TRIKAFTA- elexacaftor, tezacaftor, and ivacaftor kit
Trikafta by
Drug Labeling and Warnings
Trikafta by is a Prescription medication manufactured, distributed, or labeled by Vertex Pharmaceuticals Incorporated, Shanghai SynTheAll Pharmaceutical Co., Ltd., Hovione Limited, F.I.S. Fabbrica Italiana Sintetici S.P.A., Almac Sciences Limited, Hovione LLC, Catalent Greenville, Inc., EMSL Analytical Inc. dba MPL Laboratories, Eurofins Lancaster Laboratories, Inc., Halo Pharmaceutical Inc, Pharmaceutical Manufacturing Research Services, Inc., Almac Pharma Services LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TRIKAFTA safely and effectively. See full prescribing information for TRIKAFTA.
TRIKAFTA® (elexacaftor, tezacaftor and ivacaftor tablets; ivacaftor tablets), co-packaged for oral use
Initial U.S. Approval: 2019INDICATIONS AND USAGE
TRIKAFTA is a combination of ivacaftor, a CFTR potentiator, tezacaftor, and elexacaftor indicated for the treatment of cystic fibrosis (CF) in patients aged 12 years and older who have at least one F508del mutation in the CFTR gene.
If the patient's genotype is unknown, an FDA-cleared CF mutation test should be used to confirm the presence of at least one F508del mutation. (1)
DOSAGE AND ADMINISTRATION
- Adults and pediatric patients aged 12 years and older:
- Should not be used in patients with severe hepatic impairment. Use not recommended in patients with moderate hepatic impairment unless the benefit exceeds the risk. Reduce dose if used in patients with moderate hepatic impairment. Liver function tests should be closely monitored. (2.2, 5.1, 8.7, 12.3)
- Reduce dose when co-administered with drugs that are moderate or strong CYP3A inhibitors. (2.3, 5.3, 7.2, 12.3)
DOSAGE FORMS AND STRENGTHS
- Tablets: fixed dose combination containing elexacaftor 100 mg, tezacaftor 50 mg and ivacaftor 75 mg.
Co-packaged with:
- Tablets: ivacaftor 150 mg. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Elevated liver function tests (ALT, AST or bilirubin): Liver function tests (ALT, AST, and bilirubin) should be assessed prior to initiating TRIKAFTA, every 3 months during the first year of treatment, and annually thereafter. In patients with a history of hepatobiliary disease or liver function test elevations, more frequent monitoring should be considered. Dosing should be interrupted in patients with ALT or AST >5 × upper limit of normal (ULN) or ALT or AST >3 × ULN with bilirubin >2 × ULN. Following resolution of transaminase elevations, consider the benefits and risks of resuming treatment. (5.1, 6)
- Use with CYP3A inducers: Concomitant use with strong CYP3A inducers (e.g., rifampin, St. John's wort) significantly decrease ivacaftor exposure and are expected to decrease elexacaftor and tezacaftor exposure, which may reduce TRIKAFTA efficacy. Therefore, co-administration is not recommended. (5.2, 7.1, 12.3)
- Cataracts: Non-congenital lens opacities/cataracts have been reported in pediatric patients treated with ivacaftor-containing regimens. Baseline and follow-up examinations are recommended in pediatric patients initiating TRIKAFTA treatment. (5.4, 8.4)
ADVERSE REACTIONS
The most common adverse drug reactions to TRIKAFTA (occurring in ≥5% of patients and at a frequency higher than placebo by ≥1%) were headache, upper respiratory tract infection, abdominal pain, diarrhea, rash, alanine aminotransferase increased, nasal congestion, blood creatine phosphokinase increased, aspartate aminotransferase increased, rhinorrhea, rhinitis, influenza, sinusitis and blood bilirubin increased. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Vertex Pharmaceuticals Incorporated at 1-877-634-8789 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 1/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information in Adults and Pediatric Patients Aged 12 Years and Older
2.2 Dose Adjustment for Patients with Hepatic Impairment
2.3 Dose Adjustment for Patients Taking Drugs that are CYP3A Inhibitors
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Liver Function Test Elevations
5.2 Concomitant Use with CYP3A Inducers
5.3 Concomitant Use with CYP3A Inhibitors
5.4 Cataracts
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Inducers of CYP3A
7.2 Inhibitors of CYP3A
7.3 Ciprofloxacin
7.4 CYP2C9 Substrates
7.5 Transporters
7.6 Hormonal Contraceptives
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
8.8 Patients with Severe Lung Dysfunction
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Trial 1
14.2 Trial 2
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
TRIKAFTA is indicated for the treatment of cystic fibrosis (CF) in patients aged 12 years and older who have at least one F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene.
If the patient's genotype is unknown, an FDA-cleared CF mutation test should be used to confirm the presence of at least one F508del mutation.
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information in Adults and Pediatric Patients Aged 12 Years and Older
The recommended dose is two tablets (each containing elexacaftor 100 mg, tezacaftor 50 mg and ivacaftor 75 mg) taken in the morning and one ivacaftor tablet (containing ivacaftor 150 mg) taken in the evening, approximately 12 hours apart. TRIKAFTA is for oral use. Instruct patients to swallow the tablets whole. TRIKAFTA should be taken with fat-containing food. Examples of meals or snacks that contain fat are those prepared with butter or oils or those containing eggs, cheeses, nuts, whole milk, or meats [see Clinical Pharmacology (12.3)].
If 6 hours or less have passed since the missed morning or evening dose, the patient should take the missed dose as soon as possible and continue on the original schedule.
If more than 6 hours have passed since:
- the missed morning dose, the patient should take the missed dose as soon as possible and should not take the evening dose. The next scheduled morning dose should be taken at the usual time.
- the missed evening dose, the patient should not take the missed dose. The next scheduled morning dose should be taken at the usual time.
Morning and evening doses should not be taken at the same time.
2.2 Dose Adjustment for Patients with Hepatic Impairment
TRIKAFTA has not been studied in patients with moderate or severe hepatic impairment. Patients with severe hepatic impairment should not be treated with TRIKAFTA. Use of TRIKAFTA is not recommended in patients with moderate hepatic impairment unless the benefit exceeds the risk. If used in patients with moderate hepatic impairment, TRIKAFTA should be used with caution and at a reduced dose (see Table 1). Liver function tests should be closely monitored [see Warnings and Precautions (5.1)].
No dose adjustment is recommended for patients with mild hepatic impairment [see Use in Specific Populations (8.7), Clinical Pharmacology (12.3), and Patient Counseling Information (17)].
Table 1: Dose adjustment of TRIKAFTA in patients with hepatic impairment Mild (Child-Pugh Class A) Moderate (Child-Pugh Class B)* Severe (Child-Pugh Class C) - * Use not recommended unless the benefit exceeds the risk
Morning No dose adjustment Two tablets of elexacaftor/tezacaftor/ivacaftor Should not be used Evening No dose adjustment No ivacaftor dose Should not be used 2.3 Dose Adjustment for Patients Taking Drugs that are CYP3A Inhibitors
Table 2 describes the recommended dosage modification for TRIKAFTA when co-administered with strong (e.g., ketoconazole, itraconazole, posaconazole, voriconazole, telithromycin, and clarithromycin) or moderate (e.g., fluconazole, erythromycin) CYP3A inhibitors. Avoid food or drink containing grapefruit during TRIKAFTA treatment [see Warnings and Precautions (5.3), Drug Interactions (7.2), Clinical Pharmacology (12.3), and Patient Counseling Information (17)].
Table 2: Dosing Schedule for Concomitant Use of TRIKAFTA with Moderate and Strong CYP3A Inhibitors Moderate CYP3A Inhibitors Day 1 Day 2 Day 3 Day 4* - * Continue dosing with two elexacaftor/tezacaftor/ivacaftor tablets and one ivacaftor tablet on alternate days.
- † The evening dose of ivacaftor should not be taken.
- ‡ Continue dosing with two elexacaftor/tezacaftor/ivacaftor tablets twice a week, approximately 3 to 4 days apart.
- § The evening dose of ivacaftor tablet should not be taken.
Morning Dose Two elexacaftor/tezacaftor/ivacaftor tablets One ivacaftor tablet Two elexacaftor/tezacaftor/ivacaftor tablets One ivacaftor tablet Evening Dose† No dose Strong CYP3A Inhibitors Day 1 Day 2 Day 3 Day 4‡ Morning Dose Two elexacaftor/tezacaftor/ivacaftor tablets No dose No dose Two elexacaftor/tezacaftor/ivacaftor tablets Evening Dose§ No dose -
3 DOSAGE FORMS AND STRENGTHS
TRIKAFTA is a co-package of fixed-dose combination tablets containing elexacaftor 100 mg, tezacaftor 50 mg and ivacaftor 75 mg, and ivacaftor 150 mg tablets. The elexacaftor, tezacaftor and ivacaftor tablets are orange, capsule-shaped, and debossed with "T100" on one side and plain on the other. The ivacaftor tablets are light blue, capsule-shaped, and printed with "V 150" in black ink on one side and plain on the other.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Liver Function Test Elevations
Elevated transaminases have been observed in patients with CF treated with TRIKAFTA. Bilirubin elevations have also been observed with TRIKAFTA treatment. Assessments of liver function tests (ALT, AST, and bilirubin) are recommended for all patients prior to initiating TRIKAFTA, every 3 months during the first year of treatment, and annually thereafter. For patients with a history of hepatobiliary disease or liver function test elevations, more frequent monitoring should be considered. In the event of significant elevations in liver function tests, e.g. ALT or AST >5 × the upper limit of normal (ULN) or ALT or AST >3 × ULN with bilirubin >2 × ULN, dosing should be interrupted and laboratory tests closely followed until the abnormalities resolve. Following the resolution of liver function test elevations, consider the benefits and risks of resuming treatment [see Adverse Reactions (6.1)].
