SKYCLARYS- omaveloxolone capsule
SKYCLARYS by
Drug Labeling and Warnings
SKYCLARYS by is a Prescription medication manufactured, distributed, or labeled by Reata Pharmaceuticals, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use SKYCLARYS safely and effectively. See full prescribing information for SKYCLARYS.
SKYCLARYS® (omaveloxolone) capsules, for oral use
Initial U.S. Approval: 2023RECENT MAJOR CHANGES
Dosage and Administration (2.2) 1/2024 INDICATIONS AND USAGE
SKYCLARYS is indicated for the treatment of Friedreich's ataxia in adults and adolescents aged 16 years and older. (1)
DOSAGE AND ADMINISTRATION
- Obtain alanine aminotransferase (ALT), aspartate aminotransferase (AST), bilirubin, B-type natriuretic peptide (BNP), and lipid parameters prior to initiating SKYCLARYS and during treatment. (2.1, 5.1, 5.2, 5.3)
- Recommended dosage is 150 mg (3 capsules) taken orally once daily. (2.2)
- Administer SKYCLARYS on an empty stomach at least 1 hour before eating. (2.2)
- Swallow SKYCLARYS capsules whole or open and sprinkle the entire contents of both halves of the capsule onto applesauce and mix. Do not crush or chew capsules. (2.2)
- Moderate and Severe Hepatic Impairment: The recommended dosage of SKYCLARYS is 100 mg once daily for patients with moderate hepatic impairment. If adverse reactions emerge, further reduce the dosage to 50 mg once daily. Avoid use in patients with severe hepatic impairment. (2.5, 8.6, 12.3)
DOSAGE FORMS AND STRENGTHS
Capsules: 50 mg (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Elevation of Aminotransferases: Monitor ALT, AST, and total bilirubin prior to initiation, every month for the first 3 months of treatment, and periodically thereafter. (2.1, 5.1)
- Elevation of B-type Natriuretic Peptide (BNP): Advise patients of signs and symptoms of fluid overload. (2.1, 5.2)
- Lipid Abnormalities: Monitor cholesterol periodically during treatment. (2.1, 5.3)
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥20% and greater than placebo) are elevated liver enzymes (AST/ALT), headache, nausea, abdominal pain, fatigue, diarrhea, and musculoskeletal pain. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Reata Pharmaceuticals, Inc. at 1-800-314-3934 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 1/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Testing Before Initiating SKYCLARYS and Monitoring to Assess Safety
2.2 Recommended Dosage
2.3 Missed Doses
2.4 Recommendations for Concomitant Use with Strong or Moderate CYP3A4 Inhibitors and Inducers
2.5 Recommended Dosage for Patients with Hepatic Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Elevation of Aminotransferases
5.2 Elevation of B-Type Natriuretic Peptide
5.3 Lipid Abnormalities
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on SKYCLARYS
7.2 Effect of SKYCLARYS on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Testing Before Initiating SKYCLARYS and Monitoring to Assess Safety
Obtain ALT, AST, bilirubin, BNP, and lipid parameters prior to initiating SKYCLARYS and during treatment [see Warnings and Precautions (5.1, 5.2, 5.3)].
2.2 Recommended Dosage
The recommended dosage of SKYCLARYS is 150 mg (3 capsules) taken orally once daily.
- Administer SKYCLARYS on an empty stomach at least one hour before eating [see Clinical Pharmacology (12.3)].
- Swallow SKYCLARYS capsules whole. Do not crush or chew.
-
For patients who are unable to swallow whole capsules:
- SKYCLARYS capsules may be opened and the entire contents of both halves of the capsule sprinkled onto 2 tablespoons (30 mL) of applesauce [see Clinical Pharmacology (12.3)].
- Stir the mixture until homogenous.
- Swallow all the drug/applesauce mixture immediately.
- Do not store the mixture for future use.
- Contents of the SKYCLARYS capsules should not be mixed with milk or orange juice.
- Not for enteral feeding tube administration.
2.3 Missed Doses
If a dose of SKYCLARYS is missed, take the next dose at its scheduled time the following day. A double dose should not be taken to make up for a missed dose.
2.4 Recommendations for Concomitant Use with Strong or Moderate CYP3A4 Inhibitors and Inducers
The recommended dosage for concomitant use of SKYCLARYS with cytochrome P450 (CYP) 3A4 inhibitors and inducers are described in Table 1 [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
Table 1: Recommended Dosage of SKYCLARYS with Concomitant Use of CYP3A4 Inhibitors and Inducers Concomitant Drug Class Dosage Strong CYP3A4 inhibitor Recommended to avoid concomitant use.
