ZOMETA- zoledronic acid injection, solution, concentrate
Zometa by
Drug Labeling and Warnings
Zometa by is a Prescription medication manufactured, distributed, or labeled by Novartis Pharmaceuticals Corporation. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ZOMETA safely and effectively. See full prescribing information for ZOMETA.
ZOMETA® (zoledronic acid) injection, for intravenous use
Initial U.S. Approval: 2001
RECENT MAJOR CHANGES
Warnings and Precautions, Embryo-Fetal Toxicity (5.10)
12/2018
INDICATIONS AND USAGE
Zometa is a bisphosphonate indicated for the treatment of:
- Hypercalcemia of Malignancy (1.1)
- Patients with multiple myeloma and patients with documented bone metastases from solid tumors, in conjunction with standard antineoplastic therapy. Prostate cancer should have progressed after treatment with at least one hormonal therapy. (1.2)
Limitations of Use: The safety and efficacy of Zometa has not been established for use in hyperparathyroidism or non-tumor-related hypercalcemia.
DOSAGE AND ADMINISTRATION
Hypercalcemia of Malignancy (2.1)
- 4 mg as a single-use intravenous infusion over no less than 15 minutes
- 4 mg as retreatment after a minimum of 7 days
Multiple myeloma and Bone Metastasis of Solid Tumors (2.2)
- 4 mg as a single-use intravenous infusion over no less than 15 minutes every 3-4 weeks for patients with creatinine clearance of greater than 60 mL/min.
- Reduce the dose for patients with renal impairment.
- Coadminister oral calcium supplements of 500 mg and a multiple vitamin containing 400 international units of vitamin D daily.
Administer through a separate vented infusion line and do not allow to come in contact with any calcium or divalent cation-containing solutions. (2.3)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
Hypersensitivity to any component of Zometa (4)
WARNINGS AND PRECAUTIONS
- Patients being treated with Zometa should not be treated with Reclast®. (5.1)
- Adequately rehydrate patients with hypercalcemia of malignancy prior to administration of Zometa and monitor electrolytes during treatment. (5.2)
- Renal toxicity may be greater in patients with renal impairment. Do not use doses greater than 4 mg. Treatment in patients with severe renal impairment is not recommended. Monitor serum creatinine before each dose. (5.3)
- Osteonecrosis of the jaw (ONJ) has been reported. Preventive dental exams should be performed before starting Zometa. Avoid invasive dental procedures. (5.4)
- Severe incapacitating bone, joint, and/or muscle pain may occur. Discontinue Zometa if severe symptoms occur. (5.5)
- Atypical subtrochanteric and diaphyseal femoral fractures have been reported in patients receiving bisphosphonate therapy. These fractures may occur after minimal or no trauma. Evaluate patients with thigh or groin pain to rule out a femoral fracture. Consider drug discontinuation in patients suspected to have an atypical femur fracture. (5.6)
- Hypocalcemia: Correct before initiating Zometa. Adequately supplement patients with calcium and vitamin D. Monitor serum calcium closely with concomitant administration of other drugs known to cause hypocalcemia to avoid severe or life-threatening hypocalcemia. (5.9)
- Zometa can cause fetal harm. Advise females of reproductive potential of potential risk to a fetus and to use effective contraception. (5.10, 8.1, 8.3)
ADVERSE REACTIONS
The most common adverse events (greater than 25%) were nausea, fatigue, anemia, bone pain, constipation, fever, vomiting, and dyspnea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
- Lactation: Advise not to breastfeed (8.2)
- Females and Males of Reproductive Potential: Verify pregnancy status prior to initiation of Zometa. May impair fertility. Counsel patients on pregnancy planning and prevention (8.3)
- Pediatric Use: Not indicated for use in pediatric patients (8.4)
- Geriatric Use: Special care to monitor renal function (8.5)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2018
- Hypercalcemia of Malignancy (1.1)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Hypercalcemia of Malignancy
1.2 Multiple Myeloma and Bone Metastases of Solid Tumors
2 DOSAGE AND ADMINISTRATION
2.1 Hypercalcemia of Malignancy
2.2 Multiple Myeloma and Bone Metastases of Solid Tumors
2.3 Preparation of Solution
2.4 Method of Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Drugs With Same Active Ingredient or in the Same Drug Class
5.2 Hydration and Electrolyte Monitoring
5.3 Renal Impairment
5.4 Osteonecrosis of the Jaw
5.5 Musculoskeletal Pain
5.6 Atypical Subtrochanteric and Diaphyseal Femoral Fractures
5.7 Patients With Asthma
5.8 Hepatic Impairment
5.9 Hypocalcemia
5.10 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Aminoglycosides and Calcitonin
7.2 Loop Diuretics
7.3 Nephrotoxic Drugs
7.4 Thalidomide
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Hypercalcemia of Malignancy
14.2 Clinical Trials in Multiple Myeloma and Bone Metastases of Solid Tumors
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Hypercalcemia of Malignancy
Zometa is indicated for the treatment of hypercalcemia of malignancy defined as an albumin-corrected calcium (cCa) of greater than or equal to 12 mg/dL [3.0 mmol/L] using the formula: cCa in mg/dL = Ca in mg/dL + 0.8 (4.0 g/dL - patient albumin [g/dL]).
1.2 Multiple Myeloma and Bone Metastases of Solid Tumors
Zometa is indicated for the treatment of patients with multiple myeloma and patients with documented bone metastases from solid tumors, in conjunction with standard antineoplastic therapy. Prostate cancer should have progressed after treatment with at least one hormonal therapy.
Limitations of Use
The safety and efficacy of Zometa in the treatment of hypercalcemia associated with hyperparathyroidism or with other non–tumor-related conditions have not been established.
-
2 DOSAGE AND ADMINISTRATION
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
2.1 Hypercalcemia of Malignancy
The maximum recommended dose of Zometa in hypercalcemia of malignancy (albumin-corrected serum calcium greater than or equal to 12 mg/dL [3.0 mmol/L]) is 4 mg. The 4 mg dose must be given as a single-dose intravenous infusion over no less than 15 minutes. Patients who receive Zometa should have serum creatinine assessed prior to each treatment.
Dose adjustments of Zometa are not necessary in treating patients for hypercalcemia of malignancy presenting with mild-to-moderate renal impairment prior to initiation of therapy (serum creatinine less than 400 µmol/L or less than 4.5 mg/dL).
Patients should be adequately rehydrated prior to administration of Zometa [see Warnings and Precautions (5.2)].
Consideration should be given to the severity of, as well as the symptoms of, tumor-induced hypercalcemia when considering use of Zometa. Vigorous saline hydration, an integral part of hypercalcemia therapy, should be initiated promptly and an attempt should be made to restore the urine output to about 2 L/day throughout treatment. Mild or asymptomatic hypercalcemia may be treated with conservative measures (i.e., saline hydration, with or without loop diuretics). Patients should be hydrated adequately throughout the treatment, but overhydration, especially in those patients who have cardiac failure, must be avoided. Diuretic therapy should not be employed prior to correction of hypovolemia.
Retreatment with Zometa 4 mg may be considered if serum calcium does not return to normal or remain normal after initial treatment. It is recommended that a minimum of 7 days elapse before retreatment, to allow for full response to the initial dose. Renal function must be carefully monitored in all patients receiving Zometa and serum creatinine must be assessed prior to retreatment with Zometa [see Warnings and Precautions (5.2)].
2.2 Multiple Myeloma and Bone Metastases of Solid Tumors
The recommended dose of Zometa in patients with multiple myeloma and metastatic bone lesions from solid tumors for patients with creatinine clearance (CrCl) greater than 60 mL/min is 4 mg infused over no less than 15 minutes every 3 to 4 weeks. The optimal duration of therapy is not known.
Upon treatment initiation, the recommended Zometa doses for patients with reduced renal function (mild and moderate renal impairment) are listed in Table 1. These doses are calculated to achieve the same area under the curve (AUC) as that achieved in patients with creatinine clearance of 75 mL/min. Creatinine clearance is calculated using the Cockcroft-Gault formula [see Warnings and Precautions (5.2)].
Table 1: Reduced Doses for Patients With Baseline CrCl Less Than or Equal to 60 mL/min *Doses calculated assuming target AUC of 0.66 (mghr/L) (CrCl = 75 mL/min). Baseline Creatinine Clearance
(mL/min)Zometa Recommended Dose
(mg)*greater than 60 4 50-60 3.5 40-49 3.3 30-39 3 During treatment, serum creatinine should be measured before each Zometa dose and treatment should be withheld for renal deterioration. In the clinical studies, renal deterioration was defined as follows:
For patients with normal baseline creatinine, increase of 0.5 mg/dL
For patients with abnormal baseline creatinine, increase of 1.0 mg/dLIn the clinical studies, Zometa treatment was resumed only when the creatinine returned to within 10% of the baseline value. Zometa should be reinitiated at the same dose as that prior to treatment interruption.
Patients should also be administered an oral calcium supplement of 500 mg and a multiple vitamin containing 400 international units of vitamin D daily.
