RALOXIFENE HYDROCHLORIDE tablet, film coated
Raloxifene Hydrochloride by
Drug Labeling and Warnings
Raloxifene Hydrochloride by is a Prescription medication manufactured, distributed, or labeled by ACETRIS HEALTH, LLC, Aurobindo Pharma Limited, Aurolife Pharma LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use RALOXIFENE HYDROCHLORIDE TABLETS, USP safely and effectively. See full prescribing information for RALOXIFENE HYDROCHLORIDE TABLETS, USP.
RALOXIFENE HYDROCHLORIDE tablets, USP for oral use
Initial U.S. Approval: 1997
WARNING: INCREASED RISK OF VENOUS THROMBOEMBOLISM AND DEATH FROM STROKE
See full prescribing information for complete boxed warning.- Increased risk of deep vein thrombosis and pulmonary embolism have been reported with raloxifene hydrochloride (5.1). Women with active or past history of venous thromboembolism should not take raloxifene hydrochloride (4.1).
- Increased risk of death due to stroke occurred in a trial in postmenopausal women with documented coronary heart disease or at increased risk for major coronary events. Consider risk-benefit balance in women at risk for stroke (5.2, 14.5).
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
Raloxifene hydrochloride is an estrogen agonist/antagonist indicated for:
- Treatment and prevention of osteoporosis in postmenopausal women. (1.1)
- Reduction in risk of invasive breast cancer in postmenopausal women with osteoporosis. (1.2)
- Reduction in risk of invasive breast cancer in postmenopausal women at high risk for invasive breast cancer. (1.3)
Important Limitations: Raloxifene hydrochloride tablets USP are not indicated for the treatment of invasive breast cancer, reduction of the risk of recurrence of breast cancer, or reduction of risk of noninvasive breast cancer. (1.3)
DOSAGE AND ADMINISTRATION
60 mg tablet orally once daily. (2.1)
DOSAGE FORMS AND STRENGTHS
Tablets (not scored): 60 mg (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Venous Thromboembolism: Increased risk of deep vein thrombosis, pulmonary embolism, and retinal vein thrombosis. Discontinue use 72 hours prior to and during prolonged immobilization. (5.1, 6.1)
- Death Due to Stroke: Increased risk of death due to stroke occurred in a trial in postmenopausal women with documented coronary heart disease or at increased risk for major coronary events. No increased risk of stroke was seen in this trial. Consider risk-benefit balance in women at risk for stroke. (5.2, 14.5)
- Cardiovascular Disease: Raloxifene hydrochloride should not be used for the primary or secondary prevention of cardiovascular disease. (5.3, 14.5)
- Premenopausal Women: Use is not recommended. (5.4)
- Hepatic Impairment: Use with caution. (5.5)
- Concomitant Use with Systemic Estrogens: Not recommended. (5.6)
- Hypertriglyceridemia: If previous treatment with estrogen resulted in hypertriglyceridemia, monitor serum triglycerides. (5.7)
ADVERSE REACTIONS
Adverse reactions (>2% and more common than with placebo) include: hot flashes, leg cramps, peripheral edema, flu syndrome, arthralgia, sweating. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Lucid Pharma LLC at 1-855-224-6100 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatchDRUG INTERACTIONS
- Cholestyramine: Use with raloxifene hydrochloride is not recommended. Reduces the absorption and enterohepatic cycling of raloxifene. (7.1, 12.3)
- Warfarin: Monitor prothrombin time when starting or stopping raloxifene hydrochloride. (7.2, 12.3)
- Highly Protein-Bound Drugs: Use with raloxifene hydrochloride with caution. Highly protein-bound drugs include diazepam, diazoxide, and lidocaine. Raloxifene hydrochloride is more than 95% bound to plasma proteins. (7.3, 12.3)
USE IN SPECIFIC POPULATIONS
- Pediatric Use: Safety and effectiveness not established. (8.4)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 9/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: INCREASED RISK OF VENOUS THROMBOEMBOLISM AND DEATH FROM STROKE
1 INDICATIONS AND USAGE
1.1 Treatment and Prevention of Osteoporosis in Postmenopausal Women
1.2 Reduction in the Risk of Invasive Breast Cancer in Postmenopausal Women with Osteoporosis
1.3 Reduction in the Risk of Invasive Breast Cancer in Postmenopausal Women at High Risk of Invasive Breast Cancer
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
2.2 Recommendations for Calcium and Vitamin D Supplementation
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Venous Thromboembolism
4.2 Pregnancy, Women Who May Become Pregnant, and Nursing Mothers
5 WARNINGS AND PRECAUTIONS
5.1 Venous Thromboembolism
5.2 Death Due to Stroke
5.3 Cardiovascular Disease
5.4 Premenopausal Use
5.5 Hepatic Impairment
5.6 Concomitant Estrogen Therapy
5.7 History of Hypertriglyceridemia when Treated with Estrogens
5.8 Renal Impairment
5.9 History of Breast Cancer
5.10 Use in Men
5.11 Unexplained Uterine Bleeding
5.12 Breast Abnormalities
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Cholestyramine
7.2 Warfarin
7.3 Other Highly Protein-Bound Drugs
7.4 Systemic Estrogens
7.5 Other Concomitant Medications
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Treatment of Postmenopausal Osteoporosis
14.2 Prevention of Postmenopausal Osteoporosis
14.3 Reduction in Risk of Invasive Breast Cancer in Postmenopausal Women with Osteoporosis
14.4 Reduction in Risk of Invasive Breast Cancer in Postmenopausal Women at High Risk of Invasive Breast Cancer
14.5 Effects on Cardiovascular Disease
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
17.1 Osteoporosis Recommendations, Including Calcium and Vitamin D Supplementation
17.2 Patient Immobilization
17.3 Hot Flashes or Flushes
17.4 Reduction in Risk of Invasive Breast Cancer in Postmenopausal Women with Osteoporosis or at High Risk of Invasive Breast Cancer
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: INCREASED RISK OF VENOUS THROMBOEMBOLISM AND DEATH FROM STROKE
- Increased risk of deep vein thrombosis and pulmonary embolism have been reported with raloxifene hydrochloride [see Warnings and Precautions (5.1)]. Women with active or past history of venous thromboembolism should not take raloxifene hydrochloride [see Contraindications (4.1)].
- Increased risk of death due to stroke occurred in a trial in postmenopausal women with documented coronary heart disease or at increased risk for major coronary events. Consider risk-benefit balance in women at risk for stroke [see Warnings and Precautions (5.2) and Clinical Studies (14.5)].
-
1 INDICATIONS AND USAGE
1.1 Treatment and Prevention of Osteoporosis in Postmenopausal Women
Raloxifene hydrochloride tablets, USP are indicated for the treatment and prevention of osteoporosis in postmenopausal women [see Clinical Studies (14.1, 14.2)].
1.2 Reduction in the Risk of Invasive Breast Cancer in Postmenopausal Women with Osteoporosis
Raloxifene hydrochloride tablets, USP are indicated for the reduction in risk of invasive breast cancer in postmenopausal women with osteoporosis [see Clinical Studies (14.3)].
1.3 Reduction in the Risk of Invasive Breast Cancer in Postmenopausal Women at High Risk of Invasive Breast Cancer
Raloxifene hydrochloride tablets, USP are indicated for the reduction in risk of invasive breast cancer in postmenopausal women at high risk of invasive breast cancer [see Clinical Studies (14.4)].
The effect in the reduction in the incidence of breast cancer was shown in a study of postmenopausal women at high risk for breast cancer with a 5-year planned duration with a median follow-up of 4.3 years [see Clinical Studies (14.4)]. Twenty-seven percent of the participants received drug for 5 years. The long-term effects and the recommended length of treatment are not known.
High risk of breast cancer is defined as at least one breast biopsy showing lobular carcinoma in situ (LCIS) or atypical hyperplasia, one or more first-degree relatives with breast cancer, or a 5-year predicted risk of breast cancer ≥1.66% (based on the modified Gail model). Among the factors included in the modified Gail model are the following: current age, number of first-degree relatives with breast cancer, number of breast biopsies, age at menarche, nulliparity or age of first live birth. Healthcare professionals can obtain a Gail Model Risk Assessment Tool by dialing 1-800-545-5979. Currently, no single clinical finding or test result can quantify risk of breast cancer with certainty.
After an assessment of the risk of developing breast cancer, the decision regarding therapy with raloxifene hydrochloride tablets, USP should be based upon an individual assessment of the benefits and risks.
Raloxifene hydrochloride tablets, USP does not eliminate the risk of breast cancer. Patients should have breast exams and mammograms before starting raloxifene hydrochloride tablets, USP and should continue regular breast exams and mammograms in keeping with good medical practice after beginning treatment with raloxifene hydrochloride tablets, USP.
Important Limitations of Use for Breast Cancer Risk Reduction
There are no data available regarding the effect of raloxifene hydrochloride tablets, USP on invasive breast cancer incidence in women with inherited mutations (BRCA1, BRCA2) to be able to make specific recommendations on the effectiveness of raloxifene hydrochloride tablets, USP.
Raloxifene hydrochloride tablets, USP are not indicated for the treatment of invasive breast cancer or reduction of the risk of recurrence.
Raloxifene hydrochloride tablets, USP are not indicated for the reduction in the risk of noninvasive breast cancer. -
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
The recommended dosage is one 60 mg raloxifene hydrochloride tablet daily, which may be administered any time of day without regard to meals [see Clinical Pharmacology (12.3)].
For the indications in risk of invasive breast cancer the optimum duration of treatment is not known [see Clinical Studies (14.3, 14.4)].
2.2 Recommendations for Calcium and Vitamin D Supplementation
For either osteoporosis treatment or prevention, supplemental calcium and/or vitamin D should be added to the diet if daily intake is inadequate. Postmenopausal women require an average of 1500 mg/day of elemental calcium. Total daily intake of calcium above 1500 mg has not demonstrated additional bone benefits while daily intake above 2000 mg has been associated with increased risk of adverse effects, including hypercalcemia and kidney stones. The recommended intake of vitamin D is 400 to 800 IU daily. Patients at increased risk for vitamin D insufficiency (e.g., over the age of 70 years, nursing home bound, or chronically ill) may need additional vitamin D supplements. Patients with gastrointestinal malabsorption syndromes may require higher doses of vitamin D supplementation and measurement of 25-hydroxyvitamin D should be considered.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
4.1 Venous Thromboembolism
Raloxifene hydrochloride tablets are contraindicated in women with active or past history of venous thromboembolism (VTE), including deep vein thrombosis, pulmonary embolism, and retinal vein thrombosis [see Warnings and Precautions (5.1)].
4.2 Pregnancy, Women Who May Become Pregnant, and Nursing Mothers
Raloxifene hydrochloride tablets are contraindicated in pregnancy, in women who may become pregnant, and in nursing mothers [see Use in Specific Populations (8.1, 8.3)]. Raloxifene hydrochloride tablets may cause fetal harm when administered to a pregnant woman. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus.
