ZYDELIG- idelalisib tablet, film coated
Zydelig by
Drug Labeling and Warnings
Zydelig by is a Prescription medication manufactured, distributed, or labeled by Gilead Sciences, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ZYDELIG safely and effectively. See full prescribing information for ZYDELIG.
ZYDELIG® (idelalisib) tablets, for oral use
Initial U.S. Approval: 2014WARNING: FATAL AND SERIOUS TOXICITIES: HEPATIC, SEVERE DIARRHEA, COLITIS, PNEUMONITIS, INFECTIONS, and INTESTINAL PERFORATION
See full prescribing information for complete boxed warning.
- Fatal and/or serious hepatotoxicity occurred in 16% to 18% of Zydelig-treated patients. Monitor hepatic function prior to and during treatment. Interrupt and then reduce or discontinue Zydelig. (5.1)
- Fatal and/or serious and severe diarrhea or colitis occurred in 14% to 20% of Zydelig-treated patients. Monitor for the development of severe diarrhea or colitis. Interrupt and then reduce or discontinue Zydelig. (5.2)
- Fatal and/or serious pneumonitis occurred in 4% of Zydelig-treated patients. Monitor for pulmonary symptoms and bilateral interstitial infiltrates. Interrupt or discontinue Zydelig. (5.3)
- Fatal and/or serious infections occurred in 21% to 48% of Zydelig-treated patients. Monitor for signs and symptoms of infection. Interrupt Zydelig if infection is suspected. (5.4)
- Fatal and serious intestinal perforation can occur in Zydelig-treated patients across clinical trials. Discontinue Zydelig if intestinal perforation is suspected. (5.5)
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
Zydelig is a kinase inhibitor indicated for the treatment of patients with:
- Relapsed chronic lymphocytic leukemia (CLL), in combination with rituximab, in patients for whom rituximab alone would be considered appropriate therapy due to other co-morbidities. (1.1)
- Relapsed follicular B-cell non-Hodgkin lymphoma (FL) in patients who have received at least two prior systemic therapies. (1.2)
- Relapsed small lymphocytic lymphoma (SLL) in patients who have received at least two prior systemic therapies. (1.3)
Limitation of use:
Zydelig is not indicated and is not recommended for first-line treatment of any patient. (1.1, 1.2, 1.3)
Zydelig is not indicated and is not recommended in combination with bendamustine and/or rituximab for the treatment of FL. (1.2)
Accelerated approval was granted for FL and SLL based on overall response rate. Improvement in patient survival or disease related symptoms has not been established. Continued approval for these indications may be contingent upon verification of clinical benefit in confirmatory trials.
DOSAGE AND ADMINISTRATION
Recommended starting dose: 150 mg orally, twice daily. (2.1)
DOSAGE FORMS AND STRENGTHS
Tablets: 150 mg, 100 mg. (3)
CONTRAINDICATIONS
History of serious allergic reactions including anaphylaxis and toxic epidermal necrolysis. (4)
WARNINGS AND PRECAUTIONS
- Severe cutaneous reactions: Monitor patients for the development of severe cutaneous reactions and discontinue Zydelig. (5.6)
- Anaphylaxis: Monitor patients for anaphylaxis and discontinue Zydelig. (5.7)
- Neutropenia: monitor blood counts. (5.8)
- Embryo-fetal toxicity: Zydelig may cause fetal harm. Advise women of potential risk to a fetus and to avoid pregnancy while taking Zydelig. (5.9, 8.1, 8.3)
ADVERSE REACTIONS
The most common adverse reactions (incidence ≥20%) in patients treated with Zydelig in the monotherapy trial are diarrhea, fatigue, nausea, cough, pyrexia, abdominal pain, pneumonia, and rash. (6.1)
The most common adverse reactions (incidence ≥30%) in patients treated with Zydelig in combination trials are diarrhea, pneumonia, pyrexia, fatigue, rash, cough, and nausea. (6.1)
Common laboratory abnormalities include neutropenia, ALT elevations and AST elevations. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Gilead Sciences, Inc. at 1-800-GILEAD-5 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 10/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: FATAL AND SERIOUS TOXICITIES: HEPATIC, SEVERE DIARRHEA, COLITIS, PNEUMONITIS, INFECTIONS, and INTESTINAL PERFORATION
1 INDICATIONS AND USAGE
1.1 Relapsed Chronic Lymphocytic Leukemia
1.2 Relapsed Follicular B-cell non-Hodgkin Lymphoma
1.3 Relapsed Small Lymphocytic Lymphoma
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dose Modification
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
5.2 Severe Diarrhea or Colitis
5.3 Pneumonitis
5.4 Infections
5.5 Intestinal Perforation
5.6 Severe Cutaneous Reactions
5.7 Anaphylaxis
5.8 Neutropenia
5.9 Embryo-fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on Zydelig
7.2 Effects of Zydelig on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Relapsed Chronic Lymphocytic Leukemia
14.2 Relapsed Follicular B-cell non-Hodgkin Lymphoma
14.3 Relapsed Small Lymphocytic Lymphoma
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: FATAL AND SERIOUS TOXICITIES: HEPATIC, SEVERE DIARRHEA, COLITIS, PNEUMONITIS, INFECTIONS, and INTESTINAL PERFORATION
Fatal and/or serious hepatotoxicity occurred in 16% to 18% of Zydelig-treated patients. Monitor hepatic function prior to and during treatment. Interrupt and then reduce or discontinue Zydelig as recommended [see Dosage and Administration (2.2), Warnings and Precautions (5.1)].
Fatal and/or serious and severe diarrhea or colitis occurred in 14% to 20% of Zydelig-treated patients. Monitor for the development of severe diarrhea or colitis. Interrupt and then reduce or discontinue Zydelig as recommended [see Dosage and Administration (2.2), Warnings and Precautions (5.2)].
Fatal and/or serious pneumonitis occurred in 4% of Zydelig-treated patients. Monitor for pulmonary symptoms and bilateral interstitial infiltrates. Interrupt or discontinue Zydelig as recommended [see Dosage and Administration (2.2), Warnings and Precautions (5.3)].
Fatal and/or serious infections occurred in 21% to 48% of Zydelig-treated patients. Monitor for signs and symptoms of infection. Interrupt Zydelig if infection is suspected [see Dosage and Administration (2.2), Warnings and Precautions (5.4)].
Fatal and serious intestinal perforation can occur in Zydelig-treated patients across clinical trials. Discontinue Zydelig for intestinal perforation [see Warnings and Precautions (5.5)].
-
1 INDICATIONS AND USAGE
1.1 Relapsed Chronic Lymphocytic Leukemia
Zydelig is indicated, in combination with rituximab, for the treatment of patients with relapsed chronic lymphocytic leukemia (CLL) for whom rituximab alone would be considered appropriate therapy due to other co-morbidities.
1.2 Relapsed Follicular B-cell non-Hodgkin Lymphoma
Zydelig is indicated for the treatment of patients with relapsed follicular B-cell non-Hodgkin lymphoma (FL) who have received at least two prior systemic therapies.
Accelerated approval was granted for this indication based on Overall Response Rate [see Clinical Studies (14.2)]. An improvement in patient survival or disease related symptoms has not been established. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trials.
1.3 Relapsed Small Lymphocytic Lymphoma
Zydelig is indicated for the treatment of patients with relapsed small lymphocytic lymphoma (SLL) who have received at least two prior systemic therapies.
Accelerated approval was granted for this indication based on Overall Response Rate [see Clinical Studies (14.3)]. An improvement in patient survival or disease related symptoms has not been established. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trials.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended maximum starting dose of Zydelig is 150 mg administered orally twice daily.
