitant on Day 1 and without rolapitant on Day 8: caffeine (CYP1A2 substrate; no effect on caffeine 200 mg on Days 1 and 8), and omeprazole (CYP2C19 substrate; 14% increase in Cmax and no effect on AUC of omeprazole 40 mg on Day 1; on Day 8: 15% increase in Cmax and no effect on AUC).
No clinically significant interaction was seen on the systemic exposures of the following drugs when administered with a single oral dose of 180 mg rolapitant on Day 1: repaglinide (CYP2C8 substrate; no effect on repaglinide 0.25 mg on Day 1; on Day 8: 29% and 24% increase in Cmax and AUC, respectively), efavirenz (CYP2B6 substrate; 18% decrease in Cmax and no effect on AUC of efavirenz 600 mg on Day 1; on Day 8: no effect on Cmax and 28% increase in AUC), tolbutamide (CYP2C9 substrate; no effect on tolbutamide 500 mg on Day 1 and on Day 8), or omeprazole (CYP2C19 substrate; 44% increase in Cmax and 23% increase in AUC of omeprazole 40 mg on Day 1; on Day 8: 37% and 15% increase in Cmax and AUC, respectively).
Substrates for other transporters
In vitro studies suggest that oral rolapitant is unlikely to inhibit organic anion transporting polypeptides 1B1 and 1B3 (OATP1B1 and OATP1B3), organic anion transporters 1 and 3 (OAT1 and OAT3), organic cation transporter 2 (OCT2), and multidrug and toxin extrusion proteins 1 and 2K (MATE1 and MATE2K) in vivo.
In vitro studies suggest that intravenous rolapitant is unlikely to inhibit OATP1B3, OAT1, OAT3, OCT2, and MATE2K in vivo. However, the potential for intravenous rolapitant to inhibit OATP1B1 or MATE1 in vivo cannot be ruled out.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
The carcinogenic potential of rolapitant was assessed in 2-year carcinogenicity studies in CD-1 mice and Sprague-Dawley rats. In mice, there were no drug-related neoplastic findings at doses up to 135 mg/kg per day (approximately 3.9 times the recommended intravenous human dose on a body surface area basis). In rats, there were no drug-related neoplastic findings at doses up to 90 mg/kg per day (approximately 5.2 times the recommended intravenous human dose on a body surface area basis).
Mutagenesis
Rolapitant was not genotoxic in an Ames test, a human peripheral blood lymphocyte chromosome aberration test, and a mouse micronucleus test.
Impairment of Fertility
In a fertility and early embryonic development study in female rats, rolapitant at an oral dose of 9 mg/kg per day (approximately 0.5 times the recommended intravenous human dose on a body surface area basis) caused a transient decrease in maternal body weight gain and increases in the incidence of pre- and post-implantation loss. At a rolapitant dose of 4.5 mg/kg per day (approximately 0.3 times the recommended intravenous human dose on a body surface area basis), there were slight decreases in the number of corpora lutea and implantation sites. Rolapitant did not affect the fertility or general reproductive performance of male rats at doses up to 90 mg/kg per day (approximately 5.2 times the recommended intravenous human dose on a body surface area basis). Reversibility in female rats was demonstrated in a follow-up study.
-
14 CLINICAL STUDIES
Cisplatin-Based Highly Emetogenic Chemotherapy (HEC)
In two multicenter, randomized, double-blind, parallel group, controlled clinical studies (Study 1 and Study 2), the VARUBI regimen (VARUBI tablets, granisetron and dexamethasone) was compared with control therapy (placebo, granisetron and dexamethasone) in patients receiving a chemotherapy regimen that included cisplatin >60 mg/m2. See Table 6 for the treatment regimens.
Table 6: Treatment Regimens in Studies 1 and 2 Day 1 Day 2 to 4 - * VARUBI was administered 1 to 2 hours prior to chemotherapy treatment on Day 1.
