LYNKUET- elinzanetant capsule
LYNKUET by
Drug Labeling and Warnings
LYNKUET by is a Prescription medication manufactured, distributed, or labeled by Bayer HealthCare Pharmaceuticals Inc., Bayer AG, Esteve Quimica, S.A., Lianhe Aigen Pharma Co., Ltd., Catalent Germany Eberbach GmbH, Catalent Germany Schorndorf GmbH, Sharp Corporation. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use LYNKUET safely and effectively. See full prescribing information for LYNKUET.
LYNKUET® (elinzanetant) capsules, for oral use
Initial U.S. Approval: 2025INDICATIONS AND USAGE
LYNKUET is a neurokinin 1 (NK1) and neurokinin 3 (NK3) receptor antagonist indicated for the treatment of moderate to severe vasomotor symptoms due to menopause. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Capsules: 60 mg (3)
CONTRAINDICATIONS
- Pregnancy. (4)
WARNINGS AND PRECAUTIONS
- CNS Depressant Effect and Daytime Impairment: Advise patients about the potential for somnolence and other nervous system effects. Advise patients who experience these effects to refrain from driving or engaging in hazardous occupations or activities until the effects have resolved (5.1)
- Hepatic Transaminase Elevations: Perform bloodwork prior to initiation of LYNKUET to evaluate for hepatic function and injury. Do not start therapy if serum transaminase concentration is equal to or exceeds two times the upper limit of normal (ULN). Perform follow-up evaluations of hepatic transaminase concentration 3 months after initiation. Do not start therapy if serum transaminase concentration is equal to or exceeds two times the ULN or if the total bilirubin is equal to or exceeds two times the ULN. Advise patients to discontinue LYNKUET immediately in case of signs or symptoms suggesting liver injury. (5.2)
- Risk of pregnancy loss: May cause pregnancy loss or stillbirth when administered during pregnancy. Exclude pregnancy in females of reproductive potential prior to initiating LYNKUET. Discontinue if pregnancy is confirmed (5.3)
- Risk of seizures in patients with a history of seizures (5.4)
ADVERSE REACTIONS
The most frequently reported (≥5%) adverse reactions were headache, fatigue, dizziness and somnolence. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Bayer HealthCare Pharmaceuticals Inc. at 1-888-842-2937 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 10/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Evaluation and Testing Before Initiation of LYNKUET
2.2 Recommended Dosage
2.3 Dosage Modifications for Drug Interactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Central Nervous System (CNS) Depressant Effect and Daytime Impairment
5.2 Hepatic Transaminase Elevations
5.3 Risk of Pregnancy Loss
5.4 Risk of Seizures in Patients with History of Seizures
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on LYNKUET
7.2 Effects of LYNKUET on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Effects on Vasomotor Symptoms (VMS) in Postmenopausal Women
14.2 Effects on Endometrium in Postmenopausal Women
14.3 Special Safety Study
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Evaluation and Testing Before Initiation of LYNKUET
- Exclude pregnancy in females of reproductive potential [see Contraindications (4)], Warnings and Precautions (5.3), and Use in Specific Populations (8.3)].
- Perform baseline hepatic laboratory tests to evaluate for hepatic function and injury [including serum alanine aminotransferase (ALT), serum aspartate aminotransferase (AST), serum alkaline phosphatase (ALP), and serum bilirubin (total and direct)] before initiating treatment with LYNKUET. Do not start LYNKUET if ALT or AST is ≥ 2 times upper limit of normal (ULN) or if the total bilirubin is ≥ 2 times ULN [see Warnings and Precautions (5.2) and Use in Specific Populations (8.7)].
2.2 Recommended Dosage
The recommended dosage of LYNKUET is 120 mg (two 60 mg capsules) orally once daily at bedtime at about the same time each day. If a dose is missed at bedtime, take the next dose as scheduled on the following day. Do not take two doses on the same day to make up for a missed dose.
Take LYNKUET with or without food. Take LYNKUET with water and swallow capsules whole. Do not cut, crush or chew capsules.
2.3 Dosage Modifications for Drug Interactions
Dosage modifications for concomitant use with specific drugs are provided in Table 1 [see Drug Interactions (7.1)].
Table 1. Dosage Modifications for Drug Interactions Concomitant Drug LYNKUET Dosage Strong CYP3A4 inhibitors and grapefruit (juice) Avoid concomitant use Moderate CYP3A4 inhibitors 60 mg (one capsule) orally once daily at bedtime [see Storage and Handling (16.2)]. After discontinuation of the moderate CYP3A4 inhibitor (after 3 to 5 half-lives of the inhibitor), LYNKUET should be used at the usual dosage of 120 mg once daily. Strong and Moderate CYP3A4 inducers Avoid concomitant use - 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
LYNKUET is contraindicated in pregnancy. Exposure to LYNKUET may cause pregnancy loss or stillbirth when administered during pregnancy [see Use in Specific Populations (8.1, 8.3)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Central Nervous System (CNS) Depressant Effect and Daytime Impairment
In the three OASIS trials, nervous system effects (including somnolence, fatigue, vertigo, dizziness and presyncope) occurred in 11.9% of patients on LYNKUET compared to 3.5% on placebo. [see also Adverse Reactions (6.1)].
Advise patients about the potential for somnolence and other nervous system effects. Advise patients who experience these effects to refrain from driving or engaging in hazardous occupations or activities until the effects have resolved [see Clinical Studies (14.3)].
