ELREXFIO- elranatamab-bcmm injection, solution
Elrexfio by
Drug Labeling and Warnings
Elrexfio by is a Prescription medication manufactured, distributed, or labeled by Pfizer Laboratories Div Pfizer Inc, Wyeth BioPharma Division of Wyeth Pharmaceuticals LLC, Pharmacia & Upjohn Company LLC, PPD Development, L.P.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ELREXFIO safely and effectively. See full prescribing information for ELREXFIO.
ELREXFIO® (elranatamab-bcmm) injection, for subcutaneous use
Initial U.S. Approval: 2023WARNING: CYTOKINE RELEASE SYNDROME and NEUROLOGIC TOXICITY including IMMUNE EFFECTOR CELL-ASSOCIATED NEUROTOXICITY SYNDROME
See full prescribing information for complete boxed warning.
- Cytokine Release Syndrome (CRS), including life-threatening or fatal reactions, can occur in patients receiving ELREXFIO. Initiate treatment with ELREXFIO step-up dosing schedule to reduce risk of CRS. Withhold ELREXFIO until CRS resolves or permanently discontinue based on severity. (2.2, 2.5, 5.1)
- Neurologic toxicity, including Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS), and serious and life-threatening reactions, can occur in patients receiving ELREXFIO. Monitor patients for signs and symptoms of neurologic toxicity, including ICANS, during treatment. Withhold ELREXFIO until the neurologic toxicity resolves or permanently discontinue based on severity. (2.5, 5.2)
- ELREXFIO is available only through a restricted program called the ELREXFIO Risk Evaluation and Mitigation Strategy (REMS). (5.3)
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
ELREXFIO is a bispecific B-cell maturation antigen (BCMA)-directed CD3 T‑cell engager indicated for the treatment of adult patients with relapsed or refractory multiple myeloma who have received at least four prior lines of therapy including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody.
This indication is approved under accelerated approval based on response rate and durability of response. Continued approval for this indication may be contingent upon verification of clinical benefit in a confirmatory trial(s). (1)
DOSAGE AND ADMINISTRATION
- * Responders only week 25 onward.
- † In patients who have maintained the response following 24 weeks of treatment at the biweekly dosing schedule.
ELREXFIO Dosing Schedule (2.2)
Dosing Schedule
Day
ELREXFIO Dose
Step-up Dosing Schedule
Day 1
Step-up dose 1
12 mg
Day 4
Step-up dose 2
32 mg
Day 8
First treatment dose
76 mg
Weekly Dosing Schedule
One week after first treatment dose and weekly thereafter through week 24
Subsequent treatment doses
76 mg
Biweekly (Every 2 Week) Dosing Schedule*
Week 25 and every 2 weeks thereafter through week 48
Subsequent treatment doses
76 mg
Every 4 Week Dosing Schedule†
Week 49 and every 4 weeks thereafter
Subsequent treatment doses
76 mg
- Patients should be hospitalized for 48 hours after administration of the first step-up dose, and for 24 hours after administration of the second step-up dose. (2.1)
- For subcutaneous injection only. (2.2)
- Administer pre-treatment medications as recommended. (2.3)
- See Full Prescribing Information for instructions on preparation and administration. (2.6)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Infections: Can cause severe, life-threatening, or fatal infections. Monitor patients for signs and symptoms of infection and treat appropriately. Do not initiate treatment in patients with active infections. (5.4)
- Neutropenia: Monitor complete blood cell counts at baseline and periodically during treatment. (5.5)
- Hepatotoxicity: Can cause elevated ALT, AST, and bilirubin. Monitor liver enzymes and bilirubin at baseline and during treatment as clinically indicated. (5.6)
- Embryo-Fetal Toxicity: May cause fetal harm. Advise females of reproductive potential of the potential risk to the fetus and to use effective contraception. (5.7, 8.1. 8.3)
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥20%) are CRS, fatigue, injection site reaction, diarrhea, upper respiratory tract infection, musculoskeletal pain, pneumonia, decreased appetite, rash, cough, nausea, and pyrexia.
The most common Grade 3 to 4 laboratory abnormalities (≥30%) are decreased lymphocytes, decreased neutrophils, decreased hemoglobin, decreased white blood cells, and decreased platelets. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc. at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 2/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: CYTOKINE RELEASE SYNDROME and NEUROLOGIC TOXICITY including IMMUNE EFFECTOR CELL-ASSOCIATED NEUROTOXICITY SYNDROME
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosing Information
2.2 Recommended Dosage
2.3 Recommended Pre-treatment Medications
2.4 Restarting ELREXFIO After Dosage Delay
2.5 Dosage Modifications for Adverse Reactions
2.6 Preparation and Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Cytokine Release Syndrome (CRS)
5.2 Neurologic Toxicity, Including Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS)
5.3 ELREXFIO REMS
5.4 Infections
5.5 Neutropenia
5.6 Hepatotoxicity
5.7 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Relapsed or Refractory Multiple Myeloma
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: CYTOKINE RELEASE SYNDROME and NEUROLOGIC TOXICITY including IMMUNE EFFECTOR CELL-ASSOCIATED NEUROTOXICITY SYNDROME
- Cytokine Release Syndrome (CRS), including life-threatening or fatal reactions, can occur in patients receiving ELREXFIO. Initiate treatment with ELREXFIO step-up dosing schedule to reduce the risk of CRS. Withhold ELREXFIO until CRS resolves or permanently discontinue based on severity [see Dosage and Administration (2.2, 2.5), Warnings and Precautions (5.1)].
- Neurologic toxicity, including Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS), and serious and life-threatening reactions, can occur in patients receiving ELREXFIO. Monitor patients for signs and symptoms of neurologic toxicity, including ICANS, during treatment. Withhold ELREXFIO until the neurologic toxicity resolves or permanently discontinue based on severity [see Dosage and Administration (2.5), Warnings and Precautions (5.2)].
- Because of the risk of CRS and neurologic toxicity, including ICANS, ELREXFIO is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called ELREXFIO REMS [see Warnings and Precautions (5.3)].
-
1 INDICATIONS AND USAGE
ELREXFIO is indicated for the treatment of adult patients with relapsed or refractory multiple myeloma who have received at least four prior lines of therapy, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody.
This indication is approved under accelerated approval based on response rate and durability of response [see Clinical Studies (14)]. Continued approval for this indication may be contingent upon verification of clinical benefit in a confirmatory trial(s).
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosing Information
Administer ELREXFIO subcutaneously according to the step-up dosing schedule to reduce the incidence and severity of cytokine release syndrome (CRS).
Administer pre-treatment medications prior to each dose in the ELREXFIO step-up dosing schedule, which includes step-up dose 1, step-up dose 2, and the first treatment dose as recommended [see Dosage and Administration (2.2, 2.3)].
ELREXFIO should only be administered by a qualified healthcare professional with appropriate medical support to manage severe reactions such as CRS and neurologic toxicity, including ICANS [see Warnings and Precautions (5.1, 5.2)].
Due to the risk of CRS, patients should be hospitalized for 48 hours after administration of the first step-up dose, and for 24 hours after administration of the second step-up dose.
2.2 Recommended Dosage
For subcutaneous injection only.
The recommended dosing schedule for ELREXFIO is provided in Table 1. The recommended dosages of ELREXFIO subcutaneous injection are: step-up dose 1 of 12 mg on Day 1, step-up dose 2 of 32 mg on Day 4, followed by the first treatment dose of 76 mg on Day 8, and then 76 mg weekly thereafter through week 24.
For patients who have received at least 24 weeks of treatment with ELREXFIO and have achieved a response [partial response (PR) or better] and maintained this response for at least 2 months, the dose interval should transition to an every two-week schedule. For patients who have received at least 24 weeks of treatment with ELREXFIO at the every two-week dosing schedule and have maintained the response, the dose interval should transition to an every four-week schedule.
Continue treatment with ELREXFIO until disease progression or unacceptable toxicity.
Administer pre-treatment medications prior to each dose in the ELREXFIO step-up dosing schedule, which includes step-up dose 1, step-up dose 2, and the first treatment dose as recommended [see Dosage and Administration (2.3)].
Table 1. ELREXFIO Dosing Schedule Note: See Table 2 for recommendations on restarting ELREXFIO after dose delays. - * Administer pre-treatment medications prior to each dose in the ELREXFIO step-up dosing schedule, which includes step-up dose 1, step-up dose 2, and the first treatment dose [see Dosage and Administration (2.3)].
- † A minimum of 2 days should be maintained between step-up dose 1 (12 mg) and step-up dose 2 (32 mg).
- ‡ A minimum of 3 days should be maintained between step-up dose 2 (32 mg) and the first treatment (76 mg) dose.
- § A minimum of 6 days should be maintained between treatment doses.
Dosing Schedule
Day
ELREXFIO Dose
Step-up Dosing Schedule
Day 1*
Step-up dose 1
12 mg
Step-up dose 2
32 mg
First treatment dose
76 mg
Weekly Dosing Schedule
One week after first treatment dose and weekly thereafter§ through week 24
Subsequent treatment doses
76 mg
Biweekly (Every 2 Week) Dosing Schedule
*Responders only week 25 onward
Week 25 and every 2 weeks thereafter§
through week 48
Subsequent treatment doses
76 mg
Every 4 Week Dosing Schedule
*In patients who have maintained the response following 24 weeks of treatment at the biweekly dosing schedule
Week 49 and every 4 weeks thereafter§
Subsequent treatment doses
76 mg
2.3 Recommended Pre-treatment Medications
Administer the following pre-treatment medications approximately 1 hour before the first three doses of ELREXFIO in the step-up dosing schedule, which includes step-up dose 1, step-up dose 2, and the first treatment dose as described in Table 1 to reduce the risk of CRS [see Warnings and Precautions (5.1)]:
- acetaminophen (or equivalent) 650 mg orally
- dexamethasone (or equivalent) 20 mg orally or intravenously
- diphenhydramine (or equivalent) 25 mg orally
2.4 Restarting ELREXFIO After Dosage Delay
If a dose of ELREXFIO is delayed, restart therapy based on the recommendations listed in Table 2 and resume the dosing schedule accordingly [see Dosage and Administration (2.2)]. Administer pre-treatment medications as indicated in Table 2.
