PREDNISOLONE SODIUM PHOSPHATE- prednisolone tablet, orally disintegrating
Prednisolone Sodium Phosphate by
Drug Labeling and Warnings
Prednisolone Sodium Phosphate by is a Prescription medication manufactured, distributed, or labeled by Mylan Pharmaceuticals Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use PREDNISOLONE SODIUM PHOSPHATE ORALLY DISINTEGRATING TABLETS safely and effectively. See full prescribing information for PREDNISOLONE SODIUM PHOSPHATE ORALLY DISINTEGRATING TABLETS.
PREDNISOLONE SODIUM PHOSPHATE orally disintegrating tablets
Initial U.S. Approval: 1955RECENT MAJOR CHANGES
Warnings and Precautions, Embryo-Fetal Toxicity (5.10) 3/2020
INDICATIONS AND USAGE
Prednisolone sodium phosphate orally disintegrating tablets are a corticosteroid indicated
- as an anti-inflammatory or immunosuppressive agent for certain allergic, dermatologic, gastrointestinal, hematologic, ophthalmologic, nervous system, renal, respiratory, rheumatologic, specific infectious diseases or conditions and organ transplantation (1)
- for the treatment of certain endocrine conditions (1)
- for palliation of certain neoplastic conditions (1)
DOSAGE AND ADMINISTRATION
Individualize dosing based on disease severity and patient response:
- Initial Dose: 10 mg to 60 mg of prednisolone (as 13.4 mg to 80.6 mg of prednisolone sodium phosphate) (2)
- Maintenance Dose: Use lowest dosage that will maintain an adequate clinical response (2)
- Discontinuation: Withdraw gradually if discontinuing long-term or high-dose therapy (2)
- Take with food to avoid gastrointestinal (GI) irritation (2)
DO NOT BREAK OR USE PARTIAL PREDNISOLONE SODIUM PHOSPHATE ORALLY DISINTEGRATING TABLETS. USE AN APPROPRIATE FORMULATION OF PREDNISOLONE IF INDICATED DOSE CANNOT BE OBTAINED USING PREDNISOLONE SODIUM PHOSPHATE ORALLY DISINTEGRATING TABLETS. (2)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
- Hypersensitivity to prednisolone or any components of this product. (4)
WARNINGS AND PRECAUTIONS
- Hypothalamic-pituitary-adrenal (HPA) axis suppression, Cushing's syndrome and hyperglycemia: Monitor patients for these conditions with chronic use. Taper doses gradually for withdrawal after chronic use. (5.1)
- Infections: Increased susceptibility to new infection and increased risk of exacerbation, dissemination, or reactivation of latent infection. Signs and symptoms of infection may be masked. (5.2)
- Elevated blood pressure, salt and water retention and hypokalemia: Monitor blood pressure and sodium, potassium serum levels. (5.3)
- GI perforation: increased risk in patients with certain GI disorders. Signs and symptoms may be masked. (5.4)
- Behavioral and mood disturbances: May include euphoria, insomnia, mood swings, personality changes, severe depression, and psychosis. Existing conditions may be aggravated. (5.5)
- Decreases in bone density: Monitor bone density in patients receiving long term corticosteroid therapy. (5.6)
- Ophthalmic effects: May include cataracts, infections and glaucoma. Monitor intraocular pressure if corticosteroid therapy is continued for more than 6 weeks. (5.7)
- Live or live attenuated vaccines: Do not administer to patients receiving immunosuppressive doses of corticosteroids. (5.8)
- Negative effects on growth and development: Monitor pediatric patients on long-term corticosteroid therapy. (5.9)
- Embryo-Fetal Toxicity: Can cause fetal harm with first trimester use. Advise patients of potential harm to the fetus. (5.10)
ADVERSE REACTIONS
Common adverse reactions for corticosteroids include fluid retention, alteration in glucose tolerance, elevation in blood pressure, behavioral and mood changes, increased appetite and weight gain. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Mylan at 1-877-446-3679 (1-877-4-INFO-RX) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Anticoagulant Agents: May enhance or diminish anticoagulant effects. Monitor coagulation indices. (7)
- Antidiabetic Agents: May increase blood glucose concentrations. Dose adjustments of antidiabetic agents may be required. (7)
- CYP 3A4 inducers and inhibitors: May, respectively, increase or decrease clearance of corticosteroids, necessitating dose adjustment. (7)
- Cyclosporine: Increase in activity of both, cyclosporine and corticosteroid when administered concurrently. Convulsions have been reported with concurrent use. (7)
- NSAIDS including aspirin and salicylates: Increased risk of gastrointestinal side effects. (7)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 3/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Allergic Conditions
1.2 Dermatologic Diseases
1.3 Endocrine Conditions
1.4 Gastrointestinal Diseases
1.5 Hematologic Diseases
1.6 Neoplastic Conditions
1.7 Nervous System Conditions
1.8 Ophthalmic Conditions
1.9 Conditions Related to Organ Transplantation
1.10 Pulmonary Diseases
1.11 Renal Conditions
1.12 Rheumatologic Conditions
1.13 Specific Infectious Diseases
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
2.2 Recommended Monitoring
2.3 Corticosteroid Comparison Chart
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Alterations in Endocrine Function
5.2 Increased Risks Related to Infections
5.3 Alterations in Cardiovascular/Renal Function
5.4 Use in Patients with Gastrointestinal Disorders
5.5 Behavioral and Mood Disturbances
5.6 Decrease in Bone Density
5.7 Ophthalmic Effects
5.8 Vaccination
5.9 Effect on Growth and Development
5.10 Embryo-Fetal Toxicity
5.11 Neuromuscular Effects
5.12 Kaposi’s Sarcoma
6 ADVERSE REACTIONS
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
Prednisolone sodium phosphate orally disintegrating tablets are indicated in the treatment of the following diseases or conditions:
1.1 Allergic Conditions
Control of severe or incapacitating allergic conditions intractable to adequate trials of conventional treatment in adult and pediatric populations with:
- Atopic dermatitis
- Drug hypersensitivity reactions
- Seasonal or perennial allergic rhinitis
- Serum sickness
1.2 Dermatologic Diseases
- Bullous dermatitis herpetiformis
- Contact dermatitis
- Exfoliative erythroderma
- Mycosis fungoides
- Pemphigus
- Severe erythema multiforme (Stevens-Johnson syndrome)
1.3 Endocrine Conditions
- Congenital adrenal hyperplasia
- Hypercalcemia of malignancy
- Nonsuppurative thyroiditis
- Primary or secondary adrenocortical insufficiency: hydrocortisone or cortisone is the first choice; synthetic analogs may be used in conjunction with mineralocorticoids where applicable.