5.2 Hepatic Transaminase Elevations
Elevations in serum transaminase (ALT and/or AST) concentrations equal to or greater than three times the ULN occurred in 0.6% of patients receiving LYNKUET and 0.4% of patients receiving placebo up to 12 weeks in three clinical trials. Perform baseline bloodwork (including ALT, AST, alkaline phosphatase, and total and direct bilirubin) prior to initiation of LYNKUET to evaluate for hepatic function and injury. Do not start therapy if serum transaminase concentration is equal to or exceeds two times the ULN or if the total bilirubin is equal to or exceeds two times the ULN. Perform follow-up evaluations of hepatic transaminase concentration 3 months after initiation of therapy.
Advise patients to discontinue LYNKUET immediately and seek medical attention including hepatic laboratory tests if they experience signs or symptoms that may suggest liver injury (new onset fatigue, decreased appetite, nausea, vomiting, pruritus, jaundice, pale feces, dark urine, or abdominal pain).
Discontinue LYNKUET if transaminase elevations exceed five times the ULN or if transaminase elevations exceed three times the ULN and total bilirubin exceeds two times the ULN.
Exclude alternative causes of hepatic laboratory test elevations.
5.3 Risk of Pregnancy Loss
LYNKUET is contraindicated for use in pregnancy [see Contraindications (4)]. Findings from animal studies suggest that LYNKUET can cause pregnancy loss or stillbirth. Exclude pregnancy in females of reproductive potential prior to initiating LYNKUET. Advise females of reproductive potential to use effective contraception during treatment with LYNKUET and for 2 weeks after stopping LYNKUET [see Use in Specific Populations (8.1, 8.3)].
5.4 Risk of Seizures in Patients with History of Seizures
Seizure was reported in one patient with a history of seizures in the clinical trials of LYNKUET. In addition, convulsions were observed in studies conducted in male and female rats [see Nonclinical Toxicology (13.2)]. Use LYNKUET with caution in patients with a history of seizures or with conditions that potentially lower the seizure threshold.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Central Nervous System (CNS) Depressant Effect and Daytime Impairment [see Warnings and Precautions (5.1)]
- Hepatic Transaminase Elevations [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety of LYNKUET was evaluated in three randomized, double-blind, placebo-controlled, multicenter clinical trials (OASIS 1, OASIS 2, OASIS 3) in 1420 women. In OASIS 1 and OASIS 2 combined, 793 women received LYNKUET or placebo for 12 weeks. After the first 12 weeks, 341 women randomized to LYNKUET continued to receive LYNKUET for another 14 weeks, with a total treatment duration of up to 26 weeks. In OASIS 1 and OASIS 2 combined, 349 women received placebo for the first 12 weeks and 348 women switched to LYNKUET for the next 14 weeks. In OASIS 3, 627 women received LYNKUET or placebo for up to 52 weeks to evaluate long-term safety [see Clinical Studies (14)].
Common Adverse Reactions
In OASIS 1 and 2 combined, through the first 12 weeks, commonly reported adverse reactions in the LYNKUET group (≥2% and greater than in placebo) were headache, fatigue, gastroesophageal reflux disease, dizziness, nausea, and somnolence.
Similar adverse reactions were seen in OASIS 3. Table 2 shows adverse reactions reported in at least 2% of women and more commonly in women taking LYNKUET than placebo in OASIS 3.
Table 2. Common Adverse Reactions Reported in ≥ 2% in LYNKUET and Greater than Placebo, Weeks 1-52 (OASIS 3) Adverse Reaction LYNKUET
N=313
n (%)Placebo
N=314
n (%)- * Includes asthenia.
- † Includes balance disorder, presyncope, vertigo, vertigo CNS origin, vertigo positional, and vestibular neuronitis.
- ‡ Includes lethargy.
- § Includes abdominal discomfort, abdominal pain lower/upper.
- ¶ Includes dermatitis, urticaria.
- # Includes muscle tightness.
Headache 30 (9.6) 22 (7.0) Fatigue* 23 (7.3) 9 (2.9) Dizziness† 19 (6.1) 6 (1.9) Somnolence‡ 16 (5.1) 4 (1.3) Abdominal pain§ 14 (4.5) 8 (2.5) Rash¶ 13 (4.2) 5 (1.6) Diarrhea 12 (3.8) 3 (1.0) Muscle spasms# 10 (3.2) 2 (0.6) Adverse Reactions Leading to Discontinuation
In OASIS 3, adverse reactions leading to treatment discontinuation (≥1% in LYNKUET and greater than placebo) were abdominal pain (1.6%), fatigue (1.6%), depression (1.6%) and headache (1.3%).
Photosensitivity
In the OASIS trials, mild to moderate events of photosensitivity occurred in 0.5% of patients receiving LYNKUET and 0.1% of patients receiving placebo. Onset of photosensitivity reactions ranged from day 1 to day 290. While discontinuation occurred in one patient, photosensitivity events in other patients resolved under continued treatment with LYNKUET [see Nonclinical Toxicology (13.2)].
-
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on LYNKUET
Elinzanetant is primarily metabolized via CYP3A4 enzyme. Table 3 describes drug interactions where concomitant use of another drug affects LYNKUET.
Table 3: Drug Interactions: Concomitant Use of Other Drugs Affect the Use of LYNKUET - * See www.fda.gov/CYPandTransporterInteractingDrugs for examples of strong and moderate CYP3A4 inhibitors, and CYP3A4 inducers.