If coadministration cannot be avoided:
- Reduce the dosage of SKYCLARYS to 50 mg once daily with close monitoring for adverse reactions.
- If adverse reactions emerge, coadministration with strong CYP3A4 inhibitors should be discontinued.
Moderate CYP3A4 inhibitor Recommended to avoid concomitant use.
If coadministration cannot be avoided:
- Reduce the dosage of SKYCLARYS to 100 mg once daily with close monitoring for adverse reactions.
- If adverse reactions emerge, further reduce the dosage of SKYCLARYS to 50 mg once daily.
Strong or Moderate CYP3A4 inducer Recommended to avoid concomitant use. 2.5 Recommended Dosage for Patients with Hepatic Impairment
The recommended dosage for patients with hepatic impairment are described in Table 2 [see Use in Specific Populations (8.6)].
Table 2: Recommended Dosage in Patients with Hepatic Impairment Impairment Classification (Child-Pugh) Dosage Severe (Child-Pugh Class C) Avoid use Moderate (Child-Pugh Class B) - 100 mg once daily with close monitoring for adverse reactions
- Consider lowering to 50 mg once daily if adverse reactions emerge
Mild (Child-Pugh Class A) 150 mg once daily - 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Elevation of Aminotransferases
Treatment with SKYCLARYS can cause an elevation in hepatic transaminases (ALT and AST). In Study 1 [see Clinical Studies (14)], the incidence of elevations of ALT or AST above 5 times and 3 times the upper limit of normal (ULN) was 16% and 31%, respectively, in patients treated with SKYCLARYS. There were no cases of concomitant elevation of transaminases and total bilirubin observed in Study 1. Maximum increases in ALT and AST occurred within 12 weeks after starting SKYCLARYS. Increases in serum aminotransferases were generally asymptomatic and reversible following discontinuation of SKYCLARYS. Patients with clinically significant liver disease were excluded from Study 1.
Monitor ALT, AST, and total bilirubin prior to initiation of SKYCLARYS, every month for the first 3 months of treatment, and periodically thereafter. If transaminases increase to levels greater than 5 times the ULN, or greater than 3 times the ULN with evidence of liver dysfunction (e.g., elevated bilirubin), immediately discontinue SKYCLARYS and repeat liver function tests as soon as possible. If transaminase levels stabilize or resolve, SKYCLARYS may be reinitiated with an appropriate increased frequency of monitoring of liver function [see Adverse Reactions (6.1) and Use in Specific Populations (8.6)].
5.2 Elevation of B-Type Natriuretic Peptide
Treatment with SKYCLARYS can cause an increase in BNP, a marker of cardiac function. In Study 1, a total of 14% of patients treated with SKYCLARYS had an increase from baseline in BNP and a BNP above the ULN (100 pg/mL), compared to 4% of patients who received placebo. The incidence of elevation of BNP above 200 pg/mL was 4% in patients treated with SKYCLARYS. Cardiomyopathy and cardiac failure are common in patients with Friedreich's ataxia. Patients were excluded from Study 1 if they had BNP levels > 200 pg/mL prior to study entry, or a history of clinically significant left-sided heart disease and/or clinically significant cardiac disease, with the exception of mild to moderate cardiomyopathy associated with Friedreich's ataxia [see Adverse Reactions (6.1)]. Whether the elevations in BNP in Study 1 are related to SKYCLARYS or cardiac disease associated with Friedreich's ataxia is unclear.
Elevations in BNP may indicate cardiac failure and should prompt an evaluation of cardiac function. Check BNP prior to initiation of SKYCLARYS. Monitor patients for the signs and symptoms of fluid overload, such as sudden weight gain (3 pounds or more of weight gain in one day, or 5 pounds or more of weight gain in a week), peripheral edema, palpitations, and shortness of breath. If signs and symptoms of fluid overload develop, worsen, or require hospitalization, evaluate BNP and cardiac function, and manage appropriately. Management of fluid overload and heart failure may require discontinuation of SKYCLARYS.