2.3 Preparation of Solution
Zometa must not be mixed with calcium or other divalent cation-containing infusion solutions, such as Lactated Ringer’s solution, and should be administered as a single intravenous solution in a line separate from all other drugs.
4 mg/100 mL Single-Dose Ready-to-use Bottle
Bottles of Zometa ready-to-use solution for infusion contain overfill allowing for the administration of 100 mL of solution (equivalent to 4 mg zoledronic acid). This solution is ready-to-use and may be administered directly to the patient without further preparation. For single-use only.
To prepare reduced doses for patients with baseline CrCl less than or equal to 60 mL/min, withdraw the specified volume of the Zometa solution from the bottle (see Table 2) and replace with an equal volume of sterile 0.9% Sodium Chloride, USP, or 5% Dextrose Injection, USP. Administer the newly-prepared dose-adjusted solution to the patient by infusion. Follow proper aseptic technique. Properly discard previously withdrawn volume of ready-to-use solution - do not store or reuse.
Table 2: Preparation of Reduced Doses–Zometa Ready-to-use Bottle Remove and Discard the Following Zometa Ready-to-use Solution
(mL)Replace With the Following Volume of Sterile 0.9% Sodium Chloride, USP or 5% Dextrose Injection, USP
(mL)Dose
(mg)12.0 12.0 3.5 18.0 18.0 3.3 25.0 25.0 3.0 If not used immediately after dilution with infusion media, for microbiological integrity, the solution should be refrigerated at 2°C to 8°C (36°F–46°F). The refrigerated solution should then be equilibrated to room temperature prior to administration. The total time between dilution, storage in the refrigerator, and end of administration must not exceed 24 hours.
4 mg/5 mL Single-Dose Vial for Dilution Prior to Intravenous Infusion
Zometa 4 mg/5 mL vial for dilution prior to intravenous infusion contains an overfill to allow withdrawal of 5 mL (equivalent to 4 mg zoledronic acid). Zometa (4 mg/5 mL) should immediately be diluted in 100 mL of sterile 0.9% Sodium Chloride, USP, or 5% Dextrose Injection, USP, following proper aseptic technique, and administered to the patient by intravenous infusion. Do not store undiluted Zometa (4 mg/5 mL) in a syringe, to avoid inadvertent injection.
To prepare reduced doses for patients with baseline CrCl less than or equal to 60 mL/min, withdraw the specified volume of the Zometa (4 mg/5 mL) from the vial for the dose required (see Table 3).
Table 3: Preparation of Reduced Doses–Zometa 4 mg/5 mL Single-Dose Vial for Dilution Remove and use Zometa
Volume (mL)Dose
(mg)4.4 3.5 4.1 3.3 3.8 3.0 The withdrawn Zometa (4mg/5 mL) solution must be diluted in 100 mL of sterile 0.9% Sodium Chloride, USP, or 5% Dextrose Injection, USP.
If not used immediately after dilution with infusion media, for microbiological integrity, the solution should be refrigerated at 2°C to 8°C (36°F-46°F). The refrigerated solution should then be equilibrated to room temperature prior to administration. The total time between dilution, storage in the refrigerator, and end of administration must not exceed 24 hours.
2.4 Method of Administration
Due to the risk of clinically significant deterioration in renal function, which may progress to renal failure, single doses of Zometa should not exceed 4 mg and the duration of infusion should be no less than 15 minutes [see Warnings and Precautions (5.3)]. In the trials and in postmarketing experience, renal deterioration, progression to renal failure and dialysis, have occurred in patients, including those treated with the approved dose of 4 mg infused over 15 minutes. There have been instances of this occurring after the initial Zometa dose.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Drugs With Same Active Ingredient or in the Same Drug Class
Zometa contains the same active ingredient as found in Reclast® (zoledronic acid). Patients being treated with Zometa should not be treated with Reclast or other bisphosphonates.
5.2 Hydration and Electrolyte Monitoring
Patients with hypercalcemia of malignancy must be adequately rehydrated prior to administration of Zometa. Loop diuretics should not be used until the patient is adequately rehydrated and should be used with caution in combination with Zometa in order to avoid hypocalcemia. Zometa should be used with caution with other nephrotoxic drugs.
Standard hypercalcemia-related metabolic parameters, such as serum levels of calcium, phosphate, and magnesium, as well as serum creatinine, should be carefully monitored following initiation of therapy with Zometa. If hypocalcemia, hypophosphatemia, or hypomagnesemia occur, short-term supplemental therapy may be necessary.
5.3 Renal Impairment
Zometa is excreted intact primarily via the kidney, and the risk of adverse reactions, in particular renal adverse reactions, may be greater in patients with impaired renal function. Safety and pharmacokinetic data are limited in patients with severe renal impairment and the risk of renal deterioration is increased [see Adverse Reactions (6.1)]. Preexisting renal insufficiency and multiple cycles of Zometa and other bisphosphonates are risk factors for subsequent renal deterioration with Zometa. Factors predisposing to renal deterioration, such as dehydration or the use of other nephrotoxic drugs, should be identified and managed, if possible.
Zometa treatment in patients with hypercalcemia of malignancy with severe renal impairment should be considered only after evaluating the risks and benefits of treatment [see Dosage and Administration (2.1)]. In the clinical studies, patients with serum creatinine greater than 400 µmol/L or greater than 4.5 mg/dL were excluded.
Zometa treatment is not recommended in patients with bone metastases with severe renal impairment. In the clinical studies, patients with serum creatinine greater than 265 µmol/L or greater than 3.0 mg/dL were excluded and there were only 8 of 564 patients treated with Zometa 4 mg by 15-minute infusion with a baseline creatinine greater than 2 mg/dL. Limited pharmacokinetic data exists in patients with creatinine clearance less than 30 mL/min [see Clinical Pharmacology (12.3)].
5.4 Osteonecrosis of the Jaw
Osteonecrosis of the jaw (ONJ) has been reported predominantly in cancer patients treated with intravenous bisphosphonates, including Zometa. Many of these patients were also receiving chemotherapy and corticosteroids which may be risk factors for ONJ. The risk of ONJ may increase with duration of exposure to bisphosphonates.
Postmarketing experience and the literature suggest a greater frequency of reports of ONJ based on tumor type (advanced breast cancer, multiple myeloma), and dental status (dental extraction, periodontal disease, local trauma including poorly fitting dentures). Many reports of ONJ involved patients with signs of local infection including osteomyelitis.
Cancer patients should maintain good oral hygiene and should have a dental examination with preventive dentistry prior to treatment with bisphosphonates.
While on treatment, these patients should avoid invasive dental procedures if possible. For patients who develop ONJ while on bisphosphonate therapy, dental surgery may exacerbate the condition. For patients requiring dental procedures, there are no data available to suggest whether discontinuation of bisphosphonate treatment reduces the risk of ONJ. Clinical judgment of the treating physician should guide the management plan of each patient based on individual benefit/risk assessment [see Adverse Reactions (6.2)].
5.5 Musculoskeletal Pain
In postmarketing experience, severe and occasionally incapacitating bone, joint, and/or muscle pain has been reported in patients taking bisphosphonates, including Zometa. The time to onset of symptoms varied from one day to several months after starting the drug. Discontinue use if severe symptoms develop. Most patients had relief of symptoms after stopping. A subset had recurrence of symptoms when rechallenged with the same drug or another bisphosphonate [see Adverse Reactions (6.2)].
5.6 Atypical Subtrochanteric and Diaphyseal Femoral Fractures
Atypical subtrochanteric and diaphyseal femoral fractures have been reported in patients receiving bisphosphonate therapy, including Zometa. These fractures can occur anywhere in the femoral shaft from just below the lesser trochanter to just above the supracondylar flare and are transverse or short oblique in orientation without evidence of comminution. These fractures occur after minimal or no trauma. Patients may experience thigh or groin pain weeks to months before presenting with a completed femoral fracture. Fractures are often bilateral; therefore the contralateral femur should be examined in bisphosphonate-treated patients who have sustained a femoral shaft fracture. Poor healing of these fractures has also been reported. A number of case reports noted that patients were also receiving treatment with glucocorticoids (such as prednisone or dexamethasone) at the time of fracture. Causality with bisphosphonate therapy has not been established.
Any patient with a history of bisphosphonate exposure who presents with thigh or groin pain in the absence of trauma should be suspected of having an atypical fracture and should be evaluated. Discontinuation of Zometa therapy in patients suspected to have an atypical femur fracture should be considered pending evaluation of the patient, based on an individual benefit risk assessment. It is unknown whether the risk of atypical femur fracture continues after stopping therapy.
5.7 Patients With Asthma
While not observed in clinical trials with Zometa, there have been reports of bronchoconstriction in aspirin-sensitive patients receiving bisphosphonates.
5.8 Hepatic Impairment
Only limited clinical data are available for use of Zometa to treat hypercalcemia of malignancy in patients with hepatic insufficiency, and these data are not adequate to provide guidance on dosage selection or how to safely use Zometa in these patients.