Zydelig can be taken with or without food. Tablets should be swallowed whole.
Continue treatment until disease progression or unacceptable toxicity. The optimal and safe dosing regimen for patients who receive treatment longer than several months is unknown.
2.2 Dose Modification
See Table 1 for dose modification instructions for specific toxicities related to Zydelig. For other severe or life-threatening toxicities related to Zydelig, withhold drug until toxicity is resolved. If resuming Zydelig after interruption for other severe or life-threatening toxicities, reduce the dose to 100 mg twice daily. Discontinue Zydelig permanently for recurrence of other severe or life-threatening Zydelig-related toxicity upon rechallenge.
Table 1 Dose Modifications for Toxicities Due to Zydelig Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; BID, twice daily; ULN, upper limit of normal; CMV, cytomegalovirus; PCR: polymerase chain reaction; PJP: Pneumocystis jirovecii pneumonia - * Moderate diarrhea: increase of 4–6 stools per day over baseline; severe diarrhea: increase of ≥7 stools per day over baseline.
Pneumonitis Any symptomatic pneumonitis Discontinue Zydelig in patients with any severity of symptomatic pneumonitis ALT/AST >3–5 × ULN >5–20 × ULN >20 × ULN Maintain Zydelig dose. Monitor at least weekly until ≤1 × ULN. Withhold Zydelig.
Monitor at least weekly until ALT/AST are ≤1 × ULN, then may resume Zydelig at 100 mg BID.Discontinue Zydelig permanently. Bilirubin >1.5–3 × ULN >3–10 × ULN >10 × ULN Maintain Zydelig dose. Monitor at least weekly until ≤1 × ULN. Withhold Zydelig. Monitor at least weekly until bilirubin is ≤1 × ULN, then may resume Zydelig at 100 mg BID. Discontinue Zydelig permanently. Diarrhea* Moderate diarrhea Severe diarrhea or hospitalization Life-threatening diarrhea Maintain Zydelig dose. Monitor at least weekly until resolved. Withhold Zydelig. Monitor at least weekly until resolved, then may resume Zydelig at 100 mg BID. Discontinue Zydelig permanently. Neutropenia ANC 1.0 to <1.5 Gi/L ANC 0.5 to <1.0 Gi/L ANC <0.5 Gi/L Maintain Zydelig dose. Maintain Zydelig dose. Monitor ANC at least weekly. Interrupt Zydelig. Monitor ANC at least weekly until ANC ≥0.5 Gi/L, then may resume Zydelig at 100 mg BID. Thrombocytopenia Platelets 50 to <75 Gi/L Platelets 25 to <50 Gi/L Platelets <25 Gi/L Maintain Zydelig dose. Maintain Zydelig dose. Monitor platelet counts at least weekly. Interrupt Zydelig. Monitor platelet count at least weekly. May resume Zydelig at 100 mg BID when platelets ≥25 Gi/L. Infections Grade 3 or higher sepsis or pneumonia Interrupt Zydelig until infection has resolved. Evidence of CMV infection or viremia Interrupt Zydelig in patients with evidence of active CMV infection of any grade or viremia (positive PCR or antigen test) until the viremia has resolved. If Zydelig is resumed, monitor patients by PCR or antigen test for CMV reactivation at least monthly. Evidence of PJP infection Interrupt Zydelig in patients with suspected PJP infection of any grade. Permanently discontinue Zydelig if PJP infection is confirmed. No dose modification is required for lymphocytosis, which has been observed in some patients taking Zydelig. This observed lymphocytosis is a pharmacodynamic effect and should not be considered progressive disease in the absence of other clinical findings.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
History of serious allergic reactions including anaphylaxis and toxic epidermal necrolysis [see Warnings and Precautions (5.6, 5.7)] .
-
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
Fatal and/or serious hepatotoxicity occurred in 18% of patients treated with Zydelig monotherapy and 16% of patients treated with Zydelig in combination with rituximab or with unapproved combination therapies. Elevations in ALT or AST greater than 5 times the upper limit of normal have occurred [see Adverse Reactions (6.1)]. These findings were generally observed within the first 12 weeks of treatment and were reversible with dose interruption. After resumption of treatment at a lower dose, 26% of patients had recurrence of ALT and AST elevations. Discontinue Zydelig for recurrent hepatotoxicity.
Avoid concurrent use of Zydelig with other drugs that may cause liver toxicity.
Monitor ALT and AST in all patients every 2 weeks for the first 3 months of treatment, every 4 weeks for the next 3 months, then every 1 to 3 months thereafter. Monitor weekly for liver toxicity if the ALT or AST rises above 3 times the upper limit of normal until resolved. Withhold Zydelig if the ALT or AST is greater than 5 times the upper limit of normal, and continue to monitor AST, ALT and total bilirubin weekly until the abnormality is resolved [see Dosage and Administration (2.2)].
5.2 Severe Diarrhea or Colitis
Severe diarrhea or colitis (Grade 3 or higher) occurred in 14% of patients treated with Zydelig monotherapy and 20% of patients treated with Zydelig in combination with rituximab or with unapproved combination therapies [see Adverse Reactions (6.1)]. Diarrhea can occur at any time. Avoid concurrent use of Zydelig and other drugs that cause diarrhea. Diarrhea due to Zydelig responds poorly to antimotility agents. Median time to resolution ranged between 1 week and 1 month across trials, following interruption of Zydelig therapy and in some instances, use of corticosteroids [see Dosage and Administration (2.2)].
5.3 Pneumonitis
Fatal and serious pneumonitis occurred in patients treated with Zydelig. Clinical manifestations included interstitial infiltrates and organizing pneumonia. In randomized clinical trials of combination therapies, pneumonitis occurred in 4% of patients treated with Zydelig compared to 1% on the comparator arms. Time to onset of pneumonitis ranged from <1 to 15 months. Monitor patients on Zydelig for pulmonary symptoms. In patients taking Zydelig who present with pulmonary symptoms such as cough, dyspnea, hypoxia, interstitial infiltrates on a radiologic exam, or a decline by more than 5% in oxygen saturation, interrupt Zydelig until the etiology has been determined. If symptomatic pneumonitis or organizing pneumonia is diagnosed, initiate appropriate treatment with corticosteroids and permanently discontinue Zydelig [see Dosage and Administration (2.2)].
5.4 Infections
Fatal and/or serious infections occurred in 21% of patients treated with Zydelig monotherapy and 48% of patients treated with Zydelig in combination with rituximab or with unapproved combination therapies [see Adverse Reactions (6.1)]. The most common infections were pneumonia, sepsis, and febrile neutropenia. Treat infections prior to initiation of Zydelig therapy. Monitor patients on Zydelig for signs and symptoms of infection, and interrupt Zydelig for Grade 3 or higher infection [see Dosage and Administration (2.2)].
Serious or fatal Pneumocystis jirovecii pneumonia (PJP) or cytomegalovirus (CMV) occurred in <1% of patients treated with Zydelig. Provide PJP prophylaxis during treatment with Zydelig. Interrupt Zydelig in patients with suspected PJP infection of any grade, and permanently discontinue Zydelig if PJP infection of any grade is confirmed. Regular clinical and laboratory monitoring for CMV infection is recommended in patients with history of CMV infection or positive CMV serology at the start of treatment with Zydelig. Interrupt Zydelig in the setting of positive CMV PCR or antigen test until the viremia has resolved. If Zydelig is subsequently resumed, patients should be monitored by PCR or antigen test for CMV reactivation at least monthly [see Dosage and Administration (2.2)].