1.5 Hematologic Diseases
- Acquired (autoimmune) hemolytic anemia
- Diamond-Blackfan anemia
- Idiopathic thrombocytopenic purpura in adults
- Pure red cell aplasia
- Secondary thrombocytopenia in adults
1.7 Nervous System Conditions
- Acute exacerbations of multiple sclerosis
- Cerebral edema associated with primary or metastatic brain tumor, craniotomy or head injury
1.8 Ophthalmic Conditions
- Sympathetic ophthalmia
- Uveitis and ocular inflammatory conditions unresponsive to topical corticosteroids
1.10 Pulmonary Diseases
- Acute exacerbations of chronic obstructive pulmonary disease (COPD)
- Allergic bronchopulmonary aspergillosis
- Aspiration pneumonitis
- Asthma
- Fulminating or disseminated pulmonary tuberculosis when used concurrently with appropriate chemotherapy
- Hypersensitivity pneumonitis
- Idiopathic bronchiolitis obliterans with organizing pneumonia
- Idiopathic eosinophilic pneumonias
- Idiopathic pulmonary fibrosis Pneumocystis carinii pneumonia (PCP) associated with hypoxemia occurring in an HIV (+) individual who is also under treatment with appropriate anti-PCP antibiotics
- Symptomatic sarcoidosis
1.11 Renal Conditions
To induce a diuresis or remission of proteinuria in nephrotic syndrome, without uremia, of the idiopathic type or that due to lupus erythematosus
1.12 Rheumatologic Conditions
As adjunctive therapy for short term administration (to tide the patient over an acute episode or exacerbation) in:
- Acute gouty arthritis
During an exacerbation or as maintenance therapy in selected cases of:
- Ankylosing spondylitis
- Dermatomyositis/polymyositis
- Polymyalgia rheumatica/temporal arteritis
- Psoriatic arthritis
- Relapsing polychondritis
- Rheumatoid arthritis, including juvenile rheumatoid arthritis (selected cases may require low dose maintenance therapy)
- Sjogren’s syndrome
- Systemic lupus erythematosus
- Vasculitis
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
Dosage of prednisolone sodium phosphate orally disintegrating tablets should be individualized according to the severity of the disease and the response of the patient. For pediatric patients, the recommended dosage should be governed by the same considerations rather than strict adherence to the ratio indicated by age or body weight.
Do not break or use partial prednisolone sodium phosphate orally disintegrating tablets. Use an appropriate formulation of prednisolone if indicated dose cannot be obtained using prednisolone sodium phosphate orally disintegrating tablets. This may become important in the treatment of conditions that require tapering doses that cannot be adequately accommodated by prednisolone sodium phosphate orally disintegrating tablets, e.g., tapering the dose below 10 mg.
The initial dose of prednisolone sodium phosphate orally disintegrating tablets may vary from 10 to 60 mg (prednisolone base) per day, depending on the specific disease entity being treated. In situations of less severity, lower doses will generally suffice while in selected patients higher initial doses may be required. The initial dosage should be maintained or adjusted until a satisfactory response is noted. If after a reasonable period of time, there is a lack of satisfactory clinical response, prednisolone sodium phosphate should be discontinued and the patient placed on other appropriate therapy. IT SHOULD BE EMPHASIZED THAT DOSAGE REQUIREMENTS ARE VARIABLE AND MUST BE INDIVIDUALIZED ON THE BASIS OF THE DISEASE UNDER TREATMENT AND THE RESPONSE OF THE PATIENT. After a favorable response is noted, the proper maintenance dosage should be determined by decreasing the initial drug dosage in small decrements at appropriate time intervals until the lowest dosage that will maintain an adequate clinical response is reached. It should be kept in mind that constant monitoring is needed in regard to drug dosage. Included in the situations which may make dosage adjustments necessary are changes in clinical status secondary to remissions or exacerbations in the disease process, the patient’s individual drug responsiveness, and the effect of patient exposure to stressful situations not directly related to the disease entity under treatment; in this latter situation it may be necessary to increase the dosage of prednisolone sodium phosphate orally disintegrating tablets for a period of time consistent with the patient’s condition. If after long term therapy the drug is to be stopped, it is recommended that it be withdrawn gradually rather than abruptly.
Prednisolone sodium phosphate orally disintegrating tablets are packaged in a bottle. For patients dispensed a bottle, they should be instructed not to remove the tablet from the bottle until just prior to dosing. The bottle should then be opened, and the orally disintegrating tablet placed on the tongue, where tablets may be swallowed whole as any conventional tablet, or allowed to dissolve in the mouth, with or without the assistance of water. Orally disintegrating tablet dosage forms are friable and are not intended to be cut, split, or broken.
Multiple Sclerosis
In the treatment of acute exacerbations of multiple sclerosis, daily doses of 200 mg of prednisolone for a week followed by 80 mg every other day for one month have been shown to be effective.
Pediatric
In pediatric patients, the initial dose of prednisolone sodium phosphate may vary depending on the specific disease entity being treated. The range of initial doses is 0.14 to 2 mg/kg/day in three or four divided doses (4 to 60 mg/m2 bsa/day).
Nephrotic Syndrome
The standard regimen used to treat nephrotic syndrome in pediatric patients is 60 mg/m2/day given in three divided doses for 4 weeks, followed by 4 weeks of single dose alternate-day therapy at 40 mg/m2/day.
Asthma
The National Heart, Lung, and Blood Institute (NHLBI) recommended dosing for systemic prednisone, prednisolone or methylprednisolone in children whose asthma is uncontrolled by inhaled corticosteroids and long-acting bronchodilators is 1-2 mg/kg/day in single or divided doses.
It is further recommended that short course, or “burst” therapy, be continued until a child achieves a peak expiratory flow rate of 80% of his or her personal best or symptoms resolve. This usually requires 3 to 10 days of treatment, although it can take longer. There is no evidence that tapering the dose after improvement will prevent a relapse.
2.2 Recommended Monitoring
Blood pressure, body weight, routine laboratory studies, including serum potassium and fasting blood glucose, should be obtained at regular intervals during prolonged therapy. Appropriate diagnostic studies should be performed in patients with known or suspected peptic ulcer disease and in patients at risk for reactivation of latent tuberculosis infections.
