TUKYSA- tucatinib tablet
TUKYSA by
Drug Labeling and Warnings
TUKYSA by is a Prescription medication manufactured, distributed, or labeled by Seattle Genetics, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TUKYSA safely and effectively. See full prescribing information for TUKYSA.
TUKYSA™ (tucatinib) tablets, for oral use
Initial U.S. Approval: 2020
INDICATIONS AND USAGE
TUKYSA is a kinase inhibitor indicated in combination with trastuzumab and capecitabine for treatment of adult patients with advanced unresectable or metastatic HER2-positive breast cancer, including patients with brain metastases, who have received one or more prior anti-HER2-based regimens in the metastatic setting. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Tablets: 50 mg and 150 mg. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
-
Diarrhea: Severe diarrhea, including dehydration, acute kidney injury, and death, has been reported. Administer antidiarrheal treatment as clinically indicated. Interrupt dose, then dose reduce, or permanently discontinue TUKYSA based on severity. (2.2, 5.1)
- Hepatotoxicity: Severe hepatotoxicity has been reported on TUKYSA. Monitor ALT, AST and bilirubin prior to starting TUKYSA, every 3 weeks during treatment and as clinically indicated. Interrupt dose, then dose reduce, or permanently discontinue TUKYSA based on severity. (2.2, 5.2)
-
Embryo-Fetal Toxicity: TUKYSA can cause fetal harm. Advise patients of potential risk to a fetus and to use effective contraception. (5.3, 8.1, 8.3)
Also, refer to the Full Prescribing Information of trastuzumab and capecitabine for pregnancy and contraception information.
ADVERSE REACTIONS
The most common adverse reactions (≥20%) are diarrhea, palmar-plantar erythrodysesthesia, nausea, fatigue, hepatotoxicity, vomiting, stomatitis, decreased appetite, abdominal pain, headache, anemia, and rash. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Seattle Genetics at 1-855-4SEAGEN or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
- Strong CYP3A Inducers or Moderate CYP2C8 Inducers: Avoid concomitant use. (7.1)
- Strong CYP2C8 Inhibitors: Avoid concomitant use; reduce TUKYSA dose if concomitant use cannot be avoided. (2.4, 7.1)
- CYP3A Substrates: Avoid concomitant use with CYP3A substrates, where minimal concentration changes may lead to serious or life-threatening toxicities. (7.2)
-
P-gp Substrates: Consider reducing the dose of P-gp substrates, where minimal concentration changes may lead to serious or life-threatening toxicities. (7.2)
USE IN SPECIFIC POPULATIONS
Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 4/2020
-
Diarrhea: Severe diarrhea, including dehydration, acute kidney injury, and death, has been reported. Administer antidiarrheal treatment as clinically indicated. Interrupt dose, then dose reduce, or permanently discontinue TUKYSA based on severity. (2.2, 5.1)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dosage Modifications for Adverse Reactions
2.3 Dosage Modifications for Severe Hepatic Impairment
2.4 Dosage Modifications for Concomitant Use with Strong CYP2C8 Inhibitors
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Diarrhea
5.2 Hepatotoxicity
5.3 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on TUKYSA
7.2 Effects of TUKYSA on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 HER2-Positive Metastatic Breast Cancer
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
TUKYSA is indicated in combination with trastuzumab and capecitabine for treatment of adult patients with advanced unresectable or metastatic HER2-positive breast cancer, including patients with brain metastases, who have received one or more prior anti-HER2-based regimens in the metastatic setting.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage of TUKYSA is 300 mg taken orally twice daily in combination with trastuzumab and capecitabine until disease progression or unacceptable toxicity [see Clinical Studies (14)].
Advise patients to swallow TUKYSA tablets whole and not to chew, crush, or split prior to swallowing. Advise patients not to ingest tablet if it is broken, cracked, or not otherwise intact.
Advise patients to take TUKYSA approximately 12 hours apart and at the same time each day with or without a meal.
If the patient vomits or misses a dose of TUKYSA, instruct the patient to take the next dose at its usual scheduled time.
When given in combination with TUKYSA, the recommended dosage of capecitabine is 1000 mg/m2 orally twice daily taken within 30 minutes after a meal. TUKYSA and capecitabine can be taken at the same time. Refer to the Full Prescribing Information for trastuzumab and capecitabine for additional information.
2.2 Dosage Modifications for Adverse Reactions
The recommended TUKYSA dose reductions and dosage modifications for adverse reactions are provided in Tables 1 and 2. Refer to the Full Prescribing Information for trastuzumab and capecitabine for information about dosage modifications for these drugs.
Table 1: Recommended TUKYSA Dose Reductions for Adverse Reactions Dose Reduction Recommended TUKYSA Dosage First 250 mg orally twice daily Second 200 mg orally twice daily Third 150 mg orally twice daily Permanently discontinue TUKYSA in patients unable to tolerate 150 mg orally twice daily.
Table 2: Recommended TUKYSA Dosage Modifications for Adverse Reactions 1. Grades based on National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.03
2. Abbreviations: ULN = upper limit of normal; ALT = alanine aminotransferase; AST = aspartate aminotransferaseAdverse Reaction1 Severity TUKYSA Dosage Modification Diarrhea [see
Warnings and Precautions (5.1)]Grade 3 without anti-diarrheal treatment Initiate or intensify appropriate medical therapy. Hold TUKYSA until recovery to ≤ Grade 1, then resume TUKYSA at the same dose level. Grade 3 with anti-diarrheal treatment Initiate or intensify appropriate medical therapy. Hold TUKYSA until recovery to ≤ Grade 1, then resume TUKYSA at the next lower dose level. Grade 4 Permanently discontinue TUKYSA. Hepatotoxicity2 [see
Warnings and Precautions (5.2)]Grade 2 bilirubin (>1.5 to 3 × ULN) Hold TUKYSA until recovery to ≤ Grade 1, then resume TUKYSA at the same dose level. Grade 3 ALT or AST (> 5 to 20 × ULN)
OR
Grade 3 bilirubin (> 3 to 10 × ULN)Hold TUKYSA until recovery to ≤ Grade 1, then resume TUKYSA at the next lower dose level. Grade 4 ALT or AST (> 20 × ULN)
OR
Grade 4 bilirubin (> 10 × ULN)Permanently discontinue TUKYSA. ALT or AST > 3 × ULN
AND
Bilirubin > 2 × ULNPermanently discontinue TUKYSA. Other adverse reactions [see
Adverse Reactions (6.1) ]Grade 3 Hold TUKYSA until recovery to ≤ Grade 1, then resume TUKYSA at the next lower dose level. Grade 4 Permanently discontinue TUKYSA. 2.3 Dosage Modifications for Severe Hepatic Impairment
For patients with severe hepatic impairment (Child-Pugh C), reduce the recommended dosage to 200 mg orally twice daily [see Use in Specific Populations (8.7), Clinical Pharmacology (12.3)].
