MOVANTIK- naloxegol oxalate tablet, film coated
MOVANTIK by
Drug Labeling and Warnings
MOVANTIK by is a Prescription medication manufactured, distributed, or labeled by AstraZeneca Pharmaceuticals LP, AstraZeneca PLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use MOVANTIK safely and effectively. See full prescribing information for MOVANTIK.
MOVANTIK® (naloxegol) tablets, for oral use
Initial U.S. Approval: 2014RECENT MAJOR CHANGES
Warnings and Precautions (5.3) 04/2020
INDICATIONS AND USAGE
MOVANTIK is an opioid antagonist indicated for the treatment of opioid-induced constipation (OIC) in adult patients with chronic non-cancer pain, including patients with chronic pain related to prior cancer or its treatment who do not require frequent (e.g., weekly) opioid dosage escalation. (1)
DOSAGE AND ADMINISTRATION
Administration:
- Discontinue maintenance laxative therapy before starting MOVANTIK; may resume laxatives if patients have OIC symptoms after taking MOVANTIK for 3 days. (2.1)
- Alteration in analgesic dosing regimen prior to starting MOVANTIK is not required. (2.1)
- Patients receiving opioids for less than 4 weeks may be less responsive to MOVANTIK. (2.1)
- Take on an empty stomach at least 1 hour prior to the first meal of the day or 2 hours after the meal. (2.1)
- For patients who are unable to swallow the MOVANTIK tablet whole, the tablet can be crushed and given orally or administered via nasogastric tube, see full prescribing information. (2.1)
- Avoid consumption of grapefruit or grapefruit juice. (2.1, 7.1)
- Discontinue if treatment with the opioid pain medication is also discontinued. (2.1)
Recommended dosage:
DOSAGE FORMS AND STRENGTHS
- Tablets: 12.5 mg and 25 mg (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Opioid withdrawal: Consider the overall risk benefit in patients with disruptions to the blood-brain barrier. Monitor for symptoms of opioid withdrawal. (5.1)
- Severe abdominal pain and/or diarrhea: Monitor for the development of symptoms after initiating treatment with MOVANTIK and discontinue if severe symptoms develop. Consider restarting MOVANTIK at 12.5 mg once daily if appropriate. (5.2)
- Gastrointestinal perforation: Consider the overall risk benefit in patients with known or suspected lesions of the GI tract. Monitor for severe, persistent or worsening abdominal pain; discontinue if development of symptoms. (5.3)
ADVERSE REACTIONS
The most common adverse reactions in clinical trials (≥3%) are: abdominal pain, diarrhea, nausea, flatulence, vomiting, and headache. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AstraZeneca at 1-800-236-9933 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Moderate CYP3A4 inhibitors (e.g., diltiazem, erythromycin, verapamil): Increased naloxegol concentrations; avoid concomitant use; if unavoidable, reduce dosage to 12.5 mg once daily and monitor for adverse reactions. (2.4, 7.1)
- Strong CYP3A4 inducers (e.g., rifampin): Decreased concentrations of naloxegol; concomitant use is not recommended. (7.1)
- Other opioid antagonists: Potential for additive effect and increased risk of opioid withdrawal; avoid concomitant use. (7.1)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 4/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Administration Instructions
2.2 Adult Dosage
2.3 Dosage in Adult Patients with Renal Impairment
2.4 Dosage Recommendations due to Drug Interactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Opioid Withdrawal
5.2 Severe Abdominal Pain and/or Diarrhea
5.3 Gastrointestinal Perforation
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on MOVANTIK
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Administration Instructions
- Discontinue all maintenance laxative therapy prior to initiation of MOVANTIK. Laxative(s) can be used as needed if there is a suboptimal response to MOVANTIK after three days.
- Alteration in analgesic dosing regimen prior to initiating MOVANTIK is not required.
- Patients receiving opioids for less than 4 weeks may be less responsive to MOVANTIK [see Clinical Studies (14)].
- Take MOVANTIK on an empty stomach at least 1 hour prior to the first meal of the day or 2 hours after the meal.
- For patients who are unable to swallow the MOVANTIK tablet whole, the tablet can be crushed to a powder, mixed with 4 ounces (120 mL) of water, and drunk immediately. The glass should be refilled with 4 ounces (120 mL) of water, stirred and the contents drunk.
- MOVANTIK can also be administered via a nasogastric (NG) tube, as follows:
- 1. Flush the NG tube with 1 ounce (30 mL) of water using a 60 mL syringe.
- 2. Crush the tablet to a powder in a container and mix with approximately 2 ounces (60 mL) of water.
- 3. Draw up the mixture using the 60 mL syringe and administer the syringe contents through the NG tube.
- 4. Add approximately 2 ounces (60 mL) of water to the same container used to prepare the dose of MOVANTIK.
- 5. Draw up the water using the same 60 mL syringe and use all the water to flush the NG tube and any remaining medicine from the NG tube into the stomach.
- Avoid consumption of grapefruit or grapefruit juice during treatment with MOVANTIK.
- Discontinue MOVANTIK if treatment with the opioid pain medication is also discontinued.
2.2 Adult Dosage
The recommended MOVANTIK dosage is 25 mg once daily in the morning. If patients are not able to tolerate MOVANTIK, reduce the dosage to 12.5 mg once daily [see Clinical Pharmacology (12.2)].
2.3 Dosage in Adult Patients with Renal Impairment
The starting dosage for patients with creatinine clearance (CLcr) <60 mL/min (i.e., patients with moderate, severe, or end-stage renal impairment) is 12.5 mg once daily. If this dosage is well tolerated but OIC symptoms continue, the dosage may be increased to 25 mg once daily taking into consideration the potential for markedly increased exposures in some patients with renal impairment and the increased risk of adverse reactions with higher exposures [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
2.4 Dosage Recommendations due to Drug Interactions
Avoid concomitant use of MOVANTIK with moderate CYP3A4 inhibitor drugs (e.g., diltiazem, erythromycin, verapamil). If concurrent use is unavoidable, reduce the MOVANTIK dosage to 12.5 mg once daily and monitor for adverse reactions [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
-
3 DOSAGE FORMS AND STRENGTHS
MOVANTIK (naloxegol) is available in two strengths:
- Tablets: 12.5 mg supplied as mauve, oval, biconvex, film-coated, intagliated with “nGL” on one side and “12.5” on the other side.