5.3 Lipid Abnormalities
Treatment with SKYCLARYS can cause changes in cholesterol. In Study 1, 29% of patients treated with SKYCLARYS reported elevated cholesterol above ULN at one or more time points. Mean increases were observed within 2 weeks of initiation of SKYCLARYS and returned to baseline within 4 weeks of discontinuing treatment. A total of 16% of patients treated with SKYCLARYS had an increase in low-density lipoprotein cholesterol (LDL-C) from baseline, compared to 8% of patients who received placebo. The mean increase in LDL-C for all SKYCLARYS-treated patients was 23.5 mg/dL at 48 weeks. A total of 6% of patients treated with SKYCLARYS had decreases in high-density lipoprotein cholesterol (HDL-C) from baseline compared to 4% of patients who received placebo. The mean decrease in HDL-C for all SKYCLARYS-treated patients was 5.3 mg/dL at 48 weeks.
Assess lipid parameters prior to initiation of SKYCLARYS and monitor periodically during treatment. Manage lipid abnormalities according to clinical guidelines.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described in greater detail in other labeling sections:
- Elevation of aminotransferases [see Warnings and Precautions (5.1)]
- Elevation of BNP [see Warnings and Precautions (5.2)]
- Lipid abnormalities [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety of SKYCLARYS 150 mg once daily has been evaluated in 165 patients with Friedreich's ataxia, including 137 patients exposed for at least 48 weeks, and 125 patients exposed for at least 96 weeks.
The most common adverse reactions in Study 1 (≥20% and greater than placebo) were elevated liver enzymes (AST/ALT), headache, nausea, abdominal pain, fatigue, diarrhea, and musculoskeletal pain. Table 3 shows the adverse reactions that occurred in 10% or more of patients treated with SKYCLARYS and greater than placebo.
Table 3: Adverse Reactions Reported in 10% or More of Patients Treated with SKYCLARYS and Greater than Placebo (Study 1) Adverse Reactions SKYCLARYS
150 mg
(N = 51)
%Placebo
(N = 52)
%Elevated liver enzymes (AST/ALT) 37 2 Headache 37 25 Nausea 33 13 Abdominal pain 29 6 Fatigue 24 14 Diarrhea 20 10 Musculoskeletal pain 20 15 Oropharyngeal pain 18 6 Influenza 16 6 Vomiting 16 12 Muscle spasms 14 6 Back pain 13 8 Decreased appetite 12 4 Rash 10 4 -
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on SKYCLARYS
CYP3A4 Inhibitors
Omaveloxolone is a CYP3A4 substrate. Concomitant use of SKYCLARYS with moderate or strong CYP3A4 inhibitors is expected to result in clinically significant increased exposure of omaveloxolone [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions. Avoid concomitant use of SKYCLARYS with moderate or strong CYP3A4 inhibitors. If use cannot be avoided, dosage modifications are recommended [see Dosage and Administration (2.4)].
CYP3A4 Inducers
Omaveloxolone is a CYP3A4 substrate. Concomitant use of SKYCLARYS with moderate or strong CYP3A4 inducers may significantly decrease exposure of omaveloxolone [see Clinical Pharmacology (12.3)], which may reduce the effectiveness of SKYCLARYS. Avoid concomitant use of SKYCLARYS with moderate or strong CYP3A4 inducers.
7.2 Effect of SKYCLARYS on Other Drugs
CYP3A4 and CYP2C8 Substrates
Omaveloxolone is a weak inducer of CYP3A4 and CYP2C8. Concomitant use with SKYCLARYS can reduce the exposure of CYP3A4 and CYP2C8 substrates which may reduce the activity of these substrates [see Clinical Pharmacology (12.3)]. Refer to the prescribing information of substrates of CYP3A4 and CYP2C8 for dosing instructions if used concomitantly with SKYCLARYS and monitor for lack of efficacy of the concomitant treatment.
Hormonal Contraceptives
Omaveloxolone is a weak CYP3A4 inducer [see Clinical Pharmacology (12.3)]. Concomitant use with SKYCLARYS may reduce the efficacy of hormonal contraceptives. Advise patients to avoid concomitant use with combined hormonal contraceptives (e.g., pill, patch, ring), implants, and progestin only pills [see Use in Specific Populations (8.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate data on the developmental risks associated with the use of SKYCLARYS in pregnant women. In animal studies, administration of omaveloxolone during pregnancy or throughout pregnancy and lactation produced evidence of developmental toxicity (embryofetal mortality and growth impairment, and mortality, growth impairment, and neurobehavioral deficits in offspring) at plasma exposures similar to or less than exposures in humans (see Animal Data). In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
Oral administration of omaveloxolone (0, 1, 3, or 10 mg/kg/day) to pregnant rats throughout organogenesis resulted in no adverse effects on embryofetal development; however, in a dose range-finding study, oral administration of omaveloxolone at doses up to 30 mg/kg/day to pregnant rats throughout organogenesis produced increases in post-implantation loss and resorptions, resulting in a decrease in viable fetuses, and reduced fetal weight at the highest dose tested. At the highest dose tested in the pivotal study (10 mg/kg/day), plasma exposure (AUC) was approximately 5 times that in humans at the recommended human dose (RHD) of 150 mg/day.