2.3 Corticosteroid Comparison Chart
For the purpose of comparison, one 10 mg prednisolone sodium phosphate orally disintegrating tablet (13.4 mg prednisolone sodium phosphate) is equivalent to the following milligram dosage of the various glucocorticoids:
Betamethasone 1.75 mg
Paramethasone 4 mg
Cortisone 50 mg
Prednisolone 10 mg
Dexamethasone 1.75 mg
Prednisone 10 mg
Hydrocortisone 40 mg
Triamcinolone 8 mg
Methylprednisolone 8 mg
These dose relationships apply only to oral or intravenous administration of these compounds. When these substances or their derivatives are injected intramuscularly or into joint spaces, their relative properties may be greatly altered.
-
3 DOSAGE FORMS AND STRENGTHS
Prednisolone Sodium Phosphate Orally Disintegrating Tablets are available containing 13.4 mg, 20.2 mg or 40.3 mg of prednisolone sodium phosphate, USP equivalent to 10 mg, 15 mg or 30 mg of prednisolone.
- The 10 mg tablets are white to off-white, speckled, round, unscored tablets debossed with P10 on one side of the tablet and M on the other side.
- The 15 mg tablets are white to off-white, speckled, round, unscored tablets debossed with P15 on one side of the tablet and M on the other side.
- The 30 mg tablets are white to off-white, speckled, round, unscored tablets debossed with P30 on one side of the tablet and M on the other side.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Alterations in Endocrine Function
Hypothalamic-pituitary-adrenal (HPA) axis suppression, Cushing's syndrome, and hyperglycemia. Monitor patients for these conditions with chronic use.
Corticosteroids can produce reversible HPA axis suppression with the potential for glucocorticosteroid insufficiency after withdrawal of treatment. Drug induced secondary adrenocortical insufficiency may be minimized by gradual reduction of dosage. This type of relative insufficiency may persist for months after discontinuation of therapy; therefore, in any situation of stress occurring during that period, hormone therapy should be reinstituted.
Since mineralocorticoid secretion may be impaired, salt and/or a mineralocorticoid should be administered concurrently. Mineralocorticoid supplementation is of particular importance in infancy.
Metabolic clearance of corticosteroids is decreased in hypothyroid patients and increased in hyperthyroid patients. Changes in thyroid status of the patient may necessitate adjustment in dosage.
5.2 Increased Risks Related to Infections
Corticosteroids may increase the risks related to infections with any pathogen, including viral, bacterial, fungal, protozoan, or helminthic infections. The degree to which the dose, route and duration of corticosteroid administration correlates with the specific risks of infection is not well characterized, however, with increasing doses of corticosteroids, the rate of occurrence of infectious complications increases.
Corticosteroids may mask some signs of infection and may reduce resistance to new infections.
Corticosteroids may exacerbate infections and increase risk of disseminated infection. The use of prednisolone sodium phosphate in active tuberculosis should be restricted to those cases of fulminating or disseminated tuberculosis in which the corticosteroid is used for the management of the disease in conjunction with an appropriate antituberculous regimen.
Chickenpox and measles can have a more serious or even fatal course in non-immune children or adults on corticosteroids. In children or adults who have not had these diseases, particular care should be taken to avoid exposure. If a patient is exposed to chickenpox, prophylaxis with varicella zoster immune globulin (VZIG) may be indicated. If patient is exposed to measles, prophylaxis with pooled intramuscular immunoglobulin (IG) may be indicated. If chickenpox develops, treatment with antiviral agents may be considered.
Corticosteroids should be used with great care in patients with known or suspected Strongyloides (threadworm) infestation. In such patients, corticosteroid-induced immunosuppression may lead to Strongyloides hyperinfection and dissemination with widespread larval migration, often accompanied by severe enterocolitis and potentially fatal gram-negative septicemia.
Corticosteroids may exacerbate systemic fungal infections and therefore should not be used in the presence of such infections unless they are needed to control drug reactions.
Corticosteroids may increase risk of reactivation or exacerbation of latent infection. If corticosteroids are indicated in patients with latent tuberculosis or tuberculin reactivity, close observation is necessary as reactivation of the disease may occur. During prolonged corticosteroid therapy, these patients should receive chemoprophylaxis.
Corticosteroids may activate latent amebiasis. Therefore, it is recommended that latent or active amebiasis be ruled out before initiating corticosteroid therapy in any patient who has spent time in the tropics or in any patient with unexplained diarrhea.
Corticosteroids should not be used in cerebral malaria.
5.3 Alterations in Cardiovascular/Renal Function
Corticosteroids can cause elevation of blood pressure, salt and water retention, and increased excretion of potassium and calcium. These effects are less likely to occur with the synthetic derivatives except when used in large doses. Dietary salt restriction and potassium supplementation may be necessary. These agents should be used with caution in patients with hypertension, congestive heart failure, or renal insufficiency.
Literature reports suggest an association between use of corticosteroids and left ventricular free wall rupture after a recent myocardial infarction; therefore, therapy with corticosteroids should be used with caution in these patients.
5.4 Use in Patients with Gastrointestinal Disorders
There is an increased risk of gastrointestinal (GI) perforation in patients with certain GI disorders. Signs of GI perforation, such as peritoneal irritation, may be masked in patients receiving corticosteroids.
Corticosteroids should be used with caution if there is a probability of impending perforation, abscess or other pyogenic infections; diverticulitis; fresh intestinal anastomoses; and active or latent peptic ulcer.
5.5 Behavioral and Mood Disturbances
Corticosteroid use may be associated with central nervous system effects ranging from euphoria, insomnia, mood swings, personality changes, and severe depression, to frank psychotic manifestations. Also, existing emotional instability or psychotic tendencies may be aggravated by corticosteroids.
5.6 Decrease in Bone Density
Corticosteroids decrease bone formation and increase bone resorption both through their effect on calcium regulation (i.e., decreasing absorption and increasing excretion) and inhibition of osteoblast function. This, together with a decrease in the protein matrix of the bone secondary to an increase in protein catabolism, and reduced sex hormone production, may lead to inhibition of bone growth in children and adolescents and the development of osteoporosis at any age. Special consideration should be given to patients at increased risk of osteoporosis (e.g., postmenopausal women) before initiating corticosteroid therapy and bone density should be monitored in patients on long term corticosteroid therapy.
5.7 Ophthalmic Effects
Prolonged use of corticosteroids may produce posterior subcapsular cataracts, glaucoma with possible damage to the optic nerves, and may enhance the establishment of secondary ocular infections due to fungi or viruses.
The use of oral corticosteroids is not recommended in the treatment of optic neuritis and may lead to an increase in the risk of new episodes.