5.5 Intestinal Perforation
Fatal and serious intestinal perforation occurred in Zydelig-treated patients. At the time of perforation, some patients had moderate to severe diarrhea. Advise patients to promptly report any new or worsening abdominal pain, chills, fever, nausea, or vomiting. Discontinue Zydelig permanently in patients who experience intestinal perforation.
5.6 Severe Cutaneous Reactions
Fatal cases of Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) have occurred in patients treated with Zydelig. If SJS or TEN is suspected, interrupt Zydelig until the etiology of the reaction has been determined. If SJS or TEN is confirmed, permanently discontinue Zydelig.
Other severe or life-threatening (Grade ≥3) cutaneous reactions, including dermatitis exfoliative, rash, rash erythematous, rash generalized, rash macular, rash maculo-papular, rash papular, rash pruritic, exfoliative rash, and skin disorder, have been reported in Zydelig-treated patients. Monitor patients for the development of severe cutaneous reactions and discontinue Zydelig.
5.7 Anaphylaxis
Serious allergic reactions, including anaphylaxis, have been reported in patients on Zydelig. In patients who develop serious allergic reactions, discontinue Zydelig permanently and institute appropriate supportive measures.
5.8 Neutropenia
Treatment-emergent Grade 3 or 4 neutropenia occurred in 25% of patients treated with Zydelig monotherapy and 58% of patients treated with Zydelig in combination with rituximab or with unapproved combination therapies. Monitor blood counts at least every 2 weeks for the first 6 months of therapy, and at least weekly in patients while neutrophil counts are less than 1.0 Gi/L [see Dosage and Administration (2.2)].
5.9 Embryo-fetal Toxicity
Based on findings in animals and its mechanism of action, Zydelig may cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of idelalisib to pregnant rats during organogenesis caused decreased fetal weight and congenital malformations at systemic exposures 12 times those reported in patients at the recommended dose of 150 mg twice daily. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment and for at least 1 month after the last dose. [see Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1), and Nonclinical Toxicology (13.1)].
-
6 ADVERSE REACTIONS
The following serious adverse reactions have been associated with Zydelig in clinical trials and are discussed in greater detail in other sections of the prescribing information.
- Hepatotoxicity [see Warnings and Precautions (5.1)]
- Severe Diarrhea or Colitis [see Warnings and Precautions (5.2)]
- Pneumonitis [see Warnings and Precautions (5.3)]
- Infections [see Warnings and Precautions (5.4)]
- Intestinal Perforation [see Warnings and Precautions (5.5)]
- Severe Cutaneous Reactions [see Warnings and Precautions (5.6)]
- Anaphylaxis [see Warnings and Precautions (5.7)]
- Neutropenia [see Warnings and Precautions (5.8)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Summary of Clinical Trials in Chronic Lymphocytic Leukemia
The safety data reflect exposure to Zydelig from two randomized, double-blind clinical trials (Studies 312-0116 and 312-0115) in 634 patients with relapsed CLL [see Clinical Studies (14.1)] and one randomized, open-label trial in 259 patients with relapsed CLL (Study 312-0119).
Zydelig with Rituximab (Study 312-0116; NCT01539512)
Patients with relapsed CLL received up to 8 doses of rituximab (R) with or without Zydelig 150 mg twice daily. The median duration of exposure to Zydelig was 8 months.
Serious adverse reactions were reported in 65 (59%) patients treated with Zydelig + R The most frequent serious adverse reactions reported for patients treated with Zydelig + R were pneumonia (23%), diarrhea (10%), pyrexia (9%), sepsis (8%), and febrile neutropenia (5%). Adverse reactions that led to discontinuation of Zydelig occurred in 19 (17%) patients. The most common adverse reactions that led to treatment discontinuations were hepatotoxicity and diarrhea/colitis.
Forty-two (38%) patients had dose interruptions and sixteen (15%) patients had dose reductions due to adverse reactions or laboratory abnormalities. The most common reasons for dose interruptions or reductions were pneumonia, diarrhea or colitis, rash, and elevated transaminases.
Table 2 and Table 3 summarize common adverse reactions and laboratory abnormalities reported for Zydelig + R and placebo + R arms.
Table 2 Adverse Reactions Reported in ≥5% of Patients with CLL and Occurred at ≥2% Higher Incidence in Patients Receiving Zydelig in Study 312-0116 Zydelig + R
N=110 (%)Placebo + R
N=108 (%)Adverse Reaction Any Grade Grade ≥3 Any Grade Grade ≥3 - * Diarrhea includes the following preferred terms: diarrhea, colitis.
- † Abdominal pain includes the following preferred terms: abdominal pain, abdominal pain upper, abdominal pain lower.
- ‡ Pneumonia includes the terms: pneumonia, pneumonitis, lung infection, lung infiltration, pneumocystis jiroveci pneumonia, pneumonia legionella, lung infection pseudomonal, pneumonia fungal, respiratory tract infection, lower respiratory tract infection, and lower respiratory tract infection bacterial.
- § Rash includes the following preferred terms: dermatitis exfoliative, drug eruption, rash, rash erythematous, rash generalized, rash macular, rash maculo-papular, rash papular, rash pruritic, rash morbilliform, and exfoliative rash.
- ¶ Sepsis includes the terms: sepsis, septic shock, neutropenic sepsis, and sepsis syndrome
General disorders and administration site conditions pyrexia 44 (40) 3 (3) 20 (19) 1 (1) chills 27 (25) 2 (2) 17 (16) 0 pain 8 (7) 0 1 (1) 0 Gastrointestinal disorders diarrhea * 35 (32) 12 (11) 20 (19) 0 nausea 30 (27) 1 (1) 25 (23) 0 abdominal pain † 20 (18) 1 (1) 17 (16) 2 (2) vomiting 17 (15) 0 9 (8) 0 gastroesophageal reflux disease 11 (10) 1 (1) 0 0 stomatitis 7 (6) 2 (2) 1 (1) 0 Respiratory, thoracic, and mediastinal disorders pneumonia ‡ 33 (30) 23 (21) 20 (19) 14 (13) Skin and subcutaneous tissue disorders rash § 27 (25) 4 (4) 7 (6) 1 (1) Metabolism and Nutrition Disorders decreased appetite 18 (16) 2 (2) 12 (11) 2 (2) dehydration 7 (6) 3 (3) 0 0 Infections and infestations sepsis ¶ 10 (9) 10 (9) 4 (4) 4 (4) sinusitis 9 (8) 0 6 (6) 0 urinary tract infection 9 (8) 1 (1) 4 (4) 2 (2) bronchitis 8 (7) 1 (1) 5 (5) 1 (1) oral herpes 6 (5) 1 (1) 3 (3) 0 Psychiatric disorders insomnia 10 (9) 0 7 (6) 0 Musculoskeletal and connective tissue disorders arthralgia 9 (8) 1 (1) 4 (4) 0 Nervous system disorders lethargy 6 (5) 0 2 (2) 0 Table 3 Hematologic and Hepatic Laboratory Abnormalities Reported in ≥10% of Patients with CLL and Occurred at ≥5% Higher Incidence in Patients Receiving Zydelig in Study 312-0116 Zydelig + R
N=110 (%)Placebo + R
N=108 (%)Laboratory Parameter Any Grade Grade 3–4 Any Grade Grade 3–4 Hematology abnormalities neutropenia 71 (65) 46 (42) 61 (56) 33 (31) leukopenia 34 (31) 9 (8) 25 (23) 9 (8) lymphocytopenia 23 (21) 11 (10) 13 (12) 4 (4) Serum chemistry abnormalities ALT increased 43 (39) 10 (9) 13 (12) 1 (1) AST increased 31 (28) 6 (5) 16 (15) 0 After closure of Study 312-0116, 71 patients continued treatment with Zydelig on an extension study (Study 312-0117; NCT01539291). The median duration of exposure was 18 months. Serious adverse reactions occurred in 48 (68%) patients. The most frequent serious adverse reactions reported were pneumonia (30%), diarrhea (15%), and pyrexia (11%).