2.4 Dosage Modifications for Concomitant Use with Strong CYP2C8 Inhibitors
Avoid concomitant use of strong CYP2C8 inhibitors with TUKYSA. If concomitant use with a strong CYP2C8 inhibitor cannot be avoided, reduce the recommended dosage to 100 mg orally twice daily. After discontinuation of the strong CYP2C8 inhibitor for 3 elimination half-lives, resume the TUKYSA dose that was taken prior to initiating the inhibitor [see Drug Interactions (7.2), Clinical Pharmacology (12.3)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Diarrhea
TUKYSA can cause severe diarrhea including dehydration, hypotension, acute kidney injury, and death [see Adverse Reactions (6.1)]. In HER2CLIMB, 81% of patients who received TUKYSA experienced diarrhea, including 12% with Grade 3 diarrhea and 0.5% with Grade 4 diarrhea. Both patients who developed Grade 4 diarrhea subsequently died, with diarrhea as a contributor to death. The median time to onset of the first episode of diarrhea was 12 days and the median time to resolution was 8 days. Diarrhea led to dose reductions of TUKYSA in 6% of patients and discontinuation of TUKYSA in 1% of patients. Prophylactic use of antidiarrheal treatment was not required on HER2CLIMB.
If diarrhea occurs, administer antidiarrheal treatment as clinically indicated. Perform diagnostic tests as clinically indicated to exclude other causes of diarrhea. Based on the severity of the diarrhea, interrupt dose, then dose reduce or permanently discontinue TUKYSA [see Dosage and Administration (2.2)].
5.2 Hepatotoxicity
TUKYSA can cause severe hepatotoxicity [see Adverse Reactions (6.1)]. In HER2CLIMB, 8% of patients who received TUKYSA had an ALT increase > 5 × ULN, 6% had an AST increase > 5 × ULN, and 1.5% had a bilirubin increase > 3 × ULN (Grade ≥3). Hepatotoxicity led to dose reduction of TUKYSA in 8% of patients and discontinuation of TUKYSA in 1.5% of patients.
Monitor ALT, AST, and bilirubin prior to starting TUKYSA, every 3 weeks during treatment, and as clinically indicated. Based on the severity of hepatoxicity, interrupt dose, then dose reduce or permanently discontinue TUKYSA [see Dosage and Administration (2.2)].
5.3 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, TUKYSA can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of tucatinib to pregnant rats and rabbits during organogenesis caused embryo-fetal mortality, reduced fetal weight and fetal abnormalities at maternal exposures ≥ 1.3 times the human exposure (AUC) at the recommended dose.
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TUKYSA and for at least 1 week after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TUKYSA and for at least 1 week after the last dose [see Use in Specific Populations (8.1, 8.3)].
TUKYSA is used in combination with trastuzumab and capecitabine. Refer to the Full Prescribing Information of trastuzumab and capecitabine for pregnancy and contraception information.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Diarrhea [see Warnings and Precautions (5.1)]
- Hepatotoxicity [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
HER2-Positive Metastatic Breast Cancer
HER2CLIMB
The safety of TUKYSA in combination with trastuzumab and capecitabine was evaluated in HER2CLIMB [see Clinical Studies (14)]. Patients received either TUKYSA 300 mg twice daily plus trastuzumab and capecitabine (n=404) or placebo plus trastuzumab and capecitabine (n=197). The median duration of treatment was 5.8 months (range: 3 days, 2.9 years) for the TUKYSA arm.
Serious adverse reactions occurred in 26% of patients who received TUKYSA. Serious adverse reactions in ≥ 2% of patients who received TUKYSA were diarrhea (4%), vomiting (2.5%), nausea (2%), abdominal pain (2%), and seizure (2%). Fatal adverse reactions occurred in 2% of patients who received TUKYSA including sudden death, sepsis, dehydration, and cardiogenic shock.
Adverse reactions leading to treatment discontinuation occurred in 6% of patients who received TUKYSA. Adverse reactions leading to treatment discontinuation of TUKYSA in ≥1% of patients were hepatotoxicity (1.5%) and diarrhea (1%).
Adverse reactions leading to dose reduction occurred in 21% of patients who received TUKYSA. Adverse reactions leading to dose reduction of TUKYSA in ≥2% of patients were hepatotoxicity (8%) and diarrhea (6%).
The most common adverse reactions in patients who received TUKYSA (≥20%) were diarrhea, palmar-plantar erythrodysesthesia, nausea, fatigue, hepatotoxicity, vomiting, stomatitis, decreased appetite, abdominal pain, headache, anemia, and rash.
Table 3 summarizes the adverse reactions in HER2CLIMB.