- Tablets: 25 mg supplied as mauve, oval, biconvex, film-coated, intagliated with “nGL” on one side and “25” on the other side.
-
4 CONTRAINDICATIONS
MOVANTIK is contraindicated in:
- Patients with known or suspected gastrointestinal obstruction and patients at increased risk of recurrent obstruction, due to the potential for gastrointestinal perforation [see Warnings and Precautions (5.3)].
- Patients concomitantly using strong CYP3A4 inhibitors (e.g., clarithromycin, ketoconazole) because these medications can significantly increase exposure to naloxegol which may precipitate opioid withdrawal symptoms such as hyperhidrosis, chills, diarrhea, abdominal pain, anxiety, irritability, and yawning [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
- Patients who have had a known serious or severe hypersensitivity reaction to MOVANTIK or any of its excipients
- [see Adverse Reactions (6.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Opioid Withdrawal
Clusters of symptoms consistent with opioid withdrawal, including hyperhidrosis, chills, diarrhea, abdominal pain, anxiety, irritability, and yawning have occurred in patients treated with MOVANTIK [see Adverse Reactions (6.1)]. In addition, patients receiving methadone as therapy for their pain condition were observed in clinical trials to have a higher frequency of gastrointestinal adverse reactions that may have been related to opioid withdrawal than patients receiving other opioids [see Adverse Reactions (6.1)]. Patients having disruptions to the blood-brain barrier may be at increased risk for opioid withdrawal or reduced analgesia. Take into account the overall risk-benefit profile when using MOVANTIK in such patients. Monitor for symptoms of opioid withdrawal in such patients.
5.2 Severe Abdominal Pain and/or Diarrhea
Reports of severe abdominal pain and/or diarrhea have been reported, some of which resulted in hospitalization. Most of the cases of severe abdominal pain were reported in patients taking the 25 mg dosage. Symptoms generally occurred within a few days of initiation of MOVANTIK. Monitor patients for the development of abdominal pain and/or diarrhea with MOVANTIK and discontinue therapy if severe symptoms occur. Consider restarting MOVANTIK at 12.5 mg once daily, if appropriate.
5.3 Gastrointestinal Perforation
Cases of gastrointestinal (GI) perforation have been reported with use of peripherally acting opioid antagonists, including MOVANTIK. Postmarketing cases of GI perforation, including fatal cases, were reported when MOVANTIK was used in patients at risk of GI perforation (e.g., infiltrative gastrointestinal tract malignancy, recent gastrointestinal tract surgery, diverticular disease including diverticulitis, ischemic colitis, or concomitantly treated with bevacizumab). MOVANTIK is contraindicated in patients with known or suspected gastrointestinal obstruction or in patients at risk of recurrent obstruction [see Contraindications (4)]. Take into account the overall risk-benefit profile when using MOVANTIK in patients with these conditions or other conditions which might result in impaired integrity of the gastrointestinal tract wall (e.g., Crohn’s disease). Monitor for the development of severe, persistent or worsening abdominal pain; discontinue MOVANTIK in patients who develop this symptom.
-
6 ADVERSE REACTIONS
Serious and important adverse reactions described elsewhere in labeling include:
- Opioid withdrawal [see Warnings and Precautions (5.1)]
- Severe abdominal pain and/or diarrhea [see Warnings and Precautions (5.2)]
- Gastrointestinal perforation [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described below reflect exposure to MOVANTIK in 1497 patients in clinical trials, including 537 patients exposed for greater than six months, and 320 patients exposed for 12 months.
The safety data described in Table 1 are derived from two double-blind, placebo-controlled trials (Studies 1 and 2) in patients with OIC and non-cancer related pain [see Clinical Studies (14)]. Study 3 (n=302) was a safety extension study that allowed patients from Study 1 to continue the same blinded treatment for an additional 12 weeks. Safety data for patients in Study 3 are similar to those listed in Table 1.
Study 4 (n=844) was a Phase 3, 52-week, multi-center, open-label, randomized, parallel group, safety and tolerability study of naloxegol versus usual care treatment for OIC (as determined by the investigator and excluding peripheral opioid antagonists) in patients with non-cancer related pain. The population enrolled in Study 4 was similar to that of the other studies. Eligible patients were randomized in a 2:1 ratio to receive either naloxegol 25 mg once daily or usual care treatment for OIC. The most commonly used laxatives in the usual care group were rectal stimulants (e.g., bisacodyl), oral stimulants (e.g., senna), and oral osmotics (e.g., macrogol, magnesium). Safety data for patients in Study 4 are similar to those listed in Table 1.
Table 1 lists adverse reactions in pooled Studies 1 and 2 occurring in ≥3% of patients receiving MOVANTIK 12.5 mg or 25 mg and at an incidence greater than placebo.
Table 1. Adverse Reactions* in Patients with OIC and Non-Cancer Pain (Studies 1 and 2) - * Adverse reactions occurring in ≥3% of patients receiving MOVANTIK 12.5 mg or 25 mg and at an incidence greater than placebo.
- † Includes: abdominal pain, abdominal pain upper, abdominal pain lower, and gastrointestinal pain.
- Adverse Reaction
MOVANTIK 25 mg
(n=446)
MOVANTIK 12.5 mg
(n=441)
Placebo
(n=444)
- Abdominal Pain†
21%
12%
7%
- Diarrhea
9%
6%
5%
- Nausea
8%
7%
5%
- Flatulence
6%
3%
3%
- Vomiting
5%
3%
4%
- Headache
4%
4%
3%
- Hyperhidrosis
3%
<1%
<1%
Opioid Withdrawal
Possible opioid withdrawal, defined as at least three adverse reactions potentially related to opioid withdrawal that occurred on the same day and were not all related to the gastrointestinal system, occurred in less than 1% (1/444) of placebo subjects, 1% (5/441) receiving MOVANTIK 12.5 mg, and 3% (14/446) receiving MOVANTIK 25 mg in Studies 1 and 2 regardless of maintenance opioid treatment. Symptoms included but were not limited to hyperhidrosis, chills, diarrhea, abdominal pain, anxiety, irritability, and yawning. Patients receiving methadone as therapy for their pain condition were observed in Studies 1 and 2 to have a higher frequency of gastrointestinal adverse reactions than patients receiving other opioids [39% (7/18) vs. 26% (110/423) in the 12.5 mg group; 75% (24/32) vs. 34% (142/414) in the 25 mg group].