5.2 Concomitant Use with CYP3A Inducers
Exposure to ivacaftor is significantly decreased and exposure to elexacaftor and tezacaftor are expected to decrease by the concomitant use of strong CYP3A inducers, which may reduce the therapeutic effectiveness of TRIKAFTA. Therefore, co-administration with strong CYP3A inducers is not recommended [see Drug Interactions (7.1), Clinical Pharmacology (12.3), and Patient Counseling Information (17)].
5.3 Concomitant Use with CYP3A Inhibitors
Exposure to elexacaftor, tezacaftor and ivacaftor are increased when co-administered with strong or moderate CYP3A inhibitors. Therefore, the dose of TRIKAFTA should be reduced when used concomitantly with moderate or strong CYP3A inhibitors [see Dosage and Administration (2.3), Drug Interactions (7.2), Clinical Pharmacology (12.3), and Patient Counseling Information (17)].
5.4 Cataracts
Cases of non-congenital lens opacities have been reported in pediatric patients treated with ivacaftor-containing regimens. Although other risk factors were present in some cases (such as corticosteroid use, exposure to radiation), a possible risk attributable to treatment with ivacaftor cannot be excluded. Baseline and follow-up ophthalmological examinations are recommended in pediatric patients initiating treatment with TRIKAFTA [see Use in Specific Populations (8.4) and Patient Counseling Information (17)].
-
6 ADVERSE REACTIONS
The following adverse reaction is discussed in greater detail in other sections of the label:
- Liver Function Test Elevations [see Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety profile of TRIKAFTA is based on data from 510 CF patients in two double-blind, controlled, Phase 3 trials of 24 weeks and 4 weeks treatment duration (Trials 1 and 2). Eligible patients were also able to participate in an open-label extension safety study (up to 96 weeks of TRIKAFTA). In the two controlled Phase 3 trials, a total of 257 patients aged 12 years and older received at least one dose of TRIKAFTA.
In Trial 1, the proportion of patients who discontinued study drug prematurely due to adverse events was 1% for TRIKAFTA-treated patients and 0% for placebo-treated patients.
Serious adverse reactions that occurred more frequently in TRIKAFTA-treated patients compared to placebo were rash (1% vs <1%) and influenza (1% vs 0). There were no deaths in Trials 1 and 2.
Table 3 shows adverse reactions occurring in ≥5% of TRIKAFTA-treated patients and higher than placebo by ≥1% in the 24-week placebo-controlled, parallel-group Phase 3 trial (Trial 1).
Table 3: Incidence of Adverse Drug Reactions in ≥5% of TRIKAFTA-Treated Patients and Higher than Placebo by ≥1% Adverse Drug Reactions
(Preferred Term)TRIKAFTA
N=202
n (%)Placebo
N=201
n (%)- * Includes upper respiratory tract infection and viral upper respiratory tract infection
- † Includes abdominal pain, abdominal pain upper, abdominal pain lower
- ‡ Includes: rash, rash generalized, rash erythematous, rash macular, rash pruritic
Headache 35 (17) 30 (15) Upper respiratory tract infection* 32 (16) 25 (12) Abdominal pain† 29 (14) 18 (9) Diarrhea 26 (13) 14 (7) Rash‡ 21 (10) 10 (5) Alanine aminotransferase increased 20 (10) 7 (3) Nasal congestion 19 (9) 15 (7) Blood creatine phosphokinase increased 19 (9) 9 (4) Aspartate aminotransferase increased 19 (9) 4 (2) Rhinorrhea 17 (8) 6 (3) Rhinitis 15 (7) 11 (5) Influenza 14 (7) 3 (1) Sinusitis 11 (5) 8 (4) Blood bilirubin increased 10 (5) 2 (1) Additional adverse reactions that occurred in TRIKAFTA-treated patients at a frequency of 2 to <5% and higher than placebo by ≥1% include the following: Flatulence, abdominal distension, conjunctivitis, pharyngitis, respiratory tract infection, tonsillitis, urinary tract infection, c-reactive protein increased, hypoglycemia, dizziness, dysmenorrhea, acne, eczema, and pruritus.
Rash Events
In Trial 1, the overall incidence of rash events was 10% in TRIKAFTA-treated and 5% in placebo-treated patients (see Table 3). The incidence of rash events was higher in female TRIKAFTA-treated patients (16%) than in male TRIKAFTA-treated patients (5%).
Hormonal contraceptives may play a role in the occurrence of rash. For patients taking hormonal contraceptives who develop rash, consider interrupting TRIKAFTA and hormonal contraceptives. Following the resolution of rash, consider resuming TRIKAFTA without the hormonal contraceptives. If rash does not recur, resumption of hormonal contraceptives can be considered.
Laboratory and Vital Sign Abnormalities
Liver Function Test Elevations
In Trial 1, the incidence of maximum transaminase (ALT or AST) >8, >5, or >3 × ULN was 1%, 2%, and 8% in TRIKAFTA-treated patients and 1%, 1%, and 5% in placebo-treated patients. The incidence of adverse reactions of transaminase elevations (AST and/or ALT) was 11% in TRIKAFTA-treated patients and 4% placebo-treated patients.
In Trial 1, the incidence of maximum total bilirubin elevation >2 × ULN was 4% in TRIKAFTA-treated patients and <1% in placebo-treated patients. Maximum indirect and direct bilirubin elevations >1.5 × ULN occurred in 11% and 3% of TRIKAFTA-treated patients, respectively. No TRIKAFTA-treated patients developed maximum direct bilirubin elevation >2 × ULN.
Increased Creatine Phosphokinase
In Trial 1, the incidence of maximum creatine phosphokinase elevation >5 × ULN was 10% in TRIKAFTA-treated and 5% in placebo-treated patients. Among the TRIKAFTA-treated patients with creatine phosphokinase elevation >5 × ULN, 14% (3/21) required treatment interruption and none discontinued treatment.
Increased Blood Pressure
In Trial 1, the maximum increase from baseline in mean systolic and diastolic blood pressure was 3.5 mmHg and 1.9 mmHg, respectively for TRIKAFTA-treated patients (baseline: 113 mmHg systolic and 69 mmHg diastolic) and 0.9 mmHg and 0.5 mmHg, respectively for placebo-treated patients (baseline: 114 mmHg systolic and 70 mmHg diastolic).
The proportion of patients who had systolic blood pressure >140 mmHg and 10 mmHg increase from baseline on at least two occasions was 4% in TRIKAFTA-treated patients and 1% in placebo-treated patients. The proportion of patients who had diastolic blood pressure >90 mmHg and 5 mmHg increase from baseline on at least two occasions was 1% in TRIKAFTA-treated patients and 2% in placebo-treated patients.
With the exception of sex differences in rash, the safety profile of TRIKAFTA was generally similar across all subgroups of patients, including analysis by age, sex, baseline percent predicted FEV1 (ppFEV1), and geographic regions.
The safety profile for the CF patients enrolled in Trial 2 was similar to that observed in Trial 1.
-
7 DRUG INTERACTIONS
7.1 Inducers of CYP3A
Elexacaftor, tezacaftor and ivacaftor are substrates of CYP3A (ivacaftor is a sensitive substrate of CYP3A). Concomitant use of CYP3A inducers may result in reduced exposures and thus reduced TRIKAFTA efficacy. Co-administration of ivacaftor with rifampin, a strong CYP3A inducer, significantly decreased ivacaftor area under the curve (AUC) by 89%. Elexacaftor and tezacaftor exposures are expected to decrease during co-administration with strong CYP3A inducers. Therefore, co-administration of TRIKAFTA with strong CYP3A inducers is not recommended [see Warnings and Precautions (5.2), Clinical Pharmacology (12.3), and Patient Counseling Information (17)].
Examples of strong CYP3A inducers include:
- rifampin, rifabutin, phenobarbital, carbamazepine, phenytoin, and St. John's wort (Hypericum perforatum)
7.2 Inhibitors of CYP3A
Co-administration with itraconazole, a strong CYP3A inhibitor, increased elexacaftor AUC by 2.8-fold and tezacaftor AUC by 4.0 to 4.5-fold. When co-administered with itraconazole and ketoconazole, ivacaftor AUC increased by 15.6-fold and 8.5-fold, respectively. The dosage of TRIKAFTA should be reduced when co-administered with strong CYP3A inhibitors [see Dosage and Administration (2.3), Warnings and Precautions (5.3), Clinical Pharmacology (12.3), and Patient Counseling Information (17)].
Examples of strong CYP3A inhibitors include:
- ketoconazole, itraconazole, posaconazole, and voriconazole
- telithromycin and clarithromycin
Simulations indicated that co-administration with moderate CYP3A inhibitors may increase elexacaftor and tezacaftor AUC by approximately 1.9 to 2.3-fold and 2.1-fold, respectively. Co-administration of fluconazole increased ivacaftor AUC by 2.9-fold. The dosage of TRIKAFTA should be reduced when co administered with moderate CYP3A inhibitors [see Dosage and Administration (2.3), Warnings and Precautions (5.3), Clinical Pharmacology (12.3), and Patient Counseling Information (17)].
Examples of moderate CYP3A inhibitors include:
- fluconazole
- erythromycin
Co-administration of TRIKAFTA with grapefruit juice, which contains one or more components that moderately inhibit CYP3A, may increase exposure of elexacaftor, tezacaftor and ivacaftor; therefore, food or drink containing grapefruit should be avoided during treatment with TRIKAFTA [see Dosage and Administration (2.3), and Patient Counseling Information (17)].