In rabbit studies, abortion and a low rate of fetal heart anomalies (ventricular septal defects) occurred in rabbits at doses ≥0.1 mg/kg (≥0.04 times the human dose based on surface area, mg/m2), and hydrocephaly was observed in fetuses at doses ≥10 mg/kg (≥4 times the human dose based on surface area, mg/m2). In rat studies, retardation of fetal development and developmental abnormalities (wavy ribs, kidney cavitation) occurred at doses ≥1 mg/kg (≥0.2 times the human dose based on surface area, mg/m2). Treatment of rats at doses of 0.1 to 10 mg/kg (0.02 to 1.6 times the human dose based on surface area, mg/m2) during gestation and lactation produced effects that included delayed and disrupted parturition; decreased neonatal survival and altered physical development; sex- and age-specific reductions in growth and changes in pituitary hormone content; and decreased lymphoid compartment size in offspring. At 10 mg/kg, raloxifene disrupted parturition, which resulted in maternal and progeny death and morbidity. Effects in adult offspring (4 months of age) included uterine hypoplasia and reduced fertility; however, no ovarian or vaginal pathology was observed. -
5 WARNINGS AND PRECAUTIONS
5.1 Venous Thromboembolism
In clinical trials, raloxifene hydrochloride-treated women had an increased risk of venous thromboembolism (deep vein thrombosis and pulmonary embolism). Other venous thromboembolic events also could occur. A less serious event, superficial thrombophlebitis, also has been reported more frequently with raloxifene hydrochloride than with placebo. The greatest risk for deep vein thrombosis and pulmonary embolism occurs during the first 4 months of treatment, and the magnitude of risk appears to be similar to the reported risk associated with use of hormone therapy. Because immobilization increases the risk for venous thromboembolic events independent of therapy, raloxifene hydrochloride should be discontinued at least 72 hours prior to and during prolonged immobilization (e.g., post-surgical recovery, prolonged bed rest), and raloxifene hydrochloride therapy should be resumed only after the patient is fully ambulatory. In addition, women taking raloxifene hydrochloride should be advised to move about periodically during prolonged travel. The risk-benefit balance should be considered in women at risk of thromboembolic disease for other reasons, such as congestive heart failure, superficial thrombophlebitis, and active malignancy [see Contraindications (4.1) and Adverse Reactions (6.1)].
5.2 Death Due to Stroke
In a clinical trial of postmenopausal women with documented coronary heart disease or at increased risk for coronary events, an increased risk of death due to stroke was observed after treatment with raloxifene hydrochloride. During an average follow-up of 5.6 years, 59 (1.2%) raloxifene hydrochloride-treated women died due to a stroke compared to 39 (0.8%) placebo-treated women (22 versus 15 per 10,000 women-years; hazard ratio 1.49; 95% confidence interval, 1 to 2.24; p=0.0499). There was no statistically significant difference between treatment groups in the incidence of stroke (249 in raloxifene hydrochloride [4.9%] versus 224 placebo [4.4%]). Raloxifene hydrochloride had no significant effect on all-cause mortality. The risk-benefit balance should be considered in women at risk for stroke, such as prior stroke or transient ischemic attack (TIA), atrial fibrillation, hypertension, or cigarette smoking [see Clinical Studies (14.5 )].
5.3 Cardiovascular Disease
Raloxifene hydrochloride should not be used for the primary or secondary prevention of cardiovascular disease. In a clinical trial of postmenopausal women with documented coronary heart disease or at increased risk for coronary events, no cardiovascular benefit was demonstrated after treatment with raloxifene for 5 years [see Clinical Studies (14.5)].
5.4 Premenopausal Use
There is no indication for premenopausal use of raloxifene hydrochloride. Safety of raloxifene hydrochloride in premenopausal women has not been established and its use is not recommended.
5.5 Hepatic Impairment
Raloxifene hydrochloride should be used with caution in patients with hepatic impairment. Safety and efficacy have not been established in patients with hepatic impairment [see Clinical Pharmacology (12.3)].
5.6 Concomitant Estrogen Therapy
The safety of concomitant use of raloxifene hydrochloride with systemic estrogens has not been established and its use is not recommended.
5.7 History of Hypertriglyceridemia when Treated with Estrogens
Limited clinical data suggest that some women with a history of marked hypertriglyceridemia (>5.6 mmol/L or >500 mg/dL) in response to treatment with oral estrogen or estrogen plus progestin may develop increased levels of triglycerides when treated with raloxifene hydrochloride. Women with this medical history should have serum triglycerides monitored when taking raloxifene hydrochloride.
5.8 Renal Impairment
Raloxifene hydrochloride should be used with caution in patients with moderate or severe renal impairment. Safety and efficacy have not been established in patients with moderate or severe renal impairment [see Clinical Pharmacology (12.3)].
5.9 History of Breast Cancer
Raloxifene hydrochloride has not been adequately studied in women with a prior history of breast cancer.
5.10 Use in Men
There is no indication for the use of raloxifene hydrochloride in men. Raloxifene hydrochloride has not been adequately studied in men and its use is not recommended.
5.11 Unexplained Uterine Bleeding
Any unexplained uterine bleeding should be investigated as clinically indicated. Raloxifene hydrochloride-treated and placebo-treated groups had similar incidences of endometrial proliferation [see Clinical Studies (14.1, 14.2)].
5.12 Breast Abnormalities
Any unexplained breast abnormality occurring during raloxifene hydrochloride therapy should be investigated. Raloxifene hydrochloride does not eliminate the risk of breast cancer [see Clinical Studies (14.4)].
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described below reflect exposure to raloxifene hydrochloride in 8429 patients who were enrolled in placebo-controlled trials, including 6666 exposed for 1 year and 5685 for at least 3 years.
Osteoporosis Treatment Clinical Trial (MORE) — The safety of raloxifene in the treatment of osteoporosis was assessed in a large (7705 patients) multinational, placebo-controlled trial. Duration of treatment was 36 months, and 5129 postmenopausal women were exposed to raloxifene (2557 received 60 mg/day, and 2572 received 120 mg/day). The incidence of all-cause mortality was similar among groups: 23 (0.9%) placebo, 13 (0.5%) raloxifene hydrochloride-treated (raloxifene 60 mg), and 28 (1.1%) raloxifene 120 mg women died. Therapy was discontinued due to an adverse reaction in 10.9% of raloxifene hydrochloride-treated women and 8.8% of placebo-treated women.
Venous Thromboembolism: The most serious adverse reaction related to raloxifene hydrochloride was VTE (deep venous thrombosis, pulmonary embolism, and retinal vein thrombosis). During an average of study-drug exposure of 2.6 years, VTE occurred in about 1 out of 100 patients treated with raloxifene hydrochloride. Twenty-six raloxifene hydrochloride-treated women had a VTE compared to 11 placebo-treated women, the hazard ratio was 2.4 (95% confidence interval, 1.2, 4.5), and the highest VTE risk was during the initial months of treatment.
Common adverse reactions considered to be related to raloxifene hydrochloride therapy were hot flashes and leg cramps. Hot flashes occurred in about one in 10 patients on raloxifene hydrochloride and were most commonly reported during the first 6 months of treatment and were not different from placebo thereafter. Leg cramps occurred in about one in 14 patients on raloxifene hydrochloride.
Placebo-Controlled Osteoporosis Prevention Clinical Trials — The safety of raloxifene has been assessed primarily in 12 Phase 2 and Phase 3 studies with placebo, estrogen, and estrogen-progestin therapy control groups. The duration of treatment ranged from 2 to 30 months, and 2036 women were exposed to raloxifene (371 patients received 10 to 50 mg/day, 828 received 60 mg/day, and 837 received from 120 to 600 mg/day).
Therapy was discontinued due to an adverse reaction in 11.4% of 581 raloxifene hydrochloride-treated women and 12.2% of 584 placebo-treated women. Discontinuation rates due to hot flashes did not differ significantly between raloxifene hydrochloride and placebo groups (1.7% and 2.2%, respectively).
Common adverse reactions considered to be drug-related were hot flashes and leg cramps. Hot flashes occurred in about one in four patients on raloxifene hydrochloride versus about one in six on placebo. The first occurrence of hot flashes was most commonly reported during the first 6 months of treatment.
Table 1 lists adverse reactions occurring in either the osteoporosis treatment or in five prevention placebo-controlled clinical trials at a frequency ≥2% in either group and in more raloxifene hydrochloride-treated women than in placebo-treated women. Adverse reactions are shown without attribution of causality. The majority of adverse reactions occurring during the studies were mild and generally did not require discontinuation of therapy.
Table 1: Adverse Reactions Occurring in Placebo-Controlled Osteoporosis Clinical Trials at a Frequency ≥2% and in More Raloxifene Hydrochloride-Treated (60 mg Once Daily) Women than Placebo-Treated Womena Treatment
Prevention
Raloxifene Hydrochloride (N=2557)
%
Placebo
(N=2576)
%
Raloxifene Hydrochloride (N=581)
%
Placebo
(N=584)
%
Body as a Whole
Infection
A
A
15.1
14.6
Flu Syndrome
13.5
11.4
14.6
13.5
Headache
9.2
8.5
A
A
Leg Cramps
7
3.7
5.9
1.9
Chest Pain
A
A
4
3.6
Fever
3.9
3.8
3.1
2.6
Cardiovascular System
Hot Flashes
9.7
6.4
24.6
18.3
Migraine
A
A
2.4
2.1
Syncope
2.3
2.1
B
B
Varicose Vein
2.2
1.5
A
A
Digestive System
Nausea
8.3
7.8
8.8
8.6
Diarrhea
7.2
6.9
A
A
Dyspepsia
A
A
5.9
5.8
Vomiting
4.8
4.3
3.4
3.3
Flatulence
A
A
3.1
2.4
Gastrointestinal Disorder
A
A
3.3
2.1
Gastroenteritis
B
B
2.6
2.1
Metabolic and Nutritional
Weight Gain
A
A
8.8
6.8
Peripheral Edema
5.2
4.4
3.3
1.9
Musculoskeletal System
Arthralgia
15.5
14
10.7
10.1
Myalgia
A
A
7.7
6.2
Arthritis
A
A
4
3.6
Tendon Disorder
3.6
3.1
A
A
Nervous System
Depression
A
A
6.4
6
Insomnia
A
A
5.5
4.3
Vertigo
4.1
3.7
A
A
Neuralgia
2.4
1.9
B
B
Hypesthesia
2.1
2
B
B
Respiratory System
Sinusitis
7.9
7.5
10.3
6.5
Rhinitis
10.2
10.1
A
A
Bronchitis
9.5
8.6
A
A
Pharyngitis
5.3
5.1
7.6
7.2
Cough Increased
9.3
9.2
6
5.7
Pneumonia
A
A
2.6
1.5
Laryngitis
B
B
2.2
1.4
Skin and Appendages
Rash
A
A
5.5
3.8
Sweating
2.5
2
3.1
1.7
Special Senses
Conjunctivitis
2.2
1.7
A
A
Urogenital System
Vaginitis
A
A
4.3
3.6
Urinary Tract Infection
A
A
4
3.9
Cystitis
4.6
4.5
3.3
3.1
Leukorrhea
A
A
3.3
1.7
Uterine Disorderb, c
3.3
2.3
A
A
Endometrial Disorderb
B
B
3.1
1.9
Vaginal Hemorrhage
2.5
2.4
A
A
Urinary Tract Disorder
2.5
2.1
A
A
a A: Placebo incidence greater than or equal to raloxifene hydrochloride incidence; B: Less than 2% incidence and more frequent with raloxifene hydrochloride.
b Includes only patients with an intact uterus: Prevention Trials: raloxifene hydrochloride, n=354, Placebo, n=364; Treatment Trial: raloxifene hydrochloride, n=1948, Placebo, n=1999.
c Actual terms most frequently referred to endometrial fluid.Comparison of Raloxifene Hydrochloride and Hormone Therapy — Raloxifene hydrochloride was compared with estrogen-progestin therapy in three clinical trials for prevention of osteoporosis. Table 2 shows adverse reactions occurring more frequently in one treatment group and at an incidence ≥2% in any group. Adverse reactions are shown without attribution of causality.