- † Dexamethasone was administered 30 minutes prior to chemotherapy on Day 1. There is no drug interaction between VARUBI and dexamethasone, so no dosage adjustment for dexamethasone is required [see Clinical Pharmacology (12.3)].
- ‡ The dose of granisetron was administered 30 minutes prior to chemotherapy on Day 1.
- § VARUBI placebo was used to maintain blinding.
VARUBI Regimen Oral VARUBI* 180 mg None Oral Dexamethasone 20 mg† 8 mg twice daily Intravenous Granisetron 10 mcg/kg‡ None Control Regimen§ Oral Dexamethasone 20 mg† 8 mg twice daily Intravenous Granisetron 10 mcg/kg‡ None Study 1
A total of 532 patients were randomized to either the VARUBI regimen (N=266) or control therapy (N=266). A total of 526 patients were included in the evaluation of efficacy. Of those randomized 42% were women, 58% men, 67% White, 23% Asian, 1% Black, and 9% multiracial/other/unknown. The proportion of patients from North America was 16%. Patients in this clinical study ranged from 20 to 90 years of age, with a mean age of 57 years. In Study 1, 26% of patients were 65 years or older, with 3% of patients being 75 years or older. The mean cisplatin dose was 77 mg/m2.
During this study, 82% of the patients received a concomitant chemotherapeutic agent in addition to protocol-mandated cisplatin. The most common concomitant chemotherapeutic agents administered during Cycle 1 were: gemcitabine (17%), paclitaxel (12%), fluorouracil (11%), etoposide (10%), vinorelbine (9%), docetaxel (9%), pemetrexed (7%), doxorubicin (6%) and cyclophosphamide (5%).
Study 2
A total of 555 patients were randomized to either the VARUBI regimen (N=278) or control therapy (N=277). A total of 544 patients were included in the evaluation of efficacy. Of those randomized 32% were women, 68% men, 81% White, 14% Asian, 1% Black, and 5% multiracial/other/unknown. The proportion of patients from North America was 7%. Patients in this clinical study ranged from 18 to 83 years of age, with a mean age of 58 years. In this study, 27% of patients were 65 years or older, with 3% of patients being 75 years or older. The mean cisplatin dose was 76 mg/m2.
During this study, 85% of the patients received a concomitant chemotherapeutic agent in addition to protocol-mandated cisplatin. The most common concomitant chemotherapeutic agents administered during Cycle 1 were: vinorelbine (16%), gemcitabine (15%), fluorouracil (12%), etoposide (11%), pemetrexed (9%), docetaxel (7%), paclitaxel (7%), epirubicin (5%) and capecitabine (4%).
The primary endpoint in both studies was complete response (defined as no emetic episodes and no rescue medication) in the delayed phase (25 to 120 hours) of chemotherapy-induced nausea and vomiting.
Moderately Emetogenic Chemotherapy (MEC) and Combinations of Anthracycline and Cyclophosphamide Chemotherapy
Study 3
In Study 3, a multicenter, randomized, double-blind, parallel group, controlled clinical study in moderately emetogenic chemotherapy, the VARUBI regimen (VARUBI tablets, granisetron and dexamethasone) was compared with control therapy (placebo, granisetron and dexamethasone) in patients receiving a moderately emetogenic chemotherapy regimen that included at least 50% of patients receiving a combination of anthracycline and cyclophosphamide. The percentage of patients who received carboplatin in Cycle 1 was 30%. Treatment regimens for the VARUBI and control arms are summarized in Table 7.
Table 7: Treatment Regimens in Study 3 Day 1 Day 2 to 3 - * VARUBI was administered 1 to 2 hours prior to chemotherapy treatment on Day 1.
- † Dexamethasone was administered 30 minutes prior to chemotherapy on Day 1. There is no drug interaction between VARUBI and dexamethasone, so no dosage adjustment for dexamethasone is required [see Clinical Pharmacology (12.3)].
- ‡ The dose of granisetron was administered 30 minutes prior to chemotherapy on Day 1.
- § VARUBI placebo was used to maintain blinding.