7.3 Ciprofloxacin
Ciprofloxacin had no clinically relevant effect on the exposure of tezacaftor or ivacaftor and is not expected to affect the exposure of elexacaftor. Therefore, no dose adjustment is necessary during concomitant administration of TRIKAFTA with ciprofloxacin [see Clinical Pharmacology (12.3)].
7.4 CYP2C9 Substrates
Ivacaftor may inhibit CYP2C9; therefore, monitoring of the international normalized ratio (INR) during co administration of TRIKAFTA with warfarin is recommended. Other medicinal products for which exposure may be increased by TRIKAFTA include glimepiride and glipizide; these medicinal products should be used with caution [see Clinical Pharmacology (12.3)].
7.5 Transporters
Co-administration of ivacaftor or tezacaftor/ivacaftor with digoxin, a sensitive P-gp substrate, increased digoxin AUC by 1.3-fold, consistent with weak inhibition of P-gp by ivacaftor. Administration of TRIKAFTA may increase systemic exposure of medicinal products that are sensitive substrates of P-gp, which may increase or prolong their therapeutic effect and adverse reactions. When used concomitantly with digoxin or other substrates of P-gp with a narrow therapeutic index such as cyclosporine, everolimus, sirolimus, and tacrolimus, caution and appropriate monitoring should be used [see Clinical Pharmacology (12.3)].
Elexacaftor and M23-ELX inhibit uptake by OATP1B1 and OATP1B3 in vitro. Co-administration of TRIKAFTA may increase exposures of medicinal products that are substrates of these transporters, such as statins, glyburide, nateglinide and repaglinide. When used concomitantly with substrates of OATP1B1 or OATP1B3, caution and appropriate monitoring should be used [see Clinical Pharmacology (12.3)]. Bilirubin is an OATP1B1 and OATP1B3 substrate.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are limited and incomplete human data from clinical trials on the use of TRIKAFTA or its individual components, elexacaftor, tezacaftor and ivacaftor, in pregnant women to inform a drug-associated risk. Although there are no animal reproduction studies with the concomitant administration of elexacaftor, tezacaftor and ivacaftor, separate reproductive and developmental studies were conducted with each active component of TRIKAFTA in pregnant rats and rabbits.
In animal embryo fetal development (EFD) studies oral administration of elexacaftor to pregnant rats and rabbits during organogenesis demonstrated no teratogenicity or adverse developmental effects at doses that produced maternal exposures up to approximately 2 times the exposure at the maximum recommended human dose (MRHD) in rats and 4 times the MRHD in rabbits [based on summed AUCs of elexacaftor and its metabolite (for rat) and AUC of elexacaftor (for rabbit)]. Oral administration of tezacaftor to pregnant rats and rabbits during organogenesis demonstrated no teratogenicity or adverse developmental effects at doses that produced maternal exposures up to approximately 3 times the exposure at the MRHD in rats and 0.2 times the MRHD in rabbits (based on summed AUCs of tezacaftor and M1-TEZ). Oral administration of ivacaftor to pregnant rats and rabbits during organogenesis demonstrated no teratogenicity or adverse developmental effects at doses that produced maternal exposures up to approximately 5 and 14 times the exposure at the MRHD, respectively [based on summed AUCs of ivacaftor and its metabolites (for rat) and AUC of ivacaftor (for rabbit)]. No adverse developmental effects were observed after oral administration of elexacaftor, tezacaftor or ivacaftor to pregnant rats from the period of organogenesis through lactation at doses that produced maternal exposures approximately 1 time, approximately 1 time and 3 times the exposures at the MRHD, respectively [based on summed AUCs of parent and metabolite(s)] (see Data).
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Elexacaftor
In an EFD study in pregnant rats dosed during the period of organogenesis from gestation Days 6-17, elexacaftor was not teratogenic and did not affect fetal survival at exposures up to 9 times the MRHD (based on summed AUC for elexacaftor and its metabolite at maternal doses up to 40 mg/kg/day). Lower mean fetal body weights were observed at doses ≥25 mg/kg/day that produced maternal exposures ≥4 times the MRHD. In an EFD study in pregnant rabbits dosed during the period of organogenesis from gestation Days 7-20, elexacaftor was not teratogenic at exposures up to 4 times the MRHD (based on AUC of elexacaftor at maternal doses up to 125 mg/kg/day). In a pre- and postnatal development (PPND) study in pregnant rats dosed from gestation Day 6 through lactation Day 18, elexacaftor did not cause developmental defects in pups at maternal doses up to 10 mg/kg/day (approximately 1 time the MRHD based on summed AUCs of elexacaftor and its metabolite). Placental transfer of elexacaftor was observed in pregnant rats.
Tezacaftor
In an EFD study in pregnant rats dosed during the period of organogenesis from gestation Days 6-17 and in pregnant rabbits dosed during the period of organogenesis from gestation Days 7-20, tezacaftor was not teratogenic and did not affect fetal development or survival at exposures up to 3 and 0.2 times, respectively the MRHD (based on summed AUCs of tezacaftor and M1-TEZ). Lower fetal body weights were observed in rabbits at a maternally toxic dose that produced exposures approximately 1 time the MRHD (based on summed AUCs of tezacaftor and M1-TEZ at a maternal dose of 50 mg/kg/day). In a PPND study in pregnant rats dosed from gestation Day 6 through lactation Day 18, tezacaftor had no adverse developmental effects on pups at an exposure of approximately 1 time the MRHD (based on summed AUCs for tezacaftor and M1-TEZ at a maternal dose of 25 mg/kg/day). Decreased fetal body weights and early developmental delays in pinna detachment, eye opening, and righting reflex occurred at a maternally toxic dose (based on maternal weight loss) that produced exposures approximately 1 time the exposure at the MRHD (based on summed AUCs for tezacaftor and M1-TEZ at a maternal oral dose of 50 mg/kg/day). Placental transfer of tezacaftor was observed in pregnant rats.
Ivacaftor
In an EFD study in pregnant rats dosed during the period of organogenesis from gestation Days 7-17 and in pregnant rabbits dosed during the period of organogenesis from gestation Days 7-19, ivacaftor was not teratogenic and did not affect fetal survival at exposures up to 5 and 14 times, respectively, the MRHD [based on summed AUCs of ivacaftor and its metabolites (for rat) and AUC of ivacaftor (for rabbit)]. In a PPND study in pregnant rats dosed from gestation Day 7 through lactation Day 20, ivacaftor had no effects on delivery or growth and development of offspring at exposures up to 3 times the MRHD (based on summed AUCs for ivacaftor and its metabolites at maternal oral doses up to 100 mg/kg/day). Decreased fetal body weights were observed at a maternally toxic dose that produced exposures 5 times the MRHD (based on summed AUCs of ivacaftor and its metabolites). Placental transfer of ivacaftor was observed in pregnant rats and rabbits.
8.2 Lactation
Risk Summary
There is no information regarding the presence of elexacaftor, tezacaftor, or ivacaftor in human milk, the effects on the breastfed infant, or the effects on milk production. Elexacaftor, tezacaftor, and ivacaftor are excreted into the milk of lactating rats (see Data). The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for TRIKAFTA and any potential adverse effects on the breastfed child from TRIKAFTA or from the underlying maternal condition.
Data
Elexacaftor
Lacteal excretion of elexacaftor in rats was demonstrated following a single oral dose (10 mg/kg) of 14C-elexacaftor administered 6 to 10 days postpartum to lactating dams. Exposure of 14C-elexacaftor in milk was approximately 0.4 times the value observed in plasma (based on AUC0-72h).
8.4 Pediatric Use
The safety and effectiveness of TRIKAFTA for the treatment of CF in pediatric patients 12 years and older who have at least one F508del mutation in the CFTR gene has been established. Use of TRIKAFTA for this indication was supported by evidence from two adequate and well-controlled studies in CF patients 12 years and older (Trial 1 and Trial 2) [see Clinical Studies (14)]. In these trials, a total of 72 adolescents (aged 12 to 17 years) received TRIKAFTA, including:
- In Trial 1, 56 adolescents who had an F508del mutation on one allele and a mutation on the second allele that results in either no CFTR protein or a CFTR protein that is not responsive to ivacaftor and tezacaftor/ivacaftor [see Adverse Reactions (6) and Clinical Studies (14)].
- In Trial 2, 16 adolescents who were homozygous for the F508del mutation [see Adverse Reactions (6) and Clinical Studies (14)].
The safety and effectiveness of TRIKAFTA in patients with CF younger than 12 years of age have not been established.
Juvenile Animal Toxicity Data
Findings of cataracts were observed in juvenile rats dosed from postnatal Day 7 through 35 with ivacaftor dose levels of 10 mg/kg/day and higher (0.24 times the MRHD based on systemic exposure of ivacaftor and its metabolites). This finding has not been observed in older animals [see Warnings and Precautions (5.4) and Patient Counseling Information (17)].
8.5 Geriatric Use
Clinical studies of TRIKAFTA did not include any patients aged 65 years and older.