Table 2: Adverse Reactions Reported in the Clinical Trials for Osteoporosis Prevention with Raloxifene Hydrochloride (60 mg Once Daily) and Continuous Combined or Cyclic Estrogen Plus Progestin (Hormone Therapy) at an Incidence ≥2% in any Treatment Groupa Raloxifene Hydrochloride (N=317)
%
Hormone Therapy-
Continuous
Combinedb
(N=96)
%
Hormone Therapy-Cyclicc
(N=219)
%
Urogenital
Breast Pain
4.4
37.5
29.7
Vaginal Bleedingd
6.2
64.2
88.5
Digestive
Flatulence
1.6
12.5
6.4
Cardiovascular
Hot Flashes
28.7
3.1
5.9
Body as a Whole
Infection
11
0
6.8
Abdominal Pain
6.6
10.4
18.7
Chest Pain
2.8
0
0.5
a These data are from both blinded and open-label studies.
b Continuous Combined Hormone Therapy = 0.625 mg conjugated estrogens plus 2.5 mg medroxyprogesterone acetate.
c Cyclic Hormone Therapy = 0.625 mg conjugated estrogens for 28 days with concomitant 5 mg medroxyprogesterone acetate or 0.15 mg norgestrel on Days 1 through 14 or 17 through 28.
d Includes only patients with an intact uterus: raloxifene hydrochloride, n=290; Hormone Therapy-Continuous Combined, n=67; Hormone Therapy-Cyclic, n=217.Breast Pain — Across all placebo-controlled trials, raloxifene hydrochloride was indistinguishable from placebo with regard to frequency and severity of breast pain and tenderness. Raloxifene hydrochloride was associated with less breast pain and tenderness than reported by women receiving estrogens with or without added progestin.
Gynecologic Cancers — Raloxifene hydrochloride-treated and placebo-treated groups had similar incidences of endometrial cancer and ovarian cancer.
Placebo-Controlled Trial of Postmenopausal Women at Increased Risk for Major Coronary Events (RUTH) — The safety of raloxifene hydrochloride (60 mg once daily) was assessed in a placebo-controlled multinational trial of 10,101 postmenopausal women (age range 55 to 92) with documented coronary heart disease (CHD) or multiple CHD risk factors. Median study drug exposure was 5.1 years for both treatment groups [see Clinical Studies (14.3)]. Therapy was discontinued due to an adverse reaction in 25% of 5044 raloxifene hydrochloride-treated women and 24% of 5057 placebo-treated women. The incidence per year of all-cause mortality was similar between the raloxifene (2.07%) and placebo (2.25%) groups.
Adverse reactions reported more frequently in raloxifene hydrochloride-treated women than in placebo-treated women included peripheral edema (14.1% raloxifene versus 11.7% placebo), muscle spasms/leg cramps (12.1% raloxifene versus 8.3% placebo), hot flashes (7.8% raloxifene versus 4.7% placebo), venous thromboembolic events (2% raloxifene versus 1.4% placebo), and cholelithiasis (3.3% raloxifene versus 2.6% placebo) [see Clinical Studies (14.3, 14.5)].
Tamoxifen-Controlled Trial of Postmenopausal Women at Increased Risk for Invasive Breast Cancer (STAR) — The safety of raloxifene hydrochloride 60 mg/day versus tamoxifen 20 mg/day over 5 years was assessed in 19,747 postmenopausal women (age range 35 to 83 years) in a randomized, double-blind trial. As of 31 December 2005, the median follow-up was 4.3 years. The safety profile of raloxifene was similar to that in the placebo-controlled raloxifene trials [see Clinical Studies (14.4)].6.2 Postmarketing Experience
Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Adverse reactions reported very rarely since market introduction include retinal vein occlusion, stroke, and death associated with venous thromboembolism (VTE). -
7 DRUG INTERACTIONS
7.1 Cholestyramine
Concomitant administration of cholestyramine with raloxifene hydrochloride is not recommended. Although not specifically studied, it is anticipated that other anion exchange resins would have a similar effect. Raloxifene hydrochloride should not be co-administered with other anion exchange resins [see Clinical Pharmacology (12.3)].
7.2 Warfarin
If raloxifene hydrochloride is given concomitantly with warfarin or other warfarin derivatives, prothrombin time should be monitored more closely when starting or stopping therapy with raloxifene hydrochloride [see Clinical Pharmacology (12.3)].
7.3 Other Highly Protein-Bound Drugs
Raloxifene hydrochloride should be used with caution with certain other highly protein-bound drugs such as diazepam, diazoxide, and lidocaine. Although not examined, raloxifene hydrochloride might affect the protein binding of other drugs. Raloxifene is more than 95% bound to plasma proteins [see Clinical Pharmacology (12.3)].
7.4 Systemic Estrogens
The safety of concomitant use of raloxifene hydrochloride with systemic estrogens has not been established and its use is not recommended.
7.5 Other Concomitant Medications
Raloxifene hydrochloride can be concomitantly administered with ampicillin, amoxicillin, antacids, corticosteroids, and digoxin [see Clinical Pharmacology (12.3)].
The concomitant use of raloxifene hydrochloride and lipid-lowering agents has not been studied. -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Teratogenic Effects
Pregnancy Category X. Raloxifene hydrochloride should not be used in women who are or may become pregnant [see Contraindications (4.2)].8.3 Nursing Mothers
Raloxifene hydrochloride should not be used by lactating women [see Contraindications (4.2)]. It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when raloxifene is administered to a nursing woman.
8.5 Geriatric Use
Of the total number of patients in placebo-controlled clinical studies of raloxifene hydrochloride, 61% were 65 and over, while 15.5% were 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. Based on clinical trials, there is no need for dose adjustment for geriatric patients [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
Raloxifene hydrochloride should be used with caution in patients with moderate or severe renal impairment [see Warnings and Precautions (5.8) and Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Raloxifene hydrochloride should be used with caution in patients with hepatic impairment [see Warnings and Precautions (5.5) and Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
In an 8-week study of 63 postmenopausal women, a dose of raloxifene hydrochloride 600 mg/day was safely tolerated. In clinical trials, no raloxifene overdose has been reported.
In postmarketing spontaneous reports, raloxifene overdose has been reported very rarely (less than 1 out of 10,000 [<0.01%] patients treated). The highest overdose has been approximately 1.5 grams. No fatalities associated with raloxifene overdose have been reported. Adverse reactions were reported in approximately half of the adults who took ≥180 mg raloxifene and included leg cramps and dizziness.
Two 18-month-old children each ingested raloxifene 180 mg. In these two children, symptoms reported included ataxia, dizziness, vomiting, rash, diarrhea, tremor, and flushing, as well as elevation in alkaline phosphatase.
There is no specific antidote for raloxifene.
No mortality was seen after a single oral dose in rats or mice at 5000 mg/kg (810 times the human dose for rats and 405 times the human dose for mice based on surface area, mg/m2) or in monkeys at 1000 mg/kg (80 times the AUC in humans). -
11 DESCRIPTION
Raloxifene hydrochloride is an estrogen agonist/antagonist, commonly referred to as a selective estrogen receptor modulator (SERM) that belongs to the benzothiophene class of compounds. The chemical structure is:
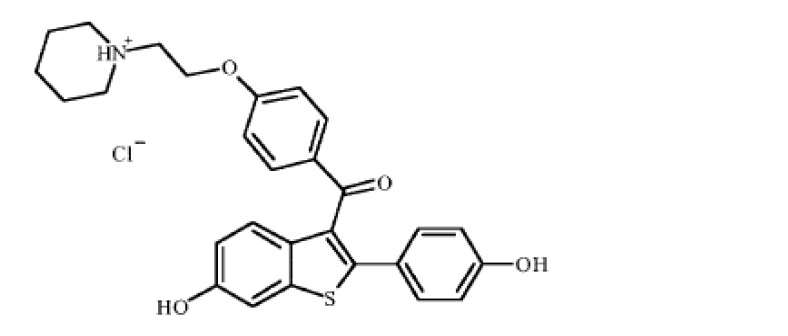
The chemical designation is methanone, [6-hydroxy-2-(4-hydroxyphenyl)benzo[b]thien-3-yl]-[4-[2-(1-piperidinyl)ethoxy]phenyl]-, hydrochloride. Raloxifene hydrochloride has the molecular formula C28H27NO4SHCl, which corresponds to a molecular weight of 510.05. Raloxifene hydrochloride USP is almost white to pale yellow powder that is very slightly soluble in water.
Raloxifene hydrochloride USP is supplied in a tablet dosage form for oral administration. Each raloxifene hydrochloride tablet, USP contains 60 mg of raloxifene hydrochloride USP, which is the molar equivalent of 55.71 mg of free base. Inactive ingredients include citric acid monohydrate, crospovidone, hypromellose, magnesium stearate, microcrystalline cellulose, polyethylene glycol, polysorbate 80, povidone, and titanium dioxide.
USP dissolution test pending. -
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Raloxifene is an estrogen agonist/antagonist, commonly referred to as a selective estrogen receptor modulator (SERM). The biological actions of raloxifene are largely mediated through binding to estrogen receptors. This binding results in activation of estrogenic pathways in some tissues (agonism) and blockade of estrogenic pathways in others (antagonism). The agonistic or antagonistic action of raloxifene depends on the extent of recruitment of coactivators and corepressors to estrogen receptor (ER) target gene promotors.