Table 3: Adverse Reactions (≥10%) in Patients Who Received TUKYSA and with a Difference Between Arms of ≥ 5% Compared to Placebo in HER2CLIMB (All Grades) - Stomatitis includes stomatitis, oropharyngeal pain, oropharyngeal discomfort, mouth ulceration, oral pain, lip ulceration, glossodynia, tongue blistering, lip blister, oral dysesthesia, tongue ulceration, and aphthous ulcer
- Rash includes rash maculo-papular, rash, dermatitis acneiform, erythema, rash macular, rash papular, rash pustular, rash pruritic, rash erythematous, skin exfoliation, urticaria, dermatitis allergic, palmar erythema, plantar erythema, skin toxicity, and dermatitis
- Hepatotoxicity includes hyperbilirubinemia, blood bilirubin increased, bilirubin conjugated increased, alanine aminotransferase increased, transaminases increased, hepatotoxicity, aspartate aminotransferase increased, liver function test increased, liver injury, and hepatocellular injury
- Anemia includes anemia, hemoglobin decreased, and normocytic anemia
- Due to inhibition of renal tubular transport of creatinine without affecting glomerular function
- Peripheral neuropathy includes peripheral sensory neuropathy, neuropathy peripheral, peripheral motor neuropathy, and peripheral sensorimotor neuropathy
Adverse Reaction TUKYSA + Trastuzumab +
Capecitabine
N = 404Placebo + Trastuzumab +
Capecitabine
N = 197All Grades% Grade 3% Grade 4% All Grades% Grade 3% Grade 4% Gastrointestinal disorders Diarrhea 81 12 0.5 53 9 0 Nausea 58 3.7 0 44 3 0 Vomiting 36 3 0 25 3.6 0 Stomatitis1 32 2.5 0 21 0.5 0 Skin and subcutaneous tissue disorders Palmar-plantar erythrodysesthesia syndrome 63 13 0 53 9 0 Rash2 20 0.7 0 15 0.5 0 Hepatobiliary disorders Hepatotoxicity3 42 9 0.2 24 3.6 0 Metabolism and nutrition disorders Decreased appetite 25 0.5 0 20 0 0 Blood and lymphatic system disorders Anemia4 21 3.7 0 13 2.5 0 Musculoskeletal and connective tissue disorders Arthralgia 15 0.5 0 4.6 0.5 0 Investigations Creatinine increased5 14 0 0 1.5 0 0 Weight decreased 13 1 0 6 0.5 0 Nervous System Disorders Peripheral neuropathy6 13 0.5 0 7 1 0 Respiratory, thoracic and mediastinal disorders Epistaxis 12 0 0 5 0 0 Table 4: Laboratory Abnormalities (≥20%) Worsening from Baseline in Patients Who Received TUKYSA and with a Difference of ≥5% Compared to Placebo in HER2CLIMB - The denominator used to calculate the rate varied from 351 to 400 in the TUKYSA arm and 173 to 197 in the control arm based on the number of patients with a baseline value and at least one post-treatment value. Grading was based on NCI-CTCAE v.4.03 for laboratory abnormalities, except for increased creatinine which only includes patients with a creatinine increase based on the upper limit of normal definition for grade 1 events (NCI CTCAE v5.0).
- Laboratory criteria for Grade 1 is identical to laboratory criteria for Grade 2.
- Due to inhibition of renal tubular transport of creatinine without affecting glomerular function.
- There is no definition for Grade 2 in CTCAE v.4.03.
TUKYSA + Trastuzumab
+ Capecitabine1Placebo + Trastuzumab
+ Capecitabine1All Grades
%Grades ≥3
%All Grades
%Grades ≥
3%Hematology Decreased hemoglobin 59 3.3 51 1.5 Chemistry Decreased phosphate 57 8 45 7 Increased bilirubin 47 1.5 30 3.1 Increased ALT 46 8 27 0.5 Increased AST 43 6 25 1 Decreased magnesium 40 0.8 25 0.5 Decreased potassium 2 36 6 31 5 Increased creatinine 3 33 0 6 0 Decreased sodium 4 28 2.5 23 2 Increased alkaline phosphatase 26 0.5 17 0 Increased Creatinine
The mean increase in serum creatinine was 32% within the first 21 days of treatment with TUKYSA. The serum creatinine increases persisted throughout treatment and were reversible upon treatment completion. Consider alternative markers of renal function if persistent elevations in serum creatinine are observed [see Clinical Pharmacology (12.3)].
-
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on TUKYSA
Table 5 summarizes the effect of other drugs on TUKYSA.
Table 5: Drug Interactions that Affect TUKYSA Strong CYP3A Inducers or Moderate CYP2C8 Inducers Clinical Impact Concomitant use of TUKYSA with a strong CYP3A or moderate CYP2C8 inducer decreased tucatinib plasma concentrations [see Clinical Pharmacology (12.3)], which may reduce TUKYSA activity. Management Avoid concomitant use of TUKYSA with a strong CYP3A inducer or a moderate CYP2C8 inducer. Strong or Moderate CYP2C8 Inhibitors Clinical Impact Concomitant use of TUKYSA with a strong CYP2C8 inhibitor increased tucatinib plasma concentrations [see Clinical Pharmacology (12.3)], which may increase the risk of TUKYSA toxicity. Management Avoid concomitant use of TUKYSA with a strong CYP2C8 inhibitor. Increase monitoring for TUKYSA toxicity with moderate CYP2C8 inhibitors. 7.2 Effects of TUKYSA on Other Drugs
Table 6 summarizes the effect of TUKYSA on other drugs.
Table 6: TUKYSA Drug Interactions that Affect Other Drugs CYP3A Substrates Clinical Impact Concomitant use of TUKYSA with a CYP3A substrate increased the plasma concentrations of CYP3A substrate [see Clinical Pharmacology (12.3)], which may increase the toxicity associated with a CYP3A substrate. Management Avoid concomitant use of TUKYSA with CYP3A substrates, where minimal concentration changes may lead to serious or life-threatening toxicities. If concomitant use is unavoidable, decrease the CYP3A substrate dosage in accordance with approved product labeling. P-glycoprotein (P-gp) Substrates Clinical Impact Concomitant use of TUKYSA with a P-gp substrate increased the plasma concentrations of P-gp substrate [see Clinical Pharmacology (12.3)], which may increase the toxicity associated with a P-gp substrate. Management Consider reducing the dosage of P-gp substrates, where minimal concentration changes may lead to serious or life-threatening toxicities. -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
TUKYSA is used in combination with trastuzumab and capecitabine. Refer to the Full Prescribing Information of trastuzumab and capecitabine for pregnancy information.
Based on findings in animals and its mechanism of action, TUKYSA can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available human data on TUKYSA use in pregnant women to inform a drug-associated risk. In animal reproduction studies, administration of tucatinib to pregnant rats and rabbits during organogenesis resulted in embryo-fetal mortality, reduced fetal weight and fetal abnormalities at maternal exposures ≥ 1.3 times the human exposure (AUC) at the recommended dose (see Data). Advise pregnant women and females of reproductive potential of the potential risk to the fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%-4% and 15%-20%, respectively.
Animal Data
In pilot embryo-fetal development studies, pregnant rats and rabbits received oral doses of tucatinib up to 150 mg/kg/day during the period of organogenesis.
In rats, oral administration of tucatinib resulted in maternal toxicity (body weight loss, reduced body weight gain, low food consumption) at doses ≥ 90 mg/kg/day. Fetal effects included reduced number of live fetuses, decreased fetal weight, and fetal abnormalities (increase in skeletal variations, incomplete ossification) at ≥ 90 mg/kg/day (approximately 3.5 times the human exposure at the recommended dose based on AUC).