5.9 Hypocalcemia
Hypocalcemia has been reported in patients treated with Zometa. Cardiac arrhythmias and neurologic adverse events (seizures, tetany, and numbness) have been reported secondary to cases of severe hypocalcemia. In some instances, hypocalcemia may be life-threatening. Caution is advised when Zometa is administered with drugs known to cause hypocalcemia, as severe hypocalcemia may develop, [see Drug Interactions (7.2)]. Serum calcium should be measured and hypocalcemia must be corrected before initiating Zometa. Adequately supplement patients with calcium and vitamin D.
5.10 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, Zometa can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of zoledronic acid to pregnant rats during organogenesis resulted in fetal malformations and embryo-fetal lethality at maternal exposures that were greater than or equal to 2.4 times the human clinical exposure based on area under the curve (AUC). Bisphosphonates, such as Zometa, are incorporated into the bone matrix, from where they are gradually released over periods of weeks to years. There may be a risk of fetal harm (e.g., skeletal and other abnormalities) if a woman becomes pregnant after completing a course of bisphosphonate therapy. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during and after Zometa treatment [see Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1)].
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Hypercalcemia of Malignancy
The safety of Zometa was studied in 185 patients with hypercalcemia of malignancy (HCM) who received either Zometa 4 mg given as a 5-minute intravenous infusion (n = 86) or pamidronate 90 mg given as a 2-hour intravenous infusion (n = 103). The population was aged 33-84 years, 60% male and 81% Caucasian, with breast, lung, head and neck, and renal cancer as the most common forms of malignancy. NOTE: pamidronate 90 mg was given as a 2-hour intravenous infusion. The relative safety of pamidronate 90 mg given as a 2-hour intravenous infusion compared to the same dose given as a 24-hour intravenous infusion has not been adequately studied in controlled clinical trials.
Renal Toxicity
Administration of Zometa 4 mg given as a 5-minute intravenous infusion has been shown to result in an increased risk of renal toxicity, as measured by increases in serum creatinine, which can progress to renal failure. The incidence of renal toxicity and renal failure has been shown to be reduced when Zometa 4 mg is given as a 15-minute intravenous infusion. Zometa should be administered by intravenous infusion over no less than 15 minutes [see Warnings and Precautions (5.3), Dosage and Administration (2.4)].
The most frequently observed adverse events were fever, nausea, constipation, anemia, and dyspnea (see Table 4).
Table 4 provides adverse events that were reported by 10% or more of the 189 patients treated with Zometa 4 mg or pamidronate 90 mg from the two HCM trials. Adverse events are listed regardless of presumed causality to study drug.
Table 4: Percentage of Patients With Adverse Events Greater Than or Equal to 10% Reported in Hypercalcemia of Malignancy Clinical Trials by Body System Zometa Pamidronate 4 mg 90 mg n (%) n (%) Patients Studied Total No. of Patients Studied 86 (100) 103 (100) Total No. of Patients with any AE 81 (94) 95 (92) Body as a Whole Fever 38 (44) 34 (33) Progression of Cancer 14 (16) 21 (20) Cardiovascular Hypotension 9 (11) 2 (2) Digestive Nausea 25 (29) 28 (27) Constipation 23 (27) 13 (13) Diarrhea 15 (17) 17 (17) Abdominal Pain 14 (16) 13 (13) Vomiting 12 (14) 17 (17) Anorexia 8 (9) 14 (14) Hemic and Lymphatic System Anemia 19 (22) 18 (18) Infections Moniliasis 10 (12) 4 (4) Laboratory Abnormalities Hypophosphatemia 11 (13) 2 (2) Hypokalemia 10 (12) 16 (16) Hypomagnesemia 9 (11) 5 (5) Musculoskeletal Skeletal Pain 10 (12) 10 (10) Nervous Insomnia 13 (15) 10 (10) Anxiety 12 (14) 8 (8) Confusion 11 (13) 13 (13) Agitation 11 (13) 8 (8) Respiratory Dyspnea 19 (22) 20 (19) Coughing 10 (12) 12 (12) Urogenital Urinary Tract Infection 12 (14) 15 (15) The following adverse events from the two controlled multicenter HCM trials (n = 189) were reported by a greater percentage of patients treated with Zometa 4 mg than with pamidronate 90 mg and occurred with a frequency of greater than or equal to 5% but less than 10%. Adverse events are listed regardless of presumed causality to study drug: asthenia, chest pain, leg edema, mucositis, dysphagia, granulocytopenia, thrombocytopenia, pancytopenia, nonspecific infection, hypocalcemia, dehydration, arthralgias, headache and somnolence.
Rare cases of rash, pruritus, and chest pain have been reported following treatment with Zometa.
Acute Phase Reaction
Within three days after Zometa administration, an acute phase reaction has been reported in patients, with symptoms including pyrexia, fatigue, bone pain and/or arthralgias, myalgias, chills, and influenza-like illness. These symptoms usually resolve within a few days. Pyrexia has been the most commonly associated symptom, occurring in 44% of patients.
Mineral and Electrolyte Abnormalities
Electrolyte abnormalities, most commonly hypocalcemia, hypophosphatemia, and hypomagnesemia, can occur with bisphosphonate use.
Grade 3 and Grade 4 laboratory abnormalities for serum creatinine, serum calcium, serum phosphorus, and serum magnesium observed in two clinical trials of Zometa in patients with HCM are shown in Table 5 and 6.
Table 5: Grade 3 Laboratory Abnormalities for Serum Creatinine, Serum Calcium, Serum Phosphorus, and Serum Magnesium in Two Clinical Trials in Patients With HCM 1Grade 3 (greater than 3x Upper Limit of Normal); Grade 4 (greater than 6x Upper Limit of Normal).
2Grade 3 (less than 7 mg/dL); Grade 4 (less than 6 mg/dL).
3Grade 3 (less than 2 mg/dL); Grade 4 (less than 1 mg/dL).
4Grade 3 (less than 0.8 mEq/L); Grade 4 (less than 0.5 mEq/L).Grade 3 Laboratory Parameter Zometa Pamidronate 4 mg 90 mg n/N (%) n/N (%) Serum Creatinine1 2/86 (2%) 3/100 (3%) Hypocalcemia2 1/86 (1%) 2/100 (2%) Hypophosphatemia3 36/70 (51%) 27/81 (33%) Hypomagnesemia4 0/71 0 0/84 0 Table 6: Grade 4 Laboratory Abnormalities for Serum Creatinine, Serum Calcium, Serum Phosphorus, and Serum Magnesium in Two Clinical Trials in Patients With HCM 1Grade 3 (greater than 3x Upper Limit of Normal); Grade 4 (greater than 6x Upper Limit of Normal).
2Grade 3 (less than 7 mg/dL); Grade 4 (less than 6 mg/dL).
3Grade 3 (less than 2 mg/dL); Grade 4 (less than 1 mg/dL).
4Grade 3 (less than 0.8 mEq/L); Grade 4 (less than 0.5 mEq/L).Grade 4 Laboratory Parameter Zometa Pamidronate 4 mg 90 mg n/N (%) n/N (%) Serum Creatinine1 0/86 0 1/100 (1%) Hypocalcemia2 0/86 0 0/100 0 Hypophosphatemia3 1/70 (1%) 4/81 (5%) Hypomagnesemia4 0/71 0 1/84 (1%) Injection-Site Reactions
Local reactions at the infusion-site, such as redness or swelling, were observed infrequently. In most cases, no specific treatment is required and the symptoms subside after 24-48 hours.
Ocular Adverse Events
Ocular inflammation such as uveitis and scleritis can occur with bisphosphonate use, including Zometa. No cases of iritis, scleritis, or uveitis were reported during these clinical trials. However, cases have been seen in postmarketing use [see Adverse Reactions (6.2)].
Multiple Myeloma and Bone Metastases of Solid Tumors
The safety analysis includes patients treated in the core and extension phases of the trials. The analysis includes the 2042 patients treated with Zometa 4 mg, pamidronate 90 mg, or placebo in the three controlled multicenter bone metastases trials, including 969 patients completing the efficacy phase of the trial, and 619 patients that continued in the safety extension phase. Only 347 patients completed the extension phases and were followed for 2 years (or 21 months for the other solid tumor patients). The median duration of exposure for safety analysis for Zometa 4 mg (core plus extension phases) was 12.8 months for breast cancer and multiple myeloma, 10.8 months for prostate cancer, and 4.0 months for other solid tumors.
Table 7 describes adverse events that were reported by 10% or more of patients. Adverse events are listed regardless of presumed causality to study drug.