Strong and Moderate CYP3A4 Inhibitors* Prevention or Management Strong CYP3A4 Inhibitors and grapefruit (juice): Avoid concomitant use. Moderate CYP3A4 Inhibitors: Reduce the LYNKUET dosage [see Dosage and Administration (2.2)]. Clinical Effect(s) Strong and moderate CYP3A4 inhibitors increase elinzanetant exposure, which may increase the risk of LYNKUET-associated adverse reactions [see Clinical Pharmacology (12.3)]. Strong and Moderate CYP3A4 Inducers* Prevention or Management Strong and moderate CYP3A4 inducers: Avoid concomitant use. Clinical Effect(s) Strong and moderate CYP3A4 inducers decrease elinzanetant exposure, which may reduce the effectiveness of LYNKUET. 7.2 Effects of LYNKUET on Other Drugs
CYP3A4 Substrates
Avoid concomitant use unless otherwise recommended in the Prescribing Information for CYP3A4 substrates where minimal concentration changes may lead to serious adverse reactions. See www.fda.gov/CYPandTransporterInteractingDrugs for examples of CYP3A4 substrates.
Elinzanetant is a weak inhibitor of CYP3A4. Concomitant use of LYNKUET increases exposure of CYP3A4 substrates [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions related to these substrates.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
LYNKUET is contraindicated in pregnancy [see Contraindications (4)]. If pregnancy occurs during the use of LYNKUET, discontinue treatment. Based on findings from animal reproduction studies, LYNKUET may cause pregnancy loss or stillbirth but not fetal malformations when administered during pregnancy. There are no data on the use of LYNKUET in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes.
In animal reproduction studies in rats, an increase in total litter loss or stillbirth and a decrease of neonatal pup viability was observed within the range of human therapeutic exposure when dams were treated orally throughout gestation and lactation [i.e., gestation day 6 to lactation day 21]. There was an increase in percentage of pre- and post-implantation embryo loss and decrease in fetal body weights at 16-fold the human therapeutic exposure when rat dams were treated orally prior to mating and through the early embryonic period [i.e., 22 days before mating to post-coitum day 6]. In rabbits, there was marked body weight loss and decreased food consumption in dams treated orally during gestation day 7 to 19 at doses equivalent to human therapeutic exposure (see Data).
Animal Data
In a fertility and early embryonic development study in female rats, once daily oral doses of elinzanetant were administered 22 days before mating through post-coitum day 6. Increased percentage of pre-implantation and post-implantation embryo loss, resulting in reduced litter size, and lower fetal body weights were seen at the dose of 100 mg/kg/day (14-fold the AUC(0-24) at the human therapeutic dose). These effects were not observed following dosing at 25 mg/kg/day (3-fold the AUC(0-24) at the human therapeutic dose).
In an embryo-fetal development study in pregnant rats, once daily oral doses of elinzanetant were administered throughout organogenesis from gestation day 6 to 17. No evidence of embryo-fetal lethality or teratogenicity occurred at doses ≤100 mg/kg/day (23-fold the AUC(0-24) at the human therapeutic dose) and no maternal toxicity occurred at doses ≤25 mg/kg/day (7-fold the AUC(0-24) at the human therapeutic dose).
In an embryo-fetal development study in pregnant rabbits, twice daily oral doses of elinzanetant were administered throughout organogenesis from gestation day 7 to 19. Maternal toxicity in the form of marked body weight loss and decreased food consumption leading to early sacrifice in 4 out of 27 females was observed at the highest tested dose of 140 mg/kg/day (equivalent to the AUC(0-24) at the human therapeutic dose). No evidence of embryo-fetal lethality or teratogenicity occurred in fetuses of surviving dams at doses ≤140 mg/kg/day (equivalent to the AUC(0-24) at the human therapeutic dose).
In a pre- and post-natal development study in rats, once daily oral doses of elinzanetant were administered from gestation day 6 to lactation day 21. A higher incidence of still born pups, an increase in total litter loss, a decrease in pup viability between postnatal days 0 and 4, and an increase in the number of neonatal pups without milk in the stomach were observed at a dose of ≥5 mg/kg/day (equivalent to the AUC(0-24) at the human therapeutic dose).
8.2 Lactation
Risk Summary
There are no data on the presence of elinzanetant or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. Elinzanetant is present in animal milk. When a drug is present in animal milk, it is likely that the drug will be present in human milk (see Data).
Animal Data
Following administration of radiolabeled elinzanetant to lactating rats, approximately 6% of the elinzanetant dose was excreted in the milk.
Principle human metabolites of elinzanetant were also detected in the plasma of pups from rat dams that were dosed once daily orally during pregnancy and lactation at ≥1.5 mg/kg/day (elinzanetant AUC(0-24) below the range of human therapeutic exposure).
8.3 Females and Males of Reproductive Potential
Based on findings from animal reproduction studies, LYNKUET may cause pregnancy loss or stillbirth when administered during pregnancy. Fetal malformations were not observed in those studies [see Use in Specific Populations (8.1)].
Pregnancy Testing
Exclude pregnancy before initiating treatment with LYNKUET. Perform pregnancy testing if pregnancy is suspected during treatment with LYNKUET, and discontinue treatment if pregnancy is confirmed [see Contraindications (4) and Warnings and Precautions (5.3)].
Contraception
Females
Pregnancy should be prevented in women of reproductive potential by using effective contraception during and for 2 weeks after stopping treatment because LYNKUET can cause pregnancy loss or stillbirth [see Use in Specific Populations (8.1)].
8.4 Pediatric Use
The efficacy and safety of LYNKUET in pediatric patients have not been established, and LYNKUET is not indicated in this population.
8.5 Geriatric Use
There have not been sufficient numbers of geriatric women involved in clinical trials utilizing LYNKUET to determine whether those over 65 years of age differ from younger women in their response to LYNKUET.