8.6 Renal Impairment
TRIKAFTA has not been studied in patients with severe renal impairment or end-stage renal disease. No dosage adjustment is recommended in patients with mild (eGFR 60 to <90 mL/min/1.73 m2) or moderate (eGFR 30 to <60 mL/min/1.73 m2) renal impairment. Use with caution in patients with severe (eGFR <30 mL/min/1.73 m2) renal impairment or end-stage renal disease [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Patients with severe hepatic impairment (Child-Pugh Class C) should not be treated with TRIKAFTA. Use of TRIKAFTA is not recommended in patients with moderate hepatic impairment (Child-Pugh Class B) unless the benefit exceeds the risk. If used in patients with moderate hepatic impairment, TRIKAFTA should be used with caution and at a reduced dose. Liver function tests should be closely monitored. No dose modification is recommended for patients with mild hepatic impairment (Child-Pugh Class A) [see Dosage and Administration (2.2), Warnings and Precautions (5.1), Clinical Pharmacology (12.3), and Patient Counseling Information (17)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
TRIKAFTA is a co-package of elexacaftor, tezacaftor and ivacaftor fixed-dose combination tablets and ivacaftor tablets. Both tablets are for oral administration.
The elexacaftor, tezacaftor and ivacaftor tablets are available as an orange, capsule-shaped, film-coated fixed-dose combination tablet containing 100 mg of elexacaftor, 50 mg of tezacaftor, 75 mg of ivacaftor, and the following inactive ingredients: hypromellose, hypromellose acetate succinate, sodium lauryl sulfate, crosscarmellose sodium, microcrystalline cellulose, and magnesium stearate. The tablet film coat contains hypromellose, hydroxypropyl cellulose, titanium dioxide, talc, iron oxide yellow, and iron oxide red.
The ivacaftor tablet is available as a light blue, capsule-shaped, film-coated tablet containing 150 mg of ivacaftor and the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, hypromellose acetate succinate, lactose monohydrate, magnesium stearate, microcrystalline cellulose, and sodium lauryl sulfate. The tablet film coat contains carnauba wax, FD&C Blue #2, PEG 3350, polyvinyl alcohol, talc, and titanium dioxide. The printing ink contains ammonium hydroxide, iron oxide black, propylene glycol, and shellac.
The active ingredients of TRIKAFTA are described below.
Elexacaftor
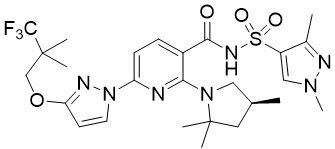
Elexacaftor is a white crystalline solid that is practically insoluble in water (<1 mg/mL). Its chemical name is N-(1,3-dimethyl-1H-pyrazole-4-sulfonyl)-6-[3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide. Its molecular formula is C26H34N7O4SF3 and its molecular weight is 597.66. Elexacaftor has the following structural formula:
Tezacaftor
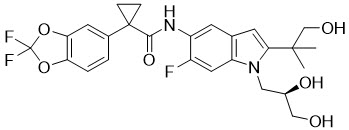
Tezacaftor is a white to off-white powder that is practically insoluble in water (<5 microgram/mL). Its chemical name of tezacaftor is 1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)-N-{1-[(2R)-2,3-dihydroxypropyl]-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl}cyclopropane-1-carboxamide. Its molecular formula is C26H27N2F3O6 and its molecular weight is 520.50. Tezacaftor has the following structural formula:
Ivacaftor
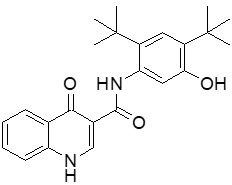
Ivacaftor is a white to off-white powder that is practically insoluble in water (<0.05 microgram/mL). Pharmacologically it is a CFTR potentiator. Its chemical name is N-(2,4-di-tert-butyl-5-hydroxyphenyl)-1,4-dihydro-4-oxoquinoline-3-carboxamide. Its molecular formula is C24H28N2O3 and its molecular weight is 392.49. Ivacaftor has the following structural formula:
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Elexacaftor and tezacaftor bind to different sites on the CFTR protein and have an additive effect in facilitating the cellular processing and trafficking of F508del-CFTR to increase the amount of CFTR protein delivered to the cell surface compared to either molecule alone. Ivacaftor potentiates the channel open probability (or gating) of the CFTR protein at the cell surface.
The combined effect of elexacaftor, tezacaftor and ivacaftor is increased quantity and function of F508del-CFTR at the cell surface, resulting in increased CFTR activity as measured by CFTR mediated chloride transport.
12.2 Pharmacodynamics
Sweat Chloride Evaluation
In Trial 1 (patients with an F508del mutation on one allele and a mutation on the second allele that results in either no CFTR protein or a CFTR protein that is not responsive ivacaftor and tezacaftor/ivacaftor), a reduction in sweat chloride was observed from baseline at Week 4 and sustained through the 24-week treatment period [see Clinical Studies (14.1)]. In Trial 2 (patients homozygous for the F508del mutation), a reduction in sweat chloride was observed from baseline at Week 4 [see Clinical Studies (14.2)].
12.3 Pharmacokinetics
The pharmacokinetics of elexacaftor, tezacaftor and ivacaftor are similar between healthy adult subjects and patients with CF. The pharmacokinetic parameters for elexacaftor, tezacaftor, and ivacaftor in patients with CF aged 12 years and older are shown in Table 4.
Table 4: Pharmacokinetic Parameters of TRIKAFTA Components Elexacaftor Tezacaftor Ivacaftor - * Based on elexacaftor 200 mg and tezacaftor 100 mg once daily/ivacaftor 150 mg every 12 hours at steady state in patients with CF aged 12 year and older.
- † AUC0-24h.
- ‡ AUC0-12h.
- § Elexacaftor, tezacaftor and ivacaftor do not partition preferentially into human red blood cells.
- ¶ Elexacaftor and tezacaftor bind primarily to albumin. Ivacaftor primarily bind to albumin, alpha 1-acid glycoprotein and human gamma-globulin.
- # Mean (SD) terminal half-lives of elexacaftor, tezacaftor and ivacaftor are approximately 24.7 (4.87) hours, 60.3 (15.7) hours and 13.1 (2.98) hours, respectively.
- Þ Following radiolabeled doses.
General Information AUC (SD), mcg∙h/mL* 162 (48.1)† 94.5 (24.0)† 11.7 (4.01)‡ Cmax, (SD), mcg/mL* 8.7 (2.1) 6.8 (1.5) 1.2 (0.3) Time to Steady State, days Within 14 days Within 8 days Within 3-5 days Accumulation Ratio 2.3 1.6 2.4 Absorption Absolute Bioavailability 80% Not determined Not determined Median Tmax (range), hours 6 (4 to 12) 3 (2 to 4) 4 (3 to 6) Effect of Food AUC increases 1.9- to 2.5-fold
(moderate-fat meal)No clinically significant effect Exposure increases 2.5- to 4-fold Distribution Mean (SD) Apparent Volume of Distribution, L§ 53.7 (17.7) 82.0 (22.3) 293 (89.8) Protein Binding¶ > 99% approximately 99% approximately 99% Elimination Mean (SD) Effective Half-Life, hours# 29.8 (10.6) 17.4 (3.66) 15.0 (3.92) Mean (SD) Apparent Clearance, L/hours 1.18 (0.29) 0.79 (0.10) 10.2 (3.13) Metabolism Primary Pathway CYP3A4/5 CYP3A4/5 CYP3A4/5 Active Metabolites M23-ELX M1-TEZ M1-IVA Metabolite Potency Relative to Parent Similar Similar approximately 1/6th of parent ExcretionÞ Primary Pathway - Feces: 87.3% (primarily as metabolites)
- Urine: 0.23%
- Feces: 72% (unchanged or as M2-TEZ)
- Urine: 14% (0.79% unchanged)
- Feces: 87.8%
- Urine: 6.6%
Specific Populations
Pediatric patients 12 to less than 18 years of age
The following conclusions about exposures between adults and the pediatric population are based on population pharmacokinetic (PK) analyses.
Following oral administration of TRIKAFTA to patients 12 to less than 18 years of age (elexacaftor 200 mg QD/tezacaftor 100 mg QD/ivacaftor 150 mg Q12h), the mean (±SD) AUCss was 149 (38.7) mcg∙h/mL, 97.1 (23.7) mcg∙h/mL and 10.6 (3.35) mcg∙h/mL, respectively for elexacaftor, tezacaftor and ivacaftor, similar to the AUCss in adult patients.
Patients with Renal Impairment
Renal excretion of elexacaftor, tezacaftor, and ivacaftor is minimal. Elexacaftor alone or in combination with tezacaftor and ivacaftor has not been studied in patients with severe (eGFR <30 mL/min/1.73 m2) renal impairment or end stage renal disease. Based on population PK analyses, the clearance of elexacaftor and tezacaftor was similar in patients with mild (eGFR 60 to <90 mL/min/1.73 m2) or moderate (eGFR 30 to <60 mL/min/1.73 m2) renal impairment relative to patients with normal renal function [see Use in Specific Populations (8.6)].
Patients with Hepatic Impairment
Elexacaftor alone or in combination with tezacaftor and ivacaftor has not been studied in subjects with hepatic impairment. Similar to tezacaftor and ivacaftor, higher exposure of elexacaftor is expected in patients with moderate (Child-Pugh Class B, score 7 to 9) and severe hepatic impairment (Child-Pugh Class C, score 10-15) [see Dosage and Administration (2.2), Use in Specific Populations (8.7), and Patient Counseling Information (17)].
Tezacaftor and ivacaftor
Following multiple doses of tezacaftor and ivacaftor for 10 days, subjects with moderately impaired hepatic function had an approximately 36% higher AUC and a 10% higher in Cmax for tezacaftor, and a 1.5-fold higher AUC but similar Cmax for ivacaftor compared with healthy subjects matched for demographics.
Drug Interactions Studies
Drug interaction studies were performed with elexacaftor, tezacaftor and/or ivacaftor and other drugs likely to be co-administered or drugs commonly used as probes for pharmacokinetic interaction studies [see Drug Interactions (7)].