Table 7: Percentage of Patients With Adverse Events Greater Than or Equal to 10% Reported in Three Bone Metastases Clinical Trials by Body System Zometa Pamidronate Placebo 4 mg 90 mg n (%) n (%) n (%) Patients Studied Total No. of Patients 1031 (100) 556 (100) 455 (100) Total No. of Patients with any AE 1015 (98) 548 (99) 445 (98) Blood and Lymphatic Anemia 344 (33) 175 (32) 128 (28) Neutropenia 124 (12) 83 (15) 35 (8) Thrombocytopenia 102 (10) 53 (10) 20 (4) Gastrointestinal Nausea 476 (46) 266 (48) 171 (38) Vomiting 333 (32) 183 (33) 122 (27) Constipation 320 (31) 162 (29) 174 (38) Diarrhea 249 (24) 162 (29) 83 (18) Abdominal Pain 143 (14) 81 (15) 48 (11) Dyspepsia 105 (10) 74 (13) 31 (7) Stomatitis 86 (8) 65 (12) 14 (3) Sore Throat 82 (8) 61 (11) 17 (4) General Disorders and Administration Site Fatigue 398 (39) 240 (43) 130 (29) Pyrexia 328 (32) 172 (31) 89 (20) Weakness 252 (24) 108 (19) 114 (25) Edema Lower Limb 215 (21) 126 (23) 84 (19) Rigors 112 (11) 62 (11) 28 (6) Infections Urinary Tract Infection 124 (12) 50 (9) 41 (9) Upper Respiratory Tract Infection 101 (10) 82 (15) 30 (7) Metabolism Anorexia 231 (22) 81 (15) 105 (23) Weight Decreased 164 (16) 50 (9) 61 (13) Dehydration 145 (14) 60 (11) 59 (13) Appetite Decreased 130 (13) 48 (9) 45 (10) Musculoskeletal Bone Pain 569 (55) 316 (57) 284 (62) Myalgia 239 (23) 143 (26) 74 (16) Arthralgia 216 (21) 131 (24) 73 (16) Back Pain 156 (15) 106 (19) 40 (9) Pain in Limb 143 (14) 84 (15) 52 (11) Neoplasms Malignant Neoplasm Aggravated 205 (20) 97 (17) 89 (20) Nervous Headache 191 (19) 149 (27) 50 (11) Dizziness (excluding vertigo) 180 (18) 91 (16) 58 (13) Insomnia 166 (16) 111 (20) 73 (16) Paresthesia 149 (15) 85 (15) 35 (8) Hypoesthesia 127 (12) 65 (12) 43 (10) Psychiatric Depression 146 (14) 95 (17) 49 (11) Anxiety 112 (11) 73 (13) 37 (8) Confusion 74 (7) 39 (7) 47 (10) Respiratory Dyspnea 282 (27) 155 (28) 107 (24) Cough 224 (22) 129 (23) 65 (14) Skin Alopecia 125 (12) 80 (14) 36 (8) Dermatitis 114 (11) 74 (13) 38 (8) Grade 3 and Grade 4 laboratory abnormalities for serum creatinine, serum calcium, serum phosphorus, and serum magnesium observed in three clinical trials of Zometa in patients with bone metastases are shown in Tables 8 and 9.
Table 8: Grade 3 Laboratory Abnormalities for Serum Creatinine, Serum Calcium, Serum Phosphorus, and Serum Magnesium in Three Clinical Trials in Patients With Bone Metastases *Serum creatinine data for all patients randomized after the 15-minute infusion amendment.
1Grade 3 (greater than 3x Upper Limit of Normal); Grade 4 (greater than 6x Upper Limit of Normal).
2Grade 3 (less than 7 mg/dL); Grade 4 (less than 6 mg/dL).
3Grade 3 (less than 2 mg/dL); Grade 4 (less than 1 mg/dL).
4Grade 3 (greater than 3 mEq/L); Grade 4 (greater than 8 mEq/L).
5Grade 3 (less than 0.9 mEq/L); Grade 4 (less than 0.7 mEq/L).Grade 3 Laboratory Parameter Zometa Pamidronate Placebo 4 mg 90 mg n/N (%) n/N (%) n/N (%) Serum Creatinine1* 7/529 (1%) 4/268 (2%) 4/241 (2%) Hypocalcemia2 6/973 (< 1%) 4/536 (< 1%) 0/415 0 Hypophosphatemia3 115/973 (12%) 38/537 (7%) 14/415 (3%) Hypermagnesemia4 19/971 (2%) 2/535 (< 1%) 8/415 (2%) Hypomagnesemia5 1/971 (< 1%) 0/535 — 1/415 (< 1%) Table 9: Grade 4 Laboratory Abnormalities for Serum Creatinine, Serum Calcium, Serum Phosphorus, and Serum Magnesium in Three Clinical Trials in Patients With Bone Metastases *Serum creatinine data for all patients randomized after the 15-minute infusion amendment.
1Grade 3 (greater than 3x Upper Limit of Normal); Grade 4 (greater than 6x Upper Limit of Normal).
2Grade 3 (less than 7 mg/dL); Grade 4 (less than 6 mg/dL).
3Grade 3 (less than 2 mg/dL); Grade 4 (less than 1 mg/dL).
4Grade 3 (greater than 3 mEq/L); Grade 4 (greater than 8 mEq/L).
5Grade 3 (less than 0.9 mEq/L); Grade 4 (less than 0.7 mEq/L).Grade 4 Laboratory Parameter Zometa Pamidronate Placebo 4 mg 90 mg n/N (%) n/N (%) n/N (%) Serum Creatinine1* 2/529 (< 1%) 1/268 (< 1%) 0/241 0 Hypocalcemia2 7/973 (< 1%) 3/536 (< 1%) 2/415 (< 1%) Hypophosphatemia3 5/973 (< 1%) 0/537 0 1/415 (< 1%) Hypermagnesemia4 0/971 0 0/535 0 2/415 (< 1%) Hypomagnesemia5 2/971 (< 1%) 1/535 (< 1%) 0/415 0 Among the less frequently occurring adverse events (less than 15% of patients), rigors, hypokalemia, influenza-like illness, and hypocalcemia showed a trend for more events with bisphosphonate administration (Zometa 4 mg and pamidronate groups) compared to the placebo group.
Less common adverse events reported more often with Zometa 4 mg than pamidronate included decreased weight, which was reported in 16% of patients in the Zometa 4 mg group compared with 9% in the pamidronate group. Decreased appetite was reported in slightly more patients in the Zometa 4 mg group (13%) compared with the pamidronate (9%) and placebo (10%) groups, but the clinical significance of these small differences is not clear.
Renal Toxicity
In the bone metastases trials, renal deterioration was defined as an increase of 0.5 mg/dL for patients with normal baseline creatinine (less than 1.4 mg/dL) or an increase of 1.0 mg/dL for patients with an abnormal baseline creatinine (greater than or equal to1.4 mg/dL). The following are data on the incidence of renal deterioration in patients receiving Zometa 4 mg over 15 minutes in these trials (see Table 10).
Table 10: Percentage of Patients With Treatment-Emergent Renal Function Deterioration by Baseline Serum Creatinine* *Table includes only patients who were randomized to the trial after a protocol amendment that lengthened the infusion duration of Zometa to 15 minutes. Patient Population/Baseline Creatinine Multiple Myeloma and Breast Cancer Zometa 4 mg Pamidronate 90 mg n/N (%) n/N (%) Normal 27/246 (11%) 23/246 (9%) Abnormal 2/26 (8%) 2/22 (9%) Total 29/272 (11%) 25/268 (9%) Solid Tumors Zometa 4 mg Placebo n/N (%) n/N (%) Normal 17/154 (11%) 10/143 (7%) Abnormal 1/11 (9%) 1/20 (5%) Total 18/165 (11%) 11/163 (7%) Prostate Cancer Zometa 4 mg Placebo n/N (%) n/N (%) Normal 12/82 (15%) 8/68 (12%) Abnormal 4/10 (40%) 2/10 (20%) Total 16/92 (17%) 10/78 (13%) The risk of deterioration in renal function appeared to be related to time on study, whether patients were receiving Zometa (4 mg over 15 minutes), placebo, or pamidronate.
In the trials and in postmarketing experience, renal deterioration, progression to renal failure, and dialysis have occurred in patients with normal and abnormal baseline renal function, including patients treated with 4 mg infused over a 15-minute period. There have been instances of this occurring after the initial Zometa dose.
6.2 Postmarketing Experience
The following adverse reactions have been reported during postapproval use of Zometa. Because these reports are from a population of uncertain size and are subject to confounding factors, it is not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Osteonecrosis of the Jaw
Cases of osteonecrosis (primarily involving the jaw but also of other anatomical sites including hip, femur and external auditory canal) have been reported predominantly in cancer patients treated with intravenous bisphosphonates including Zometa. Many of these patients were also receiving chemotherapy and corticosteroids which may be a risk factor for ONJ. Caution is advised when Zometa is administered with anti-angiogenic drugs as an increased incidence of ONJ has been observed with concomitant use of these drugs. Data suggests a greater frequency of reports of ONJ in certain cancers, such as advanced breast cancer and multiple myeloma. The majority of the reported cases are in cancer patients following invasive dental procedures, such as tooth extraction. It is therefore prudent to avoid invasive dental procedures as recovery may be prolonged [see Warnings and Precautions (5.4)].