Raloxifene appears to act as an estrogen agonist in bone. It decreases bone resorption and bone turnover, increases bone mineral density (BMD) and decreases fracture incidence. Preclinical data demonstrate that raloxifene is an estrogen antagonist in uterine and breast tissues. These results are consistent with findings in clinical trials, which suggest that raloxifene hydrochloride lacks estrogen-like effects on the uterus and breast tissue.12.2 Pharmacodynamics
Decreases in estrogen levels after oophorectomy or menopause lead to increases in bone resorption and accelerated bone loss. Bone is initially lost rapidly because the compensatory increase in bone formation is inadequate to offset resorptive losses. In addition to loss of estrogen, this imbalance between resorption and formation may be due to age-related impairment of osteoblasts or their precursors. In some women, these changes will eventually lead to decreased bone mass, osteoporosis, and increased risk for fractures, particularly of the spine, hip, and wrist. Vertebral fractures are the most common type of osteoporotic fracture in postmenopausal women.
In both the osteoporosis treatment and prevention trials, raloxifene hydrochloride therapy resulted in consistent, statistically significant suppression of bone resorption and bone formation, as reflected by changes in serum and urine markers of bone turnover (e.g., bone-specific alkaline phosphatase, osteocalcin, and collagen breakdown products). The suppression of bone turnover markers was evident by 3 months and persisted throughout the 36-month and 24-month observation periods.
In a 31-week, open-label, radiocalcium kinetics study, 33 early postmenopausal women were randomized to treatment with once-daily raloxifene hydrochloride 60 mg, cyclic estrogen/progestin (0.625 mg conjugated estrogens daily with 5 mg medroxyprogesterone acetate daily for the first 2 weeks of each month [hormone therapy]), or no treatment. Treatment with either raloxifene hydrochloride or hormone therapy was associated with reduced bone resorption and a positive shift in calcium balance (-82 mg Ca/day and +60 mg Ca/day, respectively, for raloxifene hydrochloride and -162 mg Ca/day and +91 mg Ca/day, respectively, for hormone therapy).
There were small decreases in serum total calcium, inorganic phosphate, total protein, and albumin, which were generally of lesser magnitude than decreases observed during estrogen or hormone therapy. Platelet count was also decreased slightly and was not different from estrogen therapy.12.3 Pharmacokinetics
The disposition of raloxifene has been evaluated in more than 3000 postmenopausal women in selected raloxifene osteoporosis treatment and prevention clinical trials, using a population approach. Pharmacokinetic data also were obtained in conventional pharmacology studies in 292 postmenopausal women. Raloxifene exhibits high within-subject variability (approximately 30% coefficient of variation) of most pharmacokinetic parameters. Table 3 summarizes the pharmacokinetic parameters of raloxifene.
Absorption — Raloxifene is absorbed rapidly after oral administration. Approximately 60% of an oral dose is absorbed, but presystemic glucuronide conjugation is extensive. Absolute bioavailability of raloxifene is 2%. The time to reach average maximum plasma concentration and bioavailability are functions of systemic interconversion and enterohepatic cycling of raloxifene and its glucuronide metabolites.
Administration of raloxifene hydrochloride with a standardized, high-fat meal increases the absorption of raloxifene (Cmax 28% and AUC 16%), but does not lead to clinically meaningful changes in systemic exposure. Raloxifene hydrochloride can be administered without regard to meals.
Distribution — Following oral administration of single doses ranging from 30 to 150 mg of raloxifene hydrochloride, the apparent volume of distribution is 2348 L/kg and is not dose dependent.
Raloxifene and the monoglucuronide conjugates are highly (95%) bound to plasma proteins. Raloxifene binds to both albumin and α1-acid glycoprotein, but not to sex-steroid binding globulin.
Metabolism — Biotransformation and disposition of raloxifene in humans have been determined following oral administration of 14C-labeled raloxifene. Raloxifene undergoes extensive first-pass metabolism to the glucuronide conjugates: raloxifene-4´-glucuronide, raloxifene-6-glucuronide, and raloxifene-6, 4´-diglucuronide. No other metabolites have been detected, providing strong evidence that raloxifene is not metabolized by cytochrome P450 pathways. Unconjugated raloxifene comprises less than 1% of the total radiolabeled material in plasma. The terminal log-linear portions of the plasma concentration curves for raloxifene and the glucuronides are generally parallel. This is consistent with interconversion of raloxifene and the glucuronide metabolites.
Following intravenous administration, raloxifene is cleared at a rate approximating hepatic blood flow. Apparent oral clearance is 44.1 L/kghr. Raloxifene and its glucuronide conjugates are interconverted by reversible systemic metabolism and enterohepatic cycling, thereby prolonging its plasma elimination half-life to 27.7 hours after oral dosing.
Results from single oral doses of raloxifene predict multiple-dose pharmacokinetics. Following chronic dosing, clearance ranges from 40 to 60 L/kghr. Increasing doses of raloxifene hydrochloride (ranging from 30 to 150 mg) result in slightly less than a proportional increase in the area under the plasma time concentration curve (AUC).
Excretion — Raloxifene is primarily excreted in feces, and less than 0.2% is excreted unchanged in urine. Less than 6% of the raloxifene dose is eliminated in urine as glucuronide conjugates.
Table 3: Summary of Raloxifene Pharmacokinetic Parameters in the Healthy Postmenopausal Woman Cmaxa, b
(ng/mL)/
(mg/kg)
t1/2 (hr)a
AUC0-∞a, b
(nghr/mL)/
(mg/kg)
CL/Fa
(L/kghr)
V/Fa
(L/kg)
Single Dose
Mean
0.5
27.7
27.2
44.1
2348
CVa (%)
52
10.7 to 273c
44
46
52
Multiple Dose
Mean
1.36
32.5
24.2
47.4
2853
CVa (%)
37
15.8 to 86.6c
36
41
56
a Abbreviations: Cmax = maximum plasma concentration, t1/2 = half-life, AUC = area under the curve, CL = clearance, V = volume of distribution, F = bioavailability, CV = coefficient of variation.
b Data normalized for dose in mg and body weight in kg.
c Range of observed half-life.
Special Populations
Pediatric — The pharmacokinetics of raloxifene has not been evaluated in a pediatric population [see Use in Specific Populations (8.4)].
Geriatric — No differences in raloxifene pharmacokinetics were detected with regard to age (range 42 to 84 years) [see Use in Specific Populations (8.5)].
Gender — Total extent of exposure and oral clearance, normalized for lean body weight, are not significantly different between age-matched female and male volunteers.
Race — Pharmacokinetic differences due to race have been studied in 1712 women, including 97.5% White, 1% Asian, 0.7% Hispanic, and 0.5% Black in the osteoporosis treatment trial and in 1053 women, including 93.5% White, 4.3% Hispanic, 1.2% Asian, and 0.5% Black in the osteoporosis prevention trials. There were no discernible differences in raloxifene plasma concentrations among these groups; however, the influence of race cannot be conclusively determined.
Renal Impairment — In the osteoporosis treatment and prevention trials, raloxifene concentrations in women with mild renal impairment are similar to women with normal creatinine clearance. When a single dose of 120 mg raloxifene hydrochloride was administered to 10 renally impaired males [7 moderate impairment (CrCl = 31 to 50 mL/min); 3 severe impairment (CrCl ≤30 mL/min)] and to 10 healthy males (CrCl >80 mL/min), plasma raloxifene concentrations were 122% (AUC0-∞) higher in renally impaired patients than those of healthy volunteers. Raloxifene should be used with caution in patients with moderate or severe renal impairment [see Warnings and Precautions (5.8) and Use in Specific Populations (8.6)].
Hepatic Impairment — The disposition of raloxifene was compared in 9 patients with mild (Child-Pugh Class A) hepatic impairment (total bilirubin ranging from 0.6 to 2 mg/dL) to 8 subjects with normal hepatic function following a single dose of 60 mg raloxifene hydrochloride. Apparent clearance of raloxifene was reduced 56% and the half-life of raloxifene was not altered in patients with mild hepatic impairment. Plasma raloxifene concentrations were approximately 150% higher than those in healthy volunteers and correlated with total bilirubin concentrations. The pharmacokinetics of raloxifene has not been studied in patients with moderate or severe hepatic impairment. Raloxifene should be used with caution in patients with hepatic impairment [see Warnings and Precautions (5.5) and Use in Specific Populations (8.7)].
Drug Interactions
Cholestyramine — Cholestyramine, an anion exchange resin, causes a 60% reduction in the absorption and enterohepatic cycling of raloxifene after a single dose. Although not specifically studied, it is anticipated that other anion exchange resins would have a similar effect [see Drug Interactions (7.1)].
Warfarin — In vitro, raloxifene did not interact with the binding of warfarin. The concomitant administration of raloxifene hydrochloride and warfarin, a coumarin derivative, has been assessed in a single-dose study. In this study, raloxifene had no effect on the pharmacokinetics of warfarin. However, a 10% decrease in prothrombin time was observed in the single-dose study. In the osteoporosis treatment trial, there were no clinically relevant effects of warfarin co-administration on plasma concentrations of raloxifene [see Drug Interactions (7.2)].
Other Highly Protein-Bound Drugs — In the osteoporosis treatment trial, there were no clinically relevant effects of co-administration of other highly protein-bound drugs (e.g., gemfibrozil) on plasma concentrations of raloxifene. In vitro, raloxifene did not interact with the binding of phenytoin, tamoxifen, or warfarin (see above) [see Drug Interactions (7.3)].
Ampicillin and Amoxicillin — Peak concentrations of raloxifene and the overall extent of absorption are reduced 28% and 14%, respectively, with co-administration of ampicillin. These reductions are consistent with decreased enterohepatic cycling associated with antibiotic reduction of enteric bacteria. However, the systemic exposure and the elimination rate of raloxifene were not affected. In the osteoporosis treatment trial, co-administration of amoxicillin had no discernible differences in plasma raloxifene concentrations [see Drug Interactions (7.5)].
Antacids — Concomitant administration of calcium carbonate or aluminum and magnesium hydroxide-containing antacids does not affect the systemic exposure of raloxifene [see Drug Interactions (7.5)].
Corticosteroids — The chronic administration of raloxifene in postmenopausal women has no effect on the pharmacokinetics of methylprednisolone given as a single oral dose [see Drug Interactions (7.5)].
Digoxin — Raloxifene has no effect on the pharmacokinetics of digoxin [see Drug Interactions (7.5)].
Cyclosporine — Concomitant administration of raloxifene hydrochloride with cyclosporine has not been studied.
Lipid-Lowering Agents — Concomitant administration of raloxifene hydrochloride with lipid-lowering agents has not been studied. -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis — In a 21-month carcinogenicity study in mice, there was an increased incidence of ovarian tumors in female animals given 9 to 242 mg/kg, which included benign and malignant tumors of granulosa/theca cell origin and benign tumors of epithelial cell origin. Systemic exposure (AUC) of raloxifene in this group was 0.3 to 34 times that in postmenopausal women administered a 60 mg dose. There was also an increased incidence of testicular interstitial cell tumors and prostatic adenomas and adenocarcinomas in male mice given 41 or 210 mg/kg (4.7 or 24 times the AUC in humans) and prostatic leiomyoblastoma in male mice given 210 mg/kg.