Intraocular pressure may become elevated in some individuals. If steroid therapy is continued for more than 6 weeks, intraocular pressure should be monitored.
5.8 Vaccination
Administration of live or live attenuated vaccines is contraindicated in patients receiving immunosuppressive doses of corticosteroids. Killed or inactivated vaccines may be administered; however, the response to such vaccines cannot be predicted. Immunization procedures may be undertaken in patients who are receiving corticosteroids as replacement therapy, e.g., for Addison's disease.
While on corticosteroid therapy, patients should not be vaccinated against smallpox. Other immunization procedures should not be undertaken in patients who are on corticosteroids, especially on high dose, because of possible hazards of neurological complications and a lack of antibody response.
5.9 Effect on Growth and Development
Long-term use of corticosteroids can have negative effects on growth and development in children. Growth and development of pediatric patients on prolonged corticosteroid therapy should be carefully monitored.
5.10 Embryo-Fetal Toxicity
Prednisolone can cause fetal harm when administered to a pregnant woman. Human studies suggest a small but inconsistent increased risk of orofacial clefts with use of corticosteroids during the first trimester of pregnancy. Published animal studies show prednisolone to be teratogenic in rats, rabbits, hamsters, and mice with increased incidence of cleft palate in offspring. Intrauterine growth restriction and decreased birth weight have also been reported with corticosteroid use during pregnancy, however, the underlying maternal condition may also contribute to these risks. If this drug is used during pregnancy, or if the patient becomes pregnant while using this drug, advise the patient about the potential harm to the fetus [see Use in Specific Populations (8.1)].
5.11 Neuromuscular Effects
Although controlled clinical trials have shown corticosteroids to be effective in speeding the resolution of acute exacerbations of multiple sclerosis, they do not show that they affect the ultimate outcome or natural history of the disease. The studies do show that relatively high doses of corticosteroids are necessary to demonstrate a significant effect [see Dosage and Administration (3)].
An acute myopathy has been observed with the use of high doses of corticosteroids, most often occurring in patients with disorders of neuromuscular transmission (e.g., myasthenia gravis), or in patients receiving concomitant therapy with neuromuscular blocking drugs (e.g., pancuronium). This acute myopathy is generalized, may involve ocular and respiratory muscles, and may result in quadriparesis. Elevation of creatinine kinase may occur. Clinical improvement or recovery after stopping corticosteroids may require weeks to years.
-
6 ADVERSE REACTIONS
Common adverse reactions for corticosteroids include fluid retention, alteration in glucose tolerance, elevation in blood pressure, behavioral and mood changes, increased appetite and weight gain.
Allergic Reactions: Anaphylactoid reaction, anaphylaxis, angioedema
Cardiovascular: Bradycardia, cardiac arrest, cardiac arrhythmias, cardiac enlargement, circulatory collapse, congestive heart failure, fat embolism, hypertension, hypertrophic cardiomyopathy in premature infants, myocardial rupture following recent myocardial infarction, pulmonary edema, syncope, tachycardia, thromboembolism, thrombophlebitis, vasculitis
Dermatologic: Acne, allergic dermatitis, cutaneous and subcutaneous atrophy, dry scalp, edema, facial erythema, hyper or hypo-pigmentation, impaired wound healing, increased sweating, petechiae and ecchymoses, rash, sterile abscess, striae, suppressed reactions to skin tests, thin fragile skin, thinning scalp hair, urticaria
Endocrine: Abnormal fat deposits, decreased carbohydrate tolerance, development of Cushingoid state, hirsutism, manifestations of latent diabetes mellitus and increased requirements for insulin or oral hypoglycemic agents in diabetics, menstrual irregularities, moon facies, secondary adrenocortical and pituitary unresponsiveness (particularly in times of stress, as in trauma, surgery or illness), suppression of growth in children
Fluid and Electrolyte Disturbances: Fluid retention, potassium loss, hypertension, hypokalemic alkalosis, sodium retention
Gastrointestinal: Abdominal distention; elevation in serum liver enzyme levels (usually reversible upon discontinuation); hepatomegaly, hiccups, malaise, nausea, pancreatitis; peptic ulcer with possible perforation and hemorrhage; ulcerative esophagitis
General: Increased appetite and weight gain
Metabolic: Negative nitrogen balance due to protein catabolism
Musculoskeletal: Aseptic necrosis of femoral and humeral heads; charcot-like arthropathy, loss of muscle mass; muscle weakness; osteoporosis; pathologic fracture of long bones; steroid myopathy; tendon rupture; vertebral compression fractures
Neurological: Arachnoiditis, convulsions; depression, emotional instability, euphoria, headache; increased intracranial pressure with papilledema (pseudotumor cerebri) usually following discontinuation of treatment; insomnia, meningitis, mood swings, neuritis, neuropathy, paraparesis/paraplegia, paresthesia, personality changes, sensory disturbances, vertigo
Ophthalmic: Exophthalmos; glaucoma; increased intraocular pressure; posterior subcapsular cataracts
Reproductive: Alteration in motility and number of spermatozoa
Postmarketing Experience: Adverse reactions have been identified during post approval use of prednisolone sodium phosphate orally disintegrating tablets. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. The postmarketing experience has not raised new safety concerns beyond those already established for immediate-release prednisolone.
-
7 DRUG INTERACTIONS
- Aminoglutethimide: Aminoglutethimide may lead to loss of corticosteroid-induced adrenal suppression.
- Amphotericin B: There have been cases reported in which concomitant use of Amphotericin B and hydrocortisone was followed by cardiac enlargement and congestive heart failure (see also Potassium depleting agents).
- Anticholinesterase agents: Concomitant use of anticholinesterase agents and corticosteroids may produce severe weakness in patients with myasthenia gravis. If possible, anticholinesterase agents should be withdrawn at least 24 hours before initiating corticosteroid therapy.
- Anticoagulant agents: Co-administration of corticosteroids and warfarin usually results in inhibition of response to warfarin, although there have been some conflicting reports. Therefore, coagulation indices should be monitored frequently to maintain the desired anticoagulant effect.
- Antidiabetic Agents: Because corticosteroids may increase blood glucose concentrations, dosage adjustments of antidiabetic agents may be required.
- Antitubercular drugs: Serum concentrations of isoniazid may be decreased.
- CYP 3A4 inducers (e.g., barbiturates, phenytoin, carbamazepine and rifampin): Drugs such as barbiturates, phenytoin, ephedrine, and rifampin, which induce hepatic microsomal drug metabolizing enzyme activity may enhance metabolism of prednisolone and require that the dosage of prednisolone sodium phosphate be increased.