The most frequent adverse reactions were pneumonia (51%), pyrexia (46%), and cough (45%). The most frequent Grade 3 or greater adverse reactions were pneumonia (30%), diarrhea (15%), and sepsis (10%).
Zydelig with Ofatumumab (Study 312-0119; NCT01659021)
In Study 312-0119, 259 patients with relapsed CLL received up to 12 doses of ofatumumab with or without Zydelig 150 mg twice daily. The median duration of exposure to Zydelig was 13.9 months.
Serious adverse reactions were reported in 133 (77%) patients treated with Zydelig + ofatumumab. The most frequent serious adverse reactions reported were pneumonia (14%), pyrexia (13%), and diarrhea (12%).
Adverse reactions that led to discontinuation of Zydelig occurred in 71 (41%) patients.
One hundred and ten (64%) patients had dose interruptions and 42 (24%) patients had dose reductions due to adverse reactions or laboratory abnormalities. The most common reasons for dose discontinuations, reductions, or interruptions were diarrhea and colitis. The most common adverse reactions were diarrhea (55%), pyrexia (38%), nausea (34%), and fatigue (34%).
Zydelig with Bendamustine and Rituximab (Study 312-0115; NCT01569295)
In Study 312-0115, patients with relapsed CLL received up to 6 cycles of bendamustine and rituximab (BR) with or without Zydelig 150 mg twice daily. The median duration of exposure to Zydelig was 18.2 months.
Serious adverse reactions were reported in 147 (71%) patients treated with Zydelig + BR. The most frequent serious adverse reactions reported for patients treated with Zydelig + BR were febrile neutropenia (21%), pneumonia (17%), pyrexia (12%), and diarrhea (6%).
Adverse reactions that led to discontinuation of Zydelig occurred in 68 (33%) patients. The most common adverse reactions that led to treatment discontinuations were pneumonia, diarrhea, and pyrexia.
One hundred twenty-two (59%) patients treated with Zydelig + BR had dose interruptions and 34 (16%) patients had dose reductions due to adverse reactions. The most common reasons for dose interruptions or reductions were increased ALT and diarrhea. The most common adverse reactions were neutropenia (64%), pyrexia (43%), and diarrhea (41%).
Summary of Clinical Trials in Indolent Non-Hodgkin Lymphoma
The safety data reflect exposure to Zydelig from three open-label clinical trials (Studies 101-09 (NCT01282424), 101-02 (NCT00710528), and 101-10 (NCT01306643) in 146 patients with indolent non-Hodgkin lymphoma (iNHL) treated with Zydelig 150 mg twice daily [see Clinical Studies (14.2, 14.3)]. The median duration of exposure was 6.1 months (range 0.3 to 26.4 months).
Serious adverse reactions were reported in 73 (50%) patients. The most frequent serious adverse reactions that occurred were pneumonia (15%), diarrhea (11%), and pyrexia (9%).
Adverse reactions resulted in interruption or discontinuation for 78 (53%) patients. The most common reasons for interruption or discontinuations were diarrhea (11%), pneumonia (11%), and elevated transaminases (10%).
Table 4 provides the adverse reactions occurring in at least 10% of patients receiving Zydelig monotherapy, and Table 5 provides the hematologic and hepatic laboratory abnormalities.
Table 4 Adverse Reactions Reported in ≥ 10% of Patients with Indolent NHL Treated with Zydelig 150 mg BID Zydelig Monotherapy
N=146 (%)Adverse Reaction Any Grade Grade ≥3 - * Diarrhea includes the following preferred terms: diarrhea, colitis, enterocolitis, and gastrointestinal inflammation.
- † Abdominal pain includes the following preferred terms: abdominal pain, abdominal pain upper, abdominal pain lower, and abdominal discomfort.
- ‡ Pneumonia includes the terms: pneumonia, pneumonitis, interstitial lung disease, lung infiltration, pneumonia aspiration, respiratory tract infection, atypical pneumonia, lung infection, pneumocystis jiroveci pneumonia, bronchopneumonia, pneumonia necrotizing, lower respiratory tract infection, pneumonia pneumococcal, pneumonia staphylococcal, pneumonia streptococcal, pneumonia cytomegaloviral, and respiratory syncytial virus infection.
- § Rash includes the following preferred terms: dermatitis exfoliative, rash, rash erythematous, rash macular, rash maculo-papular, rash pruritic, and exfoliative rash.
Gastrointestinal disorders diarrhea * 68 (47) 20 (14) nausea 42 (29) 2 (1) abdominal pain † 38 (26) 3 (2) vomiting 22 (15) 2 (1) General disorders and administration site conditions fatigue 44 (30) 2 (1) pyrexia 41 (28) 3 (2) asthenia 17 (12) 3 (2) peripheral edema 15 (10) 3 (2) Respiratory, thoracic, and mediastinal disorders cough 42 (29) 1 (1) pneumonia ‡ 37 (25) 23 (16) dyspnea 25 (17) 6 (4) Skin and subcutaneous disorders rash § 31 (21) 4 (3) night sweats 18 (12) 0 Metabolism and nutrition disorders decreased appetite 24 (16) 1 (1) Infections and infestations upper respiratory tract infection 18 (12) 0 Psychiatric disorders insomnia 17 (12) 0 Nervous system disorders headache 16 (11) 1 (1) Table 5 Hematologic and Hepatic Laboratory Abnormalities in Patients with Indolent non-Hodgkin Lymphoma Treated with Zydelig 150 mg BID Zydelig Monotherapy
N=146 (%)Laboratory Abnormality Any Grade Grade 3 Grade 4 Grades were obtained per CTCAE version 4.03. Serum chemistry abnormalities ALT increased 73 (50) 20 (14) 7 (5) AST increased 60 (41) 12 (8) 6 (4) Hematology abnormalities neutrophils decreased 78 (53) 20 (14) 16 (11) hemoglobin decreased 41 (28) 3 (2) 0 platelets decreased 38 (26) 4 (3) 5 (3) Summary of Discontinued Clinical Trials in First-Line CLL and Early Line iNHL
Safety data described below reflect exposure to Zydelig in three randomized, double-blind clinical trials (Studies 312-0123, 313-0124, and 313-0125) in patients with CLL and iNHL.
In Study 312-0123 (NCT01980888), 311 patients with previously untreated CLL received up to 6 cycles of BR with or without Zydelig 150 mg twice daily.
In Study 313-0124 (NCT01732913), 295 patients with previously treated iNHL received 8 doses of R with or without Zydelig 150 mg twice daily. Patients had a median of one prior therapy.
In Study 313-0125 (NCT01732926), 475 patients with previously treated iNHL received up to 6 cycles of BR with or without Zydelig 150 mg twice daily. Patients had a median of two prior therapies.
These three studies were terminated early due to a higher incidence of fatal and/or serious adverse reactions observed in patients treated with Zydelig in combination with R or BR. The most frequent serious adverse reactions were in the system organ classes of infections and infestations, blood and lymphatic system disorders, and gastrointestinal disorders.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of Zydelig. Because postmarketing reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Skin and Subcutaneous Disorders - Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN)
-
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on Zydelig
Table 6 lists the potential effects of the coadministration of strong CYP3A modulators on Zydelig.