Table 2. Recommendation for Restarting Therapy with ELREXFIO After Dosage Delay - * Administer pre-treatment medications prior to the ELREXFIO dose [see Dosage and Administration (2.3)].
- † Consider benefit-risk of restarting ELREXFIO in patients who require a dose delay of more than 56 days due to an adverse reaction.
Last Dose Administered
Time Since the Last Dose Administered
Action for Next Dose
Step-up dose 1 (12 mg)
2 weeks or less (≤14 days)
Restart ELREXFIO at step-up dose 2 (32 mg).* If tolerated, increase to 76 mg 4 days later.
Greater than 2 weeks (>14 days)
Restart ELREXFIO step-up dosing schedule at step-up dose 1 (12 mg).*
Step-up dose 2 (32 mg)
2 weeks or less (≤14 days)
Restart ELREXFIO at 76 mg.*
Greater than 2 weeks to less than or equal to 4 weeks (15 days to ≤28 days)
Restart ELREXFIO at step-up dose 2 (32 mg).* If tolerated, increase to 76 mg 1 week later.
Greater than 4 weeks (>28 days)
Restart ELREXFIO step-up dosing schedule at step-up dose 1 (12 mg).*
Any weekly treatment dose (76 mg)
8 weeks or less (≤56 days)
Restart ELREXFIO at 76 mg.
Greater than 8 weeks to less or equal to 12 weeks (57 days to ≤84 days)†
Restart ELREXFIO at step-up dose 2 (32 mg).*If tolerated, increase to 76 mg 1 week later.
Greater than 12 weeks (>84 days)†
Restart ELREXFIO step-up dosing schedule at step-up dose 1 (12 mg).*
Any biweekly or every-4-week treatment dose (76 mg)
12 weeks or less (≤84 days)
Restart ELREXFIO at 76 mg.
Greater than 12 weeks (>84 days)†
Restart ELREXFIO step-up dosing schedule at step-up dose 1 (12 mg).*
2.5 Dosage Modifications for Adverse Reactions
Dosage reductions of ELREXFIO are not recommended.
Dosage delays may be required to manage toxicities related to ELREXFIO [see Warnings and Precautions (5)]. Recommendations on restarting ELREXFIO after a dose delay are provided in Table 2.
See Table 3 and Table 4 for recommended actions for adverse reactions of CRS and ICANS, respectively. See Table 5 for recommended actions for neurologic toxicity excluding ICANS and Table 6 for recommended actions for other adverse reactions following administration of ELREXFIO. Consider further management per current practice guidelines.
Management of CRS, Neurologic Toxicity Including ICANS
Cytokine Release Syndrome (CRS)
Management recommendations for CRS are summarized in Table 3.
Identify CRS based on clinical presentation [see Warnings and Precautions (5.1)]. Evaluate and treat other causes of fever, hypoxia, and hypotension.
If CRS is suspected, withhold ELREXFIO until CRS resolves. Manage CRS according to the recommendations in Table 3 and consider further management per current practice guidelines. Administer supportive therapy for CRS, which may include intensive care for severe or life-threatening CRS. Consider laboratory testing to monitor for disseminated intravascular coagulation (DIC), hematology parameters, as well as pulmonary, cardiac, renal, and hepatic function.
Table 3. Recommendations for Management of CRS - * Based on American Society for Transplantation and Cellular Therapy (ASTCT) 2019 grading criteria for CRS.
- † Attributed to CRS. Fever may not always be present concurrently with hypotension or hypoxia as it may be masked by interventions such as antipyretics or anti-cytokine therapy.
- ‡ See Table 2 for recommendations on restarting ELREXFIO after dose delays.
- § Low-flow nasal cannula is ≤6 L/min, and high-flow nasal cannula is >6 L/min.
Grade*
Presenting Symptoms
Actions
Grade 1
Temperature ≥100.4 °F (38 °C)†
- Withhold ELREXFIO until CRS resolves.‡
- Administer pretreatment medications prior to next dose of ELREXFIO.
Grade 2
Temperature ≥100.4 °F (38 °C) with either:
- Hypotension responsive to fluid and not requiring vasopressors, and/or
- Oxygen requirement of low-flow nasal cannula§ or blow-by
- Withhold ELREXFIO until CRS resolves.‡
- Monitor patients daily for 48 hours following the next dose of ELREXFIO. Instruct patients to remain within proximity of a healthcare facility, and consider hospitalization.
- Administer pretreatment medications prior to next dose of ELREXFIO.
Grade 3
(First occurrence)
Temperature ≥100.4 °F (38 °C) with either:
- Hypotension requiring one vasopressor with or without vasopressin, and/or
- Oxygen requirement of high-flow nasal cannula§, facemask, non-rebreather mask, or Venturi mask
- Withhold ELREXFIO until CRS resolves.‡
- Provide supportive therapy, which may include intensive care.
- Patients should be hospitalized for 48 hours following the next dose of ELREXFIO.
- Administer pretreatment medications prior to next dose of ELREXFIO.
Grade 3 (Recurrent)
Temperature ≥100.4 °F (38 °C) with either:
- Hypotension requiring one vasopressor with or without vasopressin, and/or
- Oxygen requirement of high-flow nasal cannula§, facemask, non-rebreather mask, or Venturi mask
- Permanently discontinue therapy with ELREXFIO.
- Provide supportive therapy, which may include intensive care.
Grade 4
Temperature ≥100.4 °F (38 °C) with either:
- Hypotension requiring multiple vasopressors (excluding vasopressin), and/or
- Oxygen requirement of positive pressure (e.g., continuous positive airway pressure [CPAP], bilevel positive airway pressure [BiPAP], intubation, and mechanical ventilation)
- Permanently discontinue therapy with ELREXFIO.
- Provide supportive therapy, which may include intensive care.
Neurologic Toxicity Including ICANS
Management recommendations for ICANS and neurologic toxicity are summarized in Table 4 and Table 5.
At the first sign of neurologic toxicity, including ICANS, withhold ELREXFIO and consider neurology evaluation. Rule out other causes of neurologic symptoms. Provide supportive therapy, which may include intensive care, for severe or life-threatening neurologic toxicities, including ICANS [see Warnings and Precautions (5.2)]. Manage ICANS according to the recommendations in Table 4 and consider further management per current practice guidelines.
Table 4. Recommendations for Management of ICANS - * Based on American Society for Transplantation and Cellular Therapy (ASTCT) 2019 grading criteria for ICANS.
- † Management is determined by the most severe event, not attributable to any other cause.
- ‡ If patient is arousable and able to perform Immune Effector Cell-Associated Encephalopathy (ICE) Assessment, assess: Orientation (oriented to year, month, city, hospital = 4 points); Naming (name 3 objects, e.g., point to clock, pen, button = 3 points); Following Commands (e.g., “show me 2 fingers” or “close your eyes and stick out your tongue” = 1 point); Writing (ability to write a standard sentence = 1 point); and Attention (count backwards from 100 by ten = 1 point). If patient is unarousable and unable to perform ICE Assessment (Grade 4 ICANS) = 0 points.
- § Not attributable to any other cause.
- ¶ See Table 2 for recommendations on restarting ELREXFIO after dose delays.
- # All references to dexamethasone administration are dexamethasone or equivalent medications.
Grade*
Presenting Symptoms†
Actions
Grade 1
ICE score 7-9‡
Or depressed level of consciousness§: awakens spontaneously.- Withhold ELREXFIO until ICANS resolves.¶
- Monitor neurologic symptoms and consider consultation with a neurologist and other specialists for further evaluation and management.
- Consider non-sedating, anti-seizure medications (e.g., levetiracetam) for seizure prophylaxis.
Grade 2
ICE score 3-6‡
Or depressed level of consciousness§: awakens to voice.- Withhold ELREXFIO until ICANS resolves.¶
- Administer dexamethasone# 10 mg intravenously every 6 hours. Continue dexamethasone use until resolution to Grade 1 or less, then taper.
- Monitor neurologic symptoms and consider consultation with a neurologist and other specialists for further evaluation and management.
- Consider non-sedating, anti-seizure medications (e.g., levetiracetam) for seizure prophylaxis.
- Monitor patients daily for 48 hours following the next dose of ELREXFIO. Instruct patients to remain within proximity of a healthcare facility, and consider hospitalization.
Grade 3
(First occurrence)
ICE score 0-2‡
or depressed level of consciousness§: awakens only to tactile stimulus,
or seizures§, either:- any clinical seizure, focal or generalized, that resolves rapidly, or
- non-convulsive seizures on electroencephalogram (EEG) that resolve with intervention,
or raised intracranial pressure: focal/local edema on neuroimaging§
- Withhold ELREXFIO until ICANS resolves.¶
- Administer dexamethasone# 10 mg intravenously every 6 hours. Continue dexamethasone use until resolution to Grade 1 or less, then taper.
- Monitor neurologic symptoms and consider consultation with a neurologist and other specialists for further evaluation and management.
