ZILEUTON tablet, multilayer, extended release
Zileuton by
Drug Labeling and Warnings
Zileuton by is a Prescription medication manufactured, distributed, or labeled by Golden State Medical Supply, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
ZILEUTON EXTENDED-RELEASE TABLETS. These highlights do not include all the information needed to use ZILEUTON EXTENDED-RELEASE TABLETS safely and effectively. See full prescribing information for ZILEUTON EXTENDED-RELEASE TABLETS.
ZILEUTON extended-release tablets, for oral use
Initial U.S. Approval: 1996INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
Adults and children 12 years of age and older: The recommended dose of zileuton extended-release tablets is two 600 mg extended-release tablets twice daily, within one hour after morning and evening meals, for a total daily dose of 2400 mg. (2) (2)
Monitoring: Assess hepatic function enzymes prior to initiation of zileuton extended-release tablets and monitor periodically during treatment. (2, 5.1) (2)
DOSAGE FORMS AND STRENGTHS
Extended-release tablets: 600 mg. (3) (3)
CONTRAINDICATIONS
- Active liver disease or persistent hepatic function enzyme elevations ≥3 times the upper limit of normal. (4, 5.1)
- History of allergic reaction to zileuton or any of the ingredients of zileuton extended-release tablets. (4)
WARNINGS AND PRECAUTIONS
Hepatotoxicity: Elevations of one or more hepatic function enzymes and bilirubin may occur with zileuton extended-release tablets. Assess hepatic function enzymes prior to initiation of zileuton extended-release tablets, monthly for the first 3 months, every 2 to 3 months for the remainder of the first year, and periodically thereafter. Use zileuton extended-release tablets with caution in patients who consume substantial quantities of alcohol and/or have a history of liver disease. (5.1) (5)
Neuropsychiatric Events: Neuropsychiatric events, including sleep disorders and behavior changes, may occur with zileuton extended-release tablets. Instruct patients to be alert for neuropsychiatric events. Evaluate the risks and benefits of continuing treatment with zileuton extended-release tablets if such events occur. (5.2) (5)
ADVERSE REACTIONS
DRUG INTERACTIONS
- Zileuton increases theophylline levels. Reduce theophylline dose and monitor levels. (7.1)
- Zileuton increases warfarin levels. Monitor prothrombin time and adjust warfarin dose accordingly. (7.2)
- Zileuton increases propranolol levels and beta-blocker activity. Monitor appropriately. (7.3)
USE IN SPECIFIC POPULATIONS
Hepatic Impairment: Zileuton extended-release tablets are contraindicated in patients with active liver disease and in patients with elevated hepatic function enzymes ≥3 times the upper limit of normal. (4, 5, 8.7) (8)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labelling. (8)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 4/2022
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
5.2 Neuropsychiatric Events
6 ADVERSE REACTIONS
6.1 Short-Term Clinical Studies Experience
6.2 Long-Term Clinical Studies Experience
6.3 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Theophylline
7.2 Warfarin
7.3 Propranolol
7.4 Other Concomitant Drug Therapy
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.3 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
17.1 Information for Patients
17.2 FDA-Approved Patient Labeling
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
Zileuton extended-release tablets are indicated for the prophylaxis and chronic treatment of asthma in adults and children 12 years of age and older.
Zileuton extended-release tablets are not indicated for use in the reversal of bronchospasm in acute asthma attacks. Therapy with zileuton extended-release tablets can be continued during acute exacerbations of asthma.
-
2 DOSAGE AND ADMINISTRATION
The recommended dosage of Zileuton extended-release tablets for the treatment of patients with asthma is two 600 mg extended-release tablets twice daily, within one hour after morning and evening meals, for a total daily dose of 2400 mg. Tablets should not be chewed, cut or crushed. If a dose is missed, the patient should take the next dose at the scheduled time and not double the dose. Assess hepatic function enzymes prior to initiation of zileuton extended-release tablets and periodically during treatment [ see Contraindications (4), Warnings and Precautions (5), and Use in Specific Populations (8.7)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
The use of Zileuton extended-release tablets is contraindicated in patients with:
- Active liver disease or persistent hepatic function enzyme elevations greater than or equal to 3 times the upper limit of normal (≥3×ULN) [see Warnings and Precautions (5), and Use in Specific Populations (8.7)] .
- A history of allergic reaction to zileuton or any of the ingredients of zileuton extended-release tablets (e.g., rash, eosinophilia, etc.).