Potential for Elexacaftor, Tezacaftor and/or Ivacaftor to Affect Other Drugs
Based on in vitro results, elexacaftor and tezacaftor have a low potential to inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A4, whereas ivacaftor has the potential to inhibit CYP2C8, CYP2C9, and CYP3A. However, clinical studies showed that the combination regimen of tezacaftor/ivacaftor is not an inhibitor of CYP3A and ivacaftor is not an inhibitor of CYP2C8 or CYP2D6.
Based on in vitro results, elexacaftor, tezacaftor and ivacaftor are not likely to induce CYP3A, CYP1A2 and CYP2B6.
Based on in vitro results, elexacaftor and tezacaftor have a low potential to inhibit the transporter P-gp, while ivacaftor has the potential to inhibit P-gp. Co-administration of tezacaftor/ivacaftor with digoxin, a sensitive P-gp substrate, increased digoxin exposure by 1.3-fold in a clinical study. Based on in vitro results, elexacaftor and M23-ELX may inhibit OATP1B1 and OATP1B3 uptake. Tezacaftor has a low potential to inhibit BCRP, OCT2, OAT1, or OAT3. Ivacaftor is not an inhibitor of the transporters OCT1, OCT2, OAT1, or OAT3.
The effects of elexacaftor, tezacaftor and/or ivacaftor on the exposure of co-administered drugs are shown in Table 5 [see Drug Interactions (7)].
Table 5: Impact of Elexacaftor, Tezacaftor and/or Ivacaftor on Other Drugs Dose and Schedule Effect on Other Drug PK Geometric Mean Ratio (90% CI) of Other Drug
No Effect=1.0AUC Cmax ↑ = increase, ↓ = decrease, ↔ = no change. CI = Confidence interval; ELX= elexacaftor; TEZ = tezacaftor; IVA = ivacaftor; PK = Pharmacokinetics - * Effect not clinically significant [see Drug Interactions (7.6)].
Midazolam
2 mg single oral doseTEZ 100 mg qd/IVA 150 mg q12h ↔ Midazolam 1.12
(1.01, 1.25)1.13
(1.01, 1.25)Digoxin
0.5 mg single doseTEZ 100 mg qd/IVA 150 mg q12h ↑ Digoxin 1.30
(1.17, 1.45)1.32
(1.07, 1.64)Oral Contraceptive
Ethinyl estradiol 30 µg/Levonorgestrel 150 µg qdELX 200 mg qd/TEZ 100 mg qd/IVA 150 mg q12h ↑ Ethinyl estradiol* 1.33
(1.20, 1.49)1.26
(1.14, 1.39)↑ Levonorgestrel* 1.23
(1.10, 1.37)1.10
(0.985, 1.23)Rosiglitazone
4 mg single oral doseIVA 150 mg q12h ↔ Rosiglitazone 0.975
(0.897, 1.06)0.928
(0.858, 1.00)Desipramine
50 mg single doseIVA 150 mg q12h ↔ Desipramine 1.04
(0.985, 1.10)1.00
(0.939; 1.07)Potential for Other Drugs to Affect Elexacaftor, Tezacaftor and/or Ivacaftor
In vitro studies showed that elexacaftor, tezacaftor, and ivacaftor are all metabolized by CYP3A. Exposure to elexacaftor, tezacaftor and ivacaftor may be reduced by concomitant CYP3A inducers and increased by concomitant CYP3A inhibitors.
In vitro studies showed that elexacaftor and tezacaftor are substrates for the efflux transporter P-gp, but ivacaftor is not. Elexacaftor and ivacaftor are not substrates for OATP1B1 or OATP1B3; tezacaftor is a substrate for OATP1B1, but not OATP1B3. Tezacaftor is a substrate for BCRP.
The effects of co-administered drugs on the exposure of elexacaftor, tezacaftor and/or ivacaftor are shown in Table 6 [see Dosage and Administration (2.3) and Drug Interactions (7)].
Table 6: Impact of Other Drugs on Elexacaftor, Tezacaftor and/or Ivacaftor Dose and Schedule Effect on ELX, TEZ and/or IVA PK Geometric Mean Ratio (90% CI) of Elexacaftor, Tezacaftor and Ivacaftor
No Effect = 1.0AUC Cmax ↑ = increase, ↓ = decrease, ↔ = no change. CI = Confidence interval; ELX= elexacaftor; TEZ = tezacaftor; IVA = ivacaftor; PK = Pharmacokinetics - * Effect is not clinically significant [see Drug Interactions (7.3)].
Itraconazole
200 mg q12h on Day 1, followed by 200 mg qdTEZ 25 mg qd + IVA 50 mg qd ↑ Tezacaftor 4.02
(3.71, 4.63)2.83
(2.62, 3.07)↑ Ivacaftor 15.6
(13.4, 18.1)8.60
(7.41, 9.98)Itraconazole
200 mg qdELX 20 mg + TEZ 50 mg single dose ↑ Elexacaftor 2.83
(2.59, 3.10)1.05
(0.977, 1.13)↑ Tezacaftor 4.51
(3.85, 5.29)1.48
(1.33, 1.65)Ketoconazole
400 mg qdIVA 150 mg single dose ↑ Ivacaftor 8.45
(7.14, 10.0)2.65
(2.21, 3.18)Ciprofloxacin
750 mg q12hTEZ 50 mg q12h + IVA 150 mg q12h ↔ Tezacaftor 1.08
(1.03, 1.13)1.05
(0.99, 1.11)↑ Ivacaftor* 1.17
(1.06, 1.30)1.18
(1.06, 1.31)Rifampin
600 mg qdIVA 150 mg single dose ↓ Ivacaftor 0.114
(0.097, 0.136)0.200
(0.168, 0.239)Fluconazole
400 mg single dose on Day 1, followed by 200 mg qdIVA 150 mg q12h ↑ Ivacaftor 2.95
(2.27, 3.82)2.47
(1.93, 3.17) -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No studies of carcinogenicity, mutagenicity, or impairment of fertility were conducted with the combination of elexacaftor, tezacaftor and ivacaftor; however, separate studies of elexacaftor, tezacaftor and ivacaftor are described below.
Elexacaftor
A 6-month study in Tg.rasH2 transgenic mice showed no evidence of tumorigenicity at 50 mg/kg/day dose, the highest dose tested.
Elexacaftor was negative for genotoxicity in the following assays: Ames test for bacterial gene mutation, in vitro mammalian cell micronucleus assay in TK6 cells, and in vivo mouse micronucleus test.
Elexacaftor did not cause reproductive system toxicity in male rats at 55 mg/kg/day and female rats at 25 mg/kg/day, equivalent to approximately 6 times and 4 times the MRHD, respectively (based on summed AUCs of elexacaftor and its metabolite). Elexacaftor did not cause embryonic toxicity at 35 mg/kg/day which was the highest dose tested, equivalent to approximately 7 times the MHRD (based on summed AUCs of elexacaftor and its metabolite). Lower male and female fertility, male copulation, and female conception indices were observed in males at 75 mg/kg/day and females at 35 mg/kg/day, equivalent to approximately 6 times and 7 times, respectively, the MRHD (based on summed AUCs of elexacaftor and its metabolite).
Tezacaftor
A 2-year study in Sprague-Dawley rats and a 6-month study in Tg.rasH2 transgenic mice were conducted to assess the carcinogenic potential of tezacaftor. No evidence of tumorigenicity from tezacaftor was observed in male and female rats at oral doses up to 50 and 75 mg/kg/day (approximately 1 and 2 times the MRHD based on summed AUCs of tezacaftor and its metabolites in males and females, respectively). No evidence of tumorigenicity was observed in male and female Tg.rasH2 transgenic mice at tezacaftor doses up to 500 mg/kg/day.
Tezacaftor was negative for genotoxicity in the following assays: Ames test for bacterial gene mutation, in vitro chromosomal aberration assay in Chinese hamster ovary cells, and in vivo mouse micronucleus test.
There were no effects on male or female fertility and early embryonic development in rats at oral tezacaftor doses up to 100 mg/kg/day (approximately 3 times the MRHD based on summed AUC of tezacaftor and M1-TEZ).
Ivacaftor
Two-year studies were conducted in CD-1 mice and Sprague-Dawley rats to assess the carcinogenic potential of ivacaftor. No evidence of tumorigenicity from ivacaftor was observed in mice or rats at oral doses up to 200 mg/kg/day and 50 mg/kg/day, respectively (approximately equivalent to 2 and 7 times the MRHD, respectively, based on summed AUCs of ivacaftor and its metabolites).
Ivacaftor was negative for genotoxicity in the following assays: Ames test for bacterial gene mutation, in vitro chromosomal aberration assay in Chinese hamster ovary cells, and in vivo mouse micronucleus test.
Ivacaftor impaired fertility and reproductive performance indices in male and female rats at 200 mg/kg/day (approximately 7 and 5 times, respectively, the MRHD based on summed AUCs of ivacaftor and its metabolites). Increases in prolonged diestrus were observed in females at 200 mg/kg/day. Ivacaftor also increased the number of females with all nonviable embryos and decreased corpora lutea, implantations, and viable embryos in rats at 200 mg/kg/day (approximately 5 times the MRHD based on summed AUCs of ivacaftor and its metabolites) when dams were dosed prior to and during early pregnancy. These impairments of fertility and reproductive performance in male and female rats at 200 mg/kg/day were attributed to severe toxicity.