8.6 Renal Impairment
No dose adjustment is required for patients with mild to severe [estimated glomerular filtration rate (eGFR) 15 to < 90 mL/min] renal impairment. The effect of end-stage renal disease (eGFR <15 mL/min) (with or without hemodialysis) on the pharmacokinetics of LYNKUET has not been studied [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dose adjustment is required in patients with mild (Child-Pugh A) hepatic impairment. Moderate (Child-Pugh B) hepatic impairment increased the exposure of LYNKUET. LYNKUET has not been studied in individuals with severe (Child-Pugh C) hepatic impairment. LYNKUET is not recommended in patients with moderate or severe hepatic impairment [see Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
LYNKUET is an NK1 and NK3 receptor antagonist.
The chemical name of elinzanetant is 2-[3,5-bis(trifluoromethyl)phenyl]-N-{4-(4-fluoro-2-methylphenyl)-6-[(7S,9aS)-7-(hydroxymethyl)hexahydropyrazino[2,1-c][1,4]oxazin-8(1H)-yl]pyridin-3-yl}-N,2-dimethylpropanamide having a molecular formula of C33H35F7N4O3 and a molecular weight of 668.7.
The structural formula of elinzanetant is:
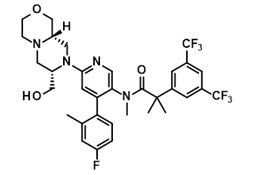
Elinzanetant is a white to yellowish powder and is practically insoluble in water and slightly soluble under acidic conditions.
Each LYNKUET (elinzanetant) capsule for oral use contains 60 mg of elinzanetant and the following inactive ingredients: all-rac-α-Tocopherol, caprylocaproyl macrogolglycerides, glycerol monocaprylocaprate, glycerol mono-oleate, and polysorbate 80. The capsule is composed of edible ink, ferric oxide red, ferric oxide yellow, gelatin, sorbitol special-glycerin, and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
LYNKUET is a neurokinin 1 (NK1) and neurokinin 3 (NK3) receptor antagonist. Inhibition of Substance P and Neurokinin B through antagonism of NK1 and NK3 receptor signaling on kisspeptin/neurokinin B/dynorphin (KNDy) neurons can modulate neuronal activity in thermoregulation associated with hot flashes.
Elinzanetant has higher affinity for human NK1 receptors (pKi values of 8.7 to 10.2) and NK3 receptors (pKi values of 8.0 to 8.8) than for human NK2 receptors (pKi values of approximately 6.0).
12.2 Pharmacodynamics
Cardiac Electrophysiology
No clinically relevant prolongation of the QTc interval was observed after single oral administration of elinzanetant at doses up to 5 times the maximum recommended dose. However, QTc prolongation effect of elinzanetant when co-administered with strong CYP3A4 inhibitors has not been sufficiently characterized.
12.3 Pharmacokinetics
Elinzanetant Cmax and AUC increased in a greater than dose-proportional manner (20% to 50%) over the dose range from 40 to 160 mg once daily (0.33 to 1.33 times the highest recommended dose).
Elinzanetant steady state plasma concentrations were reached in 5 to 14 days after daily dosing.
Elinzanetant accumulation is <2-fold at the approved recommended dosage.
Absorption
Elinzanetant median (min-max) time to maximum plasma concentration (Tmax) is 1.0 hours (1-2.5 hours) at steady state. Elinzanetant absolute bioavailability is 52% following oral administration.
Effect of Food
No clinically significant differences in elinzanetant pharmacokinetics were observed following administration with a high-calorie, high-fat meal containing approximately 1000 calories (500-600 calories from fat, 250 calories from carbohydrates, and 150 calories from protein).
Distribution
The mean volume of distribution after intravenous administration at steady state of elinzanetant is 137 L. The plasma protein binding of elinzanetant is 99.7%. The blood-to-plasma ratio is between 0.6 and 0.7.
Elimination
Elinzanetant elimination half-life was approximately 45 hours in women with vasomotor symptoms. The clearance of elinzanetant after a single intravenous dose was 8.77 L/h.
Metabolism
Elinzanetant is primarily metabolized by CYP3A4 to yield three major active metabolites, M18/21, M27, and M30/34. These metabolites have similar potency for the human NK1 and NK3 receptors as compared to elinzanetant. The ratio of these metabolites to parent in plasma is approximately 0.39.
Excretion
Following a single oral dose of radiolabeled elinzanetant in healthy subjects, approximately 90% of the dose was recovered in feces (50% unchanged) and less than 1% with urine.
Specific Populations
No clinically significant differences in the pharmacokinetics of LYNKUET were observed based on race.
Patients with Renal Impairment
In patients with mild (eGFR 60 to <90 mL/min) renal impairment, mean elinzanetant Cmax increased 1.9-fold and AUC increased 1.6-fold. In patients with moderate (eGFR 30 to <60 mL/min) renal impairment, mean elinzanetant Cmax increased 1.8-fold and AUC increased 1.7-fold. In patients with severe (eGFR< 30 mL/min) renal impairment, mean elinzanetant Cmax increased 1.2-fold and AUC increased 1.1-fold.
Population pharmacokinetic analysis of the clinical trial data indicate similar exposure of elinzanetant in patients with mild and moderate renal impairment compared to patients with normal renal function.
Elinzanetant has not been studied in patients with end-stage renal disease (eGFR <15 mL/min) [see Use in Specific Populations (8.7)].
Patients with Hepatic Impairment
In patients with Child-Pugh Class A (mild) hepatic impairment, mean elinzanetant Cmax increased 1.2-fold and AUC(0-24) increased 1.5-fold. In patients with Child-Pugh Class B (moderate) hepatic impairment, mean elinzanetant Cmax and AUC(0-24) increased by 2.3-fold.
Elinzanetant has not been studied in patients with Child-Pugh Class C (severe) hepatic impairment [see Use in Specific Populations (8.6)].