In a 2-year carcinogenicity study in rats, an increased incidence in ovarian tumors of granulosa/theca cell origin was observed in female rats given 279 mg/kg (approximately 400 times the AUC in humans). The female rodents in these studies were treated during their reproductive lives when their ovaries were functional and responsive to hormonal stimulation.
Mutagenesis— Raloxifene hydrochloride was not genotoxic in any of the following test systems: the Ames test for bacterial mutagenesis with and without metabolic activation, the unscheduled DNA synthesis assay in rat hepatocytes, the mouse lymphoma assay for mammalian cell mutation, the chromosomal aberration assay in Chinese hamster ovary cells, the in vivo sister chromatid exchange assay in Chinese hamsters, and the in vivo micronucleus test in mice.
Impairment of Fertility — When male and female rats were given daily doses ≥5 mg/kg (≥0.8 times the human dose based on surface area, mg/m2) prior to and during mating, no pregnancies occurred. In male rats, daily doses up to 100 mg/kg (16 times the human dose based on surface area, mg/m2) for at least 2 weeks did not affect sperm production or quality or reproductive performance. In female rats, at doses of 0.1 to 10 mg/kg/day (0.02 to 1.6 times the human dose based on surface area, mg/m2), raloxifene disrupted estrous cycles and inhibited ovulation. These effects of raloxifene were reversible. In another study in rats in which raloxifene was given during the preimplantation period at doses ≥0.1 mg/kg (≥0.02 times the human dose based on surface area, mg/m2), raloxifene delayed and disrupted embryo implantation, resulting in prolonged gestation and reduced litter size. The reproductive and developmental effects observed in animals are consistent with the estrogen receptor activity of raloxifene.13.2 Animal Toxicology and/or Pharmacology
The skeletal effects of raloxifene treatment were assessed in ovariectomized rats and monkeys. In rats, raloxifene prevented increased bone resorption and bone loss after ovariectomy. There were positive effects of raloxifene on bone strength, but the effects varied with time. Cynomolgus monkeys were treated with raloxifene or conjugated estrogens for 2 years. In terms of bone cycles, this is equivalent to approximately 6 years in humans. Raloxifene and estrogen suppressed bone turnover and increased BMD in the lumbar spine and in the central cancellous bone of the proximal tibia. In this animal model, there was a positive correlation between vertebral compressive breaking force and BMD of the lumbar spine.
Histologic examination of bone from rats and monkeys treated with raloxifene showed no evidence of woven bone, marrow fibrosis, or mineralization defects.
These results are consistent with data from human studies of radiocalcium kinetics and markers of bone metabolism, and are consistent with the action of raloxifene hydrochloride as a skeletal antiresorptive agent. -
14 CLINICAL STUDIES
14.1 Treatment of Postmenopausal Osteoporosis
Effect on Fracture Incidence
The effects of raloxifene hydrochloride on fracture incidence and BMD in postmenopausal women with osteoporosis were examined at 3 years in a large randomized, placebo-controlled, double-blind, multinational osteoporosis treatment trial (MORE). All vertebral fractures were diagnosed radiographically; some of these fractures also were associated with symptoms (i.e., clinical fractures). The study population consisted of 7705 postmenopausal women with osteoporosis as defined by: a) low BMD (vertebral or hip BMD at least 2.5 standard deviations below the mean value for healthy young women) without baseline vertebral fractures or b) one or more baseline vertebral fractures. Women enrolled in this study had a median age of 67 years (range 31 to 80) and a median time since menopause of 19 years.
Effect on Bone Mineral Density
Raloxifene hydrochloride, 60 mg administered once daily, increased spine and hip BMD by 2 to 3%. Raloxifene hydrochloride decreased the incidence of the first vertebral fracture from 4.3% for placebo to 1.9% for raloxifene hydrochloride (relative risk reduction = 55%) and subsequent vertebral fractures from 20.2% for placebo to 14.1% for raloxifene hydrochloride (relative risk reduction = 30%) (see Table 4). All women in the study received calcium (500 mg/day) and vitamin D (400 to 600 IU/day). Raloxifene hydrochloride reduced the incidence of vertebral fractures whether or not patients had a vertebral fracture upon study entry. The decrease in incidence of vertebral fracture was greater than could be accounted for by increase in BMD alone.
Table 4: Effect of Raloxifene Hydrochloride on Risk of Vertebral Fractures
Number of Patients
Absolute Risk
Reduction (ARR)
Relative Risk
Reduction (95% CI)
Raloxifene Hydrochloride
Placebo
Fractures diagnosed radiographically
Patients with no baseline fracturea
n=1401
n=1457
Number (%) of patients with ≥1 new vertebral fracture
27
(1.9%)
62
(4.3%)
2.4%
55%
(29%, 71%)
Patients with ≥1 baseline fracturea
n=858
n=835
Number (%) of patients with ≥1 new vertebral fracture
121
(14.1%)
169
(20.2%)
6.1%
30%
(14%, 44%)
Symptomatic vertebral fractures
All randomized patients
n=2557
n=2576
Number (%) of patients with ≥1 new clinical (painful) vertebral fracture
47
(1.8%)
81
(3.1%)
1.3%
41%
(17%, 59%)
a Includes all patients with baseline and at least one follow-up radiograph.
The mean percentage change in BMD from baseline for raloxifene hydrochloride was statistically significantly greater than for placebo at each skeletal site (see Table 5).
Table 5: Raloxifene Hydrochloride- (60 mg Once Daily) Related Increases in BMDa for the Osteoporosis Treatment Study Expressed as Mean Percentage Increase vs. Placebob, c Site
Time
12 Months
%
24 Months
%
36 Months
%
Lumbar Spine
2
2.6
2.6
Femoral Neck
1.3
1.9
2.1
Ultradistal Radius
NDd
2.2
NDd
Distal Radius
NDd
0.9
NDd
Total Body
NDd
1.1
NDd
a Note: all BMD increases were significant (p<0.001).
b Intent-to-treat analysis; last observation carried forward.
c All patients received calcium and vitamin D.
d ND = not done (total body and radius BMD were measured only at 24 months).Discontinuation from the study was required when excessive bone loss or multiple incident vertebral fractures occurred. Such discontinuation was statistically significantly more frequent in the placebo group (3.7%) than in the raloxifene hydrochloride group (1.1%).
Bone Histology
Bone biopsies for qualitative and quantitative histomorphometry were obtained at baseline and after 2 years of treatment. There were 56 paired biopsies evaluable for all indices. In raloxifene hydrochloride-treated patients, there were statistically significant decreases in bone formation rate per tissue volume, consistent with a reduction in bone turnover. Normal bone quality was maintained; specifically, there was no evidence of osteomalacia, marrow fibrosis, cellular toxicity, or woven bone after 2 years of treatment.
Effect on Endometrium
Endometrial thickness was evaluated annually in a subset of the study population (1781 patients) for 3 years. Placebo-treated women had a 0.27 mm mean decrease from baseline in endometrial thickness over 3 years, whereas the raloxifene hydrochloride-treated women had a 0.06 mm mean increase. Patients in the osteoporosis treatment study were not screened at baseline or excluded for pre-existing endometrial or uterine disease. This study was not specifically designed to detect endometrial polyps. Over the 36 months of the study, clinically or histologically benign endometrial polyps were reported in 17 of 1999 placebo-treated women, 37 of 1948 raloxifene hydrochloride-treated women, and in 31 of 2010 women treated with raloxifene hydrochloride 120 mg/day. There was no difference between raloxifene hydrochloride- and placebo-treated women in the incidences of endometrial carcinoma, vaginal bleeding, or vaginal discharge.
14.2 Prevention of Postmenopausal Osteoporosis
The effects of raloxifene hydrochloride on BMD in postmenopausal women were examined in three randomized, placebo-controlled, double-blind osteoporosis prevention trials: (1) a North American trial enrolled 544 women; (2) a European trial, 601 women; and (3) an international trial, 619 women who had undergone hysterectomy. In these trials, all women received calcium supplementation (400 to 600 mg/day). Women enrolled in these trials had a median age of 54 years and a median time since menopause of 5 years (less than 1 year up to 15 years postmenopause). The majority of the women were White (93.5%). Women were included if they had spine BMD between 2.5 standard deviations below and 2 standard deviations above the mean value for healthy young women. The mean T scores (number of standard deviations above or below the mean in healthy young women) for the three trials ranged from -1.01 to -0.74 for spine BMD and included women both with normal and low BMD. Raloxifene hydrochloride, 60 mg administered once daily, produced increases in bone mass versus calcium supplementation alone, as reflected by dual-energy x-ray absorptiometric (DXA) measurements of hip, spine, and total body BMD.
Effect on Bone Mineral Density
Compared with placebo, the increases in BMD for each of the three studies were statistically significant at 12 months and were maintained at 24 months (see Table 6). The placebo groups lost approximately 1% of BMD over 24 months.
Table 6: Raloxifene Hydrochloride- (60 mg Once Daily) Related Increases in BMDa for the Three Osteoporosis Prevention Studies Expressed as Mean Percentage Increase vs. Placebob at 24 Monthsc Site Study
NAd
%
EUd
%
INTd,e
%
Total Hip
2
2.4
1.3
Femoral Neck
2.1
2.5
1.6
Trochanter
2.2
2.7
1.3
Intertrochanter
2.3
2.4
1.3
Lumbar Spine
2
2.4
1.8
a Note: all BMD increases were significant (p≤0.001).
b All patients received calcium.
c Intent-to-treat analysis; last observation carried forward.
d Abbreviations: NA = North American, EU = European, INT = International.