- CYP 3A4 inhibitors (e.g., ketoconazole, macrolide antibiotics): Ketoconazole has been reported to decrease the metabolism of certain corticosteroids by up to 60% leading to an increased risk of corticosteroid side effects.
- Cholestyramine: Cholestyramine may increase the clearance of corticosteroids.
- Cyclosporine: Increased activity of both cyclosporine and corticosteroids may occur when the two are used concurrently. Convulsions have been reported with this concurrent use.
- Digitalis: Patients on digitalis glycosides may be at increased risk of arrhythmias due to hypokalemia.
- Estrogens, including oral contraceptives: Estrogens may decrease the hepatic metabolism of certain corticosteroids thereby increasing their effect.
- NSAIDS, including aspirin and salicylates: Concomitant use of aspirin or other non-steroidal anti-inflammatory agents and corticosteroids increases the risk of gastrointestinal side effects. Aspirin should be used cautiously in conjunction with corticosteroids in hypoprothrombinemia. The clearance of salicylates may be increased with concurrent use of corticosteroids.
- Potassium-depleting agents (e.g., diuretics, Amphotericin B): When corticosteroids are administered concomitantly with potassium-depleting agents, patients should be observed closely for development of hypokalemia.
- Skin Tests: Corticosteroids may suppress reactions to skin tests.
- Toxoids and live or inactivated Vaccines: Due to inhibition of antibody response, patients on prolonged corticosteroid therapy may exhibit a diminished response to toxoids and live or inactivated vaccines. Corticosteroids may also potentiate the replication of some organisms contained in live attenuated vaccines.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from human and animal studies, corticosteroids including prednisolone sodium phosphate, can cause fetal harm when administered to a pregnant woman (see Data) [see Warnings and Precautions (5.10)]. Published epidemiological studies suggest a small but inconsistent increased risk of orofacial clefts with use of corticosteroids during the first trimester. Intrauterine growth restriction and decreased birth weight have also been reported with maternal use of corticosteroids during pregnancy; however, the underlying maternal condition may also contribute to these risks (see Clinical Considerations). Published animal studies show prednisolone to be teratogenic in rats, rabbits, hamsters, and mice with increased incidence of cleft palate in offspring (see Data). Advise a pregnant woman about the potential harm to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriages in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Infants born to pregnant women who have received corticosteroids should be carefully monitored for signs and symptoms of hypoadrenalism [see Warnings and Precautions (5.1)].
Data
Human Data
Published epidemiological studies on the association between prednisolone and fetal outcomes have reported inconsistent findings and have important methodological limitations. Multiple cohort and case-controlled studies in humans report that maternal corticosteroid use during the first trimester increases the incidence of cleft lip with or without cleft palate from about 1/1000 infants to 3-5/1000 infants; however, a risk for orofacial clefts has not been reported in all studies. Methodological limitations of these studies include non-randomized design, retrospective data collection, and the inability to control for confounders such as underlying maternal disease and use of concomitant medications.
Two prospective case control studies showed decreased birth weight in infants exposed to maternal corticosteroids in utero. In humans, the risk of decreased birth weight appears to be dose related and may be minimized by administering lower corticosteroid doses. It is likely that underlying maternal conditions contribute to intrauterine growth restriction and decreased birth weight, but it is unclear to what extent these maternal conditions contribute to the increased risk of orofacial clefts.
Animal Data
Published literature indicates prednisolone has been shown to be teratogenic in rats, rabbits, hamsters, and mice with increased incidence of cleft palate in offspring, supportive of the clinical data. In teratogenicity studies, cleft palate along with an elevation of fetal lethality (or increase in resorptions) and reductions in fetal body weight occurred in rats at maternal doses of 30 mg/kg (equivalent to 290 mg in a 60 kg individual based on mg/m2 body surface comparison) and higher. Cleft palate was observed in mice at a maternal dose of 20 mg/kg (equivalent to 100 mg in a 60 kg individual based on mg/m2 comparison). Additionally, constriction of the ductus arteriosus was observed in fetuses of pregnant rats exposed to prednisolone.
8.2 Lactation
Risk Summary
Prednisolone is present in human milk. Published reports suggest infant daily doses are estimated to be less than 1% of the maternal daily dose. No adverse effects in the breastfed infant have been reported following maternal administration of prednisolone during breastfeeding. There are no available data on the effects of prednisolone on milk production. High doses of corticosteroids administered to lactating women for long periods could potentially produce problems in the breastfed infant including growth and development and interfere with endogenous corticosteroid production (see Clinical Considerations) [see Use in Specific Populations (8.4)]. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for prednisolone sodium phosphate and any potential adverse effects on the breastfed child from prednisolone sodium phosphate or from the mother’s underlying condition.
8.4 Pediatric Use
The efficacy and safety of prednisolone in the pediatric population are based on the well-established course of effect of corticosteroids, which is similar in pediatric and adult populations. Published studies provide evidence of efficacy and safety in pediatric patients for the treatment of nephrotic syndrome (> 2 years of age), and aggressive lymphomas and leukemias (> 1 month of age). However, some of these conclusions and other indications for pediatric use of corticosteroid, e.g., severe asthma and wheezing, are based on adequate and well-controlled trials conducted in adults, on the premises that the course of the diseases and their pathophysiology are considered to be substantially similar in both populations.
The adverse effects of prednisolone in pediatric patients are similar to those in adults [see Adverse Reactions (6)]. Like adults, pediatric patients should be carefully observed with frequent measurements of blood pressure, weight, height, intraocular pressure, and clinical evaluation for the presence of infection, psychosocial disturbances, thromboembolism, peptic ulcers, cataracts, and osteoporosis. Children, who are treated with corticosteroids by any route, including systemically administered corticosteroids, may experience a decrease in their growth velocity. This negative impact of corticosteroids on growth has been observed at low systemic doses and in the absence of laboratory evidence of HPA axis suppression (i.e., cosyntropin stimulation and basal cortisol plasma levels).
Growth velocity may therefore be a more sensitive indicator of systemic corticosteroid exposure in children than some commonly used tests of HPA axis function. The linear growth of children treated with corticosteroids by any route should be monitored, and the potential growth effects of prolonged treatment should be weighed against clinical benefits obtained and the availability of other treatment alternatives. In order to minimize the potential growth effects of corticosteroids, children should be titrated to the lowest effective dose.