-
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
Elevations of one or more hepatic function enzymes and bilirubin may occur during zileuton extended-release tablets therapy. These laboratory abnormalities may progress to clinically significant liver injury, remain unchanged, or resolve with continued treatment, usually within three weeks. The ALT (SGPT) test is considered the most sensitive indicator of liver injury for zileuton extended-release tablets.
Assess hepatic function enzymes prior to initiation of, and during therapy with, zileuton extended- release tablets. Assess serum ALT before treatment begins, once a month for the first 3 months, every 2 to 3 months for the remainder of the first year, and periodically thereafter for patients receiving long-term zileuton extended-release tablets therapy. If clinical signs and/or symptoms of liver dysfunction develop (e.g., right upper quadrant pain, nausea, fatigue, lethargy, pruritus, jaundice, or "flu-like" symptoms) or transaminase elevations ≥5×ULN occur, discontinue zileuton extended-release tablets and follow hepatic function enzymes until normal.
In controlled and open-label clinical studies involving more than 5000 patients treated with zileuton immediate-release tablets, the overall rate of ALT elevation ≥3×ULN was 3.2%. In these trials, one patient developed symptomatic hepatitis with jaundice, which resolved upon discontinuation of therapy. An additional 3 patients with transaminase elevations developed mild hyperbilirubinemia that was less than 3×ULN. There was no evidence of hypersensitivity or other alternative etiologies for these findings.
Since treatment with zileuton extended-release tablets may result in increased hepatic function enzymes and liver injury, zileuton extended-release tablets should be used with caution in patients who consume substantial quantities of alcohol and/or have a past history of liver disease.
5.2 Neuropsychiatric Events
Neuropsychiatric events have been reported in adult and adolescent patients taking zileuton, the active ingredient in zileuton extended-release tablets and zileuton immediate-release tablets. Post- marketing reports with zileuton include sleep disorders and behavior changes. The clinical details of some post-marketing reports involving zileuton appear consistent with a drug-induced effect. Patients and prescribers should be alert for neuropsychiatric events. Patients should be instructed to notify their prescriber if these changes occur. Prescribers should carefully evaluate the risks and benefits of continuing treatment with zileuton extended-release tablets if such events occur [see Adverse Reactions (6.3)].
-
6 ADVERSE REACTIONS
Hepatotoxicity: Elevations of one or more hepatic function enzymes and bilirubin may occur during zileuton extended-release tablets therapy [see Warnings and Precautions (5)].
The most commonly occurring adverse reactions (≥5%) with zileuton extended-release tablets are sinusitis, nausea, and pharyngolaryngeal pain.
6.1 Short-Term Clinical Studies Experience
The safety data described below reflect exposure to zileuton extended-release tablets in 199 patients for 12 weeks duration. In a 12-week, randomized, double-blind, placebo-controlled trial in adults and adolescents 12 years of age and older with asthma, patients received zileuton extended-release tablets two 600 mg tablets (n=199) or placebo (n=198) twice daily by mouth. Eighty-three percent of patients were white, 48% were male, and the mean age was 34 years.
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
The most commonly reported adverse reactions (occurring at a frequency of ≥5%) in zileuton extended-release tablets-treated patients and at a frequency greater than placebo-treated patients are reflected in Table 1.
Table 1. Adverse Reactions with ≥5% Incidence in a 12-Week Placebo-Controlled Trial in Patients with Asthma. Adverse Reaction
Zileuton Extended-Release Tablets 600 mg 2 Tablets Twice Daily
N=199
n (%)
Placebo 2 Tablets Twice Daily
N=198
n (%)
Sinusitis
13 (6.5)
8 (4.0)
Nausea
10 (5.0)
3 (1.5)
Pharyngolaryngeal pain
10 (5.0)
8 (4.0)
Less common adverse reactions occurring at a frequency ≥1% and more often in the zileuton extended-release tablets group than in the placebo group included gastrointestinal disorders (upper abdominal pain, diarrhea, dyspepsia, vomiting), rash, hypersensitivity, and hepatotoxicity.
There were no differences in the incidence of adverse reactions based upon gender. The clinical trials did not include sufficient numbers of patients <18 years of age or non-Caucasians to determine whether there is any difference in adverse reactions based upon age or race.