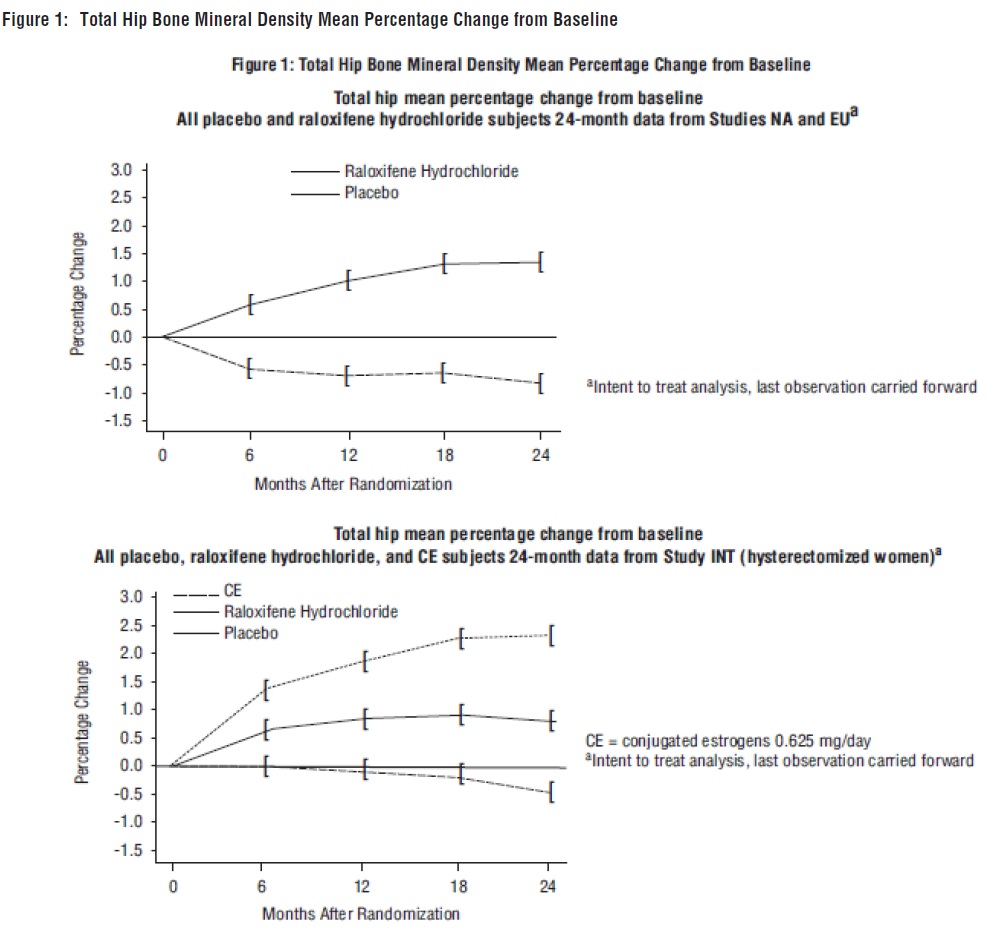
e All women in the study had previously undergone hysterectomy.Raloxifene hydrochloride also increased BMD compared with placebo in the total body by 1.3% to 2% and in Ward’s Triangle (hip) by 3.1% to 4%. The effects of raloxifene hydrochloride on forearm BMD were inconsistent between studies. In Study EU, raloxifene hydrochloride prevented bone loss at the ultradistal radius, whereas in Study NA, it did not (see Figure 1).
Effect on Endometrium
In placebo-controlled osteoporosis prevention trials, endometrial thickness was evaluated every 6 months (for 24 months) by transvaginal ultrasonography (TVU). A total of 2978 TVU measurements were collected from 831 women in all dose groups. Placebo-treated women had a 0.04 mm mean increase from baseline in endometrial thickness over 2 years, whereas the raloxifene hydrochloride-treated women had a 0.09 mm mean increase. Endometrial thickness measurements in raloxifene-treated women were indistinguishable from placebo. There were no differences between the raloxifene and placebo groups with respect to the incidence of reported vaginal bleeding.14.3 Reduction in Risk of Invasive Breast Cancer in Postmenopausal Women with Osteoporosis
MORE Trial
The effect of raloxifene hydrochloride on the incidence of breast cancer was assessed as a secondary safety endpoint in a randomized, placebo-controlled, double-blind, multinational osteoporosis treatment trial in postmenopausal women [see Clinical Studies (14.1)]. After 4 years, raloxifene hydrochloride, 60 mg administered once daily, reduced the incidence of all breast cancers by 62%, compared with placebo (HR 0.38, 95% CI 0.22 to 0.67). Raloxifene hydrochloride reduced the incidence of invasive breast cancer by 71%, compared with placebo (ARR 3.1 per 1000 women-years); this was primarily due to an 80% reduction in the incidence of ER-positive invasive breast cancer in the raloxifene hydrochloride groupcompared with placebo. Table 7 presents efficacy and selected safety outcomes.
CORE Trial
The effect of raloxifene hydrochloride on the incidence of invasive breast cancer was evaluated for 4 additional years in a follow-up study conducted in a subset of postmenopausal women originally enrolled in the MORE osteoporosis treatment trial. Women were not re-randomized; the treatment assignment from the osteoporosis treatment trial was carried forward to this study. Raloxifene hydrochloride, 60 mg administered once daily, reduced the incidence of invasive breast cancer by 56%, compared with placebo (ARR 3 per 1000 women-years); this was primarily due to a 63% reduction in the incidence of ER-positive invasive breast cancer in the raloxifene hydrochloride group compared with placebo. There was no reduction in the incidence of ER-negative breast cancer. In the osteoporosis treatment trial and the follow-up study, there was no difference in incidence of noninvasive breast cancer between the raloxifene hydrochloride and placebo groups. Table 7 presents efficacy and selected safety outcomes.
In a subset of postmenopausal women followed for up to 8 years from randomization in MORE to the end of CORE, raloxifene hydrochloride, 60 mg administered once daily, reduced the incidence of invasive breast cancer by 60% in women assigned raloxifene hydrochloride (N=1355) compared with placebo (N=1286) (HR 0.4, 95% CI 0.21, 0.77; ARR 1.95 per 1000 women-years); this was primarily due to a 65% reduction in the incidence of ER- positive invasive breast cancer in the raloxifene hydrochloride group compared with placebo.
Table 7: Raloxifene Hydrochloride (60 mg Once Daily) vs. Placebo on Outcomes in Postmenopausal Women with Osteoporosis
MORE
4 years
COREa
4 years
Outcomes
Placebo
(N=2576)
Raloxifene Hydrochloride
(N=2557)
HR
(95% CI)b
Placebo
(N=1286)
Raloxifene Hydrochloride
(N=2725)
HR
(95% CI)b
n
IRb
n
IRb
n
IRb
n
IRb
Invasivec breast cancer
38
4.36
11
1.26
0.29 (0.15, 0.56)d
20
5.41
19
2.43
0.44
(0.24, 0.83)d
ERb, c positive
29
3.33
6
0.69
0.2
(0.08, 0.49)
15
4.05
12
1.54
0.37
(0.17, 0.79)
ERb, c negative
4
0.46
5
0.57
1.23
(0.33, 4.6)
3
0.81
6
0.77
0.95
(0.24, 3.79)
ERb, c unknown
5
0.57
0
0
N/Ab
2
0.54
1
0.13
N/Ab
Noninvasivec, e breast cancer
5
0.57
3
0.34
0.59
(0.14, 2.47)
2
0.54
5
0.64
1.18
(0.23, 6.07)
Clinical vertebral fractures
107
12.27
62
7.08
0.57
(0.42, 0.78)
N/Ab
N/Ab
N/Ab
N/Ab
N/Ab
Death
36
4.13
23
2.63
0.63
(0.38, 1.07)
29
7.76
47
5.99
0.77
(0.49, 1.23)
Death due to stroke
6
0.69
3
0.34
0.49
(0.12, 1.98)
1
0.27
6
0.76
2.87
(0.35, 23.8)
Stroke
56
6.42
43
4.91
0.76
(0.51, 1.14)
14
3.75
49
6.24
1.67
(0.92, 3.03)
Deep vein thrombosis
8
0.92
20
2.28
2.5
(1.1, 5.68)
4
1.07
17
2.17
2.03
(0.68, 6.03)
Pulmonary embolism
4
0.46
11
1.26
2.76
(0.88, 8.67)
0
0
9
1.15
N/Ab
Endometrial and uterine cancerf
5
0.74
5
0.74
1.01
(0.29, 3.49)
3
1.02
4
0.65
0.64
(0.14, 2.85)
Ovarian cancer
6
0.69
3
0.34
0.49
(0.12, 1.95)
2
0.54
2
0.25
0.47
(0.07, 3.36)
Hot flashes
151
17.31
237
27.06
1.61
(1.31, 1.97)
11
2.94
26
3.31
1.12
(0.55, 2.27)
Peripheral edema
134
15.36
164
18.73
1.23
(0.98, 1.54)
30
8.03
61
7.77
0.96
(0.62, 1.49)
Cholelithiasis
45
5.16
53
6.05
1.18
(0.79, 1.75)
12
3.21
35
4.46
1.39
(0.72, 2.67)
a CORE was a follow-up study conducted in a subset of 4011 postmenopausal women who originally enrolled in MORE. Women were not re-randomized; the treatment assignment from MORE was carried forward to this study. At CORE enrollment, the raloxifene hydrochloride group included 2725 total patients with 1355 patients who were originally assigned to raloxifene 60 mg once daily and 1370 patients who were originally assigned to raloxifene 120 mg at MORE randomization.
b Abbreviations: CI = confidence interval; ER = estrogen receptor; HR = hazard ratio; IR = annual incidence rate per 1000 women; N/A = not applicable.
c Included 1274 patients in placebo and 2716 patients in raloxifene hydrochloride who were not diagnosed with breast cancer prior to CORE enrollment.
d p<0.05, obtained from the log-rank test, and not adjusted for multiple comparisons in MORE.
e All cases were ductal carcinoma in situ.
f Only patients with an intact uterus were included (MORE: placebo = 1999, raloxifene hydrochloride = 1950; CORE: placebo = 1008, raloxifene hydrochloride = 2138).RUTH Trial
The effect of raloxifene hydrochloride on the incidence of invasive breast cancer was assessed in a randomized, placebo-controlled, double-blind, multinational study in 10,101 postmenopausal women at increased risk of coronary events. Women in this study had a median age of 67.6 years (range 55 to 92) and were followed for a median of 5.6 years (range 0.01 to 7.1). Eighty-four percent were White, 9.8% of women reported a first-degree relative with a history of breast cancer, and 41.4% of the women had a 5-year predicted risk of invasive breast cancer ≥1.66%, based on the modified Gail model.
Raloxifene hydrochloride, 60 mg administered once daily, reduced the incidence of invasive breast cancer by 44% compared with placebo [absolute risk reduction (ARR) 1.2 per 1000 women-years]; this was primarily due to a 55% reduction in estrogen receptor (ER)-positive invasive breast cancer in the raloxifene hydrochloride group compared with placebo (ARR 1.2 per 1000 women-years). There was no reduction in ER-negative invasive breast cancer. Table 8 presents efficacy and selected safety outcomes.