VARUBI Regimen Oral VARUBI* 180 mg none Oral Dexamethasone 20 mg† none Oral Granisetron 2 mg‡ 2 mg once daily Control Regimen§ Oral Dexamethasone 20 mg† none Oral Granisetron 2 mg‡ 2 mg once daily A total of 1369 patients were randomized to either the VARUBI regimen (N=684) or control therapy (N=685). A total of 1332 patients were included in the evaluation of efficacy. Of those randomized 80% were women, 20% men, 77% White, 13% Asian, 4% Black, and 6% multiracial/other/unknown. The proportion of patients from North America was 33%. Patients in this clinical study ranged from 22 to 88 years of age, with a mean age of 57 years. In this study, 28% of patients were 65 years or older, with 7% of patients being 75 years or older.
The primary endpoint was complete response (defined as no emetic episodes and no rescue medication) in the delayed phase (25 to 120 hours) of chemotherapy-induced nausea and vomiting.
A summary of the study results from HEC Studies 1 and 2 and for the MEC Study 3 is shown in Table 8.
Table 8: Percent of Patients Receiving Emetogenic Chemotherapy Responding by Treatment Group for the HEC Studies 1 and 2 and for the MEC Study 3 Endpoint HEC Study 1 HEC Study 2 MEC Study 3 VARUBI*
(N=264)
Rate (%)Control*
(N=262)
Rate (%)P-Value Treatment Difference
(95% C.I.)VARUBI*
(N=271)
Rate (%)Control*
(N=273)
Rate (%)P-Value Treatment Difference
(95% C.I.)VARUBI*
(N=666)
Rate (%)Control*
(N=666)
Rate (%)P-Value Treatment Difference
(95% C.I.)- * Granisetron and dexamethasone were used as concomitant drugs.
- † Results were obtained based on the Cochran-Mantel-Haenszel test stratified by gender.
Primary Endpoint: Complete Response in the Delayed Phase 72.7 58.4 <0.001†
14.3 (6.3, 22.4)70.1 61.9 0.043†
8.2 (0.3, 16.1)71.3 61.6 <0.001†
9.8 (4.7, 14.8)Multiple-Cycle Extension: In Studies 1, 2, and 3, patients had the option of continuing into a multiple-cycle extension for up to 5 additional cycles of chemotherapy receiving the same treatment as assigned in cycle 1. At Day 6 to 8 following initiation of chemotherapy, patients were asked to recall whether they had any episode of vomiting or retching or nausea that interfered with normal daily life. The results are summarized by study and treatment group in Figure 1 below.
Figure 1: No Emesis and No Nausea Interfering with Daily Life over Cycles 2-6
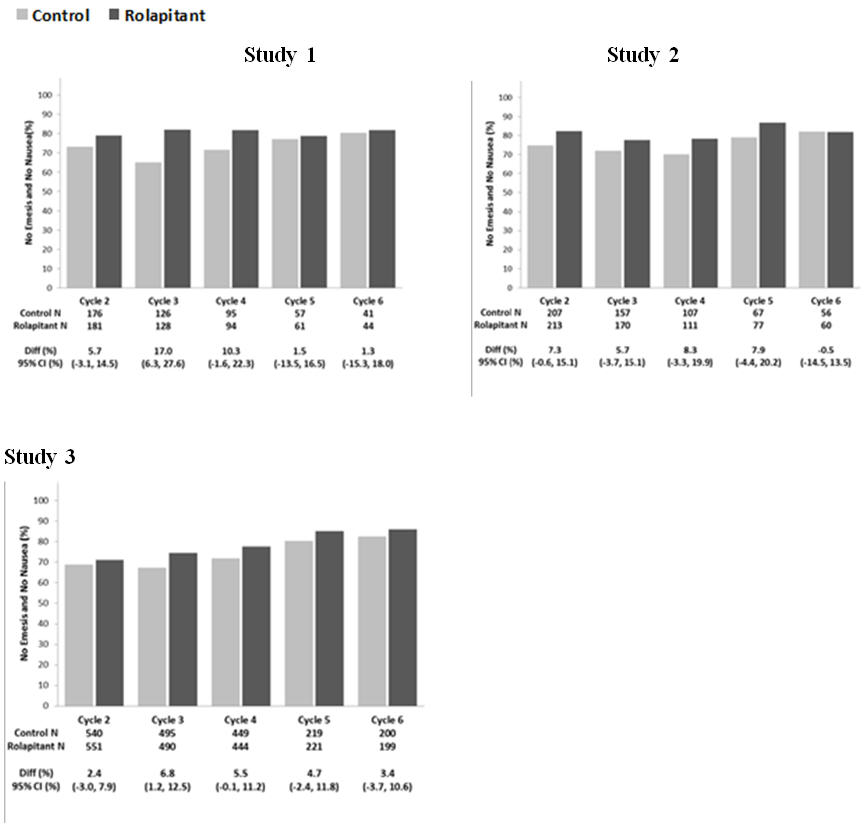
-
16 HOW SUPPLIED/STORAGE AND HANDLING
VARUBI Tablets
VARUBI tablets are available as film-coated, capsule shaped, blue tablets, debossed with T0101 on one side and 100 on the other side. Each tablet contains 90 mg rolapitant. VARUBI tablets are packaged in an Aclar blister shell with aluminum foil backing and supplied as follows:
NDC: 69656-101-02 A single dose child-resistant wallet (2 tablets as one set of twinned blisters) VARUBI Injectable Emulsion
VARUBI injectable emulsion is supplied as a sterile, translucent white homogeneous liquid that has some opalescence. Each stoppered vial delivers 166.5 mg/92.5 mL (1.8 mg/mL) rolapitant:
NDC: 69656-102-10 One single-dose vial -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Anaphylaxis, Anaphylactic Shock and Other Serious Hypersensitivity Reactions
Advise patients that hypersensitivity reactions, including anaphylaxis and anaphylactic shock, have been reported in patients taking VARUBI injectable emulsion.
Consult with patients to determine if the patient is hypersensitive to any component of the product (including soybean oil). Furthermore, as cross reactions to other allergens is possible, patients with known allergies to legumes or other related allergens should be monitored closely. Patients with a potential hypersensitivity should not be administered VARUBI injectable emulsion.
Advise patients to seek immediate medical attention if they experience signs or symptoms of a hypersensitivity reaction, such as wheezing or difficulty breathing; swelling of the face or throat; hives or flushing; itching; abdominal cramping, abdominal pain or vomiting; back pain and chest pain; hypotension or shock [see Contraindications (4) and Warnings and Precautions (5.1)].
Drug Interactions
Advise patients to tell their healthcare provider when they start or stop taking any concomitant medications. VARUBI is a moderate CYP2D6 inhibitor and can increase plasma concentrations of CYP2D6 substrates [see Contraindications (4), Warnings and Precautions (5.2), Drug Interactions (7.1)].
Infertility
Advise females of reproductive potential that VARUBI may impair fertility [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration Revised: March 2018 PATIENT INFORMATION
VARUBI® (vuh ROO bee)
(rolapitant)
tablets, for oral useVARUBI® (vuh ROO bee)
(rolapitant)
injectable emulsion, for intravenous useWhat is VARUBI?
VARUBI is a prescription medicine called an "antiemetic." VARUBI is used with other medicines in adults to help prevent nausea and vomiting that happens later with certain anti-cancer medicines (chemotherapy).
It is not known if VARUBI is safe and effective in children.
Do not take VARUBI if you:
- are allergic to rolapitant or any of the ingredients in VARUBI injectable emulsion, including soybean oil. See the end of this leaflet for a complete list of the ingredients in VARUBI.
- take thioridazine or pimozide. Taking VARUBI with thioridazine or pimozide can cause serious or life-threatening heart rhythm changes.
Before taking VARUBI, tell your doctor about all of your medical conditions, including if you:
- have liver problems.