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of MOVANTIK. Because reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate the frequency or establish a causal relationship to drug exposure.
Hypersensitivity reactions: angioedema, rash, and urticaria.
Gastrointestinal disorders: Gastrointestinal perforation [see Warnings and Precautions (5.3)].
-
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on MOVANTIK
Table 2 displays the effects of other drugs on MOVANTIK.
Table 2. Effects of Other Drugs on MOVANTIK - * The effect of grapefruit juice varies widely among brands and is concentration-, dose-, and preparation-dependent. Studies have shown that it can be classified as a “strong CYP3A inhibitor” when a certain preparation was used (e.g., high dose, double strength) or as a “moderate CYP3A inhibitor” when another preparation was used (e.g., low dose, single strength).
Concomitant Agent
Mechanism of Action
- Clinical Recommendation
CYP3A4 Inhibitors
- Strong CYP3A4 inhibitors (e.g., ketoconazole, itraconazole, clarithromycin)
- Increase plasma naloxegol concentrations and may increase the risk of adverse reactions [see Clinical Pharmacology (12.3)].
- Use with strong CYP3A4 inhibitors is contraindicated [see Contraindications (4)].
- Moderate CYP3A4 inhibitors (e.g., diltiazem, erythromycin, verapamil)
- Avoid use with moderate CYP3A4 inhibitors; if unavoidable, decrease the dosage of MOVANTIK to 12.5 mg once daily and monitor for adverse reactions [see Dosage and Administration (2.4)].
- Weak CYP3A4 inhibitors (e.g., quinidine, cimetidine)
- Clinically significant increases in naloxegol concentrations are not expected.
- No dosage adjustments are necessary.
- Grapefruit or grapefruit juice*
- Can increase plasma naloxegol concentrations.
- Avoid consumption of grapefruit or grapefruit juice during treatment with MOVANTIK [see Dosage and Administration (2.1)].
- CYP3A4 Inducers
- Strong CYP3A4 inducers (e.g., rifampin, carbamazepine, St. John’s Wort)
- Significantly decrease plasma naloxegol concentrations and may decrease the efficacy of MOVANTIK [see Clinical Pharmacology (12.3)].
- Use with strong CYP3A4 inducers is not recommended.
Other Drug Interactions
- Other opioid antagonists
- Potential for additive effect of opioid receptor antagonism and increased risk of opioid withdrawal.
- Avoid use of MOVANTIK with another opioid antagonist.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Limited available data with MOVANTIK use in pregnant women are insufficient to inform a drug associated risk of adverse developmental outcomes. MOVANTIK may precipitate opioid withdrawal in the pregnant women and the fetus (see Clinical Considerations).
In animal development studies, no effects on embryo-fetal development were observed following administration of naloxegol in pregnant rats during the period of organogenesis at doses up to 1452 times the human AUC (area under the plasma concentration-time curve) at the maximum recommended human dose. No effects on embryo-fetal development were observed following administration of naloxegol in pregnant rabbits during the period of organogenesis at doses up to 409 times the human AUC at the maximum recommended human dose.
The estimated background risk of major birth defects and miscarriage for the indicated populations are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically-recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Maternal and Fetal/Neonatal adverse reactions
The use of MOVANTIK may be associated with opioid withdrawal in the pregnant woman and the fetus.Data
Animal Data
Oral administration of up to 750 mg/kg/day naloxegol in rats (1452 times the human AUC at the maximum recommended human dose) and 450 mg/kg/day naloxegol in rabbits (409 times the human AUC at the maximum recommended human dose) during the period of organogenesis produced no adverse effects on embryo-fetal development. Oral administration of up to 500 mg/kg/day in rats (195 times the maximum recommended human dose based on body surface area) during the period of organogenesis through lactation produced no adverse effects on parturition or the offspring.
8.2 Lactation
There are no data on the presence of naloxegol in human milk, the effects in nursing infants, or the effects on milk production. Naloxegol is present in rat milk (see Data). Because of the potential for adverse reactions, including opioid withdrawal in breastfed infants, advise women that breastfeeding is not recommended during treatment with MOVANTIK.
Data
Following oral administration of naloxegol in lactating rats, concentrations of naloxegol in milk were approximately 3- to 4-fold higher than concentrations of naloxegol in maternal plasma. Naloxegol was detected in plasma from pups.
8.4 Pediatric Use
The safety and effectiveness of MOVANTIK have not been established in pediatric patients.
8.5 Geriatric Use
Of the total number of subjects in clinical studies of MOVANTIK, 11% were 65 and over, while 2% were 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
MOVANTIK exposure was higher in elderly healthy Japanese subjects compared to young subjects [see Clinical Pharmacology (12.3)]. No dosage adjustment is needed in elderly patients.
8.6 Renal Impairment
Some subjects with creatinine clearance (CLcr) values <60 mL/minute (i.e., moderate, severe, or end-stage renal disease) were shown to exhibit markedly higher systemic exposure of naloxegol compared to subjects with normal renal function. The reason for these high exposures is not understood. However, as the risk of adverse reactions increases with systemic exposure, a lower starting dosage of 12.5 mg once daily is recommended. No dosage adjustment is needed in patients with mild renal impairment [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
The effect of severe hepatic impairment (Child-Pugh Class C) on the pharmacokinetics of naloxegol has not been evaluated. Avoid use of MOVANTIK in patients with severe hepatic impairment, as the dosage in these patients has not been determined. No dosage adjustment is required for patients with mild or moderate hepatic impairment [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
In a clinical study of patients with OIC a daily dose of 50 mg (twice the recommended dosage), administered over 4 weeks, was associated with an increased incidence of GI adverse reactions, such as abdominal pain, diarrhea, and nausea. These adverse reactions frequently occurred within 1-2 days after dosing.
No antidote is known for naloxegol. Dialysis was noted to be ineffective as a means of elimination in a clinical study in patients with renal failure.
If a patient on opioid therapy receives an overdose of naloxegol, the patient should be monitored closely for potential evidence of opioid withdrawal symptoms such as chills, rhinorrhea, diaphoresis, or reversal of central analgesic effect. Base treatment on the degree of opioid withdrawal symptoms, including changes in blood pressure and heart rate, and on the need for analgesia.