- Consider non-sedating, anti-seizure medications (e.g., levetiracetam) for seizure prophylaxis.
- Provide supportive therapy, which may include intensive care.
- Patients should be hospitalized for 48 hours following the next dose of ELREXFIO.
Grade 3 (recurrent)
ICE score 0-2‡
or depressed level of consciousness§: awakens only to tactile stimulus,
or seizures§, either:- any clinical seizure, focal or generalized, that resolves rapidly, or
- non-convulsive seizures on electroencephalogram (EEG) that resolve with intervention,
or raised intracranial pressure: focal/local edema on neuroimaging§
- Permanently discontinue ELREXFIO.
- Administer dexamethasone# 10 mg intravenously every 6 hours. Continue dexamethasone use until resolution to Grade 1 or less, then taper.
- Monitor neurologic symptoms and consider consultation with a neurologist and other specialists for further evaluation and management.
- Consider non-sedating, anti-seizure medications (e.g., levetiracetam) for seizure prophylaxis.
- Provide supportive therapy, which may include intensive care.
Grade 4
ICE score 0‡
Or, depressed level of consciousness§ either:- patient is unarousable or requires vigorous or repetitive tactile stimuli to arouse, or
- stupor or coma,
or seizures§, either:
- life-threatening prolonged seizure (>5 minutes), or
- repetitive clinical or electrical seizures without return to baseline in between,
or motor findings§:
- deep focal motor weakness such as hemiparesis or paraparesis,
or raised intracranial pressure/cerebral edema§, with signs/symptoms such as:
- diffuse cerebral edema on neuroimaging, or
- decerebrate or decorticate posturing, or
- cranial nerve VI palsy, or
- papilledema, or
- Cushing’s triad
- Permanently discontinue ELREXFIO.
- Administer dexamethasone# 10 mg intravenously every 6 hours. Continue dexamethasone use until resolution to Grade 1 or less, then taper.
- Alternatively, consider administration of methylprednisolone 1,000 mg per day intravenously for 3 days.
- Monitor neurologic symptoms and consider consultation with a neurologist and other specialists for further evaluation and management.
- Consider non-sedating, anti-seizure medications (e.g., levetiracetam) for seizure prophylaxis.
- Provide supportive therapy, which may include intensive care.
Table 5. Recommendations for Management of Neurologic Toxicity, Excluding ICANS Adverse Reaction
Severity
Actions
Neurologic Toxicity (excluding ICANS)
Grade 1
- Withhold ELREXFIO until neurologic toxicity symptoms resolve or stabilize.
Grade 2
Grade 3 (First occurrence)
- Withhold ELREXFIO until neurologic toxicity symptoms improve to Grade 1 or less.
- Provide supportive therapy.
Grade 3 (Recurrent)
Grade 4
- Permanently discontinue ELREXFIO.
- Provide supportive therapy, which may include intensive care.
Table 6. Recommended Dosage Modifications for Other Adverse Reactions - * See Table 2 for recommendations on restarting ELREXFIO after dose delays.
- † Based on National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE), Version 5.0.
Adverse Reactions
Severity
Actions
Hematologic Adverse Reactions
Absolute neutrophil count less than 0.5 x 109/L
- Withhold ELREXFIO until absolute neutrophil count is 0.5 x 109/L or higher.*
Febrile neutropenia
- Withhold ELREXFIO until absolute neutrophil count is 1 x 109/L or higher and fever resolves.*
Hemoglobin less than 8 g/dL
- Withhold ELREXFIO until hemoglobin is 8 g/dL or higher.*
Platelet count less than 25,000/mcL
Platelet count between 25,000/mcL and 50,000/mcL with bleeding
- Withhold ELREXFIO until platelet count is 25,000/mcL or higher and no evidence of bleeding.*
Infections and Other Non-hematologic Adverse Reactions†
[see Warnings and Precautions (5.4, 5.6) and Adverse Reactions (6.1)]
Grade 3
- Withhold ELREXFIO until adverse reaction improves to ≤Grade 1 or baseline.*
Grade 4
- Consider permanent discontinuation of ELREXFIO.
- If ELREXFIO is not permanently discontinued, withhold subsequent treatment doses of ELREXFIO (e.g., doses administered after ELREXFIO step-up dosing schedule) until adverse reaction improves to Grade 1 or less.
2.6 Preparation and Administration Instructions
ELREXFIO is intended for subcutaneous use by a healthcare provider only.
ELREXFIO should be administered by a healthcare provider with adequate medical personnel and appropriate medical equipment to manage severe reactions, including CRS and neurologic toxicity, including ICANS [see Warnings and Precautions (5.1, 5.2)].
ELREXFIO 76 mg/1.9 mL (40 mg/mL) vial and 44 mg/1.1 mL (40 mg/mL) vial are supplied as ready-to-use solution that do not need dilution prior to administration.
ELREXFIO is a clear to slightly opalescent, and colorless to pale brown liquid solution. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not administer if solution is discolored or contains particulate matter.
Use aseptic technique to prepare and administer ELREXFIO.
Preparation
ELREXFIO vials are for one-time use in a single patient and do not contain any preservatives.
Prepare ELREXFIO following the instructions below (see Table 7) depending on the required dose.
Table 7. Injection Volumes Total Dose (mg)
Volume of Injection
12 mg
0.3 mL
32 mg
0.8 mL
76 mg
1.9 mL
Remove the appropriate strength ELREXFIO vial from refrigerated storage [2 °C to 8 °C (36 °F to 46 °F)]. Once removed from refrigerated storage, equilibrate ELREXFIO to ambient temperature [15 °C to 30 °C (59 °F to 86 °F)]. Do not warm ELREXFIO in any other way.
Withdraw the required injection volume of ELREXFIO from the vial into an appropriately sized syringe with stainless steel injection needles (30G or wider) and polypropylene or polycarbonate syringe material. Discard unused portion.
Administration
Inject the required volume of ELREXFIO into the subcutaneous tissue of the abdomen (preferred injection site). Alternatively, ELREXFIO may be injected into the subcutaneous tissue at other sites (e.g., thigh).
Do not inject into tattoos or scars or areas where the skin is red, bruised, tender, hard or not intact.
Storage of Prepared Syringe
If the prepared dosing syringe is not used immediately, the syringe may be stored refrigerated between 2 °C to 8 °C (36 °F to 46 °F) for a maximum of 72 hours or between 8 °C to 25 °C (46 °F to 77 °F) for a maximum of 24 hours. Once removed from refrigerated storage, the prepared syringe must be used or discarded.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Cytokine Release Syndrome (CRS)
ELREXFIO can cause CRS, including life-threatening or fatal reactions [see Adverse Reactions (6.1)].
In the clinical trial, CRS occurred in 58% of patients who received ELREXFIO at the recommended dosing schedule [see Dosage and Administration (2.2)], with Grade 1 CRS in 44% of patients, Grade 2 CRS in 14% of patients, and Grade 3 CRS in 0.5% of patients. Recurrent CRS occurred in 13% of patients. Most patients experienced CRS after the first step-up dose (43%) or the second step-up dose (19%), with 7% of patients having CRS after the first treatment dose and 1.6% of patients after a subsequent dose. The median time to onset of CRS was 2 (range: 1 to 9) days after the most recent dose, with a median duration of 2 (range: 1 to 19) days.
Clinical signs and symptoms of CRS may include, but are not limited to, fever, hypoxia, chills, hypotension, tachycardia, headache, and elevated liver enzymes.
Initiate therapy according to the ELREXFIO step-up dosing schedule to reduce risk of CRS and monitor patients following administration of ELREXFIO accordingly [see Dosage and Administration (2.2, 2.5)]. Administer pre-treatment medications prior to each dose in the step-up dosing schedule to reduce risk of CRS [see Dosage and Administration (2.3)].
Counsel patients to seek medical attention should signs or symptoms of CRS occur. At the first sign of CRS, evaluate patients immediately for hospitalization. Manage CRS according to the recommendations and consider further management per current practice guidelines. Withhold or permanently discontinue ELREXFIO based on severity [see Dosage and Administration (2.5)].
ELREXFIO is available only through a restricted program under a REMS [see Warnings and Precautions (5.3)].
5.2 Neurologic Toxicity, Including Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS)
ELREXFIO can cause serious or life-threatening neurologic toxicity, including ICANS [see Adverse Reactions (6.1)].
In the clinical trial, neurologic toxicity occurred in 59% of patients who received ELREXFIO at the recommended dosing schedule [see Dosage and Administration (2.2)], with Grade 3 or 4 neurologic toxicity occurring in 7% of patients. Neurologic toxicities included headache (18%), encephalopathy (15%), motor dysfunction (13%), sensory neuropathy (13%), and Guillain-Barré Syndrome (0.5%).
In the clinical trial, ICANS occurred in 3.3% of patients who received ELREXFIO at the recommended dosing schedule [see Dosage and Administration (2.2)]. Most patients had ICANS after the first step-up dose (2.7%), 1 (0.5%) patient had ICANS after the second step-up dose and 1 (0.5%) patient had ICANS after subsequent dose(s). Recurrent ICANS occurred in 1.1% of patients. The median time to onset was 3 (range: 1 to 4) days after the most recent dose, with a median duration of 2 (range: 1 to 18) days. The most frequent clinical manifestations of ICANS included a depressed level of consciousness and Grade 1 or Grade 2 Immune Effector Cell-Associated Encephalopathy (ICE) scores. The onset of ICANS can be concurrent with CRS, following resolution of CRS, or in the absence of CRS.