- are pregnant or plan to become pregnant. It is not known if VARUBI will harm your unborn baby. VARUBI may affect the ability to have a child (fertility problems) in females.
- are breastfeeding or plan to breastfeed. It is not known if VARUBI passes into your breast milk or could harm your baby.
Tell your doctor about all the medicines you take or stop taking, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Keep a list of your medicines to show your doctor and pharmacist when you get a new medicine.
VARUBI and other medicines may affect each other and could cause serious side effects.
How should I take VARUBI?
- Take VARUBI exactly as your doctor tells you to take it.
- There are 2 ways your doctor may prescribe VARUBI for you:
- 1.
VARUBI tablets:
- VARUBI tablets are taken on Day 1 of chemotherapy. Take 2 VARUBI tablets by mouth within 2 hours before you receive your anti-cancer medicine (chemotherapy).
- VARUBI tablets can be taken with or without food.
- Do not take VARUBI more than 1 time every 14 days.
- If you take too much VARUBI, call your doctor or go to the nearest hospital emergency room right away.
- 1.
VARUBI tablets:
- 2.
VARUBI injectable emulsion:
- VARUBI injectable emulsion is given on Day 1 of chemotherapy. It will be given to you in your vein (intravenously) over 30 minutes, within 2 hours before you receive your anti-cancer medicine (chemotherapy).
What are the possible side effects of VARUBI?
VARUBI may cause serious side effects, including:
- Serious allergic reactions. Allergic reactions can happen with VARUBI injectable emulsion and may be serious. Tell your doctor or nurse right away if you experience wheezing or difficulty breathing; swelling of the face or throat; hives or flushing; itching; abdominal cramping, abdominal pain or vomiting; back pain or chest pain; or feeling faint during or soon after you receive VARUBI injectable emulsion, as you may need emergency medical care.
- Change in the level of some medicines in your blood. Serious or life-threatening reactions, including heart rhythm changes, may occur if VARUBI is used with certain other medicines. You should not take VARUBI if you take pimozide.
The most common side effects of VARUBI in people who take VARUBI and receive Cisplatin chemotherapy medicine include: low white blood cell count, hiccups, and stomach (abdominal) pain.
The most common side effects of VARUBI in people who take VARUBI and receive Anthracycline and Cyclophosphamide chemotherapy medicines include: decreased appetite, low white blood cell count, dizziness, indigestion, urinary tract infection, mouth sores, and low red blood cell count.
These are not all the possible side effects of VARUBI. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store VARUBI?
VARUBI tablets come in a child-resistant package.
Store VARUBI at room temperature between 68°F to 77°F (20°C to 25°C).
Keep VARUBI and all medicines out of the reach of children.
General Information about the safe and effective use of VARUBI
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use VARUBI for a condition for which it was not prescribed. Do not give VARUBI to other people, even if they have the same symptoms that you have. It may harm them. You can ask your doctor or pharmacist for information about VARUBI that is written for health professionals.
What are the ingredients in VARUBI?
Active ingredient: rolapitant
Inactive ingredients (tablet): colloidal silicon dioxide, croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone, and pregelatinized starch. The tablets are coated in non-functional blue and clear coats. The tablet coating comprises the following inactive ingredients: FD&C Blue No. 2-Indigo Carmine Lake, polyethylene glycol, polysorbate 80, polyvinyl alcohol, talc, and titanium dioxide.
Inactive ingredients (injectable emulsion): dibasic sodium phosphate, anhydrous (2.8 mg/mL), medium chain triglycerides (11 mg/mL), polyoxyl 15 hydroxystearate (44 mg/mL), sodium chloride (6.2 mg/mL), soybean oil (6.6 mg/mL), water for injection and may contain hydrochloric acid and/or sodium hydroxide to adjust pH to 7.0 to 8.0.
Manufactured for: TESARO, Inc. 1000 Winter St., Waltham, MA 02451 For more information, go to www.tesarobio.com or call 1-844-483-7276.