Oral administration of omaveloxolone (0, 3, 10, or 30 mg/kg/day) to pregnant rabbits throughout organogenesis resulted in increased embryofetal mortality and skeletal variations and reduced fetal weight at the highest dose tested, which was associated with maternal toxicity. At the no-effect dose for adverse effects on embryofetal development (10 mg/kg/day), plasma exposure was less than that in humans at the RHD.
Oral administration of omaveloxolone (0, 1, 3, or 10 mg/kg/day) to rats throughout pregnancy and lactation resulted in an increase in stillbirths and impaired neurobehavioral function (increased locomotor activity and learning and memory deficits) in offspring at all doses, reduced body weight in offspring at all but the lowest dose tested, and delayed sexual maturation (males), increased postnatal mortality, and impaired reproductive performance in offspring at the highest dose tested. A no-effect dose for adverse effects on pre- and postnatal development was not identified. Plasma exposure (AUC) at the lowest dose tested was less than that in humans at the RHD.
8.2 Lactation
Risk Summary
There are no data on the presence of omaveloxolone or its metabolites in human milk. The effects on milk production and the breastfed infant are unknown. Omaveloxolone was excreted in the milk of lactating rats following oral administration. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for SKYCLARYS and any potential adverse effects on the breastfed infant from SKYCLARYS or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
SKYCLARYS may decrease the efficacy of hormonal contraceptives [see Drug Interactions (7.2) and Clinical Pharmacology (12.3)]. Advise patients to avoid concomitant use with combined hormonal contraceptives (e.g., pill, patch, ring), implants, and progestin only pills. Counsel females using hormonal contraceptives to use an alternative contraceptive method (e.g., non-hormonal intrauterine system) or additional non-hormonal contraceptive (e.g., condoms) during concomitant use and for 28 days after discontinuation of SKYCLARYS.
8.4 Pediatric Use
The safety and effectiveness of SKYCLARYS for the treatment of Friedreich's ataxia have been established in pediatric patients aged 16 years and older. Use of SKYCLARYS for this indication is supported by evidence from one adequate and well-controlled study (Study 1) in adults and in pediatric patients aged 16 years and older [see Clinical Studies (14)].
Safety and effectiveness of SKYCLARYS have not been established in pediatric patients less than 16 years of age.
8.5 Geriatric Use
Clinical studies of SKYCLARYS in Friedreich's ataxia did not include patients aged 65 and over. No data are available to determine whether they respond differently than younger adult patients.
8.6 Hepatic Impairment
Omaveloxolone plasma exposure is increased in patients with moderate or severe hepatic impairment (Child-Pugh Class B and C) [see Clinical Pharmacology (12.3)]. Avoid treatment with SKYCLARYS in patients with severe hepatic impairment, including those who develop severe hepatic impairment. If hepatic function improves to moderate impairment, mild impairment, or normal function, initiation of SKYCLARYS treatment at the approved recommended dosage may be considered. For patients with moderate hepatic impairment, a reduced dosage is recommended with close monitoring for adverse reactions [see Dosage and Administration (2.5)]. For patients with mild hepatic impairment (Child-Pugh Class A), no dose adjustments are recommended.
-
11 DESCRIPTION
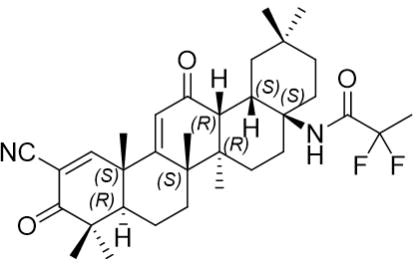
SKYCLARYS contains omaveloxolone in immediate release capsules for oral administration available in a 50 mg strength. The chemical name of omaveloxolone is N-(2-cyano-3,12-dioxo-28-noroleana-1,9(11)-dien-17-yl)-2,2-difluoro-propanamide. Omaveloxolone is a white to off-white amorphous solid. The molecular formula is C33H44F2N2O3. Molecular weight is 554.72 g/mol. The chemical structure is:
Omaveloxolone has a pKa of 7.26 and is practically insoluble in water across the physiological pH range. Capsule contents include the following inactive ingredients: croscarmellose sodium, magnesium stearate, pregelatinized starch, and silicified microcrystalline cellulose. The hard capsule shells contain FD&C Blue #1, ferric oxide yellow, hypromellose, titanium dioxide, and white ink.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The precise mechanism by which omaveloxolone exerts its therapeutic effect in patients with Friedreich's ataxia is unknown. Omaveloxolone have been shown to activate the Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) pathway in vitro and in vivo in animals and humans. The Nrf2 pathway is involved in the cellular response to oxidative stress.