Counsel patients to seek medical attention should signs or symptoms of neurologic toxicity occur. Monitor patients for signs and symptoms of neurologic toxicities during treatment with ELREXFIO. At the first sign of neurologic toxicity, including ICANS, evaluate and treat patients immediately based on severity. Withhold or permanently discontinue ELREXFIO based on severity per recommendations [see Dosage and Administration (2.5)] and consider further management per current practice guidelines.
Due to the potential for neurologic toxicity including ICANS, patients receiving ELREXFIO are at risk of depressed level of consciousness. Advise patients not to drive or operate heavy or potentially dangerous machinery for 48 hours after completing each of the 2 step-up doses and the first treatment dose within the ELREXFIO step-up dosing schedule and in the event of new onset of any neurological toxicity symptoms until symptoms resolve [see Dosage and Administration (2.2)].
ELREXFIO is available only through a restricted program under a REMS [see Warnings and Precautions (5.3)].
5.3 ELREXFIO REMS
ELREXFIO is available only through a restricted program under a REMS called the ELREXFIO REMS because of the risks of CRS and neurologic toxicity, including ICANS [see Warnings and Precautions (5.1, 5.2)].
Notable requirements of the ELREXFIO REMS include the following:
- Prescribers must be certified with the program by enrolling and completing training.
- Prescribers must counsel patients receiving ELREXFIO about the risk of CRS and neurologic toxicity, including ICANS, and provide patients with ELREXFIO Patient Wallet Card.
- Pharmacies and healthcare settings that dispense ELREXFIO must be certified with the ELREXFIO REMS program and must verify prescribers are certified through the ELREXFIO REMS program.
- Wholesalers and distributers must only distribute ELREXFIO to certified pharmacies or healthcare settings.
Further information about the ELREXFIO REMS program is available at www.ELREXFIOREMS.com or by telephone at 1‑844‑923‑7845.
5.4 Infections
ELREXFIO can cause severe, life-threatening, or fatal infections. In the clinical trial, in patients who received ELREXFIO according to the recommended dosing schedule, serious infections, including opportunistic infections, occurred in 42% of patients, with Grade 3 or 4 infections in 31%, and fatal infections in 7%. The most common serious infections reported (≥5%) were pneumonia and sepsis [see Adverse Reactions (6.1)].
Do not initiate treatment with ELREXFIO in patients with active infections. Monitor patients for signs and symptoms of infection prior to and during treatment with ELREXFIO and treat appropriately. Withhold or permanently discontinue ELREXFIO based on severity [see Dosage and Administration (2.5)]. Administer prophylactic antimicrobial and anti-viral medications according to current practice guidelines. Consider treatment with subcutaneous or intravenous immunoglobulin (IVIG) as appropriate.
5.5 Neutropenia
ELREXFIO can cause neutropenia and febrile neutropenia. In patients who received ELREXFIO at the recommended dose in the clinical trial, decreased neutrophils occurred in 62% of patients, with Grade 3 or 4 decreased neutrophils in 51%. Febrile neutropenia occurred in 2.2% of patients [see Adverse Reactions (6.1)].
Monitor complete blood cell counts at baseline and periodically during treatment. Provide supportive care according to current practice guidelines. Monitor patients with neutropenia for signs of infection. Withhold ELREXFIO based on severity [see Dosage and Administration (2.5)].
5.6 Hepatotoxicity
ELREXFIO can cause hepatotoxicity. In the clinical trial, elevated ALT occurred in 36% of patients, with Grade 3 or 4 ALT elevation occurring in 3.8%; elevated AST occurred in 40% of patients, with Grade 3 or 4 AST elevation occurring in 6%. Grade 3 or 4 total bilirubin elevations occurred in 0.5% of patients [see Adverse Reactions (6.1)]. Liver enzyme elevation can occur with or without concurrent CRS.
Monitor liver enzymes and bilirubin at baseline and during treatment as clinically indicated. Withhold ELREXFIO or consider permanent discontinuation of ELREXFIO based on severity [see Dosage and Administration (2.5)].
5.7 Embryo-Fetal Toxicity
Based on its mechanism of action, ELREXFIO may cause fetal harm when administered to a pregnant woman. Advise pregnant women of the potential risk to the fetus. Advise females of reproductive potential to use effective contraception during treatment with ELREXFIO and for 4 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed elsewhere in labeling:
- Cytokine Release Syndrome [see Warnings and Precautions (5.1)].
- Neurologic Toxicity, Including ICANS [see Warnings and Precautions (5.2)].
- Infections [see Warnings and Precautions (5.4)].
- Neutropenia [see Warnings and Precautions (5.5)].
- Hepatotoxicity [see Warnings and Precautions (5.6)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Relapsed/Refractory Multiple Myeloma
MagnetisMM-3
The safety of ELREXFIO was evaluated in MagnetisMM-3 [see Clinical Studies (14)]. The safety population described (n = 183) includes patients who received the recommended dosage regimen of 12 mg subcutaneously on Day 1, 32 mg on Day 4, and 76 mg once weekly starting on Day 8. Among patients who received ELREXFIO, 42% were exposed for 6 months or longer and 9% were exposed for one year or longer.
The median age of patients who received ELREXFIO was 68 years (range: 36 to 88 years); 48% were female; 61% were White, 10% were Hispanic/Latino, 9% were Asian, and 6% were Black or African American.
Serious adverse reactions occurred in 68% of patients who received ELREXFIO at the recommended dosing schedule. Serious adverse reactions in >2% of patients included pneumonia (25%), sepsis (13%), CRS (13%), upper respiratory tract infection (4.4%), acute kidney injury (3.8%), urinary tract infection (3.3%), COVID-19 (3.3%), encephalopathy (3.3%), pyrexia (2.2%), and febrile neutropenia (2.2%). Fatal adverse reactions occurred in 10% of patients including pneumonia (3.3%), sepsis (2.7%), acute respiratory distress syndrome (0.5%), cardio-respiratory arrest (0.5%), cardiogenic shock (0.5%), cardiopulmonary failure (0.5%), COVID-19 (0.5%), failure to thrive (0.5%), and pulmonary embolism (0.5%).
Permanent discontinuations of ELREXFIO due to an adverse reaction occurred in 17% of patients. Adverse reactions which resulted in permanent discontinuation of ELREXFIO in >2% of patients included septic shock (2.2%).
Dosage interruptions of ELREXFIO due to an adverse reaction occurred in 73% of patients. Adverse reactions which resulted in dose interruptions of ELREXFIO in >5% of patients included neutropenia, pneumonia, COVID-19, upper respiratory tract infection, thrombocytopenia, and anemia.
The most common adverse reactions (≥20%) were CRS, fatigue, injection site reaction, diarrhea, upper respiratory tract infection, musculoskeletal pain, pneumonia, decreased appetite, rash, cough, nausea, and pyrexia. The most common Grade 3 to 4 laboratory abnormalities (≥30%) were decreased lymphocytes, decreased neutrophils, decreased hemoglobin, decreased white blood cells, and decreased platelets.
Table 8 summarizes adverse reactions in MagnetisMM-3.
Table 8. Adverse Reactions (≥10%) in Patients with Relapsed or Refractory Multiple Myeloma Who Received ELREXFIO in MagnetisMM-3 Adverse reactions were graded based on CTCAE Version 5.0, with the exception of CRS, which was graded based on the ASTCT 2019 criteria. - * Only grade 3 adverse reactions occurred.
- † Includes other related terms.
- ‡ Pneumonia includes COVID-19 pneumonia, lower respiratory tract infection, lower respiratory tract infection viral, pneumocystis jirovecii pneumonia, pneumonia, pneumonia adenoviral, pneumonia bacterial, pneumonia cytomegaloviral, pneumonia fungal, pneumonia influenzal, pneumonia pseudomonal, pneumonia viral.
- § Sepsis includes bacteremia, device related bacteremia, device related sepsis, escherichia bacteremia, escherichia sepsis, klebsiella sepsis, pseudomonal sepsis, sepsis, septic shock, staphylococcal bacteremia, staphylococcal sepsis, streptococcal sepsis, urosepsis.
- ¶ Rash incudes erythema, palmar-plantar erythrodysesthesia syndrome, rash, rash erythematous, rash macular, rash maculo-papular, rash pustular, symmetrical drug-related intertriginous and flexural exanthema.
- # Encephalopathy includes agitation, altered state of consciousness, cognitive disorder, confusional state, delirium, depressed level of consciousness, disorientation, hallucination, lethargy, memory impairment, metabolic encephalopathy, somnolence, toxic encephalopathy.
- Þ Sensory neuropathy includes burning sensation, dysesthesia, hypoesthesia, neuropathy peripheral, paresthesia, parosmia, peripheral sensorimotor neuropathy, peripheral sensory neuropathy, polyneuropathy, sensory loss.
- ß Motor dysfunction includes ataxia, balance disorder, gait disturbance, motor dysfunction, muscle contracture, muscle spasms, muscular weakness, peripheral motor neuropathy, peroneal nerve palsy, tremor.