-
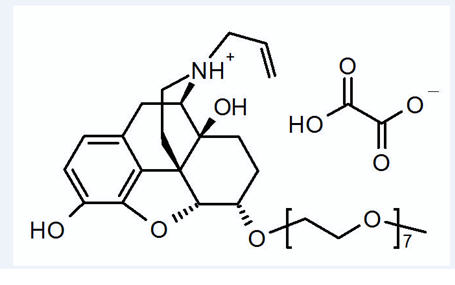
11 DESCRIPTION
MOVANTIK (naloxegol), an opioid antagonist, contains naloxegol oxalate as the active ingredient. (Naloxegol is a PEGylated derivative of naloxone.)
The chemical name for naloxegol oxalate is: (5α,6α)-17-allyl-6-(2,5,8,11,14,17,20-heptaoxadocosan-22-yloxy)-4,5-epoxymorphinan-3,14-diol oxalate. The structural formula is:
The empirical formula for naloxegol oxalate is C34H53NO11.C2H2O4 and the molecular weight is 742.
Naloxegol oxalate is a white to off-white powder, with high aqueous solubility across the physiologic pH range.
MOVANTIK (naloxegol) tablets for oral use contain 14.2 mg and 28.5 mg of naloxegol oxalate, respectively, equivalent to 12.5 mg and 25 mg of naloxegol.
Excipients in tablet core are: mannitol, cellulose microcrystalline, croscarmellose sodium, magnesium stearate, and propyl gallate.
Excipients in tablet coat are: hypromellose, titanium dioxide, polyethylene glycol, iron oxide red, and iron oxide black.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Naloxegol is an antagonist of opioid binding at the mu-opioid receptor. When administered at the recommended dose levels, naloxegol functions as a peripherally-acting mu-opioid receptor antagonist in tissues, such as the gastrointestinal tract, thereby decreasing the constipating effects of opioids.
Naloxegol is a PEGylated derivative of naloxone and is a substrate for the P-glycoprotein transporter (P-gp). Also, the presence of the PEG moiety in naloxegol reduces its passive permeability as compared with naloxone. Due to the reduced permeability and increased efflux of naloxegol across the blood-brain barrier, related to P-gp substrate properties, the CNS penetration of naloxegol is expected to be negligible at the recommended dose levels limiting the potential for interference with centrally mediated opioid analgesia.
12.2 Pharmacodynamics
Use of opioids induces slowing of gastrointestinal motility and transit. Antagonism of gastrointestinal mu-opioid receptors by naloxegol inhibits opioid-induced delay of gastrointestinal transit time.
Effect on Cardiac Repolarization
In a randomized, double-blind, 4-way cross-over thorough QTc prolongation study with moxifloxacin as a positive control, a single 25 mg therapeutic dose or a 150 mg dose (6 times the maximum recommended dosage) of naloxegol did not have an effect on the QTc interval compared to placebo. Changes in heart rate, RR, PR, and QRS ECG intervals were similar between placebo and naloxegol 25 or 150 mg.
Exposure Response Analysis
The exposure-response analysis for adverse events showed that the probability of experiencing abdominal pain increased with increasing naloxegol exposure over the dose range of 12.5 mg to 25 mg once a day. The exposure-response analysis for efficacy conducted using the definition of response in the clinical trials [see Clinical Studies (14)] indicated that response was similar over this dose range.
12.3 Pharmacokinetics
Absorption
Following oral administration, MOVANTIK is absorbed with peak concentrations (Cmax) achieved at less than 2 hours. In a majority of subjects, a secondary plasma concentration peak of naloxegol was observed approximately 0.4 to 3 hours after the first peak. Across the range of doses evaluated, peak plasma concentration and area under the plasma concentration-time curve (AUC) increased in a dose-proportional or almost dose-proportional manner. Accumulation was minimal following multiple daily doses of naloxegol.
MOVANTIK as a crushed tablet mixed in water, given orally or administered through a nasogastric tube into the stomach, provides systemic naloxegol concentrations that are comparable to the whole tablet, with a median tmax of 0.75 and 1.5 hours (range 0.25 to 5 hours) for the crushed tablet given orally and the crushed tablet given via nasogastric (NG) tube, respectively [see Dosage and Administration (2.2)].
Food Effects
A high-fat meal increased the extent and rate of naloxegol absorption. The Cmax and AUC were increased by approximately 30% and 45%, respectively. In clinical trials, naloxegol was dosed on an empty stomach approximately 1 hour prior to the first meal in the morning.
Distribution
The mean apparent volume of distribution during the terminal phase (Vz/F) in healthy volunteers ranged from 968 L to 2140 L across dosing groups and studies. Plasma protein binding of naloxegol in humans was low (~4.2%).
Metabolism
Naloxegol is metabolized primarily by the CYP3A enzyme system. In a mass balance study in humans, a total of 6 metabolites were identified in plasma, urine, and feces. These metabolites were formed via N-dealkylation, O-demethylation, oxidation, and partial loss of the PEG chain. Human metabolism data suggests absence of major metabolites. The activity of the metabolites at the opioid receptor has not been determined.
Excretion
Following oral administration of radio-labeled naloxegol, 68% and 16% of total administered dose were recovered in the feces and urine, respectively. Parent naloxegol excreted in the urine accounted for less than 6% of the total administered dose. Approximately 16% of radioactivity in feces was noted to be unchanged naloxegol, while the remaining was attributed to metabolites. Thus, renal excretion is a minor clearance pathway for naloxegol. In a clinical pharmacology study, the half-life of naloxegol at therapeutic doses ranged from 6 to 11 hours.
Specific Populations
Renal Impairment:
The effect of renal impairment on the pharmacokinetics of a 25 mg single oral dose of MOVANTIK was studied in subjects with renal impairment (RI) classified as moderate (n=8), severe (n=4), or end-stage renal disease (ESRD) not yet on dialysis (n=4), and compared with healthy subjects (n=6). Most renal impairment (RI) subjects (6 out of 8 with moderate RI, 3 out of 4 with severe RI, and 3 out of 4 with ESRD) had plasma naloxegol pharmacokinetics comparable to those in healthy subjects. The remaining individuals with renal impairment demonstrated higher naloxegol exposures (up to 10-fold) compared to the control group. The reason for these high exposures is unknown.