In rabbits, oral administration of tucatinib resulted in increased resorptions, decreased percentages of live fetuses, and skeletal, visceral, and external malformations in fetuses at doses ≥ 90 mg/kg/day (1.3 times the human exposure at the recommended dose based on AUC). Fetal abnormalities included domed head, brain dilation, incomplete ossification of frontal and parietal bones, and a hole in the parietal bone.
8.2 Lactation
Risk Summary
TUKYSA is used in combination with trastuzumab and capecitabine. Refer to the Full Prescribing Information of trastuzumab and capecitabine for lactation information.
There are no data on the presence of tucatinib or its metabolites in human or animal milk or its effects on the breastfed child or on milk production. Because of the potential for serious adverse reactions in a breastfed child, advise women not to breastfeed during treatment with TUKYSA and for at least 1 week after the last dose.
8.3 Females and Males of Reproductive Potential
TUKYSA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. TUKYSA is used in combination with trastuzumab and capecitabine. Refer to the Full Prescribing Information of trastuzumab and capecitabine for contraception and infertility information.
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating treatment with TUKYSA.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with TUKYSA and for at least 1 week after the last dose.
Males
Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TUKYSA and for at least 1 week after the last dose.
Infertility
Based on findings from animal studies, TUKYSA may impair male and female fertility [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of TUKYSA in pediatric patients have not been established.
8.5 Geriatric Use
In HER2CLIMB, 82 patients who received TUKYSA were ≥ 65 years, of whom 8 patients were ≥ 75 years. The incidence of serious adverse reactions in those receiving TUKYSA was 34% in patients ≥ 65 years compared to 24% in patients <65 years. The most frequent serious adverse reactions in patients who received TUKYSA and ≥ 65 years were diarrhea (9%), vomiting (6%), and nausea (5%). There were no observed overall differences in the effectiveness of TUKYSA in patients ≥ 65 years compared to younger patients. There were too few patients ≥75 years to assess differences in effectiveness or safety.
8.6 Renal Impairment
The use of TUKYSA in combination with capecitabine and trastuzumab is not recommended in patients with severe renal impairment (CLcr < 30 mL/min estimated by Cockcroft-Gault Equation), because capecitabine is contraindicated in patients with severe renal impairment. Refer to the Full Prescribing Information of capecitabine for additional information in severe renal impairment.
No dose adjustment is recommended for patients with mild or moderate renal impairment (creatinine clearance [CLcr] 30 to 89 mL/min).
8.7 Hepatic Impairment
Tucatinib exposure is increased in patients with severe hepatic impairment (Child-Pugh C). Reduce the dose of TUKYSA for patients with severe (Child-Pugh C) hepatic impairment [see Dosage and Administration (2.3), Clinical Pharmacology (12.3)].
No dose adjustment for TUKYSA is required for patients with mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment.
-
11 DESCRIPTION
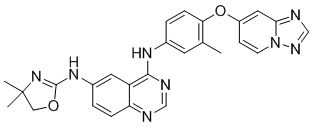
Tucatinib is a kinase inhibitor. The chemical name is (N4-(4-([1,2,4]triazolo[1,5-a]pyridin-7-yloxy)-3-methylphenyl)-N6-(4,4-dimethyl-4,5-dihydrooxazol-2-yl)quinazoline-4,6-diamine. The molecular formula is C26H24N8O2 and the molecular weight is 480.52 g/mol. The chemical structure is as follows:
TUKYSA (tucatinib) is supplied as 50 mg and 150 mg film-coated tablets for oral use and contain the following inactive ingredients:
Tablet core: copovidone, crospovidone, sodium chloride, potassium chloride, sodium bicarbonate, colloidal silicon dioxide, magnesium stearate, and microcrystalline cellulose.
Coating: yellow film coat: polyvinyl alcohol, titanium dioxide, macrogol/polyethylene glycol, talc, and yellow iron oxide non-irradiated.
Each TUKYSA 50 mg tablet contains 10.10 mg (0.258 mEq) potassium and 9.21 mg (0.401 mEq) sodium.
Each TUKYSA 150 mg tablet contains 30.29 mg (0.775 mEq) potassium and 27.64 mg (1.202 mEq) sodium.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tucatinib is a tyrosine kinase inhibitor of HER2. In vitro, tucatinib inhibits phosphorylation of HER2 and HER3, resulting in inhibition of downstream MAPK and AKT signaling and cell proliferation, and showed anti-tumor activity in HER2 expressing tumor cells. In vivo, tucatinib inhibited the growth of HER2 expressing tumors. The combination of tucatinib and trastuzumab showed increased anti-tumor activity in vitro and in vivo compared to either drug alone.
12.2 Pharmacodynamics
Exposure Response Relationship
Tucatinib exposure-response relationships and the time course of pharmacodynamics response have not been fully characterized.
Cardiac Electrophysiology
No large mean increase in QTc (i.e., > 20 ms) was detected following treatment with TUKYSA at the recommended dose of 300 mg taken orally twice daily.
12.3 Pharmacokinetics
Tucatinib AUC0-INF and Cmax increases proportionally over a dosage range from 50 mg to 300 mg (0.17 to 1 times the approved recommended dosage). Tucatinib exhibited 1.7-fold accumulation for AUC and 1.5-fold accumulation for Cmax following administration of TUKYSA 300 mg twice daily for 14 days. Time to steady state was approximately 4 days.
Absorption
The median time to peak plasma concentration of tucatinib was approximately 2 hours (range 1 to 4 hours).
Effects of Food
Following administration of a single oral dose of TUKYSA in 11 subjects after a high-fat meal (approximately 58% fat, 26% carbohydrate, and 16% protein), the mean AUC0-INF increased by 1.5-fold, the Tmax shifted from 1.5 hours to 4 hours, and Cmax was unaltered. The effect of food on the pharmacokinetics of tucatinib was not clinically meaningful.
Distribution
The geometric mean (CV%) apparent volume of distribution of tucatinib was approximately 1670 L (66%). The plasma protein binding was 97.1% at clinically relevant concentrations.
Elimination
The geometric mean (CV%) half-life of tucatinib was approximately 8.5 (21%) hours and apparent clearance was 148 L/h (55%).
Metabolism
Tucatinib is metabolized primarily by CYP2C8 and to a lesser extent via CYP3A.
Excretion
Following a single oral dose of 300 mg radiolabeled tucatinib, approximately 86% of the total radiolabeled dose was recovered in feces (16% of the administered dose as unchanged tucatinib) and 4.1% in urine with an overall total recovery of 90% within 13 days post-dose. In plasma, approximately 76% of the plasma radioactivity was unchanged, 19% was attributed to identified metabolites, and approximately 5% was unassigned.