System Organ Class
Preferred Term
ELREXFIO
N = 183
All Grades
(%)
Grade 3 or 4
(%)
Immune system disorders
Cytokine release syndrome
58
0.5*
Hypogammaglobulinemia†
13
2.2*
General disorders and site administration conditions
Fatigue†
43
6*
Injection site reaction†
37
0
Pyrexia
21
2.7*
Edema†
18
1.1*
Gastrointestinal disorders
Diarrhea
36
1.1*
Nausea
21
0
Constipation
14
0
Vomiting
14
0
Infections
Upper respiratory tract infection†
36
4.9
Pneumonia‡
32
19
Sepsis§
15
11
Urinary tract infection†
12
4.4*
Musculoskeletal and connective tissue disorders
Musculoskeletal pain†
34
2.7*
Metabolism and nutrition disorders
Decreased appetite
26
1.1*
Skin and Subcutaneous Tissue disorders
Rash¶
26
0
Dry skin
14
0
Skin exfoliation†
10
0
Respiratory, thoracic and mediastinal disorders
Cough†
24
0
Dyspnea†
15
3.3*
Nervous system disorders
Headache
18
0
Encephalopathy#
14
2.2
Sensory neuropathyÞ
13
0.5*
Motor dysfunctionß
14
1.1*
Cardiac disorders
Cardiac arrhythmia†
16
1.6
Vascular disorders
Hemorrhage†
13
1.6
Psychiatric disorders
Insomnia
13
0
Injury, poisoning and procedural complications
Fall
10
0.5*
Clinically relevant adverse reactions in <10% of patients who received ELREXFIO included ICANS, febrile neutropenia, Guillain-Barré syndrome, abdominal pain, acute kidney injury, COVID-19, cardiac failure, congestion, thrombosis, cytomegalovirus infection, and progressive multifocal leukoencephalopathy.
Table 9 summarizes laboratory abnormalities in MagnetisMM-3.
Table 9. Select Laboratory Abnormalities (≥30%) That Worsened from Baseline in Patients with Relapsed or Refractory Multiple Myeloma Who Received ELREXFIO in MagnetisMM-3* - * Laboratory tests were graded according to NCI-CTCAE Version 5.0.
- † The denominator used to calculate the rate varied from 181 to 183 based on the number of patients with a baseline value and at least one post-treatment value.
Laboratory Abnormality
ELREXFIO†
All Grades (%)
Grade 3 or 4 (%)
Hematology
Lymphocyte count decreased
91
84
White blood cell decreased
69
40
Hemoglobin decreased
68
43
Neutrophil count decreased
62
51
Platelet count decreased
61
32
Chemistry
Albumin decreased
55
6
AST increase
40
6
Creatinine increased
38
3.3
Potassium decreased
36
8
ALT increase
36
3.8
Alkaline phosphatase increased
34
1.1
Creatinine clearance decreased
32
10
-
7 DRUG INTERACTIONS
For certain CYP substrates, minimal changes in the concentration may lead to serious adverse reactions. Monitor for toxicity or drug concentrations of such CYP substrates when co-administered with ELREXFIO.
ELREXFIO causes release of cytokines [see Clinical Pharmacology (12.2)] that may suppress activity of cytochrome P450 (CYP) enzymes, resulting in increased exposure of CYP substrates. Increased exposure of CYP substrates is more likely to occur after the first dose of ELREXFIO Day 1 and up to 14 days after the 32 mg dose on Day 4 and during and after CRS [see Warnings and Precautions (5.1)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on the mechanism of action, ELREXFIO may cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data on the use of ELREXFIO in pregnant women to evaluate for a drug associated risk. No animal reproductive or developmental toxicity studies have been conducted with ELREXFIO. Elranatamab-bcmm causes T-cell activation and cytokine release; immune activation may compromise pregnancy maintenance. In addition, based on the finding of B-cell depletion in non-pregnant animals, elranatamab-bcmm can cause B-cell lymphocytopenia in infants exposed to elranatamab-bcmm in-utero. Human immunoglobulin (IgG) is known to cross the placenta after the first trimester of pregnancy; therefore, elranatamab-bcmm has the potential to be transmitted from the mother to the developing fetus. Advise women of the potential risk to the fetus.
ELREXFIO is associated with hypogammaglobulinemia, therefore, assessment of immunoglobulin levels in newborns of mothers treated with ELREXFIO should be considered.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
8.2 Lactation
Risk Summary
There are no data on the presence of elranatamab-bcmm in human milk, the effects on the breastfed child, or the effects on milk production. Maternal IgG is known to be present in human milk.
Because of the potential for serious adverse reactions in a breastfed child, advise women not to breastfeed during treatment with ELREXFIO and for 4 months after the last dose.
8.3 Females and Males of Reproductive Potential
ELREXFIO may cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating treatment with ELREXFIO.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment and for 4 months after the last dose of ELREXFIO.
8.4 Pediatric Use
The safety and effectiveness of ELREXFIO in pediatric patients have not been established.
8.5 Geriatric Use
Of the 183 patients with relapsed or refractory multiple myeloma treated with ELREXFIO in MagnetisMM-3 at the recommended dosage, 62% were 65 years of age or older, and 19% were 75 years of age or older. No overall differences in safety or effectiveness were observed in patients 65-74 years of age compared to younger patients. Clinical studies did not include sufficient numbers of patients 75 years of age or older to determine whether they respond differently from younger patients.
-
11 DESCRIPTION
Elranatamab-bcmm is a bispecific B-cell maturation antigen (BCMA)-directed CD3 T-cell engager. It is a bispecific, humanized immunoglobulin 2-alanine (IgG2Δa) kappa antibody derived from two monoclonal antibodies (mAbs), an anti-BCMA mAb and an anti-CD3 mAb. Each of these mAbs contributes one distinct heavy (H) chain and one distinct light (L) chain to the bispecific elranatamab-bcmm. The resulting 4-chain bispecific antibody is covalently linked via five inter-chain disulfide bonds. Elranatamab-bcmm is produced using two recombinant Chinese hamster ovary (CHO) cell lines, one that contains the DNA encoding the sequence for anti-BCMA monoclonal antibody (mAb) and one that contains the sequence for anti-CD3 mAb, which are grown separately in suspension culture using chemically-defined (CD), animal-derived component-free (ACF) media. The molecular weight of elranatamab-bcmm is approximately 148.5 kDa.
ELREXFIO® (elranatamab-bcmm) injection is a sterile, preservative-free, clear to slightly opalescent, and colorless to pale brown liquid solution for subcutaneous administration. ELREXFIO (elranatamab-bcmm) is supplied at a concentration of 40 mg/mL in either 76 mg/1.9 mL or 44 mg/1.1 mL single-dose vials. Each mL of solution contains 40 mg elranatamab-bcmm, edetate disodium (0.045 mg), histidine (1.12 mg), L-histidine hydrochloride monohydrate (2.67 mg), polysorbate 80 (0.2 mg), sucrose (85 mg) and Water for Injection. The pH is 5.8.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Elranatamab-bcmm is a bispecific B-cell maturation antigen (BCMA)-directed T-cell engaging antibody that binds BCMA on plasma cells, plasmablasts, and multiple myeloma cells and CD3 on T-cells leading to cytolysis of the BCMA-expressing cells. Elranatamab-bcmm activated T-cells, caused proinflammatory cytokine release, and resulted in multiple myeloma cell lysis.
12.2 Pharmacodynamics
Cytokine Concentrations
Transient elevation of circulating cytokines IL-2, IL-6, IL-8, IL-10, TNF-α, and IFN-γ was observed at dosage levels of 30 µg/kg (0.03 times the approved recommended dosage) and above. After administration of the approved recommended dosage of ELREXFIO, the highest elevation of cytokines was generally observed within 72 hours after first elranatamab-bcmm dose at 12 mg on Day 1, and generally returned to baseline prior to the administration of the first full dose 76 mg on Day 8.
12.3 Pharmacokinetics
Pharmacokinetic parameters are presented as geometric mean (coefficient of variation [CV]%) and are based upon subcutaneously administered unless otherwise specified.
Elranatamab-bcmm exhibits dose proportional pharmacokinetics over dose range from 6 to 76 mg (0.079 to 1 times the approved recommended dosage). Elranatamab-bcmm maximum concentration [33.0 mcg/mL (46%)] is achieved at the end of weekly dosing regimen (i.e., at week 24 of 76 mg weekly dosing). Pharmacokinetic exposures are summarized for the recommended dosage of ELREXFIO in Table 10.
Table 10. Pharmacokinetic Parameters of Elranatamab-bcmm in Subjects with Relapsed or Refractory Multiple Myeloma - * In patients who have achieved a response.
- † Steady state exposure of elranatamab biweekly dose is approximated at week 48.
- ‡ Steady state exposure of elranatamab once every 4 weeks dose is approximated at week 72.
Timepoint
Parameters
Cavg
(mcg/mL)
Cmax
(mcg/mL)
Ctrough
(mcg/mL)
First full 76 mg dose
3.1 (94%)
3.8 (94%)
3.3 (102%)
End of weekly dose (week 24)*
32.0 (46%)
33.0 (46%)
30.5 (48%)
17.7 (53%)
19.5 (51%)
15.1 (60%)
8.8 (58%)
11.5 (54%)
5.9 (78%)
Absorption
The mean bioavailability of elranatamab-bcmm was 56.2% when administered subcutaneously. The median (min, max) Tmax after elranatamab SC administration was 7 (3 to 7) days.
Distribution
The steady state volume of distribution of elranatamab-bcmm was 7.76 L (33%).
Elimination
The half-life of elranatamab-bcmm is 22 (64%) days at the 76 mg dosage, with clearance of 0.324 L/day (100%) following 24 weeks dosing.
Metabolism
Elranatamab-bcmm is expected to be metabolized into small peptides by catabolic pathways.
Specific Populations
No clinically significant differences in the pharmacokinetics of elranatamab-bcmm were observed based on age (36 to 89 years), sex, race (White, Asian, or Black), body weight (37 to 160 kg), mild or moderate renal impairment (estimated glomerular filtration rate [eGFR] by Modification of Diet in Renal Disease [MDRD] method: 30 to 89 mL/min), or mild hepatic impairment (total bilirubin 1 to ≤1.5 x ULN or any AST greater than ULN).