Acute Phase Reaction
Within three days after Zometa administration, an acute phase reaction has been reported, with symptoms including pyrexia, fatigue, bone pain and/or arthralgias, myalgias, chills, influenza-like illness and arthritis with subsequent joint swelling; these symptoms usually resolve within three days of onset, but resolution could take up to 7 to 14 days. However, some of these symptoms have been reported to persist for a longer duration.
Musculoskeletal Pain
Severe and occasionally incapacitating bone, joint, and/or muscle pain has been reported with bisphosphonate use [see Warnings and Precautions (5.5)].
Atypical Subtrochanteric and Diaphyseal Femoral Fractures
Atypical subtrochanteric and diaphyseal femoral fractures have been reported with bisphosphonate therapy, including Zometa [see Warnings and Precautions (5.6)].
Ocular Adverse Events
Cases of uveitis, scleritis, episcleritis, conjunctivitis, iritis, and orbital inflammation including orbital edema have been reported during postmarketing use. In some cases, symptoms resolved with topical steroids.
Hypersensitivity Reactions
There have been rare reports of allergic reaction with intravenous zoledronic acid including angioedema and bronchoconstriction. Very rare cases of anaphylactic reaction/shock have been reported. Cases of Stevens-Johnson syndrome and toxic epidermal necrolysis have also been reported.
Additional adverse reactions reported in postmarketing use include:
CNS: taste disturbance, hyperesthesia, tremor; Special Senses: blurred vision; uveitis; Gastrointestinal: dry mouth; Skin: Increased sweating; Musculoskeletal: muscle cramps; Cardiovascular: hypertension, bradycardia, hypotension (associated with syncope or circulatory collapse primarily in patients with underlying risk factors); Respiratory: bronchospasms, interstitial lung disease (ILD) with positive rechallenge; Renal: hematuria, proteinuria, acquired Fanconi syndrome; General Disorders and Administration Site: weight increase, influenza-like illness (pyrexia, asthenia, fatigue or malaise) persisting for greater than 30 days; Laboratory Abnormalities: hyperkalemia, hypernatremia, hypocalcemia (cardiac arrhythmias and neurologic adverse events including seizures, tetany, and numbness have been reported due to severe hypocalcemia).
-
7 DRUG INTERACTIONS
In vitro studies indicate that the plasma protein binding of zoledronic acid is low, with the unbound fraction ranging from 60%-77%. In vitro studies also indicate that zoledronic acid does not inhibit microsomal CYP450 enzymes. In vivo studies showed that zoledronic acid is not metabolized, and is excreted into the urine as the intact drug.
7.1 Aminoglycosides and Calcitonin
Caution is advised when bisphosphonates are administered with aminoglycosides or calcitonin, since these agents may have an additive effect to lower serum calcium level for prolonged periods. This effect has not been reported in Zometa clinical trials.
7.2 Loop Diuretics
Caution should also be exercised when Zometa is used in combination with loop diuretics due to an increased risk of hypocalcemia.
7.3 Nephrotoxic Drugs
Caution is indicated when Zometa is used with other potentially nephrotoxic drugs.
7.4 Thalidomide
No dose adjustment for Zometa 4 mg is needed when coadministered with thalidomide. In a pharmacokinetic study of 24 patients with multiple myeloma, Zometa 4 mg given as a 15-minute infusion was administered either alone or with thalidomide (100 mg once daily on days 1-14 and 200 mg once daily on Days 15-28). Coadministration of thalidomide with Zometa did not significantly change the pharmacokinetics of zoledronic acid or creatinine clearance.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action, Zometa can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data in pregnant women to inform the drug-associated risk. In animal reproduction studies, administration of zoledronic acid to pregnant rats during organogenesis resulted in fetal malformations and embryo-fetal lethality at maternal exposures that were ≥ 2.4 times the human clinical exposure based on AUC (see Data). Bisphosphonates, such as Zometa, are incorporated into the bone matrix, from where they are gradually released over periods of weeks to years. There may be a risk of fetal harm (e.g., skeletal and other abnormalities) if a woman becomes pregnant after completing a course of bisphosphonate therapy. Advise pregnant women and females of reproductive potential of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown; however, in the U.S. general population, the estimated background risk of major birth defects is 2%-4% and of miscarriage is 15%-20% of clinically recognized pregnancies.
Data
Animal Data
In female rats given subcutaneous doses of zoledronic acid of 0.01, 0.03, or 0.1 mg/kg/day beginning 15 days before mating and continuing through gestation, the number of stillbirths was increased and survival of neonates was decreased in the mid- and high-dose groups (greater than or equal to 0.2 times the human systemic exposure following an intravenous dose of 4 mg, based on an AUC comparison). Adverse maternal effects were observed in all dose groups (with a systemic exposure of greater than or equal to 0.07 times the human systemic exposure following an intravenous dose of 4 mg, based on an AUC comparison) and included dystocia and periparturient mortality in pregnant rats allowed to deliver. Maternal mortality may have been related to drug-induced inhibition of skeletal calcium mobilization, resulting in periparturient hypocalcemia. This appears to be a bisphosphonate-class effect.
In pregnant rats given a subcutaneous dose of zoledronic acid of 0.1, 0.2, or 0.4 mg/kg/day during gestation, adverse fetal effects were observed in the mid- and high-dose groups (with systemic exposures of 2.4 and 4.8 times, respectively, the human systemic exposure following an intravenous dose of 4 mg, based on an AUC comparison). These adverse effects included increases in pre- and postimplantation losses, decreases in viable fetuses, and fetal skeletal, visceral, and external malformations. Fetal skeletal effects observed in the high-dose group included unossified or incompletely ossified bones, thickened, curved, or shortened bones, wavy ribs, and shortened jaw. Other adverse fetal effects observed in the high-dose group included reduced lens, rudimentary cerebellum, reduction or absence of liver lobes, reduction of lung lobes, vessel dilation, cleft palate, and edema. Skeletal variations were also observed in the low dose group at 0.1 mg/kg/day (with systemic exposure of 1.2 times the human systemic exposure following an intravenous dose of 4 mg, based on an AUC comparison). Signs of maternal toxicity were observed in the high-dose group and included reduced body weights and food consumption, indicating that maximal exposure levels were achieved in this study.
In pregnant rabbits given subcutaneous doses of zoledronic acid of 0.01, 0.03, or 0.1 mg/kg/day during gestation (less than or equal to 0.5 times the human intravenous dose of 4 mg, based on a comparison of relative body surface areas), no adverse fetal effects were observed. Maternal mortality and abortion occurred in all treatment groups (at doses greater than or equal to 0.05 times the human intravenous dose of 4 mg, based on a comparison of relative body surface areas). Adverse maternal effects were associated with, and may have been caused by, drug-induced hypocalcemia.
8.2 Lactation
Risk Summary
After administration of Zometa, it is not known whether zoledronic acid is present in human milk, or whether it affects milk production or the breastfed child. Zoledronic acid binds to bone long term and may be released over periods of weeks to years. Because of the potential for serious adverse reactions in a breastfed child, advise a lactating woman not to breastfeed during and after Zometa treatment.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify pregnancy status of females of reproductive potential prior to initiation of Zometa.
Contraception
Females
Zometa can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Zoledronic acid binds to bone long term and may be released over periods of weeks to years. Advise females of reproductive potential to use effective contraception during and after Zometa treatment.
Infertility
Females
Based on animal studies, Zometa may impair fertility in females of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Zometa is not indicated for use in children.
The safety and effectiveness of zoledronic acid was studied in a one-year, active-controlled trial of 152 pediatric subjects (74 receiving zoledronic acid). The enrolled population was subjects with severe osteogenesis imperfecta, aged 1-17 years, 55% male, 84% Caucasian, with a mean lumbar spine bone mineral density (BMD) of 0.431 gm/cm2, which is 2.7 standard deviations below the mean for age-matched controls (BMD Z-score of -2.7). At one year, increases in BMD were observed in the zoledronic acid treatment group. However, changes in BMD in individual patients with severe osteogenesis imperfecta did not necessarily correlate with the risk for fracture or the incidence or severity of chronic bone pain. The adverse events observed with Zometa use in children did not raise any new safety findings beyond those previously seen in adults treated for hypercalcemia of malignancy or bone metastases. However, adverse reactions seen more commonly in pediatric patients included pyrexia (61%), arthralgia (26%), hypocalcemia (22%) and headache (22%). These reactions, excluding arthralgia, occurred most frequently within 3 days after the first infusion and became less common with repeat dosing. Because of long-term retention in bone, Zometa should only be used in children if the potential benefit outweighs the potential risk.
Plasma zoledronic acid concentration data was obtained from 10 patients with severe osteogenesis imperfecta (4 in the age group of 3-8 years and 6 in the age group of 9-17 years) infused with 0.05 mg/kg dose over 30 min. Mean Cmax and AUC(0-last) was 167 ng/mL and 220 ngh/mL, respectively. The plasma concentration time profile of zoledronic acid in pediatric patients represent a multi-exponential decline, as observed in adult cancer patients at an approximately equivalent mg/kg dose.