Specific Populations
Age (< 65 (n =211); ≥ 65 (n = 27)), albumin (25 to 52 g/L), creatinine clearance (creatinine clearance [CLcr] 60 to 89 mL/min (n = 89); CLcr 30 to 59 mL/min (n = 5)), body weight (41 to 138 kg), and race (White (n=168), Black (n=53), or Asian (n=10)) did not have a clinically meaningful effect on tucatinib exposure.
Renal Impairment
No clinically significant differences in the pharmacokinetics of tucatinib were observed in patients with mild to moderate renal impairment (creatinine clearance: 30 to 89 mL/min by Cockcroft-Gault). The effect of severe renal impairment (creatinine clearance: < 30 mL/min) on the pharmacokinetics of tucatinib is unknown.
Hepatic Impairment
Mild (Child-Pugh A) and moderate (Child-Pugh B) hepatic impairment had no clinically relevant effect on tucatinib exposure. Tucatinib AUC0-INF was increased by 1.6 fold in subjects with severe (Child-Pugh C) hepatic impairment compared to subjects with normal hepatic function.
Drug Interaction Studies
Clinical Studies
Table 7: Effect of Other Drugs on TUKYSA Concomitant Drug
(Dose)TUKYSA Dose Ratio (90% CI) of Tucatinib
Exposure With and Without
Concomitant DrugCmax AUC Strong CYP3A Inhibitor
Itraconazole (200 mg BID)
300 mg single dose1.3
(1.2, 1.4)1.3
(1.3, 1.4)Strong CYP3A/Moderate 2C8 Inducer
Rifampin (600 mg once daily)0.6
(0.5, 0.8)0.5
(0.4, 0.6)Strong CYP2C8 Inhibitor
Gemfibrozil (600 mg BID)1.6
(1.5, 1.8)3.0
(2.7, 3.5)Table 8: Effect of TUKYSA on Other Drugs - Tucatinib reduced the renal clearance of metformin without any effect on glomerular filtration rate (GFR) as measured by iohexol clearance and serum cystatin C.
Concomitant Drug
(Dose)TUKYSA Dose Ratio (90% CI) of Exposure
Measures of Concomitant Drug
with/without TucatinibCmax AUC CYP2C8 Substrate
Repaglinide (0.5 mg single dose)
300 mg twice daily1.7
(1.4, 2.1)1.7
(1.5, 1.9)CYP3A Substrate
Midazolam (2 mg single dose)3.0
(2.6, 3.4)5.7
(5.0, 6.5)P-gp Substrate
Digoxin (0.5 mg single dose)2.4
(1.9, 2.9)1.5
(1.3, 1.7)MATE1/2-K substratea
Metformin (850 mg single dose)1.1
(1.0, 1.2)
1.4
(1.2, 1.5)No clinically significant difference in the pharmacokinetics of tucatinib were observed when used concomitantly with omeprazole (proton pump inhibitor) or tolbutamide (sensitive CYP2C9 substrate).
In Vitro Studies
Cytochrome P450 (CYP) Enzymes: Tucatinib is a reversible inhibitor of CYP2C8 and CYP3A and a time-dependent inhibitor of CYP3A, but is not an inhibitor of CYP1A2, CYP2B6, CYP2C9, CYP2C19, or CYP2D6.
Uridine diphosphate (UDP)-glucuronosyl transferase (UGT) Enzymes: Tucatinib is not an inhibitor of UGT1A1.
Transporter Systems: Tucatinib is a substrate of P-gp and BCRP, but is not a substrate of OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, MATE1, MATE2-K, or BSEP.
Tucatinib inhibits MATE1/MATE2-K-mediated transport of metformin and OCT2/MATE1-mediated transport of creatinine. The observed serum creatinine increase in clinical studies with tucatinib is due to inhibition of tubular secretion of creatinine via OCT2 and MATE1.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with tucatinib.
Tucatinib was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay. Tucatinib was not clastogenic in either an in vitro chromosome aberration assay or an in vivo mouse bone marrow micronucleus assay.
Fertility studies in animals have not been conducted. In repeat-dose toxicity studies up to 13 weeks duration, decreased corpora lutea/corpus luteum cyst, increased interstitial cells of the ovary, atrophy of the uterus, and mucification of the vagina were observed in female rats at doses ≥ 6 mg/kg/day (approximately 0.1 times the human exposure at the recommended dose based on AUC). Atrophy and edema of the testes and oligospermia/germ cell debris in the epididymides were observed in male rats at ≥ 120 mg/kg/day (approximately 13 times the human exposure at the recommended dose based on AUC).
-
14 CLINICAL STUDIES
14.1 HER2-Positive Metastatic Breast Cancer
The efficacy of TUKYSA in combination with trastuzumab and capecitabine was evaluated in 612 patients in HER2CLIMB (NCT02614794), a randomized (2:1), double-blind, placebo-controlled trial. Patients were required to have HER2-positive, unresectable locally advanced or metastatic breast cancer, with or without brain metastases, and prior treatment with trastuzumab, pertuzumab, and ado-trastuzumab emtansine (T-DM1) separately or in combination, in the neoadjuvant, adjuvant or metastatic setting. HER2 positivity was based on archival or fresh tissue tested with an FDA-approved test at a central laboratory prior to enrollment with HER2 positivity defined as HER2 IHC 3+ or ISH positive.
Patients with brain metastases, including those with progressing or untreated lesions, were eligible provided they were neurologically stable and did not require immediate radiation or surgery. The trial excluded patients with leptomeningeal disease. Randomization was stratified by the presence or history of brain metastases (yes vs. no), Eastern Cooperative Oncology Group (ECOG) performance status (0 vs. 1), and region (U.S., Canada, or rest of world).
Patients received TUKYSA 300 mg or placebo orally twice daily with a trastuzumab loading dose of 8 mg/kg on Day 1 of Cycle 1 if needed and then a maintenance dose of 6 mg/kg on Day 1 of every 21-day cycle thereafter and capecitabine 1000 mg/m2 orally twice daily on Days 1 through 14 of every 21-day cycle. An alternate trastuzumab dosing regimen was 600 mg administered subcutaneously on Day 1 of every 21-day cycle. Patients were treated until disease progression or unacceptable toxicity. Tumor assessments, including brain-MRI in patients with presence or history of brain metastases at baseline, occurred every 6 weeks for the first 24 weeks and every 9 weeks thereafter.