The effects of severe renal impairment (eGFR 15 to 29 mL/min), end-stage renal disease (eGFR <15 mL/min), or moderate to severe hepatic impairment (total bilirubin >1.5 times ULN and any AST) on the PK of elranatamab-bcmm are unknown.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of elranatamab-bcmm or of other elranatamab products.
In the MagnetisMM-3 study, of the 168 participant who received recommended step-up and full dosage of ELREXFIO for up to 36 months and are evaluable for presence of ADA against elranatamab-bcmm, 9.5% (16/168) of patients tested positive for anti-elranatamab-bcmm-antibodies. Among the 16 patients who tested positive for ADAs, 56% (9/16) tested positive for neutralizing antibodies against elranatamab-bcmm. The effect of these antibodies on the pharmacokinetics, pharmacodynamics, safety, and/or effectiveness of ELREXFIO products is unknown.
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
14.1 Relapsed or Refractory Multiple Myeloma
The efficacy of ELREXFIO monotherapy was evaluated in patients with relapsed or refractory multiple myeloma in an open-label, single arm, multi-center study (MagnetisMM-3, NCT04649359). The study included patients who were refractory to at least one proteasome inhibitor (PI), one immunomodulatory agent (IMiD), and one anti-CD38 monoclonal antibody. MagnetisMM-3 included 123 patients naïve to prior BCMA-directed therapy (pivotal Cohort A) and 64 patients with prior BCMA-directed antibody drug conjugate (ADC) or chimeric antigen receptor (CAR) T-cell therapy (supportive Cohort B). Patients had measurable disease by International Myeloma Working Group (IMWG) criteria at enrollment. The study included patients with an Eastern Cooperative Oncology Group (ECOG) score of ≤2, adequate baseline bone marrow (absolute neutrophil count ≥1.0 x 109/L, platelet count ≥25 x 109/L, hemoglobin level ≥8 g/dL), renal (CrCL ≥ 30 mL/min), and hepatic (AST and ALT ≤2.5 x ULN, total bilirubin ≤2 x ULN) function, and left-ventricular ejection fraction ≥40%. Patients with a stem cell transplant within 12 weeks prior to enrollment and active infections were excluded from the study.
Eligible patients received subcutaneous administration of ELREXFIO at step-up doses of 12 mg on Day 1 and 32 mg on Day 4 of treatment, followed by the first treatment dose of ELREXFIO (76 mg) on Day 8 of treatment. Thereafter, patients received 76 mg once weekly. After 24 weeks, in patients who achieved an IMWG response category of partial response or better with responses persisting for at least 2 months, the dose interval was changed from every week to every 2 weeks.
The 123 patients enrolled in pivotal Cohort A had received a median of 5 prior lines of therapy (range: 2 to 22). Ninety-seven patients who were not exposed to prior BCMA-directed therapy and received at least four prior lines of therapy comprised the efficacy population. Among the 97 patients in the efficacy population, the median age was 69 (range: 46 to 89) years with 18.6% of patients ≥75 years of age. Forty percent were female; 59.8% were White, 13.4% were Asian, 7.2% were Hispanic/Latino, 5.2% were Black or African American. Disease stage (R-ISS) at study entry was 20.6% in Stage I, 53.6% in Stage II, and 17.5% in Stage III. The median time since initial diagnosis of multiple myeloma to enrollment was 79.6 (range: 16 to 228) months. 96.9% were triple-class refractory, and 94.8% were refractory to their last line of therapy. 69.1% received prior autologous stem cell transplantation, and 7.2% received prior allogenic stem cell transplantation. High-risk cytogenetics [t(4;14), t(14;16), or del(17p)] were present in 22.7% of patients. 34.0% of patients had extramedullary disease at baseline by BICR.
Efficacy was based on response rate and duration of response (DOR), as assessed by BICR based on IMWG criteria. Efficacy results from BCMA-directed therapy naïve patients are shown in Table 11.
The median (range) time to first response (TTR) was 1.22 (0.9 to 6.5) months. With a median follow-up of 11.1 months (95% CI: 10.6, 12.0) among responders, the DOR rate at 6 months was 90.4% (95% CI: 78.4%, 95.9%) and at 9 months was 82.3% (95% CI: 67.1%, 90.9%).
Table 11. Efficacy Results from BCMA-directed Therapy Naïve Patients Abbreviations: CI = Confidence interval; NR = Not reached; NE = Not estimable. - * Complete response or better = Stringent complete response (sCR) + complete response (CR).
N = 97
Objective Response Rate (ORR: sCR+CR+VGPR+PR), n (%) (95% CI)
56 (57.7%)
(47.3, 67.7)
Complete response (CR) or better*
25 (25.8%)
Very good partial response (VGPR)
25 (25.8%)
Partial response (PR)
6 (6.2%)
Duration of Response (DOR) (months)
Median (95% CI)
NR (12.0, NE)Among the 64 patients enrolled in Cohort B who previously received a PI, an IMiD, an anti-CD38 monoclonal antibody, and a BCMA-directed therapy, 63 patients received at least four prior lines of therapy. Patients had received a median of 8 prior lines of therapy (range: 4 to 19); 73% and 32% received prior BCMA-directed ADC and CAR T-cell therapy, respectively.
Confirmed ORR by BICR was 33.3% (95% CI: 22.0, 46.3). After a median (95% CI) follow-up of 10.2 (9.9, 11) months among responders, median DOR was not reached (95% CI: NE, NE) and the DOR rate at 9 months was 84.3% (95% CI: 58.7, 94.7).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
ELREXFIO® (elranatamab-bcmm) injection is a sterile, preservative-free, clear to slightly opalescent, and colorless to pale brown liquid solution supplied as follows:
- One 76 mg/1.9 mL (40 mg/mL) single-dose vial in a carton. NDC: 0069-4494-02
- One 44 mg/1.1 mL (40 mg/mL) single-dose vial in a carton. NDC: 0069-2522-02
ELREXFIO is supplied in a single-dose glass vial sealed with a rubber stopper (not made of natural rubber latex) and an aluminum seal with a flip-off cap.
Storage and Handling
Store refrigerated at 2 °C to 8 °C (36 °F to 46 °F) in the original carton until time of use to protect from light.
Do not freeze or shake the vial or carton.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Cytokine Release Syndrome (CRS)
Discuss the signs and symptoms associated with CRS, including fever, hypoxia, chills, hypotension, tachycardia, and elevated liver enzymes. Advise patients to immediately contact their healthcare provider if they experience any signs or symptoms of CRS. Advise patients that they will be hospitalized for 48 hours after administration of the first step-up dose, and for 24 hours after administration of the second step-up dose [see Dosage and Administration (2.5), Warnings and Precautions (5.1)].
Neurologic Toxicity, Including Immune Effector Cell-associated Neurotoxicity Syndrome (ICANS)
Discuss the signs and symptoms associated with neurologic toxicity, including ICANS, including headache, encephalopathy, motor dysfunction, sensory neuropathy, and Guillain-Barré Syndrome. Advise patients to immediately contact their healthcare provider if they experience any signs or symptoms of neurologic toxicity. Advise patients to refrain from driving or operating heavy or potentially dangerous machinery for 48 hours after completing each of the 2 step-up doses and the first treatment dose within the ELREXFIO step-up dosing schedule and in the event of new onset of any neurological toxicity symptoms until symptoms resolve [see Dosage and Administration (2.5), Warnings and Precautions (5.2)].
ELREXFIO REMS
ELREXFIO is available only through a restricted program called ELREXFIO REMS. Inform patients that they will be given an ELREXFIO Patient Wallet Card that they should carry with them at all times and show to all of their healthcare providers. This card describes signs and symptoms of CRS and neurologic toxicity, including ICANS which, if experienced, should prompt the patient to immediately seek medical attention [see Warnings and Precautions (5.3)].
Infections
Discuss the signs and symptoms of infection [see Dosage and Administration (2.5), Warnings and Precautions (5.4)].
Neutropenia
Discuss the signs and symptoms associated with neutropenia and febrile neutropenia [see Dosage and Administration (2.5), Warnings and Precautions (5.5)].
Hepatotoxicity
Advise patients that liver enzyme elevations may occur and that they should report symptoms that may indicate liver toxicity, including fatigue, anorexia, right upper abdominal discomfort, dark urine, or jaundice [see Warnings and Precautions (5.6)].
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider if they are pregnant or become pregnant. Advise females of reproductive potential to use effective contraception during treatment with ELREXFIO and for 4 months after the last dose [see Warnings and Precautions (5.7), Use in Specific Populations (8.1, 8.3)].
Lactation
Advise women not to breastfeed during treatment with ELREXFIO and for 4 months after the last dose [see Use in Specific Populations (8.2)].
Manufactured by:
Pfizer Inc.
NY, NY 10001
US License No. 2001

LAB-1518-4.0 -
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: Feb 2026 MEDICATION GUIDE
ELREXFIO® (el-reks-fe-o)
(elranatamab-bcmm)
injection, for subcutaneous useWhat is the most important information I should know about ELREXFIO?
ELREXFIO may cause serious side effects, including:
- Cytokine Release Syndrome (CRS). CRS is common during treatment with ELREXFIO and can also be serious, life-threatening, or can lead to death. Tell your healthcare provider or get medical help right away if you develop any signs or symptoms of CRS, including:
- o fever of 100.4 °F (38 °C) or higher
- o trouble breathing
- o chills
- o dizziness or light-headedness
- o fast heartbeat
- o headache
- o increased liver enzymes in your blood. See “What are the possible side effects of ELREXFIO?” for more information about the signs and symptoms of liver problems.