This study also included 8 ESRD patients on hemodialysis. Plasma concentrations of naloxegol in these subjects were similar to healthy volunteers with normal renal function, when MOVANTIK was administered either pre- or post-
hemodialysis [see Dosage and Administration (2.3), Use in Specific Populations (8.6) and Overdosage (10)].
Hepatic Impairment:
Slight decreases in AUC of naloxegol were observed in subjects with mild and moderate hepatic impairment (Child-Pugh Classes A and B; n=8 per group) compared to subjects with normal hepatic function (n=8), following administration of a single 25 mg oral dose of MOVANTIK. The effect of severe hepatic impairment (Child-Pugh Class C) on the pharmacokinetics of naloxegol was not evaluated [see Use in Specific Populations (8.7)].
Age:
The mean Cmax,ss and AUCτ,ss values seen in elderly healthy Japanese subjects (n=6) were approximately 45% and 54% greater than those obtained in young healthy subjects (n=6) following multiple daily doses of naloxegol (25 mg).
Gender:
There is no gender effect on the pharmacokinetics of naloxegol.
Race:
When compared to Caucasian subjects, naloxegol AUC was approximately 20% lower in Blacks and Cmax was approximately 10% lower and 30% higher in Blacks and Asians, respectively.
Drug Interaction Studies
Effect of MOVANTIK on Other Drugs
In in vitro studies at clinically relevant concentrations, naloxegol did not show a significant inhibitory effect on the activity of CYP1A2, CYP2C8, CYP2C9, CYP2D6, CYP3A4 or CYP2C19, nor a significant induction effect on the activity of CYP1A2, CYP2B6, or CYP3A4. Therefore, MOVANTIK is not expected to alter the metabolic clearance of co-administered drugs that are metabolized by these enzymes. Naloxegol is not a significant inhibitor of P-gp, BCRP, OAT1, OAT3, OCT2, OATP1B1, and OATP1B3.
In healthy subjects receiving morphine 5 mg/70 kg intravenously, single doses of MOVANTIK ranging from 8 mg to 1000 mg were given concomitantly with 5 to 6 subjects per dose cohort. With increasing MOVANTIK dose, there was no increasing or decreasing trend in morphine exposure compared to morphine administered alone. An analysis of the pooled data indicated that MOVANTIK had no meaningful impact on the systemic exposure of morphine and its major circulating metabolites.
Effect of Other Drugs on MOVANTIK
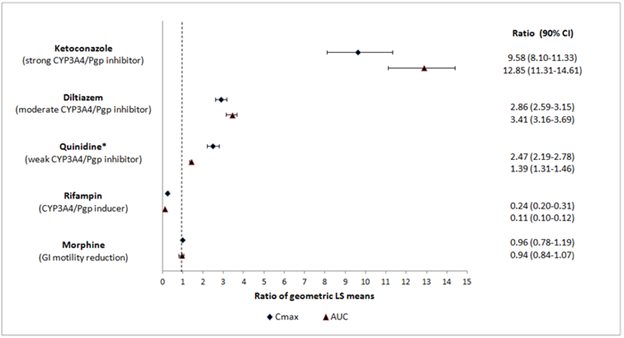
Naloxegol is metabolized mainly by CYP3A enzymes and is a substrate of P-gp transporter. The effects of co-administered drugs on the pharmacokinetics of naloxegol are summarized in Figure 1 [see Drug Interactions (7.1)].
The effects of once daily oral dosing of 400 mg ketoconazole, once daily oral dosing of 600 mg rifampicin and once daily oral dosing of 240 mg diltiazem (as an extended release formulation) on the pharmacokinetics of 25 mg MOVANTIK were studied following multiple dosing and at steady state exposure of the perpetrator drugs. The effects of 600 mg oral dosing of quinidine and intravenous morphine (5 mg/70 kg) on the pharmacokinetics of 25 mg MOVANTIK were studied following single dosing of the perpetrator drugs.
Figure 1: Effect of Co-administered Drugs on the Pharmacokinetics of Naloxegol
*Quinidine due to its effect on P-gp transporter increased naloxegol Cmax by 2.5-fold; the AUC increased by 1.4-fold; no dosage adjustment is necessary.
No drug interaction studies have been conducted for MOVANTIK with drugs that alter gastric pH (e.g., antacids, proton-pump inhibitors).
Simulations using physiologically based pharmacokinetic modeling, suggested that naloxegol exposures after co-administration of a single oral 25 mg dose of MOVANTIK with a moderate CYP3A inducer efavirenz (400 mg once a day) are similar to those after 12.5 mg MOVANTIK alone.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In a 104-week carcinogenicity study in CD-1 mice, naloxegol was not tumorigenic at oral doses up to 100 mg/kg/day in males and 160 mg/kg/day in females (43 and 27 times the human AUC at the maximum recommended human dose for male and female mice, respectively). In a carcinogenicity study in Sprague-Dawley rats, naloxegol was administered orally at doses of 40, 120, and 400 mg/kg/day for at least 93 weeks. Naloxegol did not cause an increase in tumors in female rats. In male rats, an increase in interstitial (Leydig) cell adenomas in testes was observed at 400 mg/kg/day (818 times the human AUC at the maximum recommended human dose). The no observed effect level for increased tumor incidence was 120 mg/kg/day in male and 400 mg/kg/day in female rats (246 and 1030 times the human AUC at the maximum recommended human dose for male and female rats, respectively). The Leydig cell neoplasms in rats are considered to be unlikely relevant to humans.
Mutagenesis
Naloxegol was not genotoxic in the in vitro bacterial reverse mutation (Ames) assay, mouse lymphoma TK+/- mutation assay, or the in vivo mouse micronucleus assay.
Impairment of Fertility
Naloxegol was found to have no effect on fertility or reproductive performance in male and female rats at oral doses up to 1000 mg/kg/day (greater than 1000 times the human AUC at the maximum recommended human dose).
-
14 CLINICAL STUDIES
The safety and efficacy of MOVANTIK were evaluated in two replicate, randomized, double-blind placebo-controlled trials (Study 1 and Study 2) in patients with opioid-induced constipation (OIC) and non-cancer related pain.