Table 6 Drug Interactions with Zydelig that affect Idelalisib Concentrations Strong CYP3A Inhibitors Clinical Impact - Coadministration with strong CYP3A inhibitors may increase idelalisib concentrations [see Clinical Pharmacology (12.3)].
- Increased idelalisib concentrations may increase the risk of exposure related adverse reactions.
Prevention or Management - Use other drugs that are not strong CYP3A inhibitors.
- If unable to use alternative drugs, monitor patients more frequently for Zydelig adverse reactions [see Adverse Reactions (6.1)].
Strong CYP3A Inducers Clinical Impact - Coadministration with strong CYP3A inducers may decrease idelalisib concentrations [see Clinical Pharmacology (12.3)].
- Decreased idelalisib concentrations may reduce efficacy.
Prevention or Management Avoid coadministration of Zydelig with strong CYP3A4 inducers. 7.2 Effects of Zydelig on Other Drugs
The coadministration of Zydelig with a CYP3A substrate may increase the concentrations of this CYP3A substrate. Avoid coadministration of Zydelig with sensitive CYP3A substrates [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings in animal studies (see Data) and the mechanism of action [see Clinical Pharmacology (12.1)], Zydelig may cause fetal harm when administered to a pregnant woman.
There are no available data in pregnant women to inform the drug-associated risk. In animal reproduction studies, administration of idelalisib to pregnant rats during organogenesis resulted in decreased fetal weight and congenital malformations in rats at maternal exposures (AUC) 12 times those reported in patients at the recommended dose of 150 mg twice daily (see Data).
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The background risk of major birth defects and miscarriage for the indicated population is unknown. However, the background risk of major birth defects is 2–4% and of miscarriage is 15–20% of clinically recognized pregnancies in the U.S. general population.
Data
Animal Data
In an embryo-fetal development study in rats, pregnant animals receiving oral doses of idelalisib during the period of organogenesis (implantation to closure of the hard palate), embryo-fetal toxicities were observed at the mid- and high-doses that also resulted in maternal toxicity, based on reductions in maternal body weight gain. Adverse findings at idelalisib doses ≥ 75 mg/kg/day included decreased fetal weights, external malformations (short tail), and skeletal variations (delayed ossification and/or unossification of the skull, vertebrae, and sternebrae). Additional findings were observed at 150 mg/kg/day dose of idelalisib and included urogenital blood loss, complete resorption, increased post-implantation loss, and malformations (vertebral agenesis with anury, hydrocephaly, and microphthalmia/anophthalmia). The dose of 75 and 150 mg/kg/day of idelalisib in rats resulted in exposures (AUC) of approximately 12 and 30 times, respectively, the human exposure at the recommended dose of 150 mg twice daily.
8.2 Lactation
Risk Summary
No data are available regarding the presence of idelalisib or its metabolites in human milk or its effects on the breastfed child or on milk production. Because of the potential for serious adverse reactions from Zydelig in a breastfed child, advise lactating women not to breastfeed while taking Zydelig and for at least 1 month after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy
Based on animal studies, Zydelig may cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Females of reproductive potential should have a pregnancy test prior to starting treatment with Zydelig.
Contraception
Females
Based on animal studies, Zydelig can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with Zydelig and for at least 1 month after the last dose.
Males
Based on findings in animal reproduction studies, advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 3 months after the last dose of Zydelig [see Use in Specific Populations (8.1)].
8.4 Pediatric Use
Safety and effectiveness of Zydelig in children less than 18 years of age have not been established.
8.5 Geriatric Use
In clinical trials of Zydelig in 615 patients with FL, SLL, and CLL, 327 (53%) patients were age 65 and older. No major differences in effectiveness were observed. When comparing patients 65 years of age or older to younger patients with indolent non-Hodgkin lymphoma, older patients had a higher incidence of discontinuation due to an adverse reaction (28% vs 20%), higher incidence of serious adverse reactions (64% vs 37%), and higher incidence of death (11% vs 5%). When comparing patients 65 years of age or older to younger patients with CLL, older patients had a higher incidence of discontinuation due to an adverse reaction (36% vs 28%), higher incidence of serious adverse reactions (73% vs 67%), and higher incidence of death (13% vs 9%).
8.6 Hepatic Impairment
Dose adjustment is not recommended for patients with ALT or AST or bilirubin > upper limit of normal (ULN); however, limited safety and efficacy data are available for patients with baseline AST or ALT > 2.5 × ULN or bilirubin > 1.5 × ULN. Monitor patients with baseline hepatic impairment for signs of Zydelig toxicity [see Warnings and Precautions (5)]. Follow dose modifications for adverse reactions [see Dosage and Administration (2.2)].
-
11 DESCRIPTION
Idelalisib is an inhibitor of phosphatidylinositol 3-kinase, PI3Kδ.
The chemical name for idelalisib is 5-fluoro-3-phenyl-2-[(1S)-1-(9H-purin-6-ylamino)propyl]quinazolin-4(3H)-one. It has a molecular formula of C22H18FN7O and a molecular weight of 415.42 g/mol. Idelalisib has the following structural formula:
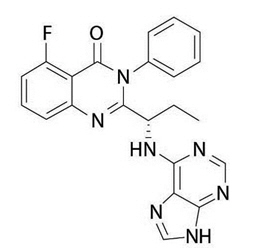
Idelalisib is a white to off-white solid with a pH-dependent aqueous solubility ranging from <0.1 mg/mL at pH 5–7 to over 1 mg/mL at pH 2 under ambient conditions.
Zydelig (idelalisib) tablets are for oral administration. Each tablet contains either 100 mg or 150 mg of idelalisib with the following inactive ingredients: microcrystalline cellulose, hydroxypropyl cellulose, croscarmellose sodium, sodium starch glycolate, magnesium stearate and a tablet coating. The tablet coating consists of polyethylene glycol, talc, polyvinyl alcohol, and titanium dioxide and of FD&C Yellow #6/Sunset Yellow FCF Aluminum Lake (for the 100 mg tablet) and red iron oxide (for the 150 mg tablet).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Idelalisib is an inhibitor of PI3Kδ kinase, which is expressed in normal and malignant B-cells. Idelalisib induced apoptosis and inhibited proliferation in cell lines derived from malignant B-cells and in primary tumor cells. Idelalisib inhibits several cell signaling pathways, including B-cell receptor (BCR) signaling and the CXCR4 and CXCR5 signaling, which are involved in trafficking and homing of B-cells to the lymph nodes and bone marrow. Treatment of lymphoma cells with idelalisib resulted in inhibition of chemotaxis and adhesion, and reduced cell viability.
12.3 Pharmacokinetics
Idelalisib exposure increased in a less than dose-proportional manner over a dose range of 50 mg to 350 mg twice daily (0.3 to 2.3 times the approved recommended dosage).
Following 150 mg twice daily administration of idelalisib, average (% coefficient of variation) maximum concentrations (Cmax) and area under the curve (AUC) at steady-state were 1861 (43%) ng/mL and 10598 (41%) ng∙h/mL for idelalisib.
Distribution
Protein binding of idelalisib is ≥ 84% with no concentration dependence.
The mean blood-to-plasma ratio was 0.7.
The apparent central volume of distribution at steady state is 23 L (%CV ~85%).
Idelalisib is a substrate of P-glycoprotein (P-gp) and BCRP in vitro.
Elimination
The population apparent systemic clearance at steady-state is 14.9 L/hr (%CV ~ 38%). The population terminal elimination half-life of idelalisib is 8.2 hours.