Hepatotoxicity
In the 12-week placebo-controlled trial, the incidence of ALT elevations (≥3×ULN) was 2.5% (5 of 199) in the zileuton extended-release tablets group, compared to 0.5% (1 of 198) in the placebo group. In the zileuton extended-release tablets group, the majority of ALT elevations (60%) occurred in the first month of treatment, and in 2 of the 5 patients in the zileuton extended-release tablets group, ALT elevations were detected 14 days after completion of the 3-month study treatment. The levels returned to <2×ULN or normal within 9 and 12 days, respectively. The ALT elevations in the other 3 patients were observed to return to <2×ULN or normal within 15, 19, and 31 days after zileuton extended-release tablets discontinuation. There appeared to be no clinically relevant relationship between the time of onset and the magnitude of the first elevation or the magnitude of first elevation and time to resolution. The hepatic function enzyme elevations attributed to zileuton extended-release tablets did not result in any cases of jaundice, development of chronic liver disease, or death in this clinical trial.
6.2 Long-Term Clinical Studies Experience
The safety of zileuton extended-release tablets was evaluated in one 6-month, randomized, double-blind, placebo-controlled clinical trial in adults and adolescents 12 years of age and older with asthma. Patients received two 600 mg zileuton extended-release tablets (n=619) or placebo (n=307) twice daily by mouth along with usual asthma care. Eighty-six percent of patients were white, 40% were male, and the overall mean age was 36.
The rate and type of adverse reactions observed in this study were comparable to the adverse reactions observed in the 12-week study. Other commonly reported adverse reactions (occurring at a frequency of ≥5%) in zileuton extended-release tablets-treated patients and at a frequency greater than placebo-treated patients included the following: headache (23%), upper respiratory tract infection (9%), myalgia (7%), and diarrhea (5%) compared to 21%, 7%, 5% and 2%, respectively, in the placebo-treated group.
ALT elevations (≥3×ULN) were observed in 1.8% of patients treated with zileuton extended-release tablets compared to 0.7% in patients treated with placebo. The majority of elevations (82%) were reported within the first 3 months of treatment and resolved within 21 days for most of these patients after discontinuation of the drug. The hepatic function enzyme elevations attributed to zileuton extended-release tablets did not result in any cases of jaundice, development of chronic liver disease, or death in this clinical trial.
Occurrences of low white blood cell (WBC) count (<3.0 × 10 9/L) were observed in 2.6% (15 of 619) of the zileuton extended-release tablets-treated patients and in 1.7% (5 of 307) of the placebo-treated patients. The WBC counts returned to normal or baseline following discontinuation of zileuton extended-release tablets. The clinical significance of these findings is not known.
6.3 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of zileuton immediate-release tablets and may be applicable to zileuton extended-release tablets. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship.
Cases of severe hepatic injury have been reported in patients taking zileuton immediate-release tablets. These cases included death, life-threatening liver injury with recovery, symptomatic jaundice, hyperbilirubinemia, and elevations of ALT >8×ULN.
Cases of sleep disorders and behavior changes have also been reported [see Warnings and Precautions (5.2)].
-
7 DRUG INTERACTIONS
The following study results were obtained using zileuton immediate-release tablets but the conclusions also apply to zileuton extended-release tablets.
7.1 Theophylline
In a drug-interaction study in 16 healthy subjects, coadministration of multiple doses of zileuton immediate-release tablets (800 mg every 12 hours) and theophylline (200 mg every 6 hours) for 5 days resulted in a significant decrease (approximately 50%) in steady-state clearance of theophylline, an approximate doubling of theophylline AUC, and an increase in theophylline C max (by 73%). The elimination half-life of theophylline was increased by 24%. Also, during coadministration, theophylline-related adverse reactions were observed more frequently than after theophylline alone. Upon initiation of zileuton extended-release tablets in patients receiving theophylline, the theophylline dosage should be reduced by approximately one-half and plasma theophylline concentrations monitored. Similarly, when initiating therapy with theophylline in a patient receiving zileuton extended-release tablets, the maintenance dose and/or dosing interval of theophylline should be adjusted accordingly and guided by serum theophylline determinations.
7.2 Warfarin
Concomitant administration of multiple doses of zileuton immediate-release tablets (600 mg every 6 hours) and warfarin (fixed daily dose obtained by titration in each subject) to 30 healthy male subjects resulted in a 15% decrease in R-warfarin clearance and an increase in AUC of 22%. The pharmacokinetics of S-warfarin were not affected. These pharmacokinetic changes were accompanied by a clinically significant increase in prothrombin times. Monitoring of prothrombin time, or other suitable coagulation tests, with the appropriate dose titration of warfarin is recommended in patients receiving concomitant zileuton extended-release tablets and warfarin therapy.