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
No clinically significant differences in elinzanetant pharmacokinetics were observed when used concomitantly with esomeprazole (proton pump inhibitor). No clinically significant differences in the pharmacokinetics of the following oral drugs were observed when used concomitantly with elinzanetant: tamoxifen (substrate of CYP2D6, CYP3A4 and P-gp) and rosuvastatin (substrate of BCRP, OATP1B1/OATP1B3, and OAT3).
Strong CYP3A4 and P-gp inhibitors: Elinzanetant Cmax increased approximately 3.3-fold and AUC increased 6.3-fold following concomitant use with itraconazole 200 mg (strong CYP3A4 and P-gp inhibitor).
Moderate CYP3A4 Inhibitors: Elinzanetant Cmax and AUC are predicted to increase 2.0-fold and 3.0-fold, respectively, following concomitant use of elinzanetant 120 mg with erythromycin (moderate CYP3A4 inhibitor). PBPK predictions after co-administration of 60 mg elinzanetant [(see Dosage and Administration (2.3)] with the moderate CYP3A4 inhibitor erythromycin showed a 1.4-fold increase of elinzanetant AUC and no increase for Cmax when compared to 120 mg, the regular recommended elinzanetant dosage.
Weak CYP3A4 Inhibitors: Elinzanetant Cmax is predicted to increase 1.3-fold and AUC increase 1.5-fold following concomitant use with cimetidine (weak CYP3A4 inhibitor).
Moderate to Strong CYP3A4 Inducers: Elinzanetant Cmax reduced by 44% and AUC reduced by 64% following concomitant use with carbamazepine (moderate to strong CYP3A4 inducer) 600 mg administered twice daily.
Sensitive CYP3A4 Substrates: Elinzanetant increased midazolam (sensitive CYP3A4 substrate) Cmax 1.5-fold and AUC 1.8-fold.
In Vitro Studies
Cytochrome P450 (CYP450) Enzymes: Elinzanetant and its metabolites M18/21, M27, and M30/M34 inhibited CYP3A4 but did not inhibit CYP1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, or 2J2, or induce CYP1A2, 2B6, 2C19, or 3A4 at clinically relevant concentrations.
Transporter systems: Elinzanetant is a substrate for P-gp but not for OATP1A2, OATP2B1, BCRP, OATP1B1, OATP1B3, or MRP4. Elinzanetant and its metabolites M18/21, M27, and M30/M34 inhibited P-gp, BCRP, and BSEP in vitro but did not inhibit OAT1, OAT3, OCT1, OCT2, MATE2-K, or MRP2.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In a 2-year carcinogenicity study in female rats, oral doses of elinzanetant ≥60 mg/kg/day (29-fold the total AUC(0-24) at the human therapeutic dose) increased the incidence of endometrial adenocarcinoma, the combination of malignant and benign endometrial tumors and squamous cell carcinoma of the uterus, and lymphomas of the hematolymphoid system. These effects were not observed at a dose representing 7-fold the total AUC(0-24) at the human therapeutic dose. The clinical significance of these findings is unknown.
In a 26-week carcinogenicity study in rasH2 transgenic mice, there was no evidence of drug-related carcinogenicity at 85 or 70 mg/kg/day in males or females, respectively.
Mutagenesis
Elinzanetant showed no genotoxic potential in bacterial mutation assay (Ames test), mouse lymphoma assay, and in-vivo bone marrow micronucleus test in rats. Additionally, the principal human metabolites of elinzanetant were negative for genotoxicity in vitro in the Ames and micronucleus test.
13.2 Animal Toxicology and/or Pharmacology
Repeat dose toxicity studies were conducted in rats and cynomolgus monkeys. The findings of clinical relevance are described below:
Reproductive organs (female)
In female rats, daily oral administration of elinzanetant for 4 weeks at 100 mg/kg (40-fold the AUC(0-24) at the human therapeutic dose) induced mucification of the vaginal epithelium, uterine atrophy, and persistent corpora lutea. In monkeys, twice daily oral administration of elinzanetant for 39 weeks at doses ≥60 mg/kg/day (2-fold the human therapeutic exposure) reduced cyclical ovarian activity.
Central Nervous System (CNS)
In male and female rats, twice daily oral administration of elinzanetant for 13 weeks induced involuntary muscle contractions starting around treatment day 24 and convulsions starting around day 34, when exposures in male and female rats were estimated to be ≥4-fold the AUC(0-24) at the human therapeutic dose.
In female rats, twice daily oral administration of elinzanetant for 2 years induced convulsions starting around treatment day 106, when exposures were estimated to be ≥17-fold the AUC(0-24) at the human therapeutic dose.
Skeletal muscle
In rats, twice daily oral administration of elinzanetant for 13 weeks at ≥100 mg/kg/day (equivalent to ≥16-fold the AUC(0-24) at human therapeutic dose) induced skeletal muscle degeneration and necrosis. These findings were not related to the convulsions presented by elinzanetant-treated rats in the same 13-week toxicity study (described under CNS above). A longer duration (26-week) toxicity study in rats as well as a follow up mechanistic study in male rats did not have any skeletal muscle findings. The clinical relevance of these findings is unknown.
Gastrointestinal system
In male and female monkeys, once daily oral administration of elinzanetant for 4 weeks at 60 mg/kg/day (4-fold the AUC(0-24) at human therapeutic exposure) caused loose/watery fecal consistency and reduced food intake. In three studies of male and female monkeys treated orally with elinzanetant for 13 to 39 weeks, gastrointestinal disturbances characterized by diarrhea were observed at doses ≥30 mg/kg/day (0.2-fold the AUC(0-24) at human therapeutic exposure). Diarrhea eventually led to dehydration, weight loss, and general ill-health in animals at doses of ≥60 mg/kg/day.