12.3 Pharmacokinetics
Absorption
The median (range) time to achieve peak plasma concentration (Tmax) was 7 to 14 (1 to 24) hours. The total plasma omaveloxolone exposure based on area under the concentration-time curve (AUC) increased in a dose-dependent and dose proportional manner over a dose range of 50 mg (0.33 times the recommended dosage) to 150 mg, but maximum omaveloxolone plasma concentration (Cmax) increased in a less than dose proportional manner over the dose range in healthy fasted subjects.
Effect of Food
Omaveloxolone Cmax and AUC0-inf increased by approximately 350% and 15%, respectively, with a high-fat meal (800-1000 calories, approximately 150, 250, and 500 to 600 calories from protein, carbohydrate, and fat, respectively) compared to fasted conditions [see Dosage and Administration (2.2)].
Omaveloxolone Cmax and AUC0-inf were similar when capsule contents were sprinkled on applesauce or when administered as intact capsules. The median Tmax of omaveloxolone was shortened from approximately 10 to 6 hours when sprinkled on applesauce [see Dosage and Administration (2.2)].
Distribution
The mean apparent volume of distribution of omaveloxolone is 7361 L (105 L/kg for a 70 kg person). Protein binding of omaveloxolone is 97%.
Elimination
The mean (range) terminal half-life of omaveloxolone is 57 hours (32 to 90 hours). The mean apparent plasma clearance of omaveloxolone is 109 L/hr.
Specific Populations
There were no clinically significant differences in the pharmacokinetics of omaveloxolone based on age (16 to 71 years of age), sex, race, or body weight (41 to 128 kg). The effect of renal impairment on the pharmacokinetics of omaveloxolone is unknown.
Patients with Hepatic Impairment
There were no clinically significant differences in the pharmacokinetics of omaveloxolone in subjects with mild hepatic impairment (Child-Pugh Class A). In subjects with moderate and severe hepatic impairment (Child-Pugh Class B and C), omaveloxolone clearance was reduced, resulting in higher plasma exposure of omaveloxolone. The omaveloxolone AUC increased up to 1.65-fold and Cmax increased up to 1.83-fold in subjects with moderate hepatic impairment. The omaveloxolone AUC increased up to 2.17-fold in subjects with severe hepatic impairment; however, this change was variable [see Use in Specific Populations (8.6)].
Drug Interaction Studies
Clinical Studies
Strong CYP3A Inhibitors: Omaveloxolone Cmax increased 3-fold and AUC 4-fold following concomitant use with itraconazole (strong CYP3A inhibitor) [see Drug Interactions (7.1)].
Moderate CYP3A Inhibitors: Omaveloxolone Cmax and AUC increased approximately 1.25-fold following concomitant use with verapamil (moderate CYP3A4 and P-gp inhibitor) [see Drug Interactions (7.1)].
Strong and Moderate CYP3A Inducers: The effect of concomitant use with moderate and strong CYP3A4 inducers is unknown; however, a significant reduction in omaveloxolone exposure is likely following concomitant use based on its metabolic pathway.
Certain CYP450 Enzymes or Transporter Substrates: Omaveloxolone decreased the AUC of midazolam (CYP3A4 substrate) by approximately 45%, AUC of repaglinide (CYP2C8 substrate) by approximately 35%, and AUC of rosuvastatin (BCRP and OATP1B1 substrate) by approximately 30% [see Drug Interactions (7.2)]. There were no clinically significant differences in the pharmacokinetics of digoxin (P-gp substrate) or metformin [(organic cation transporter (OCT)1 substrate] when co-administered with omaveloxolone.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Mutagenesis
Omaveloxolone was negative in a bacterial reverse mutation (Ames) assay, and positive in a chromosomal aberration assay in human peripheral blood lymphocytes but negative in the in vivo rat micronucleus assay and the in vitro comet assay.