Table 8: Raloxifene Hydrochloride (60 mg Once Daily) vs. Placebo on Outcomes in Postmenopausal Women at Increased Risk for Major Coronary Events Outcomes
Placeboa
(N=5057)
Raloxifene Hydrochloridea
(N=5044)
HR
(95% CI)b
n
IRb
n
IRb
Invasive breast cancer
70
2.66
40
1.5
0.56 (0.38, 0.83)c
ERb positive
55
2.09
25
0.94
0.45 (0.28, 0.72)
ERb negative
9
0.34
13
0.49
1.44 (0.61, 3.36)
ERb unknown
6
0.23
2
0.07
0.33 (0.07, 1.63)
Noninvasived breast cancer
5
0.19
11
0.41
2.17 (0.75, 6.24)
Clinical vertebral fractures
97
3.7
64
2.4
0.65 (0.47, 0.89)
Death
595
22.45
554
20.68
0.92 (0.82, 1.03)
Death due to stroke
39
1.47
59
2.2
1.49 (1, 2.24)
Stroke
224
8.6
249
9.46
1.1 (0.92, 1.32)
Deep vein thrombosis
47
1.78
65
2.44
1.37 (0.94, 1.99)
Pulmonary embolism
24
0.91
36
1.35
1.49 (0.89, 2.49)
Endometrial and uterine cancere
17
0.83
21
1.01
1.21 (0.64 to 2.3)
Ovarian cancerf
10
0.41
17
0.7
1.69 (0.78, 3.7)
Hot flashes
241
9.09
397
14.82
1.68 (1.43, 1.97)
Peripheral edema
583
22
706
26.36
1.22 (1.09, 1.36)
Cholelithiasisg
131
6.2
168
7.83
1.26 (1.01, 1.59)
a Note: There were a total of 76 breast cancer cases in the placebo group and 52 in the raloxifene hydrochloride group. For two cases, one in each treatment group, invasive status was unknown.
b Abbreviations: CI = confidence interval; ER = estrogen receptor; HR = hazard ratio; IR = annual incidence rate per 1000 women.
c p<0.05, obtained from the log-rank test, after adjusting for the co-primary endpoint of major coronary events.
d All cases were ductal carcinoma in situ.
e Only patients with an intact uterus were included (placebo = 3882, raloxifene hydrochloride = 3900).
f Only patients with at least one ovary were included (placebo = 4606, raloxifene hydrochloride = 4559).
g Only patients with an intact gallbladder at baseline were included (placebo = 4111, raloxifene hydrochloride = 4144).The effect of raloxifene hydrochloride in reducing the incidence of invasive breast cancer was consistent among women above or below age 65 or with a 5-year predicted invasive breast cancer risk, based on the modified Gail model, <1.66%, or ≥1.66%.
14.4 Reduction in Risk of Invasive Breast Cancer in Postmenopausal Women at High Risk of Invasive Breast Cancer
STAR Trial
The effects of raloxifene hydrochloride 60 mg/day versus tamoxifen 20 mg/day over 5 years on reducing the incidence of invasive breast cancer were assessed in 19,747 postmenopausal women in a randomized, double-blind trial conducted in North America by the National Surgical Adjuvant Breast and Bowel Project and sponsored by the National Cancer Institute. Women in this study had a mean age of 58.5 years (range 35 to 83), a mean 5-year predicted invasive breast cancer risk of 4.03% (range 1.66 to 23.61%), and 9.1% had a history of lobular carcinoma in situ (LCIS). More than 93% of participants were White. As of 31 December 2005, the median time of follow-up was 4.3 years (range 0.07 to 6.5 years).
Raloxifene hydrochloride was not superior to tamoxifen in reducing the incidence of invasive breast cancer. The observed incidence rates of invasive breast cancer were raloxifene hydrochloride 4.4 and tamoxifen 4.3 per 1000 women per year. The results from a noninferiority analysis are consistent with raloxifene hydrochloride potentially losing up to 35% of the tamoxifen effect on reduction of invasive breast cancer. The effect of each treatment on invasive breast cancer was consistent when women were compared by baseline age, history of LCIS, history of atypical hyperplasia, 5-year predicted risk of breast cancer by the modified Gail model, or the number of relatives with a history of breast cancer. Fewer noninvasive breast cancers occurred in the tamoxifen group compared to the raloxifene hydrochloride group. Table 9 presents efficacy and selected safety outcomes.
Table 9: Raloxifene Hydrochloride (60 mg Once Daily) vs. Tamoxifen (20 mg Once Daily) on Outcomes in Postmenopausal Women at Increased Risk for Invasive Breast Cancer Outcomes
Raloxifene Hydrochloride
(N=9751)
Tamoxifen
(N=9736)
RR
(95% CI)a
n
IRa
n
IRa
Invasive breast cancer
173
4.4
168
4.3
1.02 (0.82, 1.27)
ERa positive
115
2.93
120
3.07
0.95 (0.73, 1.24)
ERa negative
52
1.32
46
1.18
1.12 (0.74, 1.71)
ERa unknown
6
0.15
2
0.05
2.98 (0.53, 30.21)
Noninvasive breast cancerb
83
2.12
60
1.54
1.38 (0.98, 1.95)
DCISa
47
1.2
32
0.82
1.46 (0.91, 2.37)
LCISa
29
0.74
23
0.59
1.26 (0.7, 2.27)
Uterine cancerc
23
1.21
37
1.99
0.61 (0.34, 1.05)
Endometrial hyperplasiac
17
0.9
100
5.42
0.17 (0.09, 0.28)
Hysterectomyc
92
4.84
246
13.25
0.37 (0.28, 0.47)
Ovarian cancerd
18
0.66
14
0.52
1.27 (0.6, 2.76)
Ischemic heart diseasee
138
3.5
125
3.19
1.1 (0.86, 1.41)
Stroke
54
1.36
56
1.42
0.96 (0.65, 1.42)
Deep vein thrombosis
67
1.69
92
2.35
0.72 (0.52, 1)
Pulmonary embolism
38
0.96
58
1.47
0.65 (0.42, 1)
Clinical vertebral fractures
58
1.46
58
1.47
0.99 (0.68, 1.46)
Cataractsf
343
10.34
435
13.19
0.78 (0.68, 0.91)
Cataract surgeryf
240
7.17
295
8.85
0.81 (0.68, 0.96)
Death
104
2.62
109
2.76
0.95 (0.72, 1.25)
Edemag
741
18.66
664
16.83
1.11 (1, 1.23)
Hot flashes
6748
169.91
7170
181.71
0.94 (0.9, 0.97)
a Abbreviations: CI = confidence interval; DCIS = ductal carcinoma in situ; ER = estrogen receptor; IR = annual incidence rate per 1000 women; LCIS = lobular carcinoma in situ; RR = risk ratio for women in the raloxifene hydrochloride group compared with those in the tamoxifen group.
b Of the 60 noninvasive breast cases in the tamoxifen group, 5 were mixed types. Of the 83 noninvasive breast cancers in the raloxifene group, 7 were mixed types.
c Only patients with an intact uterus at baseline were included (tamoxifen = 4739, raloxifene hydrochloride = 4715).
d Only patients with at least one intact ovary at baseline were included (tamoxifen = 6813, raloxifene hydrochloride = 6787).
e Defined as myocardial infarction, severe angina, or acute ischemic syndromes.
f Only patients who were free of cataracts at baseline were included (tamoxifen = 8342, raloxifene hydrochloride = 8333).
g Peripheral edema events are included in the term edema.14.5 Effects on Cardiovascular Disease
In a randomized, placebo-controlled, double-blind, multinational clinical trial (RUTH) of 10,101 postmenopausal women with documented coronary heart disease or at increased risk for coronary events, no cardiovascular benefit was demonstrated after treatment with raloxifene hydrochloride 60 mg once daily for a median follow-up of 5.6 years. No significant increase or decrease was observed for coronary events (death from coronary causes, nonfatal myocardial infarction, or hospitalization for an acute coronary syndrome). An increased risk of death due to stroke after treatment with raloxifene hydrochloride was observed: 59 (1.2%) raloxifene hydrochloride-treated women died due to a stroke compared to 39 (0.8%) placebo-treated women (2.2 versus 1.5 per 1000 women-years; hazard ratio 1.49; 95% confidence interval, 1 to 2.24; p=0.0499). The incidence of stroke did not differ significantly between treatment groups (249 with raloxifene hydrochloride [4.9%] versus 224 with placebo [4.4%]; hazard ratio 1.1; 95% confidence interval 0.92 to 1.32; p=0.3; 9.5 versus 8.6 per 1000 women-years) [see Warnings and Precautions (5.2, 5.3)].
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Raloxifene Hydrochloride Tablets USP, 60 mg are white to off-white, elliptical, film-coated tablets debossed with ‘X’ on one side and ‘57’ on other side.
Bottles of 100 NDC: 52343-137-01
16.2 Storage and Handling
Store at 20° to 25°C (68° to 77°F) [see USP Controlled Room Temperature]. The USP defines controlled room temperature as a temperature maintained thermostatically that encompasses the usual and customary working environment of 20° to 25°C (68° to 77°F); that results in a mean kinetic temperature calculated to be not more than 25°C; and that allows for excursions between 15° and 30°C (59° and 86°F) that are experienced in pharmacies, hospitals, and warehouses.
-
17 PATIENT COUNSELING INFORMATION
See FDA-approved Medication Guide.
Physicians should instruct their patients to read the Medication Guide before starting therapy with raloxifene hydrochloride and to reread it each time the prescription is renewed.17.1 Osteoporosis Recommendations, Including Calcium and Vitamin D Supplementation
For osteoporosis treatment or prevention, patients should be instructed to take supplemental calcium and/or vitamin D if intake is inadequate. Patients at increased risk for vitamin D insufficiency (e.g., over the age of 70 years, nursing home bound, chronically ill, or with gastrointestinal malabsorption syndromes) should be instructed to take additional vitamin D if needed. Weight-bearing exercises should be considered along with the modification of certain behavioral factors, such as cigarette smoking and/or excessive alcohol consumption, if these factors exist.
17.2 Patient Immobilization
Raloxifene hydrochloride should be discontinued at least 72 hours prior to and during prolonged immobilization (e.g., post-surgical recovery, prolonged bed rest), and patients should be advised to avoid prolonged restrictions of movement during travel because of the increased risk of venous thromboembolic events [see Warnings and Precautions (5.1)].
17.3 Hot Flashes or Flushes
Raloxifene hydrochloride may increase the incidence of hot flashes and is not effective in reducing hot flashes or flushes associated with estrogen deficiency. In some asymptomatic patients, hot flashes may occur upon beginning raloxifene hydrochloride therapy.
17.4 Reduction in Risk of Invasive Breast Cancer in Postmenopausal Women with Osteoporosis or at High Risk of Invasive Breast Cancer
Use of raloxifene hydrochloride is associated with the reduction of the risk of invasive breast cancer in postmenopausal women. Raloxifene hydrochloride has not been shown to reduce the risk of noninvasive breast cancer. When considering treatment, physicians need to discuss the potential benefits and risks of raloxifene hydrochloride treatment with the patient.
Raloxifene hydrochloride is not indicated for the treatment of invasive breast cancer or reduction of the risk of recurrence.
Patients should have breast exams and mammograms before starting raloxifene hydrochloride and should continue regular breast exams and mammograms in keeping with good medical practice after beginning treatment with raloxifene hydrochloride.
Distributed by:
Acetris Health, LLC
Saddle Brook, NJ 07663
Manufactured by:
Aurolife Pharma LLC
Dayton, NJ 08810
Issued: 12/2017
-
Medication Guide
Raloxifene Hydrochloride Tablets, USP for Oral Use
(ral ox' i feen hye'' droe klor' ide)
Read the Medication Guide that comes with raloxifene hydrochloride tablets before you start taking them and each time you refill your prescription. The information may have changed. This Medication Guide does not take the place of talking with your doctor about your medical condition or treatment. Talk with your doctor about raloxifene hydrochloride tablets when you start taking them and at regular checkups.
What is the most important information I should know about raloxifene hydrochloride tablets?