8.5 Geriatric Use
Clinical studies of Zometa in hypercalcemia of malignancy included 34 patients who were 65 years of age or older. No significant differences in response rate or adverse reactions were seen in geriatric patients receiving Zometa as compared to younger patients. Controlled clinical studies of Zometa in the treatment of multiple myeloma and bone metastases of solid tumors in patients over age 65 revealed similar efficacy and safety in older and younger patients. Because decreased renal function occurs more commonly in the elderly, special care should be taken to monitor renal function.
-
10 OVERDOSAGE
Clinical experience with acute overdosage of Zometa is limited. Two patients received Zometa 32 mg over 5 minutes in clinical trials. Neither patient experienced any clinical or laboratory toxicity. Overdosage may cause clinically significant hypocalcemia, hypophosphatemia, and hypomagnesemia. Clinically relevant reductions in serum levels of calcium, phosphorus, and magnesium should be corrected by intravenous administration of calcium gluconate, potassium or sodium phosphate, and magnesium sulfate, respectively.
In an open-label study of zoledronic acid 4 mg in breast cancer patients, a female patient received a single 48-mg dose of zoledronic acid in error. Two days after the overdose, the patient experienced a single episode of hyperthermia (38°C), which resolved after treatment. All other evaluations were normal, and the patient was discharged seven days after the overdose.
A patient with non-Hodgkin’s lymphoma received zoledronic acid 4 mg daily on four successive days for a total dose of 16 mg. The patient developed paresthesia and abnormal liver function tests with increased GGT (nearly 100 unit/L, each value unknown). The outcome of this case is not known.
In controlled clinical trials, administration of Zometa 4 mg as an intravenous infusion over 5 minutes has been shown to increase the risk of renal toxicity compared to the same dose administered as a 15-minute intravenous infusion. In controlled clinical trials, Zometa 8 mg has been shown to be associated with an increased risk of renal toxicity compared to Zometa 4 mg, even when given as a 15-minute intravenous infusion, and was not associated with added benefit in patients with hypercalcemia of malignancy [see Dosage and Administration (2.4)].
-
11 DESCRIPTION
Zometa contains zoledronic acid, a bisphosphonic acid which is an inhibitor of osteoclastic bone resorption. Zoledronic acid is designated chemically as (1-Hydroxy-2-imidazol-1-yl-phosphonoethyl) phosphonic acid monohydrate and its structural formula is:
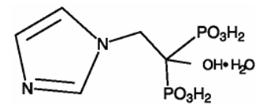
Zoledronic acid is a white crystalline powder. Its molecular formula is C5H10N2O7P2H2O and its molar mass is 290.1g/mol. Zoledronic acid is highly soluble in 0.1N sodium hydroxide solution, sparingly soluble in water and 0.1N hydrochloric acid, and practically insoluble in organic solvents. The pH of a 0.7% solution of zoledronic acid in water is approximately 2.0.
Zometa is available in 100 mL bottles as a sterile liquid ready-to-use solution for intravenous infusion and in 5 mL vials as a sterile liquid solution for dilution prior to intravenous infusion.
- Each 100 mL ready-to-use bottle contains 4.264 mg zoledronic acid monohydrate, corresponding to 4 mg zoledronic acid on an anhydrous basis, 5100 mg of mannitol, USP, water for injection, and 24 mg of sodium citrate, USP.
- Each 5 mL solution for dilution prior to intravenous infusion vial contains 4.264 mg zoledronic acid monohydrate, corresponding to 4 mg zoledronic acid on an anhydrous basis, 220 mg of mannitol, USP, water for injection, and 24 mg of sodium citrate, USP.
Inactive Ingredients: mannitol, USP, as bulking agent, water for injection, and sodium citrate, USP, as buffering agent.
- Each 100 mL ready-to-use bottle contains 4.264 mg zoledronic acid monohydrate, corresponding to 4 mg zoledronic acid on an anhydrous basis, 5100 mg of mannitol, USP, water for injection, and 24 mg of sodium citrate, USP.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The principal pharmacologic action of zoledronic acid is inhibition of bone resorption. Although the antiresorptive mechanism is not completely understood, several factors are thought to contribute to this action. In vitro, zoledronic acid inhibits osteoclastic activity and induces osteoclast apoptosis. Zoledronic acid also blocks the osteoclastic resorption of mineralized bone and cartilage through its binding to bone. Zoledronic acid inhibits the increased osteoclastic activity and skeletal calcium release induced by various stimulatory factors released by tumors.
12.2 Pharmacodynamics
Clinical studies in patients with hypercalcemia of malignancy (HCM) showed that single-dose infusions of Zometa are associated with decreases in serum calcium and phosphorus and increases in urinary calcium and phosphorus excretion.
Osteoclastic hyperactivity resulting in excessive bone resorption is the underlying pathophysiologic derangement in hypercalcemia of malignancy (HCM, tumor-induced hypercalcemia) and metastatic bone disease. Excessive release of calcium into the blood as bone is resorbed results in polyuria and gastrointestinal disturbances, with progressive dehydration and decreasing glomerular filtration rate. This, in turn, results in increased renal resorption of calcium, setting up a cycle of worsening systemic hypercalcemia. Reducing excessive bone resorption and maintaining adequate fluid administration are, therefore, essential to the management of hypercalcemia of malignancy.
Patients who have hypercalcemia of malignancy can generally be divided into two groups according to the pathophysiologic mechanism involved: humoral hypercalcemia and hypercalcemia due to tumor invasion of bone. In humoral hypercalcemia, osteoclasts are activated and bone resorption is stimulated by factors such as parathyroid hormone-related protein, which are elaborated by the tumor and circulate systemically. Humoral hypercalcemia usually occurs in squamous cell malignancies of the lung or head and neck or in genitourinary tumors such as renal cell carcinoma or ovarian cancer. Skeletal metastases may be absent or minimal in these patients.
Extensive invasion of bone by tumor cells can also result in hypercalcemia due to local tumor products that stimulate bone resorption by osteoclasts. Tumors commonly associated with locally mediated hypercalcemia include breast cancer and multiple myeloma.
Total serum calcium levels in patients who have hypercalcemia of malignancy may not reflect the severity of hypercalcemia, since concomitant hypoalbuminemia is commonly present. Ideally, ionized calcium levels should be used to diagnose and follow hypercalcemic conditions; however, these are not commonly or rapidly available in many clinical situations. Therefore, adjustment of the total serum calcium value for differences in albumin levels (corrected serum calcium, CSC) is often used in place of measurement of ionized calcium; several nomograms are in use for this type of calculation [see Dosage and Administration (2.1)].
12.3 Pharmacokinetics
Pharmacokinetic data in patients with hypercalcemia are not available.
Distribution
Single or multiple (every 28 days) 5-minute or 15-minute infusions of 2, 4, 8, or 16 mg Zometa were given to 64 patients with cancer and bone metastases. The post-infusion decline of zoledronic acid concentrations in plasma was consistent with a triphasic process showing a rapid decrease from peak concentrations at end of infusion to less than 1% of Cmax 24 hours postinfusion with population half-lives of t1/2α 0.24 hours and t1/2β 1.87 hours for the early disposition phases of the drug. The terminal elimination phase of zoledronic acid was prolonged, with very low concentrations in plasma between Days 2 and 28 postinfusion, and a terminal elimination half-life t 1/2γ of 146 hours. The area under the plasma concentration versus time curve (AUC0-24h) of zoledronic acid was dose proportional from 2-16 mg. The accumulation of zoledronic acid measured over three cycles was low, with mean AUC0-24h ratios for cycles 2 and 3 versus 1 of 1.13 ± 0.30 and 1.16 ± 0.36, respectively.
In vitro and ex vivo studies showed low affinity of zoledronic acid for the cellular components of human blood, with a mean blood to plasma concentration ratio of 0.59 in a concentration range of 30 ng/mL to 5000 ng/mL. In vitro, the plasma protein binding is low, with the unbound fraction ranging from 60% at 2 ng/mL to 77% at 2000 ng/mL of zoledronic acid.
Metabolism
Zoledronic acid does not inhibit human P450 enzymes in vitro. Zoledronic acid does not undergo biotransformation in vivo. In animal studies, less than 3% of the administered intravenous dose was found in the feces, with the balance either recovered in the urine or taken up by bone, indicating that the drug is eliminated intact via the kidney. Following an intravenous dose of 20 nCi 14C-zoledronic acid in a patient with cancer and bone metastases, only a single radioactive species with chromatographic properties identical to those of parent drug was recovered in urine, which suggests that zoledronic acid is not metabolized.
Excretion
In 64 patients with cancer and bone metastases, on average (± SD) 39 ± 16% of the administered zoledronic acid dose was recovered in the urine within 24 hours, with only trace amounts of drug found in urine post-Day 2. The cumulative percent of drug excreted in the urine over 0-24 hours was independent of dose. The balance of drug not recovered in urine over 0-24 hours, representing drug presumably bound to bone, is slowly released back into the systemic circulation, giving rise to the observed prolonged low plasma concentrations. The 0-24 hour renal clearance of zoledronic acid was 3.7 ± 2.0 L/h.