-
PRINCIPAL DISPLAY PANEL - 90 mg Tablet Blister Pack Wallet
NDC: 69656-101-02
Single dose
180 mg total dose (2 x 90 mg tablets)VARUBI®
(rolapitant) tablets90 mg* per tablet
Contains two 90 mg tablets
equal to a single dose of 180 mg*(equivalent to 100 mg rolapitant hydrochloride)
Rx only
For oral use.
Keep this and all drugs out of the reach of
children. This package is child-resistant.Store at 20°C - 25°C (68°F - 77°F);
excursions permitted to 15°C - 30°C
(59°F - 86°F) [see USP Controlled
Room Temperature].TESARO®
Manufactured for TESARO, Inc.
Waltham, MA 02451 USASee package insert for dosage information.

- PRINCIPAL DISPLAY PANEL - NDC: 69656-101-02 - Blister Backing Label
-
PRINCIPAL DISPLAY PANEL - 92.5 mL Vial Label
NDC: 69656-102-10 Rx only
VARUBI®
(rolapitant)
injectable emulsion166.5 mg/92.5 mL (1.8 mg/mL)
Each vial contains 166.5 mg rolapitant
(equivalent to 185 mg rolapitant hydrochloride)For Intravenous Use Only
One single-dose vialSterile
Ready to useNet Content: 92.5 mL
TESARO®
Manufactured for
TESARO, Inc., Waltham, MA 02451 USA
-
INGREDIENTS AND APPEARANCE
VARUBI
rolapitant tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 69656-101 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ROLAPITANT HYDROCHLORIDE (UNII: 57O5S1QSAQ) (rolapitant - UNII:NLE429IZUC) rolapitant 90 mg Inactive Ingredients Ingredient Name Strength lactose monohydrate (UNII: EWQ57Q8I5X) starch, corn (UNII: O8232NY3SJ) microcrystalline cellulose (UNII: OP1R32D61U) povidone, unspecified (UNII: FZ989GH94E) croscarmellose sodium (UNII: M28OL1HH48) silicon dioxide (UNII: ETJ7Z6XBU4) magnesium stearate (UNII: 70097M6I30) polyvinyl alcohol, unspecified (UNII: 532B59J990) titanium dioxide (UNII: 15FIX9V2JP) polyethylene glycol, unspecified (UNII: 3WJQ0SDW1A) talc (UNII: 7SEV7J4R1U) FD&C Blue NO. 2 (UNII: L06K8R7DQK) polysorbate 80 (UNII: 6OZP39ZG8H) Product Characteristics Color BLUE Score no score Shape OVAL Size 15mm Flavor Imprint Code T0101;100 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 69656-101-02 2 in 1 BLISTER PACK; Type 0: Not a Combination Product 10/07/2015 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA206500 10/07/2015 VARUBI
rolapitant injection, emulsionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 69656-102 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength rolapitant (UNII: NLE429IZUC) (rolapitant - UNII:NLE429IZUC) rolapitant 1.8 mg in 1 mL Inactive Ingredients Ingredient Name Strength peg-15 hydroxystearates (UNII: 71YMM1X75O) 44 mg in 1 mL medium-chain triglycerides (UNII: C9H2L21V7U) 11 mg in 1 mL soybean oil (UNII: 241ATL177A) 6.6 mg in 1 mL sodium chloride (UNII: 451W47IQ8X) 6.2 mg in 1 mL sodium phosphate, dibasic, anhydrous (UNII: 22ADO53M6F) 2.8 mg in 1 mL water (UNII: 059QF0KO0R) hydrochloric acid (UNII: QTT17582CB) sodium hydroxide (UNII: 55X04QC32I) Product Characteristics Color WHITE (translucent white) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 69656-102-10 1 in 1 CARTON 10/25/2017 1 92.5 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208399 10/25/2017 Labeler - Tesaro, Inc. (963385559)

Trademark Results [Varubi]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 VARUBI 86591285 4893721 Live/Registered |
Tesaro, Inc. 2015-04-08 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.