Patients receiving an opioid morphine equivalent daily dose of between 30 mg and 1,000 mg for at least four weeks before enrollment and self-reported OIC were eligible to participate. OIC was confirmed through a two-week run-in period and was defined as <3 spontaneous bowel movements (SBMs) per week on average with at least 25% of the SBMs associated with one or more of the following conditions: (1) straining, (2) hard or lumpy stools; and (3) having a sensation of incomplete evacuation. An SBM was defined as a bowel movement (BM) without rescue laxative taken within the past 24 hours. Patients with 0 BMs over the two-week run-in period or patients with an uneven distribution of SBMs across the two-week run-in period (0 SBMs in one week with ≥4 SBMs in the other week) were excluded. Throughout the studies (including the two-week run-in period), patients were prohibited from using laxatives other than bisacodyl rescue laxative (if they had not had a BM for 72 hours) and one-time use of an enema (if after 3 doses of bisacodyl, they still did not have a BM).
Patients suspected of having clinically important disruptions to the blood-brain barrier were not enrolled in these studies.
A total of 652 patients in Study 1 and 700 patients in Study 2 were randomized in a 1:1:1 ratio to receive 12.5 mg or 25 mg of MOVANTIK or placebo once daily for 12 weeks.
The mean age of the subjects in these two studies was 52 years, 10% and 13% were 65 years of age or older, 61% and 63% were women, and 78% and 80% were White in Studies 1 and 2, respectively.
Back pain was the most common reason for pain (56% and 57%); arthritis (10% and 10%) and joint pain (3% and 5%) were other prominent reasons in Studies 1 and 2, respectively. Prior to enrollment, patients had been using their current opioid for an average of 3.6 and 3.7 years. The patients who participated in Studies 1 and 2 were taking a wide range of opioids. The mean baseline opioid morphine equivalent daily dosage was 140 mg and 136 mg per day.
Use of one or more laxatives on at least one occasion within the two weeks prior to enrollment was reported by 71% of patients in both Studies 1 and 2.
The primary endpoint was response defined as: ≥3 SBMs per week and a change from baseline of ≥1 SBM per week for at least 9 out of the 12 study weeks and 3 out of the last 4 weeks.
There was a statistically significant difference for the 25 mg MOVANTIK treatment group versus placebo for the primary endpoint in Study 1 and Study 2 (see Table 3). Statistical significance for the 12.5 mg treatment group versus placebo was observed in Study 1 but not in Study 2 (see Table 3).
Table 3. Primary Endpoint: Response* (Studies 1 and 2) Study 1 Placebo
(N = 214)12.5 mg
(N = 213)25 mg
(N = 214)- * Response defined as: ≥3 SBMs per week and change from baseline of ≥1 SBM per week for at least 9 out of the 12 study weeks and 3 out of the last 4 weeks.
- † Statistically significant: p-values based on the Cochran-Mantel-Haenszel test.
Patients responding, n (%)
63 (29%)
87 (41%)
95 (44%)
Treatment Difference (MOVANTIK-Placebo)
--
11.4%
15.0%
95% Confidence Interval
--
(2.4%, 20.4%)
(5.9%, 24.0%)
p-value
--
0.015†
0.001†
Study 2
Placebo
(N = 232)12.5 mg
(N = 232)25 mg
(N = 232)Patients responding, n (%)
68 (29%)
81 (35%)
92 (40%)
Treatment Difference (MOVANTIK-Placebo)
--
5.6%
10.3%
95% Confidence Interval
--
(-2.9%, 14.1%)
(1.7%, 18.9%)
p-value
--
0.202
0.021†
One secondary endpoint in Study 1 and Study 2 was response in laxative users with OIC symptoms. This subgroup comprised 55% and 53% of total patients in these two studies, respectively. These patients (identified using an investigator-administered questionnaire), prior to enrollment, had reported using laxative(s) at least 4 out of the past 14 days with at least one of the following OIC symptoms of moderate, severe, or very severe intensity: incomplete bowel movements, hard stool, straining, or sensation of needing to pass a bowel movement but unable to do so. In this subgroup, in Studies 1 and 2, 42% and 50% reported using laxatives on a daily basis. The most frequently reported laxatives used on a daily basis were stool softeners (18% and 24%), stimulants (16% and 18%), and polyethylene glycol (6% and 5%). Use of two laxative classes was reported in 31% and 27% anytime during the 14 days prior to enrollment. The most commonly reported combination was stimulants and stool softeners (10% and 8%). In Study 1, a statistically significantly higher percentage of patients in this subgroup responded with MOVANTIK 12.5 mg compared to placebo (43% vs. 29%; p=0.03) and with MOVANTIK 25 mg compared to placebo (49% vs. 29%; p=0.002). In Study 2, a statistically significantly higher percentage of patients in this subgroup responded with MOVANTIK 25 mg compared to placebo (47% vs. 31%; p=0.01). This secondary endpoint was not tested for MOVANTIK 12.5 mg versus placebo in Study 2 because the primary endpoint was not statistically significant.
Another secondary endpoint was time to first post-dose SBM. The time to first post-dose SBM was significantly shorter with MOVANTIK 25 mg compared to placebo in both Study 1 (p <0.001) and Study 2 (p <0.001), and for MOVANTIK 12.5 mg as compared to placebo in Study 1 (p <0.001). For Study 1, the median times to first post-dose SBM were 6, 20, and 36 hours with MOVANTIK 25 mg, MOVANTIK 12.5 mg, and placebo, respectively. For Study 2, the median times to first post-dose SBM were 12 and 37 hours with MOVANTIK 25 mg and placebo, respectively. These analyses do not include the results for MOVANTIK 12.5 mg versus placebo in Study 2 because the primary endpoint was not statistically significant. In the two studies, 61-70% and 58% of patients receiving MOVANTIK 25 mg and MOVANTIK 12.5 mg, respectively, had an SBM within 24 hours of the first dose.
A third secondary endpoint was an evaluation of change from baseline between the treatment groups for mean number of days per week with at least 1 SBM but no more than 3 SBMs. There was a significant difference in number of days per week with 1 to 3 SBMs per day on average over 12 weeks between MOVANTIK 25 mg (Study 1 and Study 2) and MOVANTIK 12.5 mg (Study 1) and placebo.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
MOVANTIK (naloxegol) tablets are supplied as:
- NDC: 0310-1969-30: 12.5 mg, bottle of 30 tablets
- NDC: 0310-1969-90: 12.5 mg, bottle of 90 tablets
- NDC: 0310-1969-39: 12.5 mg, unit dose blister carton of 100 tablets (for HUD only)
- NDC: 0310-1970-30: 25 mg, bottle of 30 tablets
- NDC: 0310-1970-90: 25 mg, bottle of 90 tablets
- NDC: 0310-1970-39: 25 mg, unit dose blister carton of 100 tablets (for HUD only)
Storage
Store MOVANTIK at 20-25°C (68-77°F). Excursions permitted to 15-30°C (59-86°F) [see USP Controlled Room Temperature].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Administration
Advise patients to:
- Discontinue all maintenance laxative therapy prior to initiation of MOVANTIK. Laxative(s) can be used as needed if there is a suboptimal response to MOVANTIK after three days.