Zoledronic acid clearance was independent of dose but dependent upon the patient’s creatinine clearance. In a study in patients with cancer and bone metastases, increasing the infusion time of a 4-mg dose of zoledronic acid from 5 minutes (n = 5) to 15 minutes (n = 7) resulted in a 34% decrease in the zoledronic acid concentration at the end of the infusion ([mean ± SD] 403 ± 118 ng/mL versus 264 ± 86 ng/mL) and a 10% increase in the total AUC (378 ± 116 ng x h/mL versus 420 ± 218 ng x h/mL). The difference between the AUC means was not statistically significant.
Special Populations
Pediatrics
Zometa is not indicated for use in children [see Use in Specific Populations (8.4)].
Geriatrics
The pharmacokinetics of zoledronic acid were not affected by age in patients with cancer and bone metastases who ranged in age from 38 years to 84 years.
Race
Population pharmacokinetic analyses did not indicate any differences in pharmacokinetics among Japanese and North American (Caucasian and African American) patients with cancer and bone metastases.
Hepatic Insufficiency
No clinical studies were conducted to evaluate the effect of hepatic impairment on the pharmacokinetics of zoledronic acid.
Renal Insufficiency
The pharmacokinetic studies conducted in 64 cancer patients represented typical clinical populations with normal to moderately impaired renal function. Compared to patients with normal renal function (N = 37), patients with mild renal impairment (N = 15) showed an average increase in plasma AUC of 15%, whereas patients with moderate renal impairment (N = 11) showed an average increase in plasma AUC of 43%. Limited pharmacokinetic data are available for Zometa in patients with severe renal impairment (creatinine clearance less than 30 mL/min). Based on population PK/PD modeling, the risk of renal deterioration appears to increase with AUC, which is doubled at a creatinine clearance of 10 mL/min. Creatinine clearance is calculated by the Cockcroft-Gault formula:
CrCl = [140-age (years)] x weight (kg) {x 0.85 for female patients}
[72 x serum creatinine (mg/dL)]Zometa systemic clearance in individual patients can be calculated from the population clearance of Zometa, CL (L/h) = 6.5(CrCl/90)0.4. These formulae can be used to predict the Zometa AUC in patients, where CL = Dose/AUC0-∞. The average AUC0-24 in patients with normal renal function was 0.42 mgh/L and the calculated AUC0-∞ for a patient with creatinine clearance of 75 mL/min was 0.66 mgh/L following a 4-mg dose of Zometa. However, efficacy and safety of adjusted dosing based on these formulae have not been prospectively assessed [see Warnings and Precautions (5.3)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Standard lifetime carcinogenicity bioassays were conducted in mice and rats. Mice were given oral doses of zoledronic acid of 0.1, 0.5, or 2.0 mg/kg/day. There was an increased incidence of Harderian gland adenomas in males and females in all treatment groups (at doses greater than or equal to 0.002 times a human intravenous dose of 4 mg, based on a comparison of relative body surface areas). Rats were given oral doses of zoledronic acid of 0.1, 0.5, or 2.0 mg/kg/day. No increased incidence of tumors was observed (at doses less than or equal to 0.2 times the human intravenous dose of 4 mg, based on a comparison of relative body surface areas).
Zoledronic acid was not genotoxic in the Ames bacterial mutagenicity assay, in the Chinese hamster ovary cell assay, or in the Chinese hamster gene mutation assay, with or without metabolic activation. Zoledronic acid was not genotoxic in the in vivo rat micronucleus assay.
Female rats were given subcutaneous doses of zoledronic acid of 0.01, 0.03, or 0.1 mg/kg/day beginning 15 days before mating and continuing through gestation. Effects observed in the high-dose group (with systemic exposure of 1.2 times the human systemic exposure following an intravenous dose of 4 mg, based on AUC comparison) included inhibition of ovulation and a decrease in the number of pregnant rats. Effects observed in both the mid-dose group (with systemic exposure of 0.2 times the human systemic exposure following an intravenous dose of 4 mg, based on an AUC comparison) and high-dose group included an increase in preimplantation losses and a decrease in the number of implantations and live fetuses.
-
14 CLINICAL STUDIES
14.1 Hypercalcemia of Malignancy
Two identical multicenter, randomized, double-blind, double-dummy studies of Zometa 4 mg given as a 5-minute intravenous infusion or pamidronate 90 mg given as a 2-hour intravenous infusion were conducted in 185 patients with hypercalcemia of malignancy (HCM). NOTE: Administration of Zometa 4 mg given as a 5-minute intravenous infusion has been shown to result in an increased risk of renal toxicity, as measured by increases in serum creatinine, which can progress to renal failure. The incidence of renal toxicity and renal failure has been shown to be reduced when Zometa 4 mg is given as a 15-minute intravenous infusion. Zometa should be administered by intravenous infusion over no less than 15 minutes [see Warnings and Precautions (5.1, 5.2), Dosage and Administration (2.4)]. The treatment groups in the clinical studies were generally well balanced with regards to age, sex, race, and tumor types. The mean age of the study population was 59 years; 81% were Caucasian, 15% were Black, and 4% were of other races. Sixty percent (60%) of the patients were male. The most common tumor types were lung, breast, head and neck, and renal.
In these studies, HCM was defined as a corrected serum calcium (CSC) concentration of greater than or equal to 12.0 mg/dL (3.00 mmol/L). The primary efficacy variable was the proportion of patients having a complete response, defined as the lowering of the CSC to less than or equal to 10.8 mg/dL (2.70 mmol/L) within 10 days after drug infusion.
To assess the effects of Zometa versus those of pamidronate, the two multi-center HCM studies were combined in a preplanned analysis. The results of the primary analysis revealed that the proportion of patients that had normalization of corrected serum calcium by Day 10 were 88% and 70% for Zometa 4 mg and pamidronate 90 mg, respectively (P = 0.002) (see Figure 1). In these studies, no additional benefit was seen for Zometa 8 mg over Zometa 4 mg; however, the risk of renal toxicity of Zometa 8 mg was significantly greater than that seen with Zometa 4 mg.
Figure 1: Proportion of Complete Responders by Day 10 in Pooled HCM Studies
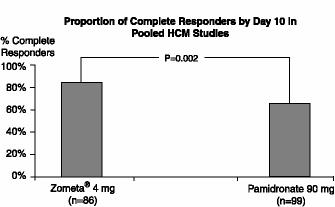
Secondary efficacy variables from the pooled HCM studies included the proportion of patients who had normalization of corrected serum calcium (CSC) by Day 4; the proportion of patients who had normalization of CSC by Day 7; time to relapse of HCM; and duration of complete response. Time to relapse of HCM was defined as the duration (in days) of normalization of serum calcium from study drug infusion until the last CSC value less than 11.6 mg/dL (less than 2.90 mmol/L). Patients who did not have a complete response were assigned a time to relapse of 0 days. Duration of complete response was defined as the duration (in days) from the occurrence of a complete response until the last CSC less than or equal to 10.8 mg/dL (2.70 mmol/L). The results of these secondary analyses for Zometa 4 mg and pamidronate 90 mg are shown in Table 11.
Table 11: Secondary Efficacy Variables in Pooled HCM Studies *P less than 0.05 versus pamidronate 90 mg. Zometa 4 mg Pamidronate 90 mg Complete Response N Response Rate N Response Rate By Day 4 86 45.3% 99 33.3% By Day 7 86 82.6%* 99 63.6% Duration of Response N Median Duration (Days) N Median Duration (Days) Time to Relapse 86 30* 99 17 Duration of Complete Response 76 32 69 18 14.2 Clinical Trials in Multiple Myeloma and Bone Metastases of Solid Tumors
Table 12 describes an overview of the efficacy population in three randomized Zometa trials in patients with multiple myeloma and bone metastases of solid tumors. These trials included a pamidronate-controlled study in breast cancer and multiple myeloma, a placebo-controlled study in prostate cancer, and a placebo-controlled study in other solid tumors. The prostate cancer study required documentation of previous bone metastases and 3 consecutive rising PSAs while on hormonal therapy. The other placebo-controlled solid tumor study included patients with bone metastases from malignancies other than breast cancer and prostate cancer, including NSCLC, renal cell cancer, small cell lung cancer, colorectal cancer, bladder cancer, GI/genitourinary cancer, head and neck cancer, and others. These trials were comprised of a core phase and an extension phase. In the solid tumor, breast cancer and multiple myeloma trials, only the core phase was evaluated for efficacy as a high percentage of patients did not choose to participate in the extension phase. In the prostate cancer trials, both the core and extension phases were evaluated for efficacy showing the Zometa effect during the first 15 months was maintained without decrement or improvement for another 9 months. The design of these clinical trials does not permit assessment of whether more than one-year administration of Zometa is beneficial. The optimal duration of Zometa administration is not known.
The studies were amended twice because of renal toxicity. The Zometa infusion duration was increased from 5 minutes to 15 minutes. After all patients had been accrued, but while dosing and follow-up continued, patients in the 8 mg Zometa treatment arm were switched to 4 mg due to toxicity. Patients who were randomized to the Zometa 8 mg group are not included in these analyses.