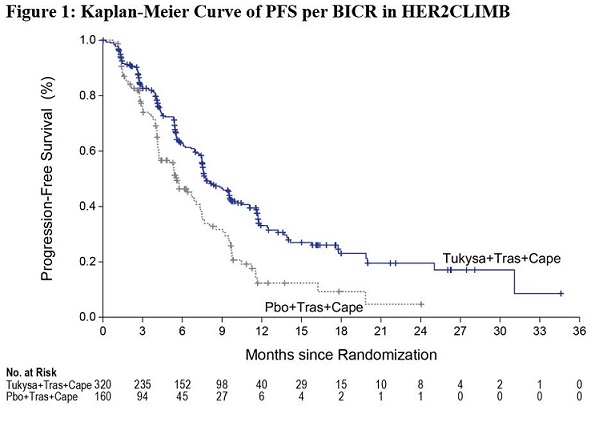
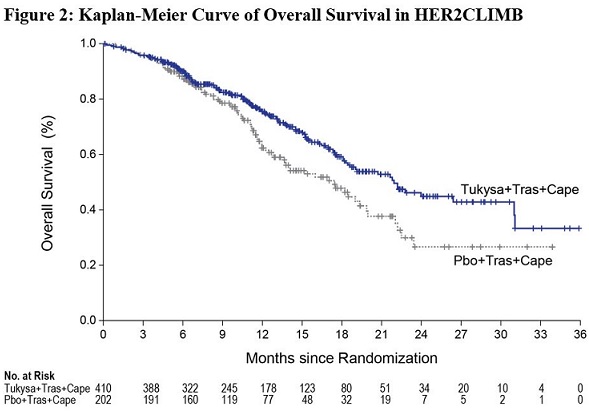
The major efficacy outcome measure was progression-free survival (PFS) in the first 480 randomized patients assessed by blinded independent central review (BICR) using Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. Additional efficacy outcome measures were evaluated in all randomized patients and included overall survival (OS), PFS among patients with a history or presence of brain metastases (PFSBrainMets), and confirmed objective response rate (ORR).
The median age was 54 years (range: 22 - 82); 116 (19%) patients were age 65 or older. The majority were White (73%) and female (99%) and 51% had an ECOG performance status of 1. Sixty percent had estrogen and/or progesterone receptor-positive disease. Forty-eight percent had a presence or history of brain metastases; of these patients, 23% had untreated brain metastases, 40% had treated but stable brain metastases, and 37% had treated but radiographically progressing brain metastases. Seventy-four percent of patients had visceral metastases. Patients had received a median of 4 (range, 2 to 17) prior lines of systemic therapy and a median of 3 (range, 1 to 14) prior lines of systemic therapy in the metastatic setting. All patients received prior trastuzumab and T-DM1 and all but two patients had prior pertuzumab.
Efficacy results are summarized in Table 9 and Figure 1 and 2. Efficacy results were consistent across patient subgroups defined by stratification factors (presence or history of brain metastases, ECOG status, region of world) and hormone receptor status.
Table 9: Efficacy Results in HER2CLIMB BICR=blinded independent central review; CI=confidence interval; PFS=progression-free survival; OS=overall survival; ORR=objective response rate; CR=complete response; PR=partial response; DOR=duration of response.
1. Primary PFS analysis conducted in first 480 randomized patients.
2. Hazard ratio and 95% confidence intervals are based on stratified Cox proportional
hazards regression model controlling for stratification factors (presence or history of brain
metastases, ECOG status, and region of world)
3. Two-sided p-value based on re-randomization procedure (Rosenberger and Lachin
2002) controlling for stratification factors, compared with the allocated alpha of 0.05
4. Two-sided p-value based on re-randomization procedure (Rosenberger and Lachin 2002)
controlling for stratification factors, compared with the allocated alpha of 0.0074 for this
interim analysis (with 60% of the planned number of events for final analysis)
5. Analysis includes patients with history or presence of parenchymal brain metastases at
baseline, including target and non-target lesions. Does not include patients with dural
lesions only.
6. Two-sided p-value based on re-randomization procedure (Rosenberger and Lachin 2002)
controlling for stratification factors, compared with the allocated alpha of 0.0080 for this
interim analysis (with 71% of the planned number of events for final analysis)
7. Two-sided 95% exact confidence interval, computed using the Clopper-Pearson method (1934)
8. Calculated using the complementary log-log transformation method (Collett, 1994)TUKYSA + Trastuzumab +
CapecitabinePlacebo + Trastuzumab +
CapecitabinePFS1 N=320 N=160 Number of events (%) 178 (56) 97 (61) Median, months (95% CI) 7.8 (7.5, 9.6) 5.6 (4.2, 7.1) Hazard ratio (95% CI)2 0.54 (0.42, 0.71) P-value3 <0.00001 OS N=410 N=202 Number of deaths (%) 130 (32) 85 (42) Median, months (95% CI) 21.9 (18.3, 31.0) 17.4 (13.6, 19.9) Hazard ratio (95% CI)2 0.66 (0.50, 0.87) P-value4 0.00480 PFSBrainMets5 N=198 N=93 Number of events (%) 106 (53.5) 51 (54.8) Median, months (95% CI) 7.6 (6.2, 9.5) 5.4 (4.1, 5.7) Hazard ratio (95% CI)2 0.48 (0.34, 0.69) P-value6 <0.00001 Confirmed ORR for Patients with Measurable Disease N=340 N=171 ORR (95% CI)7 40.6 (35.3, 46.0) 22.8 (16.7, 29.8) CR (%) 3 (0.9) 2 (1.2) PR (%) 135 (39.7) 37 (21.6) P-value3 0.00008 DOR Median, months (95% CI)8 8.3 (6.2, 9.7) 6.3 (5.8, 8.9) -
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
TUKYSA 50 mg tablets are supplied as yellow, film-coated, round tablets containing 50 mg of tucatinib. Each tablet is debossed with “TUC” on one side and “50” on the other side, and is packaged as follows:
50 mg tablets: 60 count in 75 cc bottle: NDC: 51144-001-60
TUKYSA 150 mg tablets are supplied as yellow, film-coated, oval-shaped tablets containing 150 mg of tucatinib. Each tablet is debossed with “TUC” on one side and “150” on the other side, and is packaged as follows:
150 mg tablets: 60 count in 75 cc bottle: NDC: 51144-002-60
150 mg tablets: 120 count in 150 cc bottle: NDC: 51144-002-12
Store at controlled room temperature, 20ºC to 25ºC (68ºF to 77ºF); excursions permitted from 15ºC to 30ºC (59ºF to 86ºF) [see USP Controlled Room Temperature].