Photosensitivity
An in-vitro study demonstrated that elinzanetant has some potential for photosensitization. While studies of photosensitivity in animals were not conducted, there is evidence that elinzanetant distributes to and accumulates in melanin-containing tissues in the rats.
-
14 CLINICAL STUDIES
14.1 Effects on Vasomotor Symptoms (VMS) in Postmenopausal Women
The efficacy of LYNKUET for the treatment of moderate to severe vasomotor symptoms due to menopause was demonstrated in the first 12 weeks of two randomized, double-blind, placebo-controlled, multicenter clinical trials. In OASIS 1 (NCT05042362) and OASIS 2 (NCT05099159), 796 menopausal women were randomized 1:1 to receive LYNKUET 120 mg or placebo once daily at bedtime for 12 weeks. In each of these two trials, after the first 12 weeks, women on placebo were switched over to LYNKUET and all women were then treated with LYNKUET for a 14-week extension for up to 26 weeks total exposure. Women who had at least 50 moderate to severe hot flashes (HFs), including night-time HFs, per week, were enrolled in OASIS 1 and 2. In these trials, postmenopausal status was defined as at least 12 months of spontaneous amenorrhea, or at least 6 months of spontaneous amenorrhea with serum follicle stimulating hormone levels >40 mIU/mL and a serum estradiol concentration of <30 pg/mL, or at least 6 months after hysterectomy with serum follicle-stimulating hormone >40 mIU/mL and serum estradiol <30 pg/mL, or at least 6 weeks post-surgical bilateral oophorectomy with or without hysterectomy.
In OASIS 1 and OASIS 2, the mean age of women was 54.6 years (range 40-65). The racial distribution was 80.4% White, 17.1% Black or African American, and 0.5% Asian. About 8.5 % women were of Hispanic or Latino ethnicity. The study population included women with prior hysterectomy (38.8%), prior uni-/bilateral oophorectomy (20.6%), or prior menopausal hormone therapy (MHT) use (31.4%).
The primary efficacy endpoints in both trials were the mean change in frequency and severity of moderate to severe vasomotor symptoms from baseline to Weeks 4 and 12, including day and night HFs measured using the Hot Flash Daily Diary (HFDD). In both trials, LYNKUET treatment groups showed statistically significant and clinically meaningful reduction (≥ 2 hot flashes over 24 hours) in the frequency of moderate to severe HFs from baseline to Weeks 4 and 12 compared to placebo. In both trials, LYNKUET treatment groups showed a statistically significant reduction in severity of moderate to severe vasomotor symptoms from baseline to Weeks 4 and 12 compared to placebo. Results of the co-primary endpoints for change from baseline to Weeks 4 and 12 in mean frequency and severity of moderate to severe vasomotor symptoms over 24 hours from OASIS 1 and OASIS 2 are shown in Tables 4 and 5.
Table 4: Mean Baseline and Mean Change in Frequency of Moderate to Severe VMS over 24 hours from Baseline to Weeks 4 and 12 (OASIS 1 and 2) OASIS 1 OASIS 2 Parameter LYNKUET 120 mg
(N= 199)Placebo
(N= 197)LYNKUET 120 mg
(N= 200)Placebo
(N= 200)CI = Confidence Interval, LS-Means = Least Squares Means estimated from a mixed model for repeated measures analysis of covariance, SD = standard deviation, SE = Standard Error - * - one-sided p-value.
Baseline Mean (SD) 13.38 (6.57) 14.26 (13.94) 14.66 (11.08) 16.16 (11.15) Change from Baseline to Week 4 LS-Means (SE)
Difference vs. Placebo (95% CI)
P-value*-7.60 (0.43)
-3.29
(-4.47, -2.10)
<0.0001-4.31 (0.43) -8.58 (0.49)
-3.04
(-4.40, -1.68)
<0.0001-5.54 (0.49) Change from Baseline to Week 12 LS-Means (SE)
Difference vs. Placebo (95% CI)
P-value*-8.66 (0.58)
-3.22
(-4.81, -1.63)
<0.0001-5.44 (0.59) -9.72 (0.50)
-3.24
(-4.60, -1.88)
<0.0001-6.48 (0.49) Table 5: Mean Baseline and Mean Change in Severity of Moderate to Severe VMS over 24 hours from Baseline to Weeks 4 and 12 (OASIS 1 and 2) Trial 1 Trial 2 Parameter LYNKUET 120 mg
(N= 199)Placebo
(N= 197)LYNKUET 120 mg
(N= 200)Placebo
(N= 200)CI = Confidence Interval, LS-Means = Least Squares Means estimated from a mixed model for repeated measures analysis of covariance, SD = standard deviation, SE = Standard Error - * - one-sided p-value.
Baseline Mean (SD) 2.56 (0.22) 2.53 (0.23) 2.53 (0.24) 2.54 (0.24) Change from Baseline to Week 4 LS-Means (SE)
Difference vs. Placebo (95% CI)
P-value*-0.73 (0.04)
-0.33
(-0.44, -0.23)
<0.0001-0.40 (0.04) -0.75 (0.04)
-0.22
(-0.34, -0.09)
0.0003-0.53 (0.04) Change from Baseline to Week 12 LS-Means (SE)
Difference vs. Placebo (95% CI)
P-value*-0.92 (0.05)
-0.40
(-0.54, -0.25)
<0.0001-0.52 (0.05) -0.91 (0.06)
-0.29
(-0.44, -0.14)
<0.0001-0.62 (0.05) The results in reduction of frequency and severity of moderate to severe vasomotor symptoms were consistent across various patient subgroups such as race, ethnicity, BMI and smoking status.