Impairment of Fertility
Oral administration of omaveloxolone (0, 1, 3, and 10 mg/kg/day) to male and females rats prior to and during mating and continuing in females to gestation day 7 produced an increase in pre-and post-implantation loss and resorptions, resulting in a decrease in viable embryos at the highest dose tested. The no-effect dose (3 mg/kg/day) for adverse effects on fertility and reproductive function was associated with plasma exposures (AUC) approximately 2 times that in humans at the recommended human dose of 150 mg/day.
-
14 CLINICAL STUDIES
The efficacy of SKYCLARYS was evaluated in a 48-week, randomized, double-blind, placebo-controlled study in patients 16 to 40 years of age with Friedreich's Ataxia (Study 1; NCT02255435).
A total of 103 patients were randomized (1:1) to receive SKYCLARYS 150 mg once daily (n=51) or placebo (n=52).
Enrolled patients had to have a stable modified Friedreich's Ataxia Rating Scale (mFARS) score between 20 and 80, be able to complete maximal exercise testing, and have a left ventricular ejection fraction of at least 40%. In Study 1, 53% of enrolled patients were male, 97% were White, and the mean age was 24 years at study entry. Patients with or without pes cavus were included in Study 1. Pes cavus was defined as having a loss of lateral support and was determined if light from a flashlight could be seen under the patient's arch when barefoot and weight bearing.
The prespecified primary analysis was the change from baseline in the mFARS score compared to placebo at Week 48 in the Full Analysis Population of patients without pes cavus (n=82). The mFARS is a clinical assessment tool to assess patient function, which consists of 4 domains to evaluate bulbar function, upper limb coordination, lower limb coordination, and upright stability. The mFARS has a maximum score of 99, with a lower score on the mFARS signifying lesser physical impairment.
Treatment with SKYCLARYS resulted in statistically significant lower mFARS scores (less impairment) relative to placebo (see Table 4) at Week 48.
Table 4: Primary Analysis in Full Analysis Population: mFARS Least Squares (LS) Mean Change from Baseline at Week 48 SD = Standard Deviation; LS = Least Squares; CI = Confidence Interval
Mean (SD) Baseline mFARS Total Score LS Mean
Change from Baseline at Week 48Treatment difference
SKYCLARYS-placebo (95% CI)
p-valueSKYCLARYS
(n = 40)
40.95 (10.39) -1.56 -2.41
(-4.32, -0.51)
0.0138 Placebo
(n = 42)38.78 (11.03) 0.85 The All Randomized Population (N=103), which included all patients regardless of pes cavus status, demonstrated similar results to the Full Analysis Population of lower mFARS scores in patients treated with SKYCLARYS compared to placebo, with a nominally significant least squares mean difference between treatment groups of ‑1.94 (95% CI: -3.71, -0.16, p=0.0331).
In a post hoc, propensity-matched analysis, lower mFARS scores were observed in patients treated with SKYCLARYS after 3 years relative to a matched set of untreated patients from a natural history study. These exploratory analyses should be interpreted cautiously given the limitations of data collected outside of a controlled study, which may be subject to confounding.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
SKYCLARYS (omaveloxolone) capsules, 50 mg, are supplied as opaque, hard capsules having a light green body and blue cap imprinted with “RTA 408” in white ink on the body and “50” in white ink on the cap. SKYCLARYS is supplied in high density polyethylene bottles that contain 90 capsules, with a foil induction seal and child-resistant closure (NDC: 73179-250-90).
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Elevation of Aminotransferases
Inform patients that elevation in aminotransferases have occurred in patients treated with SKYCLARYS. Liver function tests will be performed prior to initiating SKYCLARYS, every month for the first 3 months of treatment, and periodically thereafter as needed [see Warnings and Precautions (5.1)].
Fluid Overload
Inform patients that elevations in BNP, a marker of cardiac function, have occurred in patients treated with SKYCLARYS. BNP will be performed prior to initiating SKYCLARYS and if signs and symptoms of fluid overload occur, such as sudden weight gain, peripheral edema, palpitations, and shortness of breath. Advise patients to contact their healthcare provider if signs and symptoms of fluid overload develop [see Warnings and Precautions (5.2)].
Lipid Abnormalities
Inform patients that treatment with SKYCLARYS has been associated with increases in LDL cholesterol and decreases in HDL cholesterol. Cholesterol will be assessed prior to starting SKYCLARYS and monitored periodically during treatment [see Warnings and Precautions (5.3)].