-
14 CLINICAL STUDIES
Efficacy:
The efficacy of TRIKAFTA in patients with CF aged 12 years and older was evaluated in two Phase 3, double blind, controlled trials (Trials 1 and 2).
Trial 1 was a 24-week, randomized, double-blind, placebo-controlled study in patients who had an F508del mutation on one allele and a mutation on the second allele that results in either no CFTR protein or a CFTR protein that is not responsive to ivacaftor and tezacaftor/ivacaftor. An interim analysis was planned when at least 140 patients completed Week 4 and at least 100 patients completed Week 12.
Trial 2 was a 4-week, randomized, double-blind, active-controlled study in patients who are homozygous for the F508del mutation. Patients received tezacaftor 100 mg qd/ivacaftor 150 mg q12hr during a 4-week open-label run-in period and were then randomized and dosed to receive TRIKAFTA or tezacaftor 100 mg qd/ivacaftor 150 mg q12hr during a 4-week double-blind treatment period.
Patients in Trials 1 and 2 had a confirmed diagnosis of CF and at least one F508del mutation. Patients discontinued any previous CFTR modulator therapies,
but continued on their other standard-of-care CF therapies (e.g., bronchodilators, inhaled antibiotics, dornase alfa, and hypertonic saline). Patients had a ppFEV1 at screening between 40-90%. Patients with a history of colonization with organisms associated with a more rapid decline in pulmonary status, including but not limited to Burkholderia cenocepacia, Burkholderia dolosa, or Mycobacterium abscessus, or who had an abnormal liver function test at screening (ALT, AST, ALP, or GGT ≥3 × ULN, or total bilirubin ≥2 × ULN), were excluded from the trials. Patients in Trials 1 and 2 were eligible to roll over into a 96-week open-label extension study.
14.1 Trial 1
Trial 1 evaluated 403 patients (200 TRIKAFTA, 203 placebo) with CF aged 12 years and older (mean age 26.2 years). The mean ppFEV1 at baseline was 61.4% (range: 32.3%, 97.1%). The primary endpoint assessed at the time of interim analysis was mean absolute change in ppFEV1 from baseline at Week 4. The final analysis tested all key secondary endpoints in the 403 patients who completed the 24-week study participation, including absolute change in ppFEV1 from baseline through Week 24; absolute change in sweat chloride from baseline at Week 4 and through Week 24; number of pulmonary exacerbations through Week 24; absolute change in BMI from baseline at Week 24, and absolute change in CFQ-R Respiratory Domain Score (a measure of respiratory symptoms relevant to patients with CF, such as cough, sputum production, and difficulty breathing) from baseline at Week 4 and through Week 24.
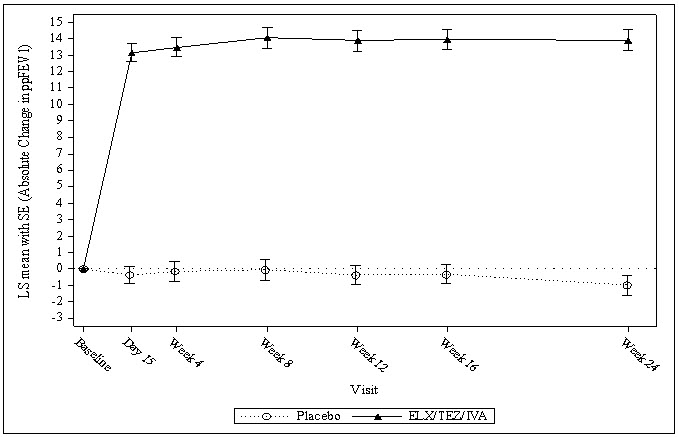
Of the 403 patients included in the interim analysis, the treatment difference between TRIKAFTA and placebo for the mean absolute change from baseline in ppFEV1 at Week 4 was 13.8 percentage points (95% CI: 12.1, 15.4; P<0.0001).
The treatment difference between TRIKAFTA and placebo for mean absolute change in ppFEV1 from baseline through Week 24 was 14.3 percentage points (95% CI: 12.7, 15.8; P<0.0001). Mean improvement in ppFEV1 was observed at the first assessment on Day 15 and sustained through the 24-week treatment period (see Figure 1). Improvements in ppFEV1 were observed regardless of age, baseline ppFEV1, sex, and geographic region.
See Table 7 for a summary of primary and key secondary outcomes in Trial 1.
Table 7: Primary and Key Secondary Efficacy Analyses (Trial 1) Analysis Statistic Treatment Difference* for TRIKAFTA (N=200) vs Placebo (N=203) ppFEV1: percent predicted forced expiratory volume in 1 second; CI: confidence interval; CFQ-R: Cystic Fibrosis Questionnaire-Revised; BMI: body mass index. - * Treatment difference provided as the outcome measure for changes in ppFEV1, sweat chloride, CFQ-R and BMI; Rate ratio provided as the outcome measure for the number of pulmonary exacerbations.
- † Primary endpoint was based on interim analysis in 403 patients.
- ‡ Key secondary endpoints were tested at the final analysis in 403 patients.
- § A pulmonary exacerbation was defined as a change in antibiotic therapy (IV, inhaled, or oral) as a result of 4 or more of 12 pre-specified sino-pulmonary signs/symptoms.
- ¶ Number of pulmonary exacerbation events (event rate per year calculated based on 48 weeks per year) in the TRIKAFTA group were 41 (0.37) and 113 (0.98) in the placebo group.
Primary (Interim Full Analysis Set)† Absolute change in ppFEV1 from baseline at Week 4 (percentage points) Treatment difference (95% CI)
P value13.8 (12.1, 15.4)
P<0.0001Key Secondary (Full Analysis Set)‡ Absolute change in ppFEV1 from baseline through Week 24 (percentage points) Treatment difference (95% CI)
P value14.3 (12.7, 15.8)
P<0.0001Number of pulmonary exacerbations from baseline through Week 24§¶ Rate ratio (95% CI)
P value0.37 (0.25, 0.55)
P<0.0001Absolute change in Sweat Chloride from baseline through Week 24 (mmol/L) Treatment difference (95% CI)
P value-41.8 (-44.4, -39.3)
P<0.0001Absolute change in CFQ-R respiratory domain score from baseline through Week 24 (points) Treatment difference (95% CI)
P value20.2 (17.5, 23.0)
P<0.0001Absolute change in BMI from baseline at Week 24 (kg/m2) Treatment difference (95% CI)
P value1.04 (0.85, 1.23)
P<0.0001Absolute change in Sweat Chloride from baseline at Week 4 (mmol/L) Treatment difference (95% CI)
P value-41.2 (-44.0, -38.5)
P<0.0001Absolute change in CFQ-R respiratory domain score from baseline at Week 4 (points) Treatment difference (95% CI)
P value20.1 (16.9, 23.2)
P<0.0001Figure 1: Absolute Change from Baseline in Percent Predicted FEV1 at Each Visit in Trial 1
14.2 Trial 2
Trial 2 evaluated 107 patients with CF aged 12 years and older (mean age 28.4 years). The mean ppFEV1 at baseline, following the 4-week open-label run-in period with tezacaftor/ivacaftor was 60.9% (range: 35.0%, 89.0%). The primary endpoint was mean absolute change in ppFEV1 from baseline at Week 4 of the double-blind treatment period. The key secondary efficacy endpoints were absolute change in sweat chloride and CFQ-R Respiratory Domain Score from baseline at Week 4. Treatment with TRIKAFTA compared to tezacaftor/ivacaftor resulted in a statistically significant improvement in ppFEV1 of 10.0 percentage points (95% CI: 7.4, 12.6; P<0.0001). Mean improvement in ppFEV1 was observed at the first assessment on Day 15. Improvements in ppFEV1 were observed regardless of age, sex, baseline ppFEV1, and geographic region. See Table 8 for a summary of primary and key secondary outcomes.
Table 8: Primary and Key Secondary Efficacy Analyses, Full Analysis Set (Trial 2) Analysis* Statistic Treatment Difference for TRIKAFTA (N=55) vs Tezacaftor/Ivacaftor† (N=52) ppFEV1: percent predicted forced expiratory volume in 1 second; CI: confidence interval; CFQ-R: Cystic Fibrosis Questionnaire-Revised. - * Baseline for primary and key secondary endpoints is defined as the end of the 4-week tezacaftor/ivacaftor run-in period.
- † Regimen of tezacaftor 100 mg qd/ivacaftor 150 mg q12hr.