- Take MOVANTIK on an empty stomach at least 1 hour prior to the first meal of the day or 2 hours after the meal.
- Discontinue MOVANTIK if treatment with the opioid pain medication is also discontinued.
- Avoid consumption of grapefruit or grapefruit juice during treatment with MOVANTIK.
- Inform their healthcare provider if their opioid pain medication is discontinued.
- Inform their healthcare provider if they are unable to tolerate MOVANTIK, so a dosage adjustment can be considered.
If patients are unable to swallow the MOVANTIK tablet whole, the tablet can be crushed to a powder, mixed with water and administered orally or via a nasogastric (NG) tube, as described in the Medication Guide.
Drug Interactions
Advise patients to tell their healthcare provider when they start or stop taking any concomitant medications. Strong CYP3A4 inhibitors (e.g., clarithromycin, ketoconazole) are contraindicated with MOVANTIK, and other CYP3A4 enzyme modulating drugs can alter MOVANTIK exposure [see Contraindications (4) and Drug Interactions (7.1)].
Opioid Withdrawal
Advise patients that clusters of symptoms consistent with opioid withdrawal may occur while taking MOVANTIK, including sweating, chills, diarrhea, abdominal pain, anxiety, irritability, and yawning. Inform patients taking methadone as therapy for their pain condition that they may be more likely to have gastrointestinal adverse reactions such as abdominal pain and diarrhea that may be related to opioid withdrawal, than patients receiving other opioids [see Warnings and Precautions (5.1)].
Severe Abdominal Pain and/or Diarrhea
Advise patients that symptoms may occur after starting treatment. The patient should discontinue MOVANTIK and contact their healthcare provider if they develop severe abdominal pain and/or diarrhea [see Warnings and Precautions (5.2)].
Gastrointestinal Perforation
Advise patients to discontinue MOVANTIK and promptly seek medical attention if they develop unusually severe, persistent or worsening abdominal pain [see Warnings and Precautions (5.3)].
Pregnancy
Advise females of reproductive potential, who become pregnant or are planning to become pregnant that the use of MOVANTIK during pregnancy may precipitate opioid withdrawal in the pregnant women and the fetus [see Use in Specific Populations (8.1)].
Lactation
Advise females that breastfeeding is not recommended during treatment with MOVANTIK [see Use in Specific Populations (8.2)].
MOVANTIK is a registered trademark of the AstraZeneca group of companies.
Distributed by: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
© AstraZeneca 2020
-
MEDICATION GUIDE
MOVANTIK® (mo-van-tic)
(naloxegol) tablets, for oral use
Read this Medication Guide before you start taking MOVANTIK and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or your treatment.
What is the most important information I should know about MOVANTIK?
MOVANTIK may cause serious side effects, including:
- Opioid withdrawal. You may have symptoms of opioid withdrawal during treatment with MOVANTIK including sweating, chills, diarrhea, stomach pain, anxiety, irritability, and yawning. If you take methadone to treat your pain, you may be more likely to have stomach pain and diarrhea than people who do not take methadone. Tell your health care provider if you have any of these symptoms.
- Severe stomach pain or diarrhea, or both severe stomach pain and diarrhea. Severe stomach pain and diarrhea can happen when you take MOVANTIK. These problems can happen within a few days after you start taking MOVANTIK and can lead to hospitalization. Stop taking MOVANTIK and call your healthcare provider right away if you have severe stomach pain or diarrhea, or both severe stomach pain and diarrhea.
- Tear in your stomach or intestinal wall (perforation). Stomach pain that is severe can be a sign of a serious medical condition. If you have stomach pain that gets worse or does not go away, stop taking MOVANTIK and get emergency medical help right away.
What is MOVANTIK?
MOVANTIK is a prescription medicine used to treat constipation that is caused by prescription pain medicines called opioids, in adults with long-lasting (chronic) pain that is not caused by active cancer.
It is not known if MOVANTIK is safe and effective in children.
Who should not take MOVANTIK?
Do not take MOVANTIK if you:
- have a bowel blockage (intestinal obstruction) or have a history of bowel blockage.
- are allergic to MOVANTIK or any of the ingredients in MOVANTIK. See the end of this Medication Guide for a complete list of ingredients in MOVANTIK.
MOVANTIK can interact with certain other medicines and cause side effects, including opioid withdrawal symptoms such as sweating, chills, diarrhea, stomach pain, anxiety, irritability, and yawning. Tell your healthcare provider or pharmacist before you start or stop any medicines during treatment with MOVANTIK.
What should I tell my healthcare provider before taking MOVANTIK?
Before you take MOVANTIK, tell your healthcare provider about all of your medical conditions, including if you:
- have any stomach or bowel (intestines) problems, including inflammation in parts of the large intestine (diverticulitis), inflammation and injury of the intestines caused by reduced blood flow (ischemic colitis), or cancer of the stomach or bowel.
- have had recent surgery on the stomach or intestines.
- have kidney problems.
- have liver problems.
- are pregnant or plan to become pregnant. Taking MOVANTIK during pregnancy may cause opioid withdrawal symptoms in you or your unborn baby. Tell your healthcare provider right away if you become pregnant during treatment with MOVANTIK.
- are breastfeeding or plan to breastfeed. It is not known if MOVANTIK passes into your breast milk.
- taking MOVANTIK while you are breastfeeding may cause opioid withdrawal in your baby. You and your healthcare provider should decide if you will take MOVANTIK or breastfeed. You should not breastfeed if you take MOVANTIK.
Tell your healthcare provider about all of the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Other medicines may affect the way MOVANTIK works.
Especially tell your healthcare provider if you take a medicine called bevacizumab.
How should I take MOVANTIK?
- Take MOVANTIK exactly as your healthcare provider tells you.