Serious and life-threatening side effects can occur while taking raloxifene hydrochloride tablets. These include blood clots and dying from stroke:
- Increased risk of blood clots in the legs (deep vein thrombosis) and lungs (pulmonary embolism) have been reported with raloxifene hydrochloride tablets. Women who have or have had blood clots in the legs, lungs, or eyes should not take raloxifene hydrochloride tablets.
- Women who have had a heart attack or are at risk for a heart attack may have an increased risk of dying from stroke when taking raloxifene hydrochloride tablets.
1. Before starting raloxifene hydrochloride tablets, tell your doctor if you have had blood clots in your legs, lungs, or eyes, a stroke, mini-stroke (transient ischemic attack), or have an irregular heartbeat.
2. Stop taking raloxifene hydrochloride tablets and call your doctor if you have:
- leg pain or a feeling of warmth in the lower leg (calf).
- swelling of the legs, hands, or feet.
- sudden chest pain, shortness of breath, or coughing up blood.
- sudden change in your vision, such as loss of vision or blurred vision.
3. Being still for a long time (such as sitting still during a long car or airplane trip or being in bed after surgery) can increase your risk of blood clots. (See “What should I avoid while taking raloxifene hydrochloride tablets?”)
What are raloxifene hydrochloride tablets?
Raloxifene hydrochloride tablets are a type of prescription medicine called a Selective Estrogen Receptor Modulator (SERM). Raloxifene hydrochloride tablets are for women after menopause, and has more than one use:
- Osteoporosis: Raloxifene hydrochloride tablets treat and prevent osteoporosis by helping make your bones stronger and less likely to break.
-
Invasive Breast Cancer: If you have osteoporosis or are at high risk for breast cancer, raloxifene hydrochloride tablets can be used to lower your chance of getting invasive breast cancer. Raloxifene hydrochloride tablets will not totally get rid of your chance of getting breast cancer. Your doctor can estimate your risk of breast cancer by asking you about risk factors, including:
- your age (getting older).
- family history of breast cancer in your mother, sister, or daughter.
- a history of any breast biopsy, especially an abnormal biopsy.
You and your doctor should talk about whether the possible benefit of raloxifene hydrochloride in lowering your chance of getting invasive breast cancer is greater than its possible risks.
Raloxifene hydrochloride tablets are not for use in premenopausal women (women who have not passed menopause).
Who should not take raloxifene hydrochloride tablets?
Do not take raloxifene hydrochloride tablets if you:
- have or have had blood clots in your legs, lungs, or eyes. Taking raloxifene hydrochloride tablets may increase the risk of getting blood clots.
- are pregnant or could become pregnant. Raloxifene hydrochloride tablets could harm your unborn child.
- are nursing a baby. It is not known if raloxifene hydrochloride passes into breast milk or what effect it might have on the baby.
What should I tell my doctor before taking raloxifene hydrochloride tablets?
Raloxifene hydrochloride tablets may not be right for you. Before taking raloxifene hydrochloride tablets, tell your doctor about all your medical conditions, including if you:
- have had blood clots in your legs, lungs, or eyes, a stroke, mini-stroke (TIA/transient ischemic attack), or a type of irregular heartbeat (atrial fibrillation).
- have had breast cancer. Raloxifene hydrochloride tablets have not been fully studied in women who have a history of breast cancer.
- have liver or kidney problems.
- have taken estrogen in the past and had a high increase of triglycerides (a kind of fat in the blood).
- are pregnant, planning to become pregnant, or breast-feeding (see “Who should not take raloxifene hydrochloride tablets?”).
Tell your doctor about all medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements. Know the medicines you take. Keep a list of them and show it to your doctor and pharmacist each time you get a new medicine. Especially tell your doctor if you take*:
- warfarin (Coumadin®, Jantoven®)
If you are taking warfarin or other coumarin blood thinners, your doctor may need to do a blood test when you first start or if you need to stop taking raloxifene hydrochloride tablets. Names for this test include “prothrombin time”, “pro-time”, or “INR”. Your doctor may need to adjust the dose of your warfarin or other coumarin blood thinner.
- cholestyramine
- estrogens
Raloxifene hydrochloride tablets should not be taken with cholestyramine or estrogens.
How should I take raloxifene hydrochloride tablets?
- Take raloxifene hydrochloride tablets exactly how your doctor tells you to.
- Keep taking raloxifene hydrochloride tablets for as long as your doctor prescribes them for you. It is not known how long you should keep taking raloxifene hydrochloride tablets to lower your chance of getting invasive breast cancers.
- It is important to get your refills on time so you do not run out of the medicine.
- Take one raloxifene hydrochloride tablet each day.
- Take raloxifene hydrochloride tablets at any time of the day, with or without food.
- To help you remember to take raloxifene hydrochloride tablets, it may be best to take them at about the same time each day.
- Calcium and vitamin D may be taken at the same time as raloxifene hydrochloride tablets. It is important to take calcium and vitamin D, as directed by your physician, to prevent or treat osteoporosis.
- If you miss a dose, take it as soon as you remember. However, if it is almost time for your next dose, skip the missed dose and take only your next regularly scheduled dose. Do not take two doses at the same time.
What should I avoid while taking raloxifene hydrochloride tablets?
- Being still for a long time (such as during long trips or being in bed after surgery) can increase the risk of blood clots. Raloxifene hydrochloride tablets may add to this risk. If you will need to be still for a long time, talk with your doctor about ways to reduce the risk of blood clots. On long trips, move around periodically. Stop taking raloxifene hydrochloride tablets at least 3 days before a planned surgery or before you plan on being still for a long time. You should start taking raloxifene hydrochloride tablets again when you return to your normal activities.
- Some medicines should not be taken with raloxifene hydrochloride tablets (see “What should I tell my doctor before taking raloxifene hydrochloride tablets?”).
What are the possible side effects of raloxifene hydrochloride tablets?
Serious and life-threatening side effects can occur while taking raloxifene hydrochloride tablets. These include blood clots and dying from stroke:
- Increased risk of blood clots in the legs (deep vein thrombosis) and lungs (pulmonary embolism) have been reported with raloxifene hydrochloride tablets. Women who have or have had blood clots in the legs, lungs, or eyes should not take raloxifene hydrochloride tablets.
- Women who have had a heart attack or are at risk for a heart attack may have an increased risk of dying from stroke when taking raloxifene hydrochloride tablets.
See “What is the most important information I should know about raloxifene hydrochloride tablets?”
The most common side effects of raloxifene hydrochloride tablets are hot flashes, leg cramps, swelling of the feet, ankles, and legs, flu syndrome, joint pain, and sweating. Hot flashes are more common during the first 6 months after starting treatment.
These are not all the side effects of raloxifene hydrochloride tablets. Tell your doctor about any side effect that bothers you or that does not go away. If you have any problems or questions that concern you while taking raloxifene hydrochloride tablets, ask your doctor or pharmacist for more information.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
What else should I know about raloxifene hydrochloride tablets?
- Do not use raloxifene hydrochloride tablets to prevent heart disease, heart attack, or strokes.
- To get the calcium and vitamin D you need, your doctor may advise you to change your diet and/or take supplemental calcium and vitamin D. Your doctor may suggest other ways to help treat or prevent osteoporosis, in addition to taking raloxifene hydrochloride tablets and getting the calcium and vitamin D you need. These may include regular exercise, stopping smoking, and drinking less alcohol.
- Women who have hot flashes can take raloxifene hydrochloride tablets. Raloxifene hydrochloride tablets do not treat hot flashes, and they may cause hot flashes in some women. (See “What are the possible side effects of raloxifene hydrochloride tablets?”)
- Raloxifene hydrochloride tablets have not been found to cause breast tenderness or enlargement. If you notice any changes in your breasts, call your doctor to find out the cause. Before starting and while taking raloxifene hydrochloride tablets you should have breast exams and mammograms, as directed by your doctor. Because raloxifene hydrochloride tablets do not eliminate the chance of developing breast cancers, you need these examinations to find any breast cancers as early as possible.
- Raloxifene hydrochloride tablets should not cause spotting or menstrual-type bleeding. If you have any vaginal bleeding, call your doctor to find out the cause. Raloxifene hydrochloride tablets have not been found to increase the risk for cancer of the lining of the uterus.
- Women in clinical trials have taken raloxifene hydrochloride tablets for up to eight years.
How should I store raloxifene hydrochloride tablets?
- Store raloxifene hydrochloride tablets at 20° to 25°C (68° to 77°F).
- Keep raloxifene hydrochloride tablets and all medicines out of the reach of children.
General Information about the safe and effective use of raloxifene hydrochloride tablets
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use raloxifene hydrochloride tablets for a condition for which it was not prescribed. Do not give your raloxifene hydrochloride tablets to other people, even if they have the same symptoms you have. They may harm them.
This Medication Guide is a summary of the most important information about raloxifene hydrochloride tablets. If you would like more information about raloxifene hydrochloride tablets, talk with your doctor. You can ask your doctor or pharmacist for information about raloxifene hydrochloride tablets that is written for health professionals. For more information, call Lucid Pharma LLC at 1-855-244-6100. (toll-free).
What are the ingredients in raloxifene hydrochloride tablets?
Active Ingredient: raloxifene hydrochloride
Inactive Ingredients: citric acid monohydrate, crospovidone, hypromellose, magnesium stearate, microcrystalline cellulose, polyethylene glycol, polysorbate 80, povidone, and titanium dioxide.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
*All brands listed are the trademarks of their respective owners and are not trademarks of Aurobindo Pharma Limited.
Distributed by:
Acetris Health, LLC
Saddle Brook, NJ 07663
Manufactured by:
Aurolife Pharma LLC
Dayton, NJ 08810
Issued: 12/2017
- PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 60 mg (100 Tablets Bottle)
-
INGREDIENTS AND APPEARANCE
RALOXIFENE HYDROCHLORIDE
raloxifene hydrochloride tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 52343-137 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RALOXIFENE HYDROCHLORIDE (UNII: 4F86W47BR6) (RALOXIFENE - UNII:YX9162EO3I) RALOXIFENE HYDROCHLORIDE 60 mg Inactive Ingredients Ingredient Name Strength CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) CROSPOVIDONE (UNII: 68401960MK) HYPROMELLOSE 2910 (5 MPA.S) (UNII: R75537T0T4) MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) POLYSORBATE 80 (UNII: 6OZP39ZG8H) POVIDONE K30 (UNII: U725QWY32X) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color WHITE ((White to Off-white)) Score no score Shape OVAL ((Elliptical)) Size 12mm Flavor Imprint Code X;57 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 52343-137-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 08/28/2015 05/01/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA204310 08/28/2015 05/01/2020 Labeler - ACETRIS HEALTH, LLC (080500964) Establishment Name Address ID/FEI Business Operations Aurobindo Pharma Limited 918917626 API MANUFACTURE(52343-137) Establishment Name Address ID/FEI Business Operations Aurolife Pharma LLC 829084461 ANALYSIS(52343-137) , MANUFACTURE(52343-137)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.