Primary Absolute change in ppFEV1 from baseline at Week 4 (percentage points) Treatment difference (95% CI)
P value10.0 (7.4, 12.6)
P<0.0001Key Secondary Absolute change in Sweat Chloride from baseline at Week 4 (mmol/L) Treatment difference (95% CI)
P value-45.1 (-50.1, -40.1)
P<0.0001Absolute change in CFQ-R respiratory domain score from baseline at Week 4 (points) Treatment difference (95% CI)
P value17.4 (11.8, 23.0)
P<0.0001 -
16 HOW SUPPLIED/STORAGE AND HANDLING
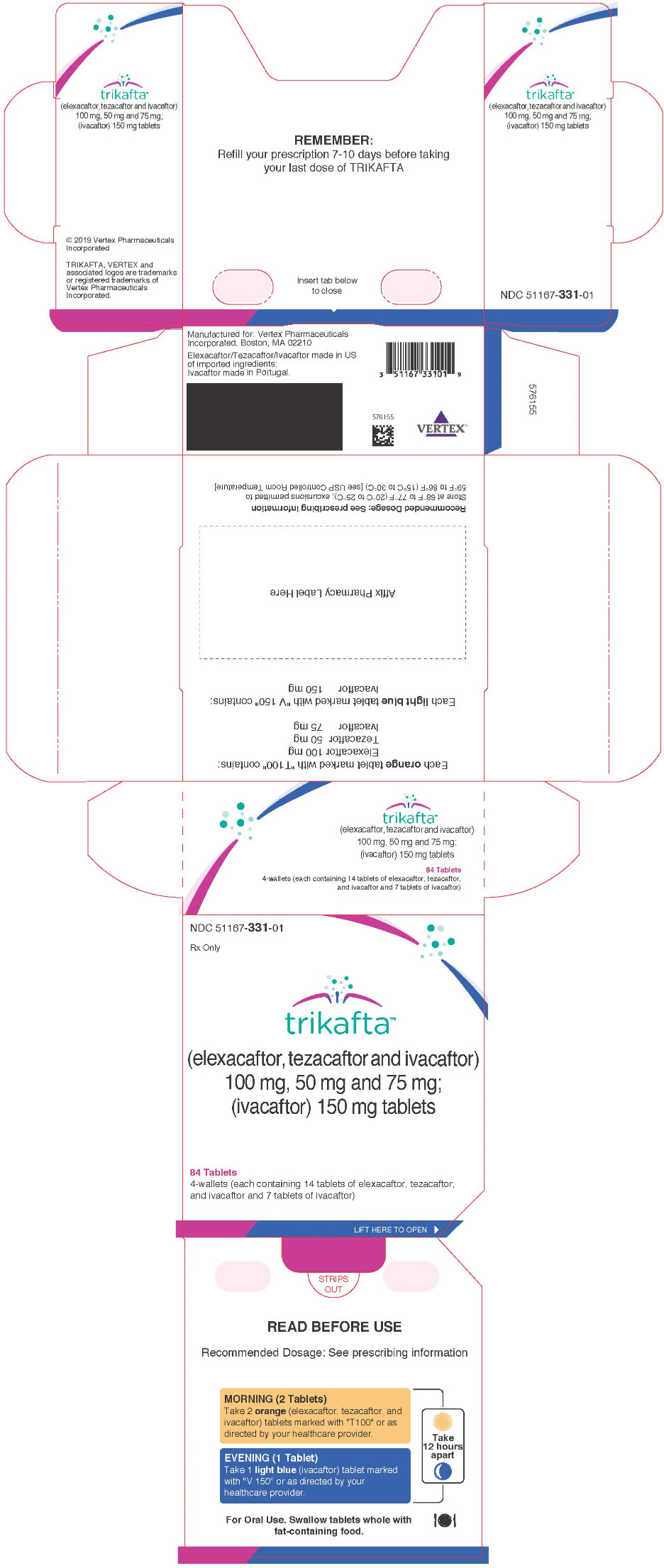
TRIKAFTA is supplied as a co-packaged blister pack sealed in to a printed wallet, containing elexacaftor, tezacaftor and ivacaftor fixed-dose combination tablets and ivacaftor tablets. Four such wallets are placed in a printed outer carton. The elexacaftor, tezacaftor and ivacaftor tablets are supplied as orange, capsule-shaped tablets; each containing 100 mg of elexacaftor, 50 mg of tezacaftor and 75 mg of ivacaftor. Each tablet is debossed with "T100" on one side and plain on the other. Ivacaftor tablets are supplied as light blue, film-coated, capsule-shaped tablets; each containing 150 mg of ivacaftor. Each tablet is printed with the characters "V 150" in black ink on one side and plain on the other. TRIKAFTA is supplied as:
84-count tablet carton NDC 51167-331-01 (4 wallets, each wallet containing 14 tablets of elexacaftor, tezacaftor and ivacaftor and 7 tablets of ivacaftor) -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Liver Function Test Elevations and Monitoring
Inform patients that elevation in transaminases has occurred in patients treated with TRIKAFTA. Elevations in bilirubin have also been observed with TRIKAFTA treatment. Liver function tests (ALT, AST, and bilirubin) should be assessed prior to initiating TRIKAFTA, every 3 months during the first year of treatment, and annually thereafter. More frequent monitoring should be considered in patients with a history of hepatobiliary disease or liver function test elevations [see Warnings and Precautions (5.1)].
Drug Interactions with CYP3A Inducers and Inhibitors
Ask patients to tell you all the medications they are taking including any herbal supplements or vitamins. Co-administration of TRIKAFTA with strong CYP3A inducers (e.g., rifampin, St. John's wort) is not recommended, as they may reduce the efficacy of TRIKAFTA. Dose reduction to two elexacaftor/tezacaftor/ivacaftor tablets twice a week, taken approximately 3 to 4 days apart is recommended when co-administered with strong CYP3A inhibitors, such as ketoconazole. Advise the patient not to take the evening dose of ivacaftor. Dose reduction to two elexacaftor/tezacaftor/ivacaftor tablets and one ivacaftor tablet taken on alternate days is recommended when co-administered with moderate CYP3A inhibitors, such as fluconazole. Advise the patient not to take the evening dose of ivacaftor. Food or drink containing grapefruit should be avoided [see Dosage and Administration (2), Warnings and Precautions (5), Drug Interactions (7), and Clinical Pharmacology (12.3)].
Use in Patients with Hepatic Impairment
TRIKAFTA has not been studied in patients with moderate or severe hepatic impairment. Inquire and/or assess whether patients have liver impairment. Patients with severe hepatic impairment (Child-Pugh Class C, score 10-15) should not be treated with TRIKAFTA. Use of TRIKAFTA is not recommended in patients with moderate hepatic impairment (Child-Pugh Class B, score 7-9) unless the benefit exceeds the risk. If used in patients with moderate hepatic impairment, TRIKAFTA should be used with caution and at a reduced dose. Liver function tests should be closely monitored. No dose adjustment is recommended for patients with mild hepatic impairment (Child-Pugh Class A, score 5-6) [see Dosage and Administration (2.2), Warnings and Precautions (5.1), Use in Specific Populations (8.7), and Clinical Pharmacology (12.3)].
Cataracts
Inform patients that abnormality of the eye lens (cataract) has been noted in some children and adolescents receiving ivacaftor-containing regimens. Baseline and follow-up ophthalmological examinations should be performed in pediatric patients initiating treatment with TRIKAFTA [see Warnings and Precautions (5.4) and Use in Specific Populations (8.4)].
Administration
Inform patients that TRIKAFTA is best absorbed by the body when taken with food that contains fat. A typical CF diet will satisfy this requirement. Examples include eggs, butter, peanut butter, whole-milk dairy products (such as whole milk, cheese, and yogurt), etc. [see Dosage and Administration (2.1) and Clinical Pharmacology (12.3)].
Patients should be informed about what to do in the event they miss a dose of elexacaftor/tezacaftor/ivacaftor tablets or ivacaftor tablet:
- If 6 hours or less have passed since the missed morning or evening dose is usually taken, patients should be instructed to take the prescribed dose with fat-containing food as soon as possible.
- If more than 6 hours have passed since:
- the time the morning dose is usually taken, patients should be instructed to take the morning dose as soon as possible, and not take the evening dose. Patients should take the next scheduled morning dose at the usual time.
- the time the evening dose is usually taken, patients should be instructed to not take the missed evening dose. Patients should take the next scheduled morning dose at the usual time.
- Patients should be instructed to not take the morning and evening doses at the same time.
- Patients should be advised to contact their health care provider if they have questions.
-
SPL UNCLASSIFIED SECTION
Manufactured for
Vertex Pharmaceuticals Incorporated
50 Northern Avenue
Boston, MA 02210Approved January 2020
TRIKAFTA, VERTEX and associated logos are registered trademarks of Vertex Pharmaceuticals Incorporated.
All other trademarks referenced herein are the property of their respective owners.
©2019 Vertex Pharmaceuticals Incorporated
ALL RIGHTS RESERVED -
PATIENT PACKAGE INSERT
Patient Information is perforated for dispensing to the patient.
This Patient Information has been approved by the U.S. Food and Drug Administration. Approved January 2020 Patient Information
TRIKAFTA (tri-KAF-tuh)
(elexacaftor/tezacaftor/ivacaftor tablets; ivacaftor tablets)
for oral useWhat is TRIKAFTA? - TRIKAFTA is a prescription medicine used for the treatment of cystic fibrosis (CF) in people aged 12 years and older who have at least one copy of the F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene.
- Talk to your doctor to learn if you have an indicated CF gene mutation.
Who should not take TRIKAFTA?
Do not take TRIKAFTA if you take certain medicines or herbal supplements such as:- antibiotics such as rifampin (RIFAMATE®, RIFATER®) or rifabutin (MYCOBUTIN®)
- seizure medicines such as phenobarbital, carbamazepine (TEGRETOL®, CARBATROL®, EQUETRO®), or phenytoin (DILANTIN®, PHENYTEK®)
- St. John's wort
Before taking TRIKAFTA, tell your doctor about all of your medical conditions, including if you: - have kidney problems.
- have or have had liver problems.
- are pregnant or plan to become pregnant. It is not known if TRIKAFTA will harm your unborn baby. You and your doctor should decide if you will take TRIKAFTA while you are pregnant.
- are breastfeeding or planning to breastfeed. It is not known if TRIKAFTA passes into your breast milk. You and your doctor should decide if you will take TRIKAFTA while you are breastfeeding.
Especially tell your doctor if you take:- antifungal medicines including ketoconazole (such as NIZORAL®), itraconazole (such as SPORANOX®), posaconazole (such as NOXAFIL®), voriconazole (such as VFEND®), or fluconazole (such as DIFLUCAN®)
- antibiotics including telithromycin (such as KETEK®), clarithromycin (such as BIAXIN®), or erythromycin (such as ERY-TAB®)
- other medicines including rifampin, rifabutin, phenobarbital, carbamazepine, phenytoin, and St. John's wort (see 'Who should not take TRIKAFTA' section).