14.2 Effects on Endometrium in Postmenopausal Women
In all three OASIS trials, endometrial biopsies were performed to evaluate endometrial safety. A total of 477 patients who received elinzanetant 120 mg underwent end-of-treatment endometrial biopsies. Among these, 140 received elinzanetant for up to 52 weeks, 162 received elinzanetant for up to 26 weeks, and 175 received elinzanetant for up to 14 weeks. In addition, 132 women in the placebo arm in OASIS 3 also had end-of-treatment endometrial biopsies.
No endometrial malignancies were identified. One case of endometrial hyperplasia with atypia was identified in OASIS 1 and two cases of endometrial hyperplasia without atypia were seen in OASIS 3. Also, one case of endometrial glandular hyperplastic polyp was identified in OASIS 2. The rate of endometrial abnormalities was 0.8% (4 in 477) in patients who received elinzanetant, in line with the expected background rate.
14.3 Special Safety Study
Ability to Operate Vehicles or Machinery
Driving performance was assessed at 9 hours after bedtime administration of LYNKUET 120 mg and 240 mg (two times the recommended dose) in a randomized, double-blind, placebo-and active-controlled, four-period crossover study in 64 healthy women (mean age 52.1 years) using a computer-based driving simulation. The primary outcome measure was the difference from placebo in the Standard Deviation of Lateral Position (SDLP). Driving performance was evaluated using a validated threshold established in a population with blood alcohol concentration of 0.05%. Although the mean SDLP did not reach the threshold for driving impairment after administration of LYNKUET 120 or 240 mg, compared to placebo, driving ability was impaired in some subjects taking LYNKUET 120 or 240 mg after the initial dose. For a smaller percentage of subjects driving ability was impaired after 5-days of consecutive dosing.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
LYNKUET 60 mg capsules are supplied as opaque red, oblong, soft gelatin capsules, marked with white printing of "EZN60." LYNKUET capsules are available in the following package size:
- 60-count carton containing 5 blister cards (5 × 12 capsules)
NDC: 50419-475-05 16.2 Storage and Handling
Store at 20°C to 25°C (68°F to 77°F) with excursions permitted from 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
For patients requiring a dosage modification (i.e., one 60 mg capsule once daily), instruct them to partially peel back the foil covering of the blister cell exposing only one of the two capsules. Store the remaining capsule in the original blister card in the carton until the next dose.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
CNS Depressant Effect and Daytime Impairment
Advise patients who experience somnolence and other nervous system effects to refrain from driving or engaging in hazardous occupations or activities until the effects have resolved [see Warnings and precaution (5.1) and Clinical Studies (14.3)].
Hepatic Transaminase Elevations
Advise patients that bloodwork to evaluate for hepatic functions and injury will be obtained at baseline and three months after initiating therapy.
Advise patients to discontinue LYNKUET immediately and seek medical attention including hepatic laboratory tests if they experience signs or symptoms that may suggest liver injury (new onset fatigue, decreased appetite, nausea, vomiting, pruritus, jaundice, pale feces, dark urine, or abdominal pain). [see Warnings and Precautions (5.2)].
Females of Reproductive Potential
Exclude pregnancy in females of reproductive potential prior to initiating LYNKUET. Advise females of reproductive potential to use effective contraception during treatment with LYNKUET and for two weeks after discontinuing treatment and to discontinue LYNKUET if pregnancy is confirmed [see Warnings and Precautions (5.3) and Use in Specific Populations (8.1, 8.3)].
Risk of Seizures in Patients with History of Seizures
Advise patients to report any history of seizures or conditions that potentially lower the seizure threshold. [see Warnings and Precautions (5.4)].
Important Administration Instructions
Advise patients to take LYNKUET once daily at bedtime with water and swallow capsules whole. Advise patients not to cut, crush or chew capsules [see Dosage and Administration (2.1)].
Instruct patients requiring a dosage modification (i.e., one 60 mg capsule once daily) to partially peel back the foil covering the blister cell to expose only one of the two capsules. Instruct patients to store the remaining capsule in the original blister card in the carton until the next dose [see Storage and Handling (16.2)].
Drug Interactions
Advise patients to report their use of any other prescription or nonprescription medications or dietary supplements [see Drug Interactions (7.1)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
LYNKUET® (lin kew et')
(elinzanetant)
capsules, for oral useThis Patient Information has been approved by the U.S. Food and Drug Administration. Issued: 10/2025 What is LYNKUET?
LYNKUET is a prescription medicine used to reduce moderate to severe hot flashes (also known as vasomotor symptoms) due to menopause. LYNKUET is not a hormone. Hot flashes are feelings of warmth in the face, neck, and chest, or sudden intense feelings of heat and sweating.Do not take LYNKUET if you: - are pregnant.
Before you use LYNKUET, tell your healthcare provider about all of your medical conditions, including if you: - have liver problems.
- have a history of seizures.
- are pregnant or planning to become pregnant. LYNKUET may harm your unborn baby. Women who can become pregnant should talk to their healthcare provider to exclude pregnancy before starting treatment with LYNKUET and use effective birth control during and for 2 weeks after stopping treatment.
How should I take LYNKUET? - Take LYNKUET exactly as your healthcare provider tells you to take it.
- Take 2 LYNKUET capsules by mouth, with or without food, about the same time each day at bedtime.
- Swallow the LYNKUET capsules whole with water. Do not cut, crush, or chew capsules.
- If you miss a dose of LYNKUET at bedtime, take the next dose as scheduled the following day. Do not take more than 2 capsules on the same day to make up for a missed dose.
- If you take certain medicines, your healthcare provider may reduce your dose to 1 LYNKUET capsule. If so, partially peel back the foil of the blister card and leave the remaining capsule inside until your next dose.
What should I avoid while taking LYNKUET? - Avoid eating grapefruit or drinking grapefruit juice during treatment with LYNKUET.