Drug Interactions
Advise patients to discuss all medications they are taking, including other prescription medications, non-prescription medications, or herbal products (e.g., St. John's Wort) with their healthcare provider [see Drug Interactions (7)].
Pregnancy
Advise women to notify their healthcare provider if they become pregnant or intend to become pregnant during SKYCLARYS therapy [see Use in Specific Populations (8.1)].
Females of Reproductive Potential
Counsel females using hormonal contraceptives to use an alternative contraceptive method (e.g., non-hormonal intrauterine system) or additional non-hormonal contraceptive (e.g., condoms) during concomitant use and for 28 days after discontinuation of SKYCLARYS [see Drug Interactions (7.2) and Use in Specific Populations (8.3)].
Administration
Advise patients to take SKYCLARYS on an empty stomach at least 1 hour before eating [see Dosage and Administration (2.2)].
Advise patients to swallow SKYCLARYS capsules whole or to open the capsules and sprinkle the entire contents of both halves onto 2 tablespoons (30 mL) of applesauce, then stir the mixture. If sprinkled, advise patients to swallow the drug/applesauce mixture immediately and not to store for future use. Do not crush or chew the capsules. Inform patients that the contents of the SKYCLARYS capsules should not be mixed with milk or orange juice. Advise patients to not administer SKYCLARYS by an enteral feeding tube [see Dosage and Administration (2.2)].
Advise patients that if a dose of SKYCLARYS is missed to not to double their dose or take more than the prescribed dose [see Dosage and Administration (2.3)].
Advise patients to avoid grapefruit juice and grapefruit while they are taking SKYCLARYS [see Drug Interactions (7.1)].
FPI-0006-02
Manufactured for Reata Pharmaceuticals, Inc., a wholly owned subsidiary of Biogen, Cambridge, MA 02142 USA
SKYCLARYS is a trademark of Reata Pharmaceuticals, Inc., Cambridge, MA 02142 USA
Copyright© 2023, Reata Pharmaceuticals, Inc., Cambridge, MA 02142 USA
All rights reserved -
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration
Approved: 1/2024
PATIENT INFORMATION
SKYCLARYS® (skye klar' is)
(omaveloxolone)
capsules, for oral useWhat is SKYCLARYS?
SKYCLARYS is used for the treatment of Friedreich's ataxia in adults and children aged 16 years and older.
It is not known if SKYCLARYS is safe and effective for use in children younger than 16 years of age.Before taking SKYCLARYS, tell your healthcare provider about all of your medical conditions, including if you:
- have liver problems.
- have a history of heart problems, including heart failure.
- have a high level of fat in your blood (high blood cholesterol).
- are pregnant or plan to become pregnant.
- It is not known if SKYCLARYS will harm your unborn baby.
- Women who use hormonal birth control should use another form of birth control such as a non-hormonal intrauterine system or an extra non-hormonal birth control such as condoms while using SKYCLARYS and for 28 days after stopping SKYCLARYS.
- are breastfeeding or plan to breastfeed. It is not known if SKYCLARYS passes into your breast milk. Talk to your healthcare provider about the best way to feed your baby if you take SKYCLARYS.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements such as St. John's Wort.
Taking SKYCLARYS with other medicines can cause serious side effects.
SKYCLARYS may affect the way other medicines work, and other medicines may affect how SKYCLARYS works.
Know the medicines you take. Keep a list of them to show to your healthcare provider and pharmacist when you get a new medicine.How should I take SKYCLARYS?
- Take SKYCLARYS exactly as your healthcare provider tells you to take it.
- Take SKYCLARYS capsules on an empty stomach at least 1 hour before eating.
- Swallow SKYCLARYS capsules whole. Do not crush or chew.
- If SKYCLARYS capsules cannot be swallowed whole, the capsules may be opened and the entire contents of both halves sprinkled onto 2 tablespoonfuls (30 mL) of applesauce.
- Stir the mixture.
- Swallow all the mixture of medicine and applesauce right away. Do not store the mixture of medicine and applesauce to use at a later time.
- The contents of the SKYCLARYS capsules should not be mixed with milk or orange juice.
- Do not administer SKYCLARYS by an enteral feeding tube.
- If you miss a dose, then you should skip the missed dose and take the next dose at the regular time the next day. Do not double your next dose or take more than the prescribed dose.
What should I avoid while taking SKYCLARYS?
Do not drink grapefruit juice or eat grapefruit. These may change the amount of SKYCLARYS in your blood.What are the possible side effects of SKYCLARYS?