-
Due to the risk of CRS, you will receive ELREXFIO on a “step-up dosing schedule” and should be hospitalized for 48 hours after the first “step-up” dose and for 24 hours after the second “step-up” dose of ELREXFIO.
- o
During the step-up dosing schedule:
- ▪ for your first dose, you will receive a smaller “step-up” dose of ELREXFIO on Day 1 of your treatment
- ▪ for your second dose, you will receive a larger “step-up” dose of ELREXFIO, which is usually given on Day 4 of your treatment
- ▪ for your third dose, you will receive the first full “treatment” dose of ELREXFIO, which is usually given on Day 8 of your treatment
- o If your dose of ELREXFIO is delayed for any reason, you may need to repeat the step-up dosing schedule.
- o Before each step-up dose and the first full treatment dose of ELREXFIO, you will receive medicines to help reduce your risk of CRS. Your healthcare provider will decide if you need to receive medicines to help reduce your risk of CRS with future doses.
- o See “How will I receive ELREXFIO?” for more information about how you will receive ELREXFIO.
- o
During the step-up dosing schedule:
- Neurologic problems. ELREXFIO can cause neurologic problems that can be serious or life-threatening. Tell your healthcare provider or get medical help right away if you develop any signs or symptoms of neurologic problems, including:
- o headache
- o agitation, trouble staying awake, confusion or disorientation, seeing or hearing things that are not real (hallucinations)
- o trouble speaking, thinking, remembering things, paying attention, or understanding things
- o problems walking, muscle weakness, shaking (tremors), loss of balance, or muscle spasms
- o numbness and tingling (feeling like “pins and needles”)
- o burning, throbbing, or stabbing pain
- o changes in your handwriting
- ELREXFIO is available only through the ELREXFIO Risk Evaluation and Mitigation Strategy (REMS) due to the risk of CRS and neurologic problems. You will receive an ELREXFIO Patient Wallet Card from your healthcare provider. Carry the ELREXFIO Patient Wallet Card with you at all times and show it to all of your healthcare providers. The ELREXFIO Patient Wallet Card lists symptoms of CRS and neurologic problems. Get medical help right away if you develop any of the symptoms listed on the ELREXFIO Patient Wallet Card. You may need to be treated in a hospital.
Your healthcare provider will monitor you for signs and symptoms of CRS and neurologic problems during treatment with ELREXFIO, as well as other side effects, and will treat you if needed. Your healthcare provider may temporarily stop or completely stop your treatment with ELREXFIO if you develop CRS, neurologic problems, or any other side effects that are severe.
If you have any questions about ELREXFIO, ask your healthcare provider.
See “What are possible side effects of ELREXFIO?” for more information about side effects.
What is ELREXFIO?
ELREXFIO is a prescription medicine used to treat adults with multiple myeloma who:
- have already received at least 4 treatment regimens, including a proteasome inhibitor, an immunomodulatory agent and an anti-CD38 monoclonal antibody to treat their multiple myeloma, and
- their cancer has come back or did not respond to prior treatment.
It is not known if ELREXFIO is safe and effective in children.
Before receiving ELREXFIO, tell your healthcare provider about all of your medical conditions, including if you:
- have an infection.
-
are pregnant or plan to become pregnant. ELREXFIO may harm your unborn baby.
Females who are able to become pregnant:- o Your healthcare provider should do a pregnancy test before you start treatment with ELREXFIO.
- o You should use effective birth control (contraception) during treatment and for 4 months after your last dose of ELREXFIO.
- o Tell your healthcare provider right away if you become pregnant or think that you may be pregnant during treatment with ELREXFIO.
- are breastfeeding or plan to breastfeed. It is not known if ELREXFIO passes into your breast milk. Do not breastfeed during treatment and for 4 months after your last dose of ELREXFIO.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
- ELREXFIO will be given to you by your healthcare provider as an injection under your skin (subcutaneous injection), usually in your stomach-area (abdomen). Your thigh or another area of your body may also be used.
- See “What is the most important information I should know about ELREXFIO?” for more information about how you will receive ELREXFIO.
- After you receive your first full “treatment” dose, ELREXFIO is usually given 1 time each week through Week 24.
- Starting on Week 25, your future doses will usually be given 1 time every 2 weeks.
- Starting on Week 49, your future doses will usually be given 1 time every 4 weeks.
- Your healthcare provider will decide the time between doses and how long you will receive treatment with ELREXFIO.
If you miss any appointments, call your healthcare provider as soon as possible to reschedule your appointment. It is important for you to be monitored closely for side effects during treatment with ELREXFIO.
What should I avoid while receiving ELREXFIO?
Do not drive, operate heavy or potentially dangerous machinery, or do other dangerous activities during treatment with ELREXFIO:
- for 48 hours after completing each of the 2 doses of ELREXFIO that are part of the “step-up dosing schedule” and your first full treatment dose, and
- at any time during treatment with ELREXFIO if you develop any new neurologic symptoms such as dizziness, confusion, shaking (tremors), sleepiness, or any other symptom that impairs consciousness, until the symptoms go away.
See “What is the most important information I should know about ELREXFIO?” for more information about signs and symptoms of neurologic problems.
What are the possible side effects of ELREXFIO?
ELREXFIO may cause serious side effects, including:
- See “What is the most important information I should know about ELREXFIO?”
-
Infections. Upper respiratory tract infections and pneumonia are common during treatment with ELREXFIO. ELREXFIO can cause bacterial and viral infections that are severe, life-threatening, or that may lead to death.
- o Your healthcare provider may prescribe medicines for you to help prevent infections and treat you as needed if you develop an infection during treatment with ELREXFIO.
- o Tell your healthcare provider right away if you develop any signs or symptoms of an infection during treatment with ELREXFIO, including:
- ▪ fever of 100.4 °F (38 °C) or higher
- ▪ chills
- ▪ cough
- ▪ shortness of breath
- ▪ chest pain
- ▪ sore throat
- ▪ pain during urination
- ▪ feeling weak or generally unwell
- Decreased white blood cell counts. Decreased white blood cell counts are common during treatment with ELREXFIO and can also be severe. Fever can happen with low white blood cell counts and may be a sign that you have an infection. Your healthcare provider will treat you as needed.
- Liver problems. ELREXFIO can cause increased liver enzymes and bilirubin in your blood. These increases can happen with or without you also having CRS. Tell your healthcare provider if you develop any of the following signs or symptoms of liver problems:
- o tiredness
- o loss of appetite
- o pain in your right upper stomach-area (abdomen)
- o dark urine
- o yellowing of your skin or the white part of your eyes
Your healthcare provider will check your blood and monitor you for signs and symptoms of these serious side effects before you start and during treatment with ELREXFIO and may temporarily or completely stop treatment with ELREXFIO if you develop certain side effects.
The most common side effects of ELREXFIO include:
- tiredness
- injection site reaction, such as redness, itching, pain, bruising, rash, swelling, tenderness
- diarrhea
- muscle and bone pain
- decreased appetite
- rash
- cough
- nausea
- fever
The most common severe abnormal blood test results with ELREXFIO include decreased white blood cells, red blood cells, and platelets.
These are not all of the possible side effects of ELREXFIO.Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
General information about the safe and effective use of ELREXFIO.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your pharmacist or healthcare provider for more information about ELREXFIO that is written for health professionals.
What are the ingredients in ELREXFIO?