- LYNKUET may cause you to feel drowsy, if you experience this avoid driving and other hazardous activities until these effects go away. See "What are the possible side effects of LYNKUET?".
What are the possible side effects of LYNKUET?
LYNKUET can cause serious side effects, including:- Central nervous system (CNS) effects and daytime impairment. LYNKUET can cause difficulty staying awake (somnolence) and other nervous system effects including fatigue, having a spinning feeling (vertigo), dizziness, and feeling faint (presyncope). If you experience these effects, you should not drive or do hazardous activities until these effects go away.
- Increased liver blood test values. LYNKUET may cause increased liver enzymes. Your healthcare provider will do a blood test to check your liver before you start and 3 months after taking LYNKUET. Stop taking LYNKUET and tell your healthcare provider right away if you have the following signs or symptoms that suggest liver problems:
- feeling more tired than you do usually
- decreased appetite
- nausea
- vomiting
- itching
- yellowing of the eyes or skin (jaundice)
- pale feces
- dark urine
- pain in the stomach (abdomen)
- Risk of pregnancy loss. Taking LYNKUET while pregnant may cause loss of pregnancy or stillbirth. If you think you are pregnant, stop taking LYNKUET and tell your healthcare provider right away.
- Risk of seizures in people with a history of seizures. Seek medical attention right away if you have loss of consciousness or seizure.
- headache
- fatigue
- dizziness
- feeling drowsy or sleepy
- stomach (abdominal) pain
- rash
- diarrhea
- muscle spasms
Tell your healthcare provider if you have any side effects that bother you or do not go away. These are not all the possible side effects of LYNKUET.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store LYNKUET? - Store LYNKUET at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep LYNKUET and all medicines out of the reach of children.
General information about the safe and effective use of LYNKUET.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use LYNKUET for a condition for which it was not prescribed. Do not give LYNKUET to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about LYNKUET that is written for healthcare professionals.What are the ingredients in LYNKUET?
Active ingredient: elinzanetant
Inactive ingredients: all-rac-α-Tocopherol, caprylocaproyl macrogolglycerides, glycerol monocaprylocaprate, glycerol mono-oleate, and polysorbate 80. The capsule is composed of edible ink, ferric oxide red, ferric oxide yellow, gelatin, sorbitol special-glycerin, and titanium dioxide.Distributed by Bayer HealthCare Pharmaceuticals Inc., Whippany, NJ 07981 USA
For more information, go to www.lynkuet.com or call 1-888-842-2937. -
PRINCIPAL DISPLAY PANEL - 60 mg Capsule Blister Pack
BAYER
NDC: 50419-475-01
Lynkuet®
(elinzanetant) capsules
60 mg per capsule
Rx onlyLOT:
EXP:XXXXXXX
YYYY/MMM
-
PRINCIPAL DISPLAY PANEL - 60 mg Capsule Blister Pack Carton
NDC: 50419-475-05
Rx onlyLynkuet®
(elinzanetant) capsules
60 mg per capsule60 capsules
5 blister cards of 12 capsules eachBAYER
Swallow capsule whole

-
INGREDIENTS AND APPEARANCE
LYNKUET
elinzanetant capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 50419-475 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ELINZANETANT (UNII: NZW2BOW35N) (ELINZANETANT - UNII:NZW2BOW35N) ELINZANETANT 60 mg Inactive Ingredients Ingredient Name Strength .ALPHA.-TOCOPHEROL (UNII: H4N855PNZ1) CAPRYLOCAPROYL POLYOXYLGLYCERIDES, UNSPECIFIED (UNII: MJ9J2BSO5V) GLYCERYL MONOOLEATE (UNII: C4YAD5F5G6) GLYCERYL MONOCAPRYLOCAPRATE (UNII: G7515SW10N) POLYSORBATE 80 (UNII: 6OZP39ZG8H) FERRIC OXIDE RED (UNII: 1K09F3G675) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) SORBITOL (UNII: 506T60A25R) GLYCERIN (UNII: PDC6A3C0OX) TITANIUM (UNII: D1JT611TNE) Product Characteristics Color RED (Opague) Score no score Shape CAPSULE Size 24mm Flavor Imprint Code EZN60 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 50419-475-05 5 in 1 CARTON 10/24/2025 1 NDC: 50419-475-01 12 in 1 BLISTER PACK; Type 0: Not a Combination Product 2 NDC: 50419-475-72 2 in 1 CARTON 10/24/2025 2 NDC: 50419-475-71 12 in 1 BLISTER PACK; Type 0: Not a Combination Product 3 NDC: 50419-475-73 1 in 1 CARTON 05/07/2026 3 NDC: 50419-475-71 12 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA219469 10/24/2025 Labeler - Bayer HealthCare Pharmaceuticals Inc. (005436809) Registrant - Bayer AG (314947622) Establishment Name Address ID/FEI Business Operations Esteve Quimica, S.A. 633485529 API MANUFACTURE(50419-475) Establishment Name Address ID/FEI Business Operations Lianhe Aigen Pharma Co., Ltd. 542992986 API MANUFACTURE(50419-475) Establishment Name Address ID/FEI Business Operations Catalent Germany Eberbach GmbH 318612223 ANALYSIS(50419-475) , MANUFACTURE(50419-475) Establishment Name Address ID/FEI Business Operations Catalent Germany Schorndorf GmbH 315732628 PACK(50419-475) Establishment Name Address ID/FEI Business Operations Sharp Corporation 143696495 PACK(50419-475) , LABEL(50419-475)
Trademark Results [LYNKUET]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 LYNKUET 97488452 not registered Live/Pending |
Bayer Aktiengesellschaft 2022-07-05 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.