Dispense to patient in original container only. Store in original container to protect from moisture. Replace cap securely each time after opening. Do not discard desiccant.
Once opened, use within 3 months. Discard any unused tablets 3 months after opening the bottle.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Diarrhea
- Inform patients that TUKYSA has been associated with severe diarrhea. Instruct patients on how to manage diarrhea and to inform their healthcare provider immediately if there is any change in bowel patterns [see Warnings and Precautions (5.1)].
Hepatotoxicity
- Inform patients that TUKYSA has been associated with severe hepatotoxicity and that they should report signs and symptoms of liver dysfunction to their healthcare provider immediately [see Warnings and Precautions (5.2)].
Embryo-Fetal Toxicity
- Inform pregnant women and females of reproductive potential of the risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.3) and Use in Specific Populations (8.1)].
- Advise females of reproductive potential to use effective contraception during treatment with TUKYSA and for at least 1 week after the last dose [see Use in Specific Populations (8.3)].
- Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TUKYSA and for at least 1 week after the last dose [see Use in Specific Populations (8.3)].
- Refer to the Full Prescribing Information of trastuzumab and capecitabine for pregnancy and contraception information.
Lactation
- Advise women not to breastfeed during treatment with TUKYSA and for at least 1 week after the last dose [see Use in Specific Populations (8.2)]. Refer to the Full Prescribing Information of trastuzumab and capecitabine for lactation information.
Infertility
- Advise males and females of reproductive potential that TUKYSA may impair fertility [see Use in Specific Populations (8.3)]. Refer to the Full Prescribing Information of trastuzumab and capecitabine for infertility information.

Manufactured by:
Seattle Genetics, Inc.
Bothell, WA 98021
1-855-4SEAGEN
TUKYSA is a trademark, and Seattle Genetics and
 are US registered trademarks of Seattle Genetics, Inc.
are US registered trademarks of Seattle Genetics, Inc.
© 2020 Seattle Genetics, Inc., Bothell, WA 98021. All rights reserved.
-
PATIENT PACKAGE INSERT
This Patient information has been approved by the U.S. Food and Drug Administration. Issued 04/2020 PATIENT INFORMATION
TUKYSATM (too-KYE-sah)
(tucatinib)
tabletsImportant information: TUKYSA is used with the medicines trastuzumab and capecitabine, also read the Medication Guide that comes with capecitabine. What is TUKYSA?
TUKYSA is a prescription medicine used with the medicines trastuzumab and capecitabine to treat adults with:- human epidermal growth factor receptor-2 (HER2) positive breast cancer that has spread to other parts of the body such as the brain (metastatic), or that cannot be removed by surgery, and
- who have received one or more anti-HER2 breast cancer treatments.
Before taking TUKYSA, tell your healthcare provider about all of your medical conditions, including if you: - have liver problems.
- are pregnant or plan to become pregnant. TUKYSA can harm your unborn baby.
Females who are able to become pregnant:- Your healthcare provider will do a pregnancy test before you start treatment with TUKYSA.
- You should use effective birth control (contraception) during treatment with TUKYSA and for at least 1 week after the last dose of TUKYSA. Talk with your healthcare provider about forms of birth control that you can use during this time.
- Tell your healthcare provider right away if you become pregnant or think you may be pregnant during treatment with TUKYSA.
- Your healthcare provider will do a pregnancy test before you start treatment with TUKYSA.
- are breastfeeding or plan to breastfeed. It is not known if TUKYSA passes into your breast milk. Do not breastfeed during treatment with TUKYSA and for at least 1 week after the last dose of TUKYSA.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. TUKYSA may affect the way your other medicines work, and other medicines may affect the way TUKYSA works.
Know the medicines you take. Keep a list of all the medicines you take and show it to your healthcare provider and pharmacist every time you get a new medicine.How should I take TUKYSA?
- Take TUKYSA exactly as your healthcare provider tells you.
- TUKYSA is used with the medicines trastuzumab and capecitabine. Your healthcare provider will tell you the dose of trastuzumab and capecitabine you will take and how you will receive them.
- Take TUKYSA 2 times a day, with or without a meal.
- Take TUKYSA about 12 hours apart or at the same times every day.
- Swallow TUKYSA tablets whole. Do not chew, crush, or split TUKYSA tablets before swallowing. Do not take TUKYSA tablets if they are broken, cracked, or damaged.
- If you vomit or miss a dose of TUKYSA, take your next dose at your regular time.
What are the possible side effects of TUKYSA?
TUKYSA may cause serious side effects, including:-
Diarrhea. Diarrhea is common with TUKYSA and can sometimes be severe. Tell your healthcare provider if you have a change in your bowel movements or severe diarrhea. Severe diarrhea can lead to loss of too much body fluids (dehydration), low blood pressure, kidney problems and death. Your healthcare provider may prescribe medicines to treat your diarrhea during treatment with TUKYSA.
-
Liver Problems. TUKYSA can cause severe liver problems. Your healthcare provider will do blood tests to check your liver function before and every 3 weeks during treatment with TUKYSA, or as needed. Tell your healthcare provider right away if you have any signs and symptoms of liver problems including:
- itching
- yellowing of your skin or eyes
- dark or brown urine (tea-colored)
- pain in the upper right side of your stomach-area (abdomen)
- feel very tired
- decreased appetite
- bleeding or bruising more easily than normal
The most common side effects of TUKYSA: - diarrhea
- rash, redness, pain, swelling or blisters on the palms of your hands or soles of your feet
- nausea
- tiredness
- increased liver function blood tests
- vomiting
- mouth sores (stomatitis)
- decreased appetite
- stomach-area (abdomen) pain
- headache
- low red blood cell counts (anemia)
- rash
Your healthcare provider may change your dose of TUKYSA, temporarily stop, or permanently stop treatment with TUKYSA if you have certain side effects.
TUKYSA may cause fertility problems in males and females, which may affect the ability to have children. Talk to your healthcare provider if you have concerns about fertility.
These are not all of the possible side effects of TUKYSA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store TUKYSA? - Store TUKYSA at room temperature 68°F to 77°F (20ºC to 25ºC).
- Keep TUKYSA in its original container. The TUKYSA bottle contains a desiccant packet to help keep your tablets dry (protect from moisture). Keep the desiccant in the bottle.