8.5 Geriatric Use
No overall differences in safety or effectiveness were observed between elderly subjects and younger subjects, and other reported clinical experience with prednisolone has not identified differences in responses between the elderly and younger patients. However, the incidence of corticosteroid-induced side effects may be increased in geriatric patients and appear to be dose-related. Osteoporosis is the most frequently encountered complication, which occurs at a higher incidence rate in corticosteroid-treated geriatric patients as compared to younger populations and in age-matched controls. Losses of bone mineral density appear to be greatest early on in the course of treatment and may recover over time after steroid withdrawal or use of lower doses (i.e., ≤ 5 mg/day). Prednisolone doses of 7.5 mg/day or higher, have been associated with an increased relative risk of both vertebral and nonvertebral fractures, even in the presence of higher bone density compared to patients with involutional osteoporosis.
Routine screening of geriatric patients, including regular assessments of bone mineral density and institution of fracture prevention strategies, along with regular review of prednisolone sodium phosphate indication should be undertaken to minimize complications and keep the prednisolone sodium phosphate dose at the lowest acceptable level. Co-administration of bisphosphonates has been shown to retard the rate of bone loss in corticosteroid-treated males and postmenopausal females, and these agents are recommended in the prevention and treatment of corticosteroid-induced osteoporosis.
It has been reported that equivalent weight-based doses yield higher total and unbound prednisolone plasma concentrations and reduced renal and non-renal clearance in elderly patients compared to younger populations. However, it is not clear whether dosing reductions would be necessary in elderly patients, since these pharmacokinetic alterations may be offset by age-related differences in responsiveness of target organs and/or less pronounced suppression of adrenal release of cortisol. Dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
-
10 OVERDOSAGE
The effects of accidental ingestion of large quantities of prednisolone over a very short period of time have not been reported, but prolonged use of the drug can produce mental symptoms, moon face, abnormal fat deposits, fluid retention, excessive appetite, weight gain, hypertrichosis, acne, striae, ecchymosis, increased sweating, pigmentation, dry scaly skin, thinning scalp hair, increased blood pressure, tachycardia, thrombophlebitis, decreased resistance to infection, negative nitrogen balance with delayed bone and wound healing, headache, weakness, menstrual disorders, accentuated menopausal symptoms, neuropathy, fractures, osteoporosis, peptic ulcer, decreased glucose tolerance, hypokalemia, and adrenal insufficiency. Hepatomegaly and abdominal distention have been observed in children.
Treatment of acute overdosage is by immediate gastric lavage or emesis followed by supportive and symptomatic therapy. For chronic overdosage in the face of severe disease requiring continuous steroid therapy, the dosage of prednisolone may be reduced only temporarily, or alternate day treatment may be introduced.
-
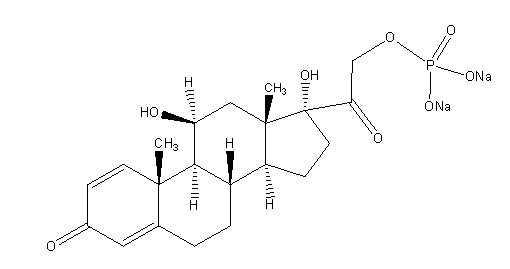
11 DESCRIPTION
Prednisolone sodium phosphate is a sodium salt of the phosphoester of the glucocorticoid prednisolone. Glucocorticoids are adrenocortical steroids, both naturally occurring and synthetic, which are readily absorbed from the gastrointestinal tract.
Prednisolone sodium phosphate, USP occurs as white or slightly yellow, friable granules or powder. It is freely soluble in water; soluble in methanol; slightly soluble in alcohol and in chloroform; and very slightly soluble in acetone and in dioxane.
The chemical name of prednisolone sodium phosphate is 11β,17,21-Trihydroxypregna-1,4-diene-3,20-dione 21-(disodium phosphate).
The molecular formula is C21H27Na2O8P; the molecular weight is 484.39. Its chemical structure is:
Each orally disintegrating tablet also contains the following inactive ingredients: amino methacrylate copolymer, colloidal silicon dioxide, corn starch, crospovidone, hypromellose, lemon oil, mannitol, microcrystalline cellulose, sodium stearyl fumarate, sucralose and sugar spheres (corn starch and sucrose).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Prednisolone is a synthetic adrenocortical steroid drug with predominantly glucocorticoid properties. Some of these properties reproduce the physiological actions of endogenous glucocorticosteroids, but others do not necessarily reflect any of the adrenal hormones’ normal functions; they are seen only after administration of large therapeutic doses of the drug. The pharmacological effects of prednisolone which are due to its glucocorticoid properties include: promotion of gluconeogenesis; increased deposition of glycogen in the liver; inhibition of the utilization of glucose; anti-insulin activity; increased catabolism of protein; increased lipolysis; stimulation of fat synthesis and storage; increased glomerular filtration rate and resulting increase in urinary excretion of urate (creatinine excretion remains unchanged); and increased calcium excretion. Depressed production of eosinophils and lymphocytes occurs, but erythropoiesis and production of polymorphonuclear leukocytes are stimulated. Inflammatory processes (edema, fibrin deposition, capillary dilatation, migration of leukocytes and phagocytosis) and the later stages of wound healing (capillary proliferation, deposition of collagen, cicatrization) are inhibited. Prednisolone can stimulate secretion of various components of gastric juice. Suppression of the production of corticotropin may lead to suppression of endogenous corticosteroids. Prednisolone has slight mineralocorticoid activity, whereby entry of sodium into cells and loss of intracellular potassium is stimulated. This is particularly evident in the kidney, where rapid ion exchange leads to sodium retention and hypertension.
12.3 Pharmacokinetics
Absorption
Oral administration of single doses of 30 mg prednisolone base equivalent of prednisolone sodium phosphate orally disintegrating tablets and prednisolone sodium phosphate solution to 21 adult volunteers yielded comparable pharmacokinetic data:
Table 1. Comparison of Mean Pharmacokinetic Parameters (%CV) in Healthy Volunteers Following a Single Dose of 30 mg Prednisolone Sodium Phosphate Orally Disintegrating Tablets and Prednisolone Sodium Phosphate Solution - * Administered under fasting conditions.
- † Mean values of 21 normal volunteers
Dose*
(30 mg prednisolone base equivalent)
AUC0-∞ (nghr/mL)
(± S.D.)
Cmax (nghr/mL)†
(± S.D.)
Prednisolone Sodium Phosphate Solution
2426.1 (360.0)
461.33 (77.94)
Prednisolone Sodium Phosphate Orally Disintegrating Tablets
2408.1 (361.5)
420.91 (78.28)
Distribution
Prednisolone is 70-90% protein bound in the plasma and the volume of distribution is reported as 0.22-0.7 L/kg.