- Take your prescribed dose of MOVANTIK 1 time each day, on an empty stomach, at least 1 hour before your first meal of the day or 2 hours after the meal.
-
If you cannot swallow MOVANTIK tablets whole, MOVANTIK can be mixed with water and taken by mouth or given through a nasogastric (NG) tube. To take MOVANTIK by mouth:
- ∘ crush the tablet to a powder
- ∘ place your dose of MOVANTIK in a glass that contains 4 ounces (120 mL) of water and stir
- ∘ swallow MOVANTIK and water mixture right away
- ∘ add 4 more ounces (120 mL) of water to the glass and drink right away, to make sure that you take your full dose of MOVANTIK.
-
If you cannot swallow MOVANTIK tablets and have a nasogastric (NG) tube, MOVANTIK may be given as follows:
- ∘ draw up 1 ounce (30 mL) of water into a 60 mL syringe and flush the NG tube
- ∘ crush the tablet to a powder
- ∘ place your dose of MOVANTIK in a container and mix with approximately 2 ounces (60 mL) of water
- ∘ draw up the MOVANTIK and water into the 60 mL syringe and give the mixture through the NG tube
- ∘ add approximately 2 ounces (60 mL) of water to the same container you used to prepare your dose of MOVANTIK
- ∘ draw up the water using the same 60 mL syringe and use all the water to flush the NG tube and any remaining medicine from the NG tube into the stomach
- Stop taking other laxatives before you start treatment with MOVANTIK. Your healthcare provider may prescribe other laxatives if MOVANTIK does not work after 3 days of treatment.
- MOVANTIK has been shown to be effective in people who have taken opioid pain medicines for at least 4 weeks.
- Tell your healthcare provider if you stop taking your opioid pain medicine. If you stop taking your opioid pain medicine, you should also stop taking MOVANTIK.
- If you take too much MOVANTIK, call your healthcare provider or go to the nearest emergency room right away.
What should I avoid while taking MOVANTIK?
- Avoid eating grapefruit or drinking grapefruit juice during treatment with MOVANTIK.
What are the possible side effects of MOVANTIK?
See “What is the most important information I should know about MOVANTIK?”
The most common side effects of MOVANTIK include: stomach (abdomen) pain, diarrhea, nausea, gas, vomiting, and headache.
Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all the possible side effects of MOVANTIK.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store MOVANTIK?
- Store MOVANTIK at room temperature between 68°F to 77°F (20°C to 25°C).
- Safely throw away medicine that is out of date or that you no longer need.
Keep MOVANTIK and all medicines out of the reach of children.
General information about the safe and effective use of MOVANTIK
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use MOVANTIK for a condition for which it was not prescribed. Do not give MOVANTIK to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about MOVANTIK that is written for health professionals.
What are the ingredients in MOVANTIK?
Active ingredient: naloxegol oxalate
Inactive ingredients: The tablet core contains mannitol, cellulose microcrystalline, croscarmellose sodium, magnesium stearate, and propyl gallate. The tablet coat contains hypromellose, titanium dioxide, polyethylene glycol, iron oxide red, and iron oxide black.
Distributed by: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
MOVANTIK is a registered trademark of the AstraZeneca Group of companies. © 2018 AstraZeneca Pharmaceuticals LP. All rights reserved. For more information, go to www.MOVANTIK.com or call 1-800-236-9933.
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 04/2020
-
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL – 12.5
NDC: 0310-1969-30 30 Tablets
movantik®
(naloxegol) Tablets
12.5 mg*
*Each tablet contains 14.2 mg
naloxegol oxalate
Dispense the accompanying
Medication Guide to each patient.
Rx only
AstraZeneca
-
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL – 25 mg
NDC: 0310-1970-30 30 Tablets
movantik®
(naloxegol) Tablets
25 mg*
*Each tablet contains 28.5 mg
naloxegol oxalate
Dispense the accompanying
Medication Guide to each patient.
Rx only
AstraZeneca
-
INGREDIENTS AND APPEARANCE
MOVANTIK
naloxegol oxalate tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0310-1969 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength NALOXEGOL OXALATE (UNII: 65I14TNM33) (NALOXEGOL - UNII:44T7335BKE) NALOXEGOL 12.5 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) PROPYL GALLATE (UNII: 8D4SNN7V92) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) FERRIC OXIDE RED (UNII: 1K09F3G675) FERROSOFERRIC OXIDE (UNII: XM0M87F357) WATER (UNII: 059QF0KO0R) Product Characteristics Color PINK (mauve) Score no score Shape OVAL (biconvex) Size 10mm Flavor Imprint Code nGL;12;5 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0310-1969-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 03/06/2015 2 NDC: 0310-1969-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 03/06/2015 3 NDC: 0310-1969-39 10 in 1 CARTON 03/06/2015 3 10 in 1 BLISTER PACK; Type 0: Not a Combination Product 4 NDC: 0310-1969-95 1 in 1 CARTON 08/01/2018 4 3 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA204760 03/06/2015 MOVANTIK
naloxegol oxalate tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0310-1970 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength NALOXEGOL OXALATE (UNII: 65I14TNM33) (NALOXEGOL - UNII:44T7335BKE) NALOXEGOL 25 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) PROPYL GALLATE (UNII: 8D4SNN7V92) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) FERRIC OXIDE RED (UNII: 1K09F3G675) FERROSOFERRIC OXIDE (UNII: XM0M87F357) WATER (UNII: 059QF0KO0R) Product Characteristics Color PINK (mauve) Score no score Shape OVAL (biconvex) Size 13mm Flavor Imprint Code nGL;25 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0310-1970-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 03/06/2015 2 NDC: 0310-1970-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 03/06/2015 3 NDC: 0310-1970-39 10 in 1 CARTON 03/06/2015 3 10 in 1 BLISTER PACK; Type 0: Not a Combination Product 4 NDC: 0310-1970-95 1 in 1 CARTON 03/06/2015 4 3 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA204760 03/06/2015 Labeler - AstraZeneca Pharmaceuticals LP (054743190) Registrant - AstraZeneca PLC (230790719)
Trademark Results [MOVANTIK]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 MOVANTIK 86205691 4782846 Live/Registered |
AstraZeneca AB 2014-02-27 |
 MOVANTIK 86089235 4600146 Live/Registered |
AstraZeneca AB 2013-10-11 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.