- Tightly close the bottle of TUKYSA after you take your dose.
- TUKYSA must be used within 3 months after opening the bottle. Throw away (discard) any unused tablets 3 months after opening the bottle.
Keep TUKYSA and all medicines out of reach of children. General information about the safe and effective use of TUKYSA.
Medicines are sometimes prescribed for conditions not listed in the Patient Information. Do not use TUKYSA for a condition for which it was not prescribed. Do not give TUKYSA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about TUKYSA that is written for healthcare professionals.What are the ingredients in TUKYSA?
Active ingredient: tucatinib
Inactive ingredients:
Tablet core: copovidone, crospovidone, sodium chloride, potassium chloride, sodium bicarbonate, colloidal silicon dioxide, magnesium stearate, and microcrystalline cellulose.
Tablet coating: yellow film coat: polyvinyl alcohol, titanium dioxide, macrogol/polyethylene glycol, talc, and yellow iron oxide non-irradiated.
Manufactured by Seattle Genetics, Inc., Bothell, WA 98021
TUKYSA TM is a trademark owned by Seattle Genetics, Inc.
©2020 Seattle Genetics, Inc.
For more information, call 1-855-473-2436 (1-855-4SEAGEN) or go to www.TUKYSA.com. - human epidermal growth factor receptor-2 (HER2) positive breast cancer that has spread to other parts of the body such as the brain (metastatic), or that cannot be removed by surgery, and
-
PRINCIPAL DISPLAY PANEL
NDC 51144-001-60
Rx OnlyTM
TUKYSATM
(tucatinib) tablets
50 mg*
Attention: Dispense and store Tukysa
in original container to protect from moisture.
SeattleGenetics®
60 Tablets
* Each tablet contains 50 mg tucatinib.
Recommended Dosage: See prescribing information
Store at controlled room temperature,
20°C to 25°C (68° to 77°F); excursions
permitted from 15°C to 30°C (59°F to 86°F)
[see USP Controlled Room Temperature].
KEEP OUT OF REACH OF CHILDREN
Discard unused tablets 3 months after opening.
Mfg by Seattle Genetics, Inc.
Bothell, WA 98021 USA
TUKYSA and its logo are trademarks of
Seattle Genetics, Inc.
©2020 Seattle Genetics, Inc.
All rights reserved. Printed in USA
1208-00
NDC 51144-002-60
Rx Only
TUKYSATM
(tucatinib) tablets
150 mg*
Attention: Dispense and store Tukysa
in original container to protect from moisture.
SeattleGenetics®
60 Tablets
* Each tablet contains 150 mg tucatinib.
Recommended Dosage: See prescribing information
Store at controlled room temperature,
20°C to 25°C (68° to 77°F); excursions
permitted from 15°C to 30°C (59°F to 86°F)
[see USP Controlled Room Temperature].
KEEP OUT OF REACH OF CHILDREN
Discard unused tablets 3 months after opening.
Mfg by Seattle Genetics, Inc.
Bothell, WA 98021 USA
TUKYSA and its logo are trademarks of
Seattle Genetics, Inc.
©2020 Seattle Genetics, Inc.
All rights reserved. Printed in USA
1200-00
-
INGREDIENTS AND APPEARANCE
TUKYSA
tucatinib tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 51144-001 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Tucatinib (UNII: 234248D0HH) (Tucatinib - UNII:234248D0HH) Tucatinib 50 mg Inactive Ingredients Ingredient Name Strength Copovidone K25-31 (UNII: D9C330MD8B) Crospovidone (120 .Mu.M) (UNII: 68401960MK) Sodium Chloride (UNII: 451W47IQ8X) Potassium Chloride (UNII: 660YQ98I10) Sodium Bicarbonate (UNII: 8MDF5V39QO) Silicon Dioxide (UNII: ETJ7Z6XBU4) Magnesium Stearate (UNII: 70097M6I30) Microcrystalline Cellulose 102 (UNII: PNR0YF693Y) Polyvinyl Alcohol, Unspecified (UNII: 532B59J990) Titanium Dioxide (UNII: 15FIX9V2JP) Polyethylene Glycol, Unspecified (UNII: 3WJQ0SDW1A) Talc (UNII: 7SEV7J4R1U) Ferric Oxide Yellow (UNII: EX438O2MRT) Product Characteristics Color YELLOW Score no score Shape ROUND Size 8mm Flavor Imprint Code TUC;50 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 51144-001-60 60 in 1 BOTTLE; Type 0: Not a Combination Product 04/17/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA213411 04/17/2020 TUKYSA
tucatinib tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 51144-002 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Tucatinib (UNII: 234248D0HH) (Tucatinib - UNII:234248D0HH) Tucatinib 150 mg Inactive Ingredients Ingredient Name Strength Copovidone K25-31 (UNII: D9C330MD8B) Crospovidone (120 .Mu.M) (UNII: 68401960MK) Sodium Chloride (UNII: 451W47IQ8X) Potassium Chloride (UNII: 660YQ98I10) Sodium Bicarbonate (UNII: 8MDF5V39QO) Silicon Dioxide (UNII: ETJ7Z6XBU4) Magnesium Stearate (UNII: 70097M6I30) Microcrystalline Cellulose 102 (UNII: PNR0YF693Y) Polyvinyl Alcohol, Unspecified (UNII: 532B59J990) Titanium Dioxide (UNII: 15FIX9V2JP) Polyethylene Glycol, Unspecified (UNII: 3WJQ0SDW1A) Talc (UNII: 7SEV7J4R1U) Ferric Oxide Yellow (UNII: EX438O2MRT) Product Characteristics Color YELLOW Score no score Shape OVAL Size 18mm Flavor Imprint Code TUC;150 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 51144-002-60 60 in 1 BOTTLE; Type 0: Not a Combination Product 04/17/2020 2 NDC: 51144-002-12 120 in 1 BOTTLE; Type 0: Not a Combination Product 04/17/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA213411 04/17/2020 Labeler - Seattle Genetics, Inc. (028484371) Establishment Name Address ID/FEI Business Operations Seattle Genetics, Inc. 028484371 manufacture(51144-001, 51144-002)
Trademark Results [TUKYSA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 TUKYSA 88682720 not registered Live/Pending |
Seattle Genetics, Inc. 2019-11-06 |
 TUKYSA 88209362 not registered Live/Pending |
Seattle Genetics, Inc. 2018-11-28 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.