Metabolism
Prednisolone is reported to be metabolized mainly in the liver and excreted in the urine as sulfate and glucuronide conjugates.
Excretion
Prednisolone is eliminated from the plasma with a mean (± SD) half-life of 2.6 (± 0.27) hours.
Special Populations
The systemic availability, metabolism and elimination of prednisolone after administration of single weight-based doses (0.8 mg/kg) of intravenous (IV) prednisolone and oral prednisone were reported in a small study of 19 younger (23 to 34 years) and 12 geriatric (65 to 89 years) subjects. Results showed that the systemic availability of total and unbound prednisolone, as well as interconversion between prednisolone and prednisone were independent of age. The mean unbound fraction of prednisolone was higher, and the steady-state volume of distribution (Vss) of unbound prednisolone was reduced in elderly patients. Plasma prednisolone concentrations were higher in elderly subjects, and the higher AUCs of total and unbound prednisolone were most likely reflective of an impaired metabolic clearance, evidenced by reduced fractional urinary clearance of 6b-hydroxyprednisolone. Despite these findings of higher total and unbound prednisolone concentrations, elderly subjects had higher AUCs of cortisol, suggesting that the elderly population is less sensitive to suppression of endogenous cortisol or their capacity for hepatic inactivation of cortisol is diminished.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Prednisolone sodium phosphate was not formally evaluated in carcinogenicity studies. Review of the published literature identified the potential for malignancy at doses within the therapeutic range. In a 2-year study, male Sprague-Dawley rats administered prednisolone in drinking water at an estimated continuous daily prednisolone consumption of 368 mcg/kg/day (equivalent to 3.5 mg/day in a 60 kg individual based on an mg/m2 body surface area comparison) developed increased incidences of hepatic adenomas. However infrequent administration of prednisolone did not result in malignancy. In an 18-month study, intermittent (1, 2, 4.5 or 9 times per month) oral gavage of 3 mg/kg prednisolone did not induce tumors in female Sprague-Dawley rats (equivalent to 29 mg in a 60 kg individual based on a mg/m2 body surface area comparison).
Prednisolone sodium phosphate was not formally evaluated for genotoxicity. However, in published studies prednisolone was not mutagenic with or without metabolic activation in the Ames bacterial reverse mutation assay using Salmonella typhimurium and Escherichia coli, or in a mammalian cell gene mutation assay using mouse lymphoma L5178Y cells, according to current evaluation standards. In a published chromosomal aberration study in Chinese Hamster Lung (CHL) cells, a slight increase was seen in the incidence of structural chromosomal aberrations with metabolic activation at the highest concentration tested, however, the effect appears to be equivocal.
Prednisolone sodium phosphate was not formally evaluated in fertility studies. However, alterations in motility and numbers of spermatozoa, and menstrual irregularities have been described with clinical use [see Adverse Reactions (6)].
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Prednisolone Sodium Phosphate Orally Disintegrating Tablets are available containing 13.4 mg, 20.2 mg or 40.3 mg of prednisolone sodium phosphate, USP equivalent to 10 mg, 15 mg or 30 mg of prednisolone.
The 10 mg tablets are white to off-white, speckled, round, unscored tablets debossed with P10 on one side of the tablet and M on the other side. They are available as follows:
NDC: 0378-4710-22
bottles of 48 tabletsThe 15 mg tablets are white to off-white, speckled, round, unscored tablets debossed with P15 on one side of the tablet and M on the other side. They are available as follows:
NDC: 0378-4715-22
bottles of 48 tabletsThe 30 mg tablets are white to off-white, speckled, round, unscored tablets debossed with P30 on one side of the tablet and M on the other side. They are available as follows:
NDC: 0378-4730-22
bottles of 48 tabletsStore at 20° to 25°C (68° to 77°F). [See USP Controlled Room Temperature.] Protect from moisture.
Do not break or use partial prednisolone sodium phosphate orally disintegrating tablets. Keep out of the reach of children.
Dispense in a tight, light-resistant container as defined in the USP using a child-resistant closure.
-
17 PATIENT COUNSELING INFORMATION
Advise patients not to discontinue the use of prednisolone sodium phosphate abruptly or without medical supervision, to advise any healthcare provider that they are taking it, and to seek medical advice at once should they develop fever or other signs of infection. Inform patients to take prednisolone sodium phosphate exactly as prescribed, follow the instructions on the prescription label, and not stop taking prednisolone sodium phosphate without first checking with their health-care providers, as there may be a need for gradual dose reduction.
Patients should discuss with their physician if they have had recent or ongoing infections or if they have recently received a vaccine.
Warn patients who are on immunosuppressant doses of corticosteroids to avoid exposure to chickenpox or measles. Advise patients that if they are exposed, to seek medical advice without delay.
There are a number of medicines that can interact with prednisolone sodium phosphate. Patients should inform their healthcare provider of all the medicines they are taking, including over-the counter and prescription medicines (such as phenytoin, diuretics, digitalis or digoxin, rifampin, amphotericin B, cyclosporine, insulin or diabetes medicines, ketoconazole, estrogens including birth control pills and hormone replacement therapy, blood thinners such as warfarin, aspirin or other NSAIDS, barbiturates), dietary supplements, and herbal products. If patients are taking any of these drugs, alternate therapy, dosage adjustment, and/or a special test may be needed during the treatment.
For missed doses, inform patients to take the missed dose as soon as they remember. If it is almost time for the next dose, the missed dose should be skipped and the medicine taken at the next regularly scheduled time. Advise patients not to take an extra dose to make up for the missed dose.
Inform patients to take prednisolone sodium phosphate with food to avoid GI irritation.
Advise patients of common adverse reactions that could occur with prednisolone sodium phosphate use to include fluid retention, alteration in glucose tolerance, elevation in blood pressure, behavioral and mood changes, increased appetite and weight gain.
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females to inform their healthcare provider of a known or a suspected pregnancy [see Warnings and Precautions (5.10) and Use in Specific Populations (8.1)].
Prednisolone sodium phosphate orally disintegrating tablets are packaged in a bottle. Patients dispensed a bottle should be instructed not to remove the tablet from the bottle until just prior to dosing. The bottle should then be opened, and the orally disintegrating tablet placed on the tongue, where the tablets may be swallowed whole as any conventional tablet, or allowed to dissolve in the mouth, with or without the assistance of water. Orally disintegrating tablet dosage forms are friable and are not intended to be cut, split, or broken.