SKYCLARYS may cause serious side effects, including:
-
increase in blood liver enzymes. Some people taking SKYCLARYS have had an increase in the level of liver enzymes in their blood. Your healthcare provider will do liver function tests
- before you start taking SKYCLARYS
- every month for the first 3 months after starting your treatment with SKYCLARYS
- during certain times as needed while taking SKYCLARYS
If your liver enzymes increase, your healthcare provider may change your dose during treatment, stop treatment for some time, or completely stop treatment with SKYCLARYS.
-
increase in a blood protein called B-Type Natriuretic Peptide (BNP). BNP tells how well your heart is working. Your healthcare provider will check your BNP levels before your treatment with SKYCLARYS. Tell your healthcare provider if you have signs and symptoms of your heart not working well such as too much fluid in your body (fluid overload). Signs and symptoms may include:
- sudden weight gain (3 pounds or more of weight gain in 1 day, or 5 pounds or more of weight gain in 1 week)
- swelling in your arms, hands, legs, or feet (peripheral edema)
- fast heartbeat (palpitations)
- shortness of breath
If you have symptoms of fluid overload that is considered a side effect of SKYCLARYS, your healthcare provider may stop treatment with SKYCLARYS. - changes in cholesterol levels. Increases in low density lipoprotein cholesterol (LDL-C) or bad cholesterol and decreases in high density lipoprotein cholesterol (HDL-C) or good cholesterol have happened during treatment with SKYCLARYS.Your healthcare provider will check your cholesterol levels before and during your treatment with SKYCLARYS.
The most common side effects of SKYCLARYS include:
- increased liver enzymes (ALT/AST)
- headache
- nausea
- stomach pain
- tiredness
- diarrhea
- muscle pain
These are not all the possible side effects of SKYCLARYS.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store SKYCLARYS?
- Store SKYCLARYS at room temperature between 68°F to 77°F (20°C to 25°C)
Keep SKYCLARYS and all medicines out of the reach of children. General information about the safe and effective use of SKYCLARYS.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use SKYCLARYS for a condition for which it was not prescribed. Do not give SKYCLARYS to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about SKYCLARYS that is written for health professionals.What are the ingredients in SKYCLARYS?
Active ingredient: omaveloxolone
Inactive ingredients: croscarmellose sodium, magnesium stearate, pregelatinized starch, silicified microcrystalline cellulose. The hard capsule shells contain FD&C Blue #1, ferric oxide yellow, hypromellose, titanium dioxide, and white edible ink.
Manufactured for Reata Pharmaceuticals, Inc., a wholly owned subsidiary of Biogen, Cambridge, MA 02142 USA
SKYCLARYS is a trademark of Reata Pharmaceuticals, Inc., Cambridge, MA 02142 USA
Copyright© 2023, Reata Pharmaceuticals, Inc., Cambridge, MA 02142 USA
All rights reserved
For more information about SKYCLARYS®, go to www.SKYCLARYS.com or call Reata Pharmaceuticals, Inc.at 1-800-314-3934. - PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
SKYCLARYS
omaveloxolone capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 73179-250 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength omaveloxolone (UNII: G69Z98951Q) (omaveloxolone - UNII:G69Z98951Q) omaveloxolone 50 mg Inactive Ingredients Ingredient Name Strength croscarmellose sodium (UNII: M28OL1HH48) magnesium stearate (UNII: 70097M6I30) starch, corn (UNII: O8232NY3SJ) microcrystalline cellulose (UNII: OP1R32D61U) FD&C Blue No. 1 (UNII: H3R47K3TBD) ferric oxide yellow (UNII: EX438O2MRT) hypromellose, unspecified (UNII: 3NXW29V3WO) titanium dioxide (UNII: 15FIX9V2JP) Product Characteristics Color GREEN (light green body and blue cap) Score no score Shape CAPSULE (CAPSULE) Size 22mm Flavor Imprint Code RTA;408;50 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 73179-250-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 02/28/2023 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA216718 02/28/2023 Labeler - Reata Pharmaceuticals, Inc. (128566457)

Trademark Results [SKYCLARYS]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 SKYCLARYS 97877325 not registered Live/Pending |
Reata Pharmaceuticals, Inc. 2023-04-07 |
 SKYCLARYS 97611495 not registered Live/Pending |
Reata Pharmaceuticals, Inc. 2022-09-28 |
 SKYCLARYS 88601903 not registered Live/Pending |
Reata Pharmaceuticals, Inc. 2019-09-03 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.