Specific Populations
Age (18 to 91 years), sex, race (White, and non-Whites), renal impairment (creatinine clearance ≥ 15 mL/min) and weight (38 to 148 kg) had no effect on idelalisib exposure.
Patients with Hepatic Impairment
The mean AUC increased up to 1.7-fold in patients with hepatic impairment (defined as ALT or AST or bilirubin values ≥ ULN) compared to patients with normal hepatic function. There is limited information on idelalisib exposure in patients with baseline AST or ALT > 2.5 × ULN or bilirubin > 1.5 × ULN [see Specific Populations (8.6)].
Drug Interaction Studies
Effect of Other Drugs on Idelalisib
The coadministration of rifampin (strong CYP3A inducer and P-gp inducer) to healthy subjects decreased the mean idelalisib AUC by 75% and the mean Cmax by 58% [see Drug Interactions (7.1)].
The coadministration of ketoconazole (strong CYP3A inhibitor and P-gp inhibitor) to healthy subjects increased the mean idelalisib AUC by 1.8-fold. No changes in the mean Cmax were observed [see Drug Interactions (7.1)].
In vitro studies suggest that idelalisib inhibits CYP2C8, CYP2C19, and UGT1A1 and induces CYP2B6.
Effect of Idelalisib on Other Drugs
The mean Cmax of midazolam increased by 2.4-fold and the mean AUC of midazolam increased by 5.4-fold when midazolam (sensitive CYP3A substrate) was coadministered with Zydelig [see Drug Interactions (7.2)].
No changes in exposure to rosuvastatin (OATP1B1 and OATP1B3 substrate) or digoxin (P-glycoprotein substrate) were observed when coadministered with Zydelig.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Idelalisib was not carcinogenic in a 26-week study in transgenic mice when administered daily by oral gavage at doses up to 500 mg/kg/day in males and 1000 mg/kg/day in females. Idelalisib was not carcinogenic in a 2-year study in rats when administered daily by oral gavage at exposures 0.40/0.62-fold (male/female), compared to the exposure in patients with hematologic malignancies administered the recommended dose of 150 mg twice daily.
Idelalisib did not induce mutations in the bacterial mutagenesis (Ames) assay and was not clastogenic in the in vitro chromosome aberration assay using human peripheral blood lymphocytes. Idelalisib was genotoxic in males in the in vivo rat micronucleus study at a high dose of 2000 mg/kg.
Idelalisib may impair fertility in humans. In a fertility study, treated male rats (25, 50, or 100 mg/kg/day of idelalisib) were mated with untreated females. Decreased epididymidal and testicular weights were observed at all dose levels and reduced sperm concentration at the mid- and high doses; however, there were no adverse effects on fertility parameters. The low dose in males resulted in an exposure (AUC) that is approximately 50% of the exposure in patients at the recommended dose of 150 mg twice daily.
In a separate fertility study, treated female rats (25, 50, or 100 mg/kg/day of idelalisib) were mated with untreated males. There were no adverse effects on fertility parameters; however, there was a decrease in the number of live embryos at the high dose. The high dose in females resulted in an exposure (AUC) that is approximately 17-fold the exposure in patients at the recommended dose of 150 mg twice daily.
13.2 Animal Toxicology and/or Pharmacology
Toxicities observed in animals and not reported in patients include cardiac toxicity (cardiomyopathy, inflammation, and increased heart weight) and pancreatic findings (inflammation, hemorrhage, and low-incidence acinar degeneration and hyperplasia). These findings were observed in Sprague-Dawley rats in toxicology studies at exposures (AUCs) higher than those reported in patients at the recommended dose of 150 mg twice daily. Cardiac inflammation was mainly seen in a 28-day study in rats, the other findings were observed in the 13-week and/or 6-month studies.
-
14 CLINICAL STUDIES
14.1 Relapsed Chronic Lymphocytic Leukemia
Study 312-0116
Zydelig was evaluated in a randomized, double-blind, placebo-controlled study GS-US-312-0116 (referred to as 312-0116) (NCT01539512) in 220 patients with relapsed CLL who required treatment and were unable to tolerate standard chemoimmunotherapy due to coexisting medical conditions, reduced renal function as measured by creatinine clearance < 60 mL/min, or NCI CTCAE Grade ≥ 3 neutropenia or Grade ≥ 3 thrombocytopenia resulting from myelotoxic effects of prior therapy with cytotoxic agents. Patients were randomized 1:1 to receive 8 doses of rituximab (first dose at 375 mg/m2, subsequent doses at 500 mg/m2 every 2 weeks for 4 infusions and every 4 weeks for an additional 4 infusions) in combination with either an oral placebo twice daily or with Zydelig 150 mg taken twice daily until disease progression or unacceptable toxicity.
In Study 312-0116, the median age was 71 years (range 47, 92) with 78% over 65, 66% were male, and 90% were Caucasian. The median time since diagnosis was 8.5 years. The median number of prior therapies was 3. Nearly all (96%) patients had received prior anti-CD20 monoclonal antibodies. The most common (>15%) prior regimens were: bendamustine + rituximab (BR) (98 patients, 45%), fludarabine + cyclophosphamide + rituximab (75 patients, 34%), single-agent rituximab (67 patients, 31%), fludarabine + rituximab (37 patients, 17%), and chlorambucil (36 patients, 16%). The median CIRS (Cumulative Illness Rating Scale) score was 8 (range 0–17), and 85% of patients had a score of >6. Median Karnofsky score was 80. Median estimated Creatinine Clearance (eCrCl) was 63.6 mL/min, with 41% of patients having an eCrCl of <60 mL/min. At screening, 19.5% of patients had a platelet count of <50 × 109/L, and 13.2% had an absolute neutrophil count (ANC) of <1 × 109/L.
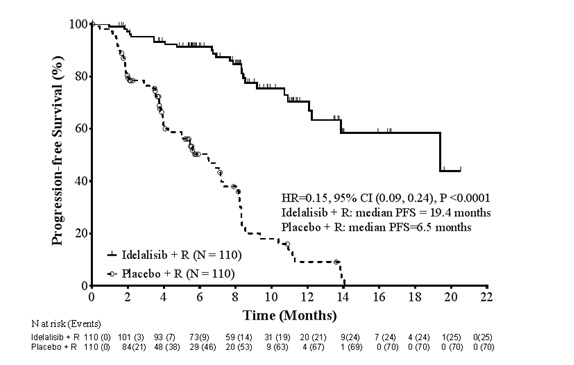
The efficacy of Zydelig was evaluated by progression free survival (PFS), as assessed by an independent review committee (IRC). The trial was stopped for efficacy following the first pre-specified interim analysis. Results of a second interim analysis continued to show a statistically significant improvement for Zydelig + R over placebo + R for the primary endpoint of PFS (HR: 0.18, 95% CI [0.10, 0.32], p < 0.0001).
At the final analysis, with a median follow-up of 8.3 months for the Zydelig + R group, and 5.6 months for the placebo + R group, the median PFS for the Zydelig + R group was 19.4 months (95% CI: 12.3, Not Reached) versus 6.5 months (95% CI: 4.0, 7.3) for the placebo + R group (HR: 0.15, 95% CI [0.09, 0.24], p < 0.0001).
Updated efficacy results are shown in Table 7, and the Kaplan-Meier curve for PFS is shown in Figure 1.
Table 7 Efficacy Results from Study 312-0116 Zydelig + R
N=110Placebo + R
N=110PFS: progression-free survival; NR: not reached; ORR: overall response rate; PR: partial response; DOR: duration of response - * The p value for PFS was based on stratified log-rank test.
- † ORR defined as the proportion of patients who achieved a complete response (CR) or PR. All PRs achieved; none of the patients achieved a CR.