Table 12: Overview of Efficacy Population for Phase III Studies *Patients who were randomized to the 8 mg Zometa group are not included in any of the analyses in this package insert. Patient Population No. of Patients Zometa Dose Control Median Duration (Planned Duration) Zometa 4 mg Multiple myeloma or metastatic breast cancer 1,648 4 and 8* mg
Q3-4 weeksPamidronate 90 mg
Q3-4 weeks12.0 months
(13 months)Metastatic prostate cancer 643 4 and 8* mg
Q3 weeksPlacebo 10.5 months
(15 months)Metastatic solid tumor other than breast or prostate cancer 773 4 and 8* mg
Q3 weeksPlacebo 3.8 months
(9 months)Each study evaluated skeletal-related events (SREs), defined as any of the following: pathologic fracture, radiation therapy to bone, surgery to bone, or spinal cord compression. Change in antineoplastic therapy due to increased pain was a SRE in the prostate cancer study only. Planned analyses included the proportion of patients with a SRE during the study and time to the first SRE. Results for the two Zometa placebo-controlled studies are given in Table 13.
Table 13: Zometa Compared to Placebo in Patients With Bone Metastases from Prostate Cancer or Other Solid Tumors 1SRE = Skeletal-Related Event.
2Difference for the proportion of patients with a SRE of Zometa 4 mg versus placebo.
3Hazard ratio for the first occurrence of a SRE of Zometa 4 mg versus placebo.I. Analysis of Proportion of Patients with a SRE1 II. Analysis of Time to the First SRE Study Study Arm &
Patient NumberProportion Difference2
& 95% CIP-value Median
(Days)Hazard Ratio3
& 95% CIP-value Prostate Cancer Zometa 4 mg
(n = 214)33% -11%
(-20%, -1%)0.02 Not Reached 0.67
(0.49, 0.91)0.011 Placebo
(n = 208)44% 321 Solid Tumors Zometa 4 mg
(n = 257)38% -7%
(-15%, 2%)0.13 230 0.73
(0.55, 0.96)0.023 Placebo
(n = 250)44% 163 In the breast cancer and myeloma trial, efficacy was determined by a noninferiority analysis comparing Zometa to pamidronate 90 mg for the proportion of patients with a SRE. This analysis required an estimation of pamidronate efficacy. Historical data from 1,128 patients in three pamidronate placebo-controlled trials demonstrated that pamidronate decreased the proportion of patients with a SRE by 13.1% (95% CI = 7.3%, 18.9%). Results of the comparison of treatment with Zometa compared to pamidronate are given in Table 14.
Table 14: Zometa Compared to Pamidronate in Patients With Multiple Myeloma or Bone Metastases from Breast Cancer 1SRE = Skeletal-Related Event.
2Difference for the proportion of patients with a SRE of Zometa 4 mg versus pamidronate 90 mg.
3Hazard ratio for the first occurrence of a SRE of Zometa 4 mg versus pamidronate 90 mg.I. Analysis of Proportion of Patients with a SRE1 II. Analysis of Time to the First SRE Study Study Arm &
Patient NumberProportion Difference2
& 95% CIP-value Median
(Days)Hazard Ratio3
& 95% CIP-value Multiple Myeloma
& Breast CancerZometa 4 mg
(n = 561)
Pamidronate
(n = 555)44%
46%-2%
(-7.9%, 3.7%)0.46 373
3630.92
(0.77, 1.09)0.32 -
16 HOW SUPPLIED/STORAGE AND HANDLING
4 mg/100 mL (0.04 mg/mL) single-dose ready-to-use bottle
Carton of 1 bottle……………………………………………………………………………...NDC 0078-0590-61
Store at 25°C (77°F); excursions permitted to 15°C to 30°C (59°F-86°F) [see USP Controlled Room Temperature]. Discard unused portion.
4 mg/5 mL (0.8 mg/mL) single-dose vial for dilution prior to intravenous infusion
Carton of 1 vial……………………………………………………………………………..NDC 0078-0387-25
Store at 25°C (77°F); excursions permitted to 15°C to 30°C (59°F-86°F) [see USP Controlled Room Temperature]. Discard unused portion.
-
17 PATIENT COUNSELING INFORMATION
Drugs With Same Active Ingredient or in the Same Drug Class
Inform patients not to take Reclast or other bisphosphonates during the course of their Zometa therapy [see Warnings and Precautions (5.1)].
Renal Impairment
- Instruct patients to tell their doctor if they have kidney problems before being given Zometa.
- Inform patients of the importance of getting their blood tests (serum creatinine) during the course of their Zometa therapy [see Warnings and Precautions (5.3)].
Osteonecrosis of the Jaw (ONJ)
- Advise patients to have a dental examination prior to treatment with Zometa and to avoid invasive dental procedures during treatment.
- Inform patients of the importance of good dental hygiene, routine dental care, and regular dental check-ups.
- Advise patients to immediately tell their doctor about any oral symptoms such as loosening of a tooth, pain, swelling, or non-healing of sores or discharge during treatment with Zometa [see Warnings and Precautions (5.4)].
Musculoskeletal Pain
Advise patients to immediately tell their doctor about any severe bone, joint, and/or muscle pain [see Warnings and Precautions (5.5)].
Atypical Subtrochanteric and Diaphyseal Femoral Fracture
Advise patients to report any thigh, hip, or groin pain. It is unknown whether the risk of atypical femur fracture continues after stopping therapy [see Warnings and Precautions (5.6)].
Patients With Asthma
There have been reports of bronchoconstriction in aspirin-sensitive patients receiving bisphosphonates, including zoledronic acid. Before being given zoledronic acid, instruct patients to tell their doctor if they are aspirin-sensitive [see Warnings and Precautions (5.7)].
Hypocalcemia
Advise patients with multiple myeloma and bone metastasis of solid tumors to take an oral calcium supplement of 500 mg and a multiple vitamin containing 400 international units of vitamin D daily [see Warnings and Precautions (5.9)].
Embryo-Fetal Toxicity
- Zometa should not be given if the patient is pregnant or plans to become pregnant. Inform female patients of the risk to a fetus and potential loss of the pregnancy [see Warnings and Precautions (5.10)].
- Advise females of reproductive potential to use effective contraception during and after treatment with Zometa [see Warnings and Precautions (5.10), Use in Specific Populations (8.1, 8.3)].
Lactation
Advise lactating women not to breastfeed during and after treatment with Zometa [see Use in Specific Populations (8.2)].
Common Adverse Reactions
Advise patients that the most common side effects of ZOMETA are nausea, fatigue, anemia, bone pain, constipation, fever, vomiting, and dyspnea.
Distributed by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936© Novartis
T2018-137
December 2018 -
PRINCIPAL DISPLAY PANEL
NDC: 0078-0387-25
Zometa®
(zoledronic acid)
Injection
4 mg / 5 mL
(0.8 mg / mL)For Intravenous Infusion after Dilution
Single-dose
Discard unsed portion.
One 5 mL vial
Sterile
Not for direct injection. Dose must be diluted.
See package insert for Preparation of Solution.
Do not mix with calcium-containing infusion solutions.
Rx only
NOVARTIS

-
PRINCIPAL DISPLAY PANEL
NDC: 0078-0590-61
ZOMETA®
(zoledronic acid) Injection
Sterile Solution
100 mL bottle
4 mg/100 mL
(0.04 mg/mL)Solution for Intravenous Infusion
Single-dose
Discard unsed portion.
For more information visit
www.us.zometa.com
or call
1-866-4-ZOMETA
(1-866-496-6382)
NOVARTIS
Rx only

-
INGREDIENTS AND APPEARANCE
ZOMETA
zoledronic acid injection, solution, concentrateProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-0387 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ZOLEDRONIC ACID (UNII: 6XC1PAD3KF) (ZOLEDRONIC ACID ANHYDROUS - UNII:70HZ18PH24) ZOLEDRONIC ACID ANHYDROUS 4 mg in 5 mL Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) 220 mg in 5 mL SODIUM CITRATE (UNII: 1Q73Q2JULR) 24 mg in 5 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0387-25 1 in 1 CARTON 08/20/2001 11/30/2020 1 5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021223 08/20/2001 11/30/2020 ZOMETA
zoledronic acid injection, solution, concentrateProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-0590 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ZOLEDRONIC ACID (UNII: 6XC1PAD3KF) (ZOLEDRONIC ACID ANHYDROUS - UNII:70HZ18PH24) ZOLEDRONIC ACID ANHYDROUS 4 mg in 100 mL Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) 5100 mg in 100 mL SODIUM CITRATE (UNII: 1Q73Q2JULR) 24 mg in 100 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0590-61 1 in 1 CARTON 08/20/2001 01/31/2020 1 100 mL in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021223 08/20/2001 01/31/2020 Labeler - Novartis Pharmaceuticals Corporation (002147023)
Trademark Results [Zometa]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ZOMETA 75411654 2393885 Live/Registered |
NOVARTIS AG 1997-12-29 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.