Mylan Pharmaceuticals Inc.
Morgantown, WV 26505 U.S.A.Revised: 3/2020
PRED:R3 -
PRINCIPAL DISPLAY PANEL – 10 mg
NDC: 0378-4710-22
PrednisoLONE
Sodium Phosphate
Orally Disintegrating
Tablets
10 mg*Lemon Line Flavor
Rx only 48 Tablets
*Each orally disintegrating tablet
contains 13.4 mg of prednisolone
sodium phosphate, USP equivalent
to 10 mg of prednisolone.Dispense in a tight, light-resistant
container as defined in the USP
using a child-resistant closure.Keep container tightly closed.
Keep this and all medication
out of the reach of children.Store at 20° to 25°C (68° to 77°F).
[See USP Controlled Room
Temperature.]Protect from moisture.
Do not break or use partial ODT
tablets.Do not remove the tablet from the
bottle until just prior to dosing.Usual Dosage: See accompanying
prescribing information.Mylan Pharmaceuticals Inc.
Morgantown, WV 26505 U.S.A.RM4710BX
-
PRINCIPAL DISPLAY PANEL – 15 mg
NDC: 0378-4715-22
PrednisoLONE
Sodium Phosphate
Orally Disintegrating
Tablets
15 mg*Lemon Line Flavor
Rx only 48 Tablets
*Each orally disintegrating tablet
contains 20.2 mg of prednisolone
sodium phosphate, USP equivalent
to 15 mg of prednisolone.Dispense in a tight, light-resistant
container as defined in the USP
using a child-resistant closure.Keep container tightly closed.
Keep this and all medication
out of the reach of children.Store at 20° to 25°C (68° to 77°F).
[See USP Controlled Room
Temperature.]Protect from moisture.
Do not break or use partial ODT
tablets.Do not remove the tablet from the
bottle until just prior to dosing.Usual Dosage: See accompanying
prescribing information.Mylan Pharmaceuticals Inc.
Morgantown, WV 26505 U.S.A.RM4715BX
-
PRINCIPAL DISPLAY PANEL – 30 mg
NDC: 0378-4730-22
PrednisoLONE
Sodium Phosphate
Orally Disintegrating
Tablets
30 mg*Lemon Line Flavor
Rx only 48 Tablets
*Each orally disintegrating tablet
contains 40.3 mg of prednisolone
sodium phosphate, USP equivalent
to 30 mg of prednisolone.Dispense in a tight, light-resistant
container as defined in the USP
using a child-resistant closure.Keep container tightly closed.
Keep this and all medication
out of the reach of children.Store at 20° to 25°C (68° to 77°F).
[See USP Controlled Room
Temperature.]Protect from moisture.
Do not break or use partial ODT
tablets.Do not remove the tablet from the
bottle until just prior to dosing.Usual Dosage: See accompanying
prescribing information.Mylan Pharmaceuticals Inc.
Morgantown, WV 26505 U.S.A.RM4730BX
-
INGREDIENTS AND APPEARANCE
PREDNISOLONE SODIUM PHOSPHATE
prednisolone tablet, orally disintegratingProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0378-4710 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PREDNISOLONE SODIUM PHOSPHATE (UNII: IV021NXA9J) (PREDNISOLONE - UNII:9PHQ9Y1OLM) PREDNISOLONE 21-PHOSPHATE 10 mg Inactive Ingredients Ingredient Name Strength DIMETHYLAMINOETHYL METHACRYLATE - BUTYL METHACRYLATE - METHYL METHACRYLATE COPOLYMER (UNII: 905HNO1SIH) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) STARCH, CORN (UNII: O8232NY3SJ) CROSPOVIDONE, UNSPECIFIED (UNII: 2S7830E561) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) LEMON OIL (UNII: I9GRO824LL) MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE 102 (UNII: PNR0YF693Y) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) SUCROSE (UNII: C151H8M554) SUCRALOSE (UNII: 96K6UQ3ZD4) Product Characteristics Color WHITE (white to off white, speckled) Score no score Shape ROUND Size 8mm Flavor Imprint Code M;P10 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0378-4710-22 48 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 12/08/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA202179 12/08/2014 PREDNISOLONE SODIUM PHOSPHATE
prednisolone tablet, orally disintegratingProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0378-4715 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PREDNISOLONE SODIUM PHOSPHATE (UNII: IV021NXA9J) (PREDNISOLONE - UNII:9PHQ9Y1OLM) PREDNISOLONE 21-PHOSPHATE 15 mg Inactive Ingredients Ingredient Name Strength DIMETHYLAMINOETHYL METHACRYLATE - BUTYL METHACRYLATE - METHYL METHACRYLATE COPOLYMER (UNII: 905HNO1SIH) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) STARCH, CORN (UNII: O8232NY3SJ) CROSPOVIDONE, UNSPECIFIED (UNII: 2S7830E561) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) LEMON OIL (UNII: I9GRO824LL) MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE 102 (UNII: PNR0YF693Y) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) SUCROSE (UNII: C151H8M554) SUCRALOSE (UNII: 96K6UQ3ZD4) Product Characteristics Color WHITE (white to off white, speckled) Score no score Shape ROUND Size 10mm Flavor Imprint Code M;P15 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0378-4715-22 48 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 12/08/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA202179 12/08/2014 PREDNISOLONE SODIUM PHOSPHATE
prednisolone tablet, orally disintegratingProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0378-4730 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PREDNISOLONE SODIUM PHOSPHATE (UNII: IV021NXA9J) (PREDNISOLONE - UNII:9PHQ9Y1OLM) PREDNISOLONE 21-PHOSPHATE 30 mg Inactive Ingredients Ingredient Name Strength DIMETHYLAMINOETHYL METHACRYLATE - BUTYL METHACRYLATE - METHYL METHACRYLATE COPOLYMER (UNII: 905HNO1SIH) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) STARCH, CORN (UNII: O8232NY3SJ) CROSPOVIDONE, UNSPECIFIED (UNII: 2S7830E561) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) LEMON OIL (UNII: I9GRO824LL) MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE 102 (UNII: PNR0YF693Y) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) SUCROSE (UNII: C151H8M554) SUCRALOSE (UNII: 96K6UQ3ZD4) Product Characteristics Color WHITE (white to off white, speckled) Score no score Shape ROUND Size 13mm Flavor Imprint Code M;P30 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0378-4730-22 48 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 12/08/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA202179 12/08/2014 Labeler - Mylan Pharmaceuticals Inc. (059295980)


© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.