PFS Median (months) (95% CI) 19.4 (12.3, NR) 6.5 (4.0, 7.3) Hazard ratio (95% CI) 0.15 (0.09, 0.24) P-value < 0.0001 * ORR† (All PRs) 92 (83.6%) 17 (15.5%) 95% CI 75.4, 90.0 9.3, 23.6 Odds Ratio (95% CI) 27.8 (13.4, 57.5) P-value <0.0001 DOR Median (months) (95% CI) NR (12, NR) 6.2 (2.8, 6.5) Figure 1 Kaplan-Meier Plot of IRC-Assessed PFS for Study 312-0116
14.2 Relapsed Follicular B-cell non-Hodgkin Lymphoma
Study 101-09
The safety and efficacy of Zydelig in patients with FL was evaluated in a single-arm, multicenter study 101-09 (NCT01282424) which included 72 patients with follicular B-cell non-Hodgkin lymphoma who had relapsed within 6 months following rituximab and an alkylating agent and had received at least 2 prior treatments. The median age was 62 years (range 33 to 84), 54% were male, and 90% were Caucasian. At enrollment, 92% of patients had a baseline ECOG performance status of 0 or 1. The median time since diagnosis was 4.7 years and the median number of prior treatments was 4 (range 2 to 12). The most common prior combination regimens were R-CHOP (49%), BR (50%), and R-CVP (28%). At baseline, 33% of patients had extranodal involvement and 26% had bone marrow involvement.
Patients received 150 mg of Zydelig orally twice daily until evidence of disease progression or unacceptable toxicity. Tumor response was assessed according to the revised International Working Group response criteria for malignant lymphoma. The primary endpoint was Independent Review Committee-assessed overall response rate (ORR). Efficacy results are summarized in Table 8.
Table 8 Overall Response Rate (ORR) and Duration of Response (DOR) in Patients with Relapsed Follicular Lymphoma N=72 CI = confidence interval; CR = complete response; PR = partial response - * Kaplan-Meier estimate
ORR 39 (54%) 95% CI (42, 66%) CR 6 (8%) PR 33 (46%) Median* DOR, months (range) median not evaluable (0.0+, 14.8+) The median time to response was 1.9 months (range 1.6–8.3).
14.3 Relapsed Small Lymphocytic Lymphoma
Study 101-09
The safety and efficacy of Zydelig in patients with SLL was evaluated in a single-arm, multicenter study 101-09 (NCT01282424) which included 26 patients with small lymphocytic lymphoma who had relapsed within 6 months following rituximab and an alkylating agent and had received at least 2 prior treatments. The median age was 65 years (range 50 to 87), 73% were male, and 81% were Caucasian. At enrollment, 96% of patients had a baseline ECOG performance status of 0 or 1. The median time since diagnosis was 6.7 years and the median number of prior treatments was 4 (range 2 to 9). The most common prior combination regimens were BR (81%), FCR (62%) and R-CHOP (35%). At baseline, 27% of patients had extranodal involvement.
Patients received 150 mg of Zydelig orally twice daily until evidence of disease progression or unacceptable toxicity. Tumor response was assessed according to the revised International Working Group response criteria for malignant lymphoma. The primary endpoint was Independent Review Committee-assessed overall response rate (ORR). Efficacy results are summarized in Table 9.
Table 9 Overall Response Rate (ORR) and Duration of Response (DOR) in Patients with Relapsed Small Lymphocytic Lymphoma N=26 CI = confidence interval; CR = complete response; PR = partial response - * Kaplan-Meier estimate
ORR 15 (58%) 95% CI (37, 77%) CR 0 PR 15 (58%) Median* DOR, months (range) 11.9 (0.0+, 14.7+) The median time to response was 1.9 months (range 1.6–8.3).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Zydelig tablets supplied as follows:
Tablet Strength Package Configuration NDC No. Description of Tablet; Debossed on Tablet 150 mg High density polyethylene (HDPE) bottle with a polyester fiber coil, capped with a child-resistant closure. Each bottle contains 60 film-coated tablets. 61958-1702-1 Oval shaped; pink; "150" on one side and "GSI" on the other side 100 mg 61958-1701-1 Oval-shaped; orange; "100" on one side and "GSI" on the other side -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Physicians and health care professionals are advised to discuss the following with patients prior to treatment with Zydelig:
- Hepatotoxicity
Advise patients that Zydelig can cause significant elevations in liver enzymes, and that serial testing of serum liver tests (ALT, AST, and bilirubin) are recommended while taking Zydelig [see Warnings and Precautions (5.1)]. Advise patients to report symptoms of liver dysfunction including jaundice, bruising, abdominal pain, or bleeding.
- Severe Diarrhea or Colitis
Advise patients that Zydelig may cause severe diarrhea or colitis and to notify their healthcare provider immediately if the number of bowel movements in a day increases by six or more [see Warnings and Precautions (5.2)].
- Pneumonitis
Advise patients of the possibility of pneumonitis, and to report any new or worsening respiratory symptoms including cough or dyspnea [see Warnings and Precautions (5.3)].
- Infections
Advise patients that Zydelig can cause serious infections that may be fatal. Advise patients to immediately report symptoms of infection (e.g. pyrexia) [see Warnings and Precautions (5.4)].
- Intestinal Perforation
Advise patients of the possibility for intestinal perforation and to notify their healthcare provider immediately if they experience severe abdominal pain [see Warnings and Precautions (5.5)].
- Severe Cutaneous Reactions
Advise patients that Zydelig may cause severe cutaneous reactions and to notify their healthcare provider immediately if they develop a severe skin reaction [see Warnings and Precautions (5.6)].
- Anaphylaxis
Advise patients that anaphylaxis can occur during treatment with Zydelig and to notify their healthcare provider immediately if they experience symptoms of anaphylaxis [see Warnings and Precautions (5.7)].
- Neutropenia
Advise patients of the need for periodic monitoring of blood counts. Advise patients to notify their healthcare provider immediately if they develop a fever or any signs of infection [see Warnings and Precautions (5.8)].
- Embryo-Fetal Toxicity
Advise females to inform their healthcare provider if they are pregnant or become pregnant. Inform female patients of the risk to a fetus and potential loss of the pregnancy [see Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment and for 1 month after receiving the last dose of Zydelig [see Warnings and Precautions (5.9) and Use in Specific Populations (8.1, 8.3)].
Advise lactating women not to breastfeed during treatment with Zydelig and for at least 1 month after the last dose [see Use in Specific Populations (8.2)].
- Instructions for Taking Zydelig
Advise patients to take Zydelig exactly as prescribed and not to change their dose or to stop taking Zydelig unless they are told to do so by their healthcare provider. Zydelig may be taken with or without food. Zydelig tablets should be swallowed whole. Advise patients that if a dose of Zydelig is missed by less than 6 hours, to take the missed dose right away and take the next dose as usual. If a dose of Zydelig is missed by more than 6 hours, advise patients to wait and take the next dose at the usual time.
-
SPL UNCLASSIFIED SECTION
Manufactured and distributed by:
Gilead Sciences, Inc.
Foster City, CA 94404
GSI and Zydelig are trademarks or registered trademarks of Gilead Sciences, Inc., or its related companies. All other trademarks referenced herein are the property of their respective owners.
© 2018 Gilead Sciences, Inc. All rights reserved.
205858-GS-004-PI
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 01 2018 MEDICATION GUIDE
ZYDELIG® (zye-DEL-ig)
(idelalisib)
tabletsWhat is the most important information I should know about Zydelig?