How should I take TRIKAFTA? - Take TRIKAFTA exactly as your doctor tells you to take it.
- Take TRIKAFTA by mouth only.
- TRIKAFTA consists of 2 different tablets.
- The orange tablet is marked with 'T100' and each tablet contains the medicines elexacaftor, tezacaftor and ivacaftor. Take 2 orange tablets in the morning.
- The light blue tablet is marked with 'V 150' and contains the medicine ivacaftor. Take 1 light blue tablet in the evening.
- Take the orange tablets and the light blue tablet about 12 hours apart.
- Always take TRIKAFTA with food that contains fat. Examples of fat-containing foods include butter, peanut butter, eggs, nuts, meat, and whole-milk dairy products such as whole milk, cheese, and yogurt.
- If you miss a dose of TRIKAFTA and:
- it is 6 hours or less from the time you usually take the orange tablets in the morning or the light blue tablet in the evening, take the missed dose with food that contains fat as soon as you can. Then take your next dose at your usual time.
- it is more than 6 hours from the time you usually take the orange tablets in the morning, take the missed dose with food that contains fat as soon as you can. Do not take the light blue tablet in the evening.
- it is more than 6 hours from the time you usually take the light blue tablet in the evening, do not take the missed dose. Take your next dose of orange tablets at the usual time with food that contains fat.
- Do not take more than your usual dose of TRIKAFTA to make up for a missed dose.
What should I avoid while taking TRIKAFTA? - TRIKAFTA can cause dizziness in some people who take it. Do not drive a car, use machinery, or do anything that needs you to be alert until you know how TRIKAFTA affects you.
- Avoid food or drink that contains grapefruit while you are taking TRIKAFTA.
What are the possible side effects of TRIKAFTA?
TRIKAFTA can cause serious side effects, including:-
High liver enzymes in the blood is a common side effect in people treated with TRIKAFTA. These can be serious and may be a sign of liver injury. Your doctor will do blood tests to check your liver:
- before you start TRIKAFTA
- every 3 months during your first year of taking TRIKAFTA
- then every year while you are taking TRIKAFTA
Call your doctor right away if you have any of the following symptoms of liver problems:- pain or discomfort in the upper right stomach (abdominal) area
- yellowing of your skin or the white part of your eyes
- loss of appetite
- nausea or vomiting
- dark, amber-colored urine
- Abnormality of the eye lens (cataract) in some children and adolescents treated with TRIKAFTA. If you are a child or adolescent, your doctor should perform eye examinations before and during treatment with TRIKAFTA to look for cataracts.
- headache
- diarrhea
- upper respiratory tract infection (common cold) including stuffy and runny nose
- stomach (abdominal) pain
- inflamed sinuses
- increase in liver enzymes
- increase in a certain blood enzyme called creatine phosphokinase
- rash
- flu (influenza)
- increase in blood bilirubin
These are not all the possible side effects of TRIKAFTA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store TRIKAFTA? - Store TRIKAFTA at room temperature between 68ºF to 77ºF (20ºC to 25ºC).
- Do not use TRIKAFTA after the expiration date on the package.
General information about the safe and effective use of TRIKAFTA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use TRIKAFTA for a condition for which it was not prescribed. Do not give TRIKAFTA to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or doctor for information about TRIKAFTA that is written for health professionals.What are the ingredients in TRIKAFTA?
Elexacaftor/tezacaftor/ivacaftor tablets:
Active ingredients: elexacaftor, tezacaftor and ivacaftor
Inactive ingredients: hypromellose, hypromellose acetate succinate, sodium lauryl sulfate, croscarmellose sodium, microcrystalline cellulose, magnesium stearate, hydroxypropyl cellulose, titanium dioxide, talc, iron oxide yellow and iron oxide red.
ivacaftor tablets:
Active ingredients: ivacaftor
Inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, hypromellose acetate succinate, lactose monohydrate, magnesium stearate, microcrystalline cellulose, sodium lauryl sulfate, carnauba wax, FD&C Blue #2, PEG 3350, polyvinyl alcohol, talc, titanium dioxide, ammonium hydroxide, iron oxide black, propylene glycol, and shellac.Manufactured for: Vertex Pharmaceuticals Incorporated; 50 Northern Avenue, Boston, MA 02210
TRIKAFTA, VERTEX and associated logos are registered trademarks of Vertex Pharmaceuticals Incorporated.
All other trademarks referenced herein are the property of their respective owners.
©2019 Vertex Pharmaceuticals Incorporated
For more information, go to www.trikafta.com or call 1-877-752-5933. -
PRINCIPAL DISPLAY PANEL - Kit Carton
NDC: 51167-331-01
Rx Onlytrikafta™
(elexacaftor, tezacaftor and ivacaftor)
100 mg, 50 mg and 75 mg;
(ivacaftor) 150 mg tablets84 Tablets
4-wallets (each containing 14 tablets of elexacaftor, tezacaftor,
and ivacaftor and 7 tablets of ivacaftor)LIFT HERE TO OPEN ►
-
INGREDIENTS AND APPEARANCE
TRIKAFTA
elexacaftor, tezacaftor, and ivacaftor kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 51167-331 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 51167-331-01 4 in 1 CARTON 10/21/2019 1 1 in 1 BLISTER PACK Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 BLISTER PACK 14 Part 2 1 BLISTER PACK 7 Part 1 of 2 ELEXACAFTOR, TEZACAFTOR, AND IVACAFTOR
elexacaftor, tezacaftor, and ivacaftor tablet, film coatedProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Elexacaftor (UNII: RRN67GMB0V) (Elexacaftor - UNII:RRN67GMB0V) Elexacaftor 100 mg Tezacaftor (UNII: 8RW88Y506K) (Tezacaftor - UNII:8RW88Y506K) Tezacaftor 50 mg Ivacaftor (UNII: 1Y740ILL1Z) (Ivacaftor - UNII:1Y740ILL1Z) Ivacaftor 75 mg Inactive Ingredients Ingredient Name Strength hypromellose acetate succinate 06081224 (3 mm2/s) (UNII: 6N003M473W) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) sodium lauryl sulfate (UNII: 368GB5141J) croscarmellose sodium (UNII: M28OL1HH48) microcrystalline cellulose (UNII: OP1R32D61U) magnesium stearate (UNII: 70097M6I30) hypromellose 2910 (6 mpa.s) (UNII: 0WZ8WG20P6) hydroxypropyl cellulose, unspecified (UNII: 9XZ8H6N6OH) titanium dioxide (UNII: 15FIX9V2JP) talc (UNII: 7SEV7J4R1U) ferric oxide yellow (UNII: EX438O2MRT) ferric oxide red (UNII: 1K09F3G675) Product Characteristics Color ORANGE Score no score Shape CAPSULE Size 15mm Flavor Imprint Code T100 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 14 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212273 10/21/2019 Part 2 of 2 IVACAFTOR
ivacaftor tablet, film coatedProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Ivacaftor (UNII: 1Y740ILL1Z) (Ivacaftor - UNII:1Y740ILL1Z) Ivacaftor 150 mg Inactive Ingredients Ingredient Name Strength silicon dioxide (UNII: ETJ7Z6XBU4) hypromellose acetate succinate 06081224 (3 mm2/s) (UNII: 6N003M473W) lactose monohydrate (UNII: EWQ57Q8I5X) Magnesium stearate (UNII: 70097M6I30) microcrystalline cellulose (UNII: OP1R32D61U) sodium lauryl sulfate (UNII: 368GB5141J) carnauba wax (UNII: R12CBM0EIZ) FD&C Blue No. 2 (UNII: L06K8R7DQK) polyethylene glycol 3350 (UNII: G2M7P15E5P) polyvinyl alcohol, unspecified (UNII: 532B59J990) talc (UNII: 7SEV7J4R1U) titanium dioxide (UNII: 15FIX9V2JP) Ammonia (UNII: 5138Q19F1X) ferrosoferric oxide (UNII: XM0M87F357) propylene glycol (UNII: 6DC9Q167V3) shellac (UNII: 46N107B71O) Product Characteristics Color BLUE (light blue) Score no score Shape CAPSULE Size 17mm Flavor Imprint Code V;150 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 7 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212273 10/21/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212273 10/21/2019 Labeler - Vertex Pharmaceuticals Incorporated (602478257) Establishment Name Address ID/FEI Business Operations Shanghai SynTheAll Pharmaceutical Co., Ltd. 545342792 API MANUFACTURE(51167-331) , ANALYSIS(51167-331) Establishment Name Address ID/FEI Business Operations Hovione Limited 985003840 MANUFACTURE(51167-331) , ANALYSIS(51167-331) Establishment Name Address ID/FEI Business Operations F.I.S. Fabbrica Italiana Sintetici S.P.A. 431189117 API MANUFACTURE(51167-331) , ANALYSIS(51167-331) Establishment Name Address ID/FEI Business Operations Almac Sciences Limited 232665666 ANALYSIS(51167-331)
Trademark Results [Trikafta]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 TRIKAFTA 88031776 not registered Live/Pending |
Vertex Pharmaceuticals Incorporated 2018-07-10 |
 TRIKAFTA 87924202 5938974 Live/Registered |
Vertex Pharmaceuticals Incorporated 2018-05-16 |
 TRIKAFTA 87893193 5938914 Live/Registered |
Vertex Pharmaceuticals Incorporated 2018-04-25 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.