Active ingredient: elranatamab-bcmm
Inactive ingredients: edetate disodium, histidine, L-histidine hydrochloride monohydrate, polysorbate 80, sucrose, and Water for Injection
Manufactured by: Pfizer Inc., NY, NY 10001
US License No. 2001

LAB-1551-3.0
For more information on ELREXFIO, go to www.ELREXFIO.com
For more information on Pfizer, go to www.Pfizer.com or call 1-800-438-1985
-
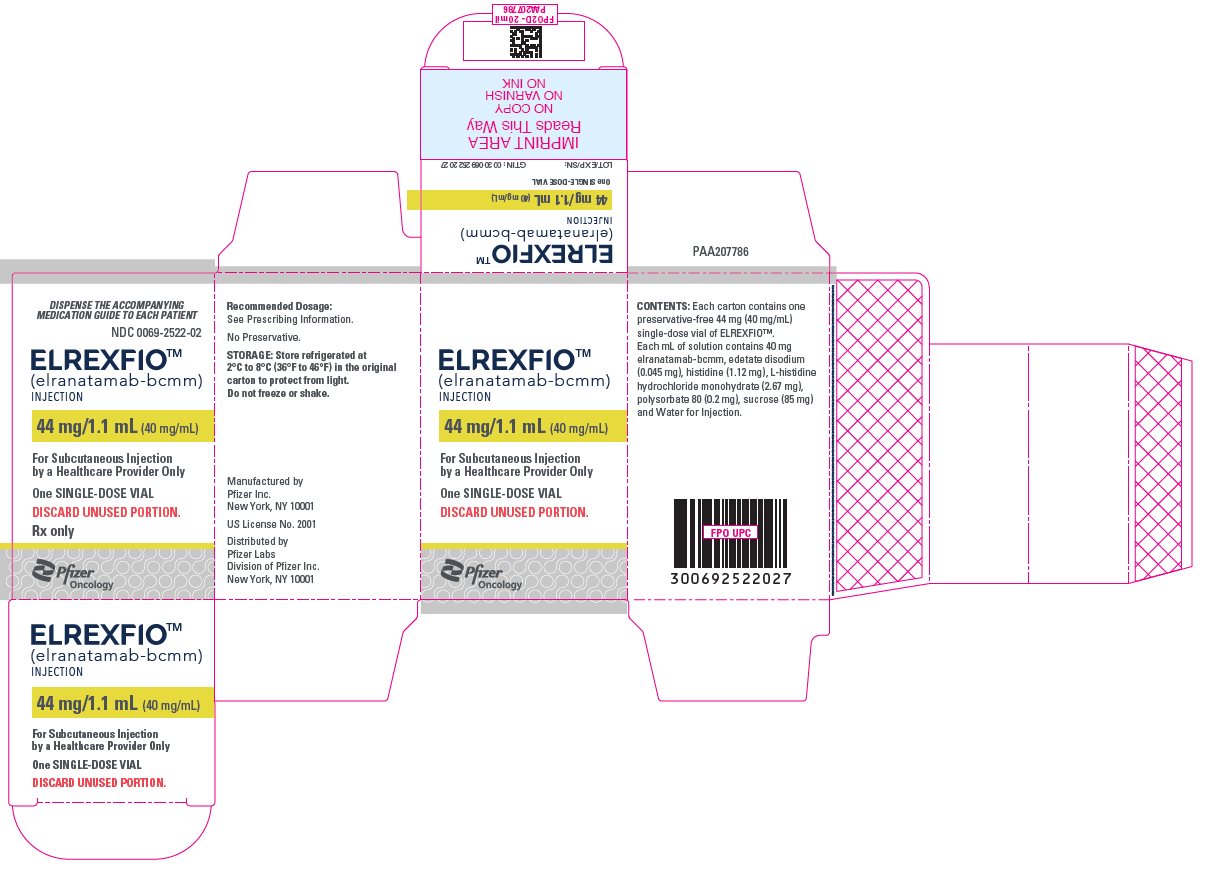
PRINCIPAL DISPLAY PANEL – 44 mg/1.1 mL Vial
DISPENSE THE ACCOMPANYING
MEDICATION GUIDE TO EACH PATIENTNDC: 0069-2522-01
Rx onlyELREXFIOTM
(elranatamab-bcmm)
INJECTION44 mg/1.1 mL (40 mg/mL)
FOR SUBCUTANEOUS USE
One SINGLE-DOSE VIAL
DISCARD UNUSED PORTION.Pfizer
-
PRINCIPAL DISPLAY PANEL – 44 mg/1.1 mL Vial Carton
DISPENSE THE ACCOMPANYING
MEDICATION GUIDE TO EACH PATIENTNDC: 0069-2522-02
ELREXFIOTM
(elranatamab- bcmm)
INJECTION44 mg/1.1 mL (40 mg/mL)
For Subcutaneous Injection
by a Healthcare Provider OnlyOne SINGLE-DOSE VIAL
DISCARD UNUSED PORTION.
Rx only
Pfizer
Oncology -
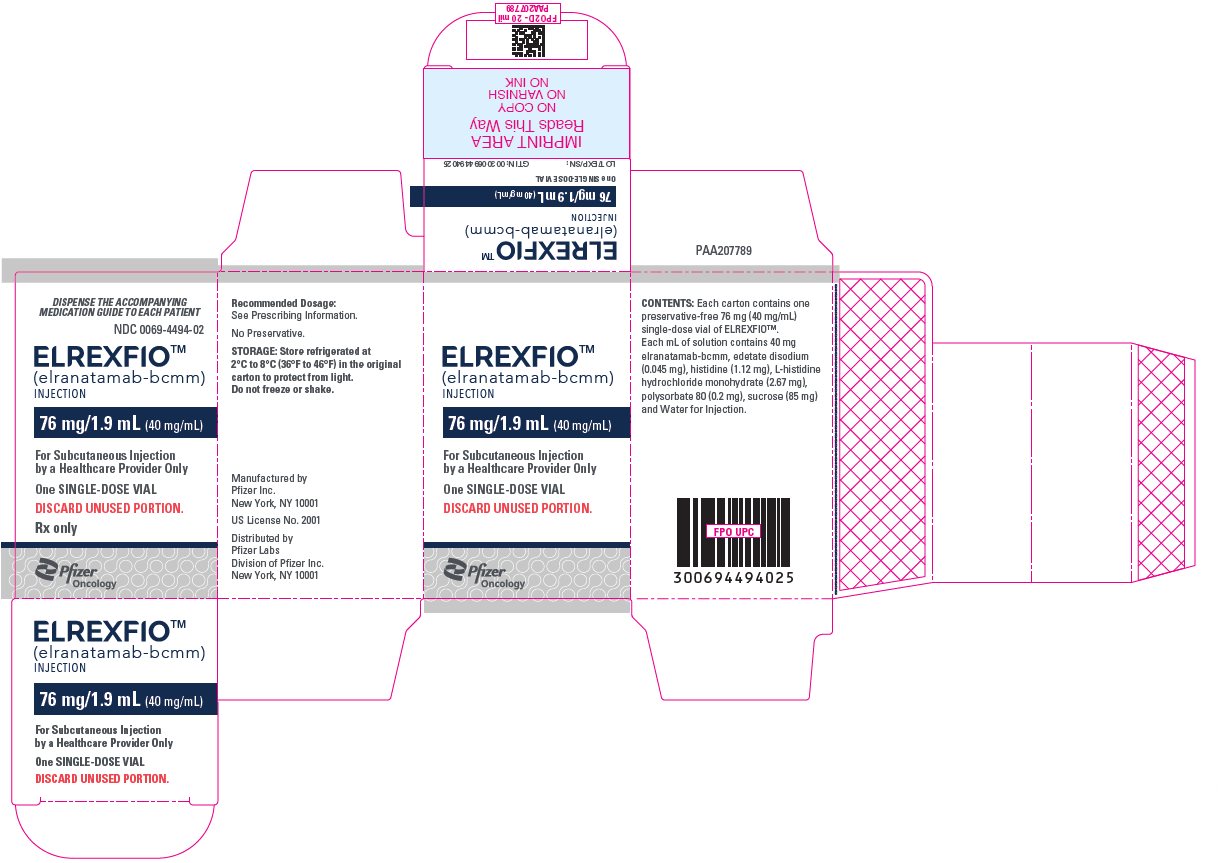
PRINCIPAL DISPLAY PANEL – 76 mg/1.9 mL Vial
DISPENSE THE ACCOMPANYING
MEDICATION GUIDE TO EACH PATIENTNDC: 0069-4494-01
Rx onlyELREXFIOTM
(elranatamab- bcmm)
INJECTION76 mg/1.9 mL (40 mg/mL)
FOR SUBCUTANEOUS USE
One SINGLE-DOSE VIAL
DISCARD UNUSED PORTION.Pfizer
-
PRINCIPAL DISPLAY PANEL – 76 mg/1.9 mL Vial Carton
DISPENSE THE ACCOMPANYING
MEDICATION GUIDE TO EACH PATIENTNDC: 0069-4494-02
ELREXFIOTM
(elranatamab- bcmm)
INJECTION76 mg/1.9 mL (40 mg/mL)
For Subcutaneous Injection
by a Healthcare Provider OnlyOne SINGLE-DOSE VIAL
DISCARD UNUSED PORTION.
Rx only
Pfizer
Oncology -
INGREDIENTS AND APPEARANCE
ELREXFIO
elranatamab-bcmm injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0069-2522 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ELRANATAMAB (UNII: L0HR9A577V) (ELRANATAMAB - UNII:L0HR9A577V) ELRANATAMAB 44 mg in 1.1 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) 1.23 mg in 1.1 mL HISTIDINE MONOHYDROCHLORIDE MONOHYDRATE (UNII: X573657P6P) 2.94 mg in 1.1 mL EDETATE DISODIUM (UNII: 7FLD91C86K) 0.06 mg in 1.1 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.2 mg in 1.1 mL SUCROSE (UNII: C151H8M554) 94 mg in 1.1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0069-2522-02 1 in 1 CARTON 08/15/2023 1 NDC: 0069-2522-01 1.1 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761345 08/15/2023 ELREXFIO
elranatamab-bcmm injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0069-4494 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ELRANATAMAB (UNII: L0HR9A577V) (ELRANATAMAB - UNII:L0HR9A577V) ELRANATAMAB 76 mg in 1.9 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) 2.13 mg in 1.9 mL HISTIDINE MONOHYDROCHLORIDE MONOHYDRATE (UNII: X573657P6P) 5.07 mg in 1.9 mL EDETATE DISODIUM (UNII: 7FLD91C86K) 0.1 mg in 1.9 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.4 mg in 1.9 mL SUCROSE (UNII: C151H8M554) 162 mg in 1.9 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0069-4494-02 1 in 1 CARTON 08/15/2023 1 NDC: 0069-4494-01 1.9 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761345 08/15/2023 Labeler - Pfizer Laboratories Div Pfizer Inc (134489525) Establishment Name Address ID/FEI Business Operations Wyeth BioPharma Division of Wyeth Pharmaceuticals LLC 174350868 API MANUFACTURE(0069-2522, 0069-4494) , ANALYSIS(0069-2522, 0069-4494) Establishment Name Address ID/FEI Business Operations Pharmacia & Upjohn Company LLC 618054084 ANALYSIS(0069-2522, 0069-4494) , MANUFACTURE(0069-2522, 0069-4494) , PACK(0069-2522, 0069-4494) , LABEL(0069-2522, 0069-4494) Establishment Name Address ID/FEI Business Operations PPD Development, L.P. 838082055 ANALYSIS(0069-2522, 0069-4494)


Trademark Results [Elrexfio]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ELREXFIO 90651029 not registered Live/Pending |
Pfizer Inc. 2021-04-16 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.