Zydelig can cause serious side effects that can lead to death, including:-
Liver problems. Abnormal liver blood test results are common during treatment with Zydelig. Zydelig can cause severe liver problems. Your doctor will do blood tests before and during your treatment with Zydelig to check for liver problems. Tell your doctor right away if you get any of the following symptoms of liver problems:
- yellowing of your skin or the white part of your eyes (jaundice)
- dark or brown (tea colored) urine
- pain in the upper right side of your stomach area (abdomen)
- bleeding or bruising more easily than normal
- Severe diarrhea. Diarrhea is common during treatment with Zydelig and can sometimes be severe. Tell your doctor right away if the number of bowel movements you have in a day increases by six or more. Ask your doctor about medicines you can take to treat your diarrhea.
- Lung problems (pneumonitis). Your doctor may do tests to check your lungs if you have breathing problems during treatment with Zydelig. Tell your doctor right away if you get new or worsening cough, shortness of breath, difficulty breathing, or wheezing. Your doctor may treat you with a corticosteroid medicine if you develop lung problems.
- Infections. Zydelig can cause serious infections that may lead to death. Tell your doctor right away if you have a fever or any signs of an infection during treatment with Zydelig.
- Tear in intestinal wall (perforation). Tell your doctor or get medical help right away if you get new or worsening stomach area (abdomen) pain, chills, fever, nausea, or vomiting.
-
Severe skin reactions. Tell your doctor right away if you get any of the following symptoms during treatment with Zydelig:
- painful sores or ulcers on your skin, lips, or in your mouth
- severe rash with blisters or peeling skin
- rash with itching
See "What are the possible side effects of Zydelig?" for more information about side effects.What is Zydelig?
Zydelig is a prescription medicine used to treat people with:- Chronic Lymphocytic Leukemia (CLL) in combination with rituximab when CLL comes back after prior cancer treatment and when rituximab treatment alone may be used due to other health problems.
- Follicular B-cell non-Hodgkin Lymphoma (FL) when the disease has come back after treatment with at least two prior medicines.
- Small Lymphocytic Lymphoma (SLL) when the disease comes back after treatment with at least two prior medicines.
Zydelig should not be used in combination with bendamustine and/or rituximab to treat people with FL.
It is not known if Zydelig is safe and effective in children less than 18 years of age.Do not take Zydelig if you have a history of serious allergic reactions including a severe skin reaction called toxic epidermal necrolysis (TEN). Before taking Zydelig, tell your doctor about all of your medical conditions, including if you: - have liver problems
- have lung problems
- have an infection
- are pregnant or plan to become pregnant. Zydelig may harm your unborn baby. Females who are able to become pregnant should have a pregnancy test before starting treatment with Zydelig.
- Females who are able to become pregnant should use effective birth control (contraception) during treatment with Zydelig and for at least 1 month after the last dose of Zydelig. Talk to your doctor about birth control methods that may be right for you. Tell your doctor right away if you become pregnant or think you are pregnant during treatment with Zydelig.
- Males with female partners who are able to become pregnant should use effective birth control (contraception) during treatment with Zydelig and for 3 months after the last dose.
- are breastfeeding or plan to breastfeed. It is not known if Zydelig passes into your breast milk. Do not breastfeed during your treatment with Zydelig and for at least 1 month after the last dose.
How should I take Zydelig? - Take Zydelig exactly as your doctor tells you to take it.
- Your doctor may change your dose of Zydelig or tell you to stop taking Zydelig. Do not change your dose or stop taking Zydelig without first talking to your doctor.
- Take Zydelig 2 times a day.
- You may take Zydelig with or without food.
- Take Zydelig tablets whole.
- Do not miss a dose of Zydelig. If you miss a dose of Zydelig by less than 6 hours, take the missed dose right away. Then take your next dose as usual. If you miss a dose of Zydelig by more than 6 hours, wait and take the next dose of Zydelig at your usual time.
What are the possible side effects of Zydelig?
Zydelig can cause serious side effects, including:- See "What is the most important information I should know about Zydelig?"
- Anaphylaxis. Tell your doctor or get medical help right away if you have a serious allergic reaction during treatment with Zydelig.
- Low white blood cell count (neutropenia). Neutropenia is common during treatment with Zydelig and can sometimes be severe. Your doctor will check your blood counts regularly during treatment with Zydelig. Tell your doctor right away if you have a fever or any signs of an infection during treatment with Zydelig.
The most common side effects of Zydelig when used alone include: - tiredness
- nausea
- cough
- fever
- stomach area (abdomen) pain
- pneumonia
- rash
The most common side effects of Zydelig when used in combination with rituximab include: - pneumonia
- fever
- tiredness
- rash
- cough
- nausea
Tell your doctor if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of Zydelig. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store Zydelig? - Store Zydelig between 68°F to 86°F (20°C to 30°C).
- Keep Zydelig in its original container.
- Do not use Zydelig if the seal over the bottle opening is broken or missing.
General information about the safe and effective use of Zydelig.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use Zydelig for a condition for which it was not prescribed. Do not give Zydelig to other people, even if they have the same symptoms you have. It may harm them. You can ask your doctor or pharmacist for information about Zydelig that is written for health professionals.What are the ingredients in Zydelig?
Active ingredient: idelalisib
Inactive ingredients: microcrystalline cellulose, hydroxypropyl cellulose, croscarmellose sodium, sodium starch glycolate, and magnesium stearate. The tablet coating contains polyethylene glycol, talc, polyvinyl alcohol, titanium dioxide and FD&C Yellow #6 or Sunset Yellow FCF Aluminum Lake (for the 100 mg tablet) and red iron oxide (for the 150 mg tablet).
Manufactured and distributed by: Gilead Sciences, Inc. Foster City, CA 94404
©2018 Gilead Sciences, Inc. All rights reservedFor more information, call 1-800-445-3235 or go to www.Zydelig.com. 205858-GS-003-MG -
Liver problems. Abnormal liver blood test results are common during treatment with Zydelig. Zydelig can cause severe liver problems. Your doctor will do blood tests before and during your treatment with Zydelig to check for liver problems. Tell your doctor right away if you get any of the following symptoms of liver problems:
- PRINCIPAL DISPLAY PANEL - 100 mg Tablet Bottle Label
- PRINCIPAL DISPLAY PANEL - 150 mg Tablet Bottle Label
-
INGREDIENTS AND APPEARANCE
ZYDELIG
idelalisib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 61958-1701 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength IDELALISIB (UNII: YG57I8T5M0) (IDELALISIB - UNII:YG57I8T5M0) IDELALISIB 100 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) ALUMINUM OXIDE (UNII: LMI26O6933) Product Characteristics Color orange Score no score Shape OVAL Size 10mm Flavor Imprint Code GSI;100 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 61958-1701-1 60 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 07/23/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205858 07/23/2014 ZYDELIG
idelalisib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 61958-1702 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength IDELALISIB (UNII: YG57I8T5M0) (IDELALISIB - UNII:YG57I8T5M0) IDELALISIB 150 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color pink Score no score Shape OVAL Size 10mm Flavor Imprint Code GSI;150 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 61958-1702-1 60 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 07/23/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205858 07/23/2014 Labeler - Gilead Sciences, Inc. (185049848)


Trademark Results [Zydelig]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ZYDELIG 86346343 4780443 Live/Registered |
GILEAD SCIENCES IRELAND UC 2014-07-23 |
 ZYDELIG 86346340 4780442 Live/Registered |
GILEAD SCIENCES IRELAND UC 2014-07-23 |
 ZYDELIG 85773807 4364273 Live/Registered |
GILEAD SCIENCES IRELAND UC 2012-11-07 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.