7.3 Propranolol
Coadministration of zileuton immediate-release tablets and propranolol results in a significant increase in propranolol concentrations. Administration of a single 80 mg dose of propranolol in 16 healthy male subjects who received zileuton immediate-release tablets 600 mg every 6 hours for 5 days resulted in a 42% decrease in propranolol clearance. This resulted in an increase in propranolol C max, AUC, and elimination half-life by 52%, 104%, and 25%, respectively. There was an increase in β-blockade as shown by a decrease in heart rate associated with the coadministration of these drugs. Patients concomitantly on zileuton extended-release tablets and propranolol should be closely monitored and the dose of propranolol reduced as necessary. No formal drug-drug interaction studies between zileuton and other beta-adrenergic blocking agents (i.e., β-blockers) have been conducted. It is reasonable to employ appropriate clinical monitoring when these drugs are coadministered with zileuton extended-release tablets.
7.4 Other Concomitant Drug Therapy
Drug-drug interaction studies conducted in healthy subjects between zileuton immediate-release tablets and prednisone and ethinyl estradiol (oral contraceptive), drugs known to be metabolized by the CYP3A4 isoenzyme, have shown no significant interaction. However, no formal drug-drug interaction studies between zileuton and CYP3A4 inhibitors, such as ketaconazole, have been conducted. It is reasonable to employ appropriate clinical monitoring when these drugs are coadministered with zileuton extended-release tablets.
Drug-drug interaction studies in healthy subjects have been conducted with zileuton immediate-release tablets and digoxin, phenytoin, sulfasalazine, and naproxen. There was no significant interaction between zileuton and any of these drugs.
-
8 USE IN SPECIFIC POPULATIONS
Information on specific populations is based on studies conducted with zileuton immediate-release tablets and is applicable to zileuton extended-release tablets.
8.1 Pregnancy
There are no adequate human data on zileuton extended-release tablets use in pregnant women to inform a drug associated risk. In animal studies, oral administration of zileuton to pregnant rats and rabbits during organogenesis produced adverse developmental outcomes. Structural abnormalities (cleft palate) were observed in rabbits at a dose similar to the maximum recommended human daily oral dose (MRHD), and alterations to growth (reduced fetal body weight and increased skeletal variations) were observed in rats at maternal plasma exposures 20 times greater than at the MRHD [see Data]. In a pre- and post-natal development study, oral administration of zileuton to pregnant rats from organogenesis through weaning at maternal plasma exposures 20 times greater than the MRHD resulted in reduced pup survival and body weights. Zileuton and/or its metabolites cross the placental barrier of rats; therefore, zileuton extended-release tablets may be transmitted from the mother to the developing fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to asthma medications during pregnancy. For more information, contact the MothersToBaby Pregnancy Studies conducted by the Organization of Teratology Information Specialists at 1-877- 311-8972 or visit http://mothertobaby.org/pregnancy-studies/.
Data
Animal Data
In a fertility and general reproductive performance study in rats, 0, 15, 75, 150 or 300 mg/kg/day zileuton was administered orally to male and female rats. The treated males were dosed daily for 100 days prior to mating with the treated females and 80 days prior to mating with untreated females, and throughout the mating periods. The treated females were dosed for 14 days before mating with untreated males and dosing continued throughout gestation, and in 1/3 of the females through parturition and lactation period. Maternal body weight gain was reduced at 150 and 300 mg/kg/day groups (9 to12% differences in body weight relative to controls).
During fetal evaluation, zileuton produced lower litter size (7.1 pup/dams at 300 mg/kg/day vs. 9.6 pup/dams at 150 mg/kg/day vs. 13.5 pup/dams in control group), lower fetal weights (-9%), decreased viable fetuses, and increased in unossification of fetal skeletal structure at 300 mg/kg at exposures greater than 20 times the MRHD (on an AUC basis with data obtained from the comparable doses of 3-month general toxicity study). There were no embryofetal effects at 150 mg/kg/day.
During post-natal development evaluation, zileuton produced decrease in pup viability (-16% at 150 mg/kg/day and -43.5% at 300 mg/kg/day on lactation Day 4) as well as depression of body weight gain in pups at ≥ 150 mg/kg/day at exposures close to 20 times the MRHD (on an AUC basis with data obtained from the comparable doses of 1-year general toxicity study). Observations of lower pup weight and survival rate at 300 mg/kg/day group were confirmed in a peri- & post-natal study administered with the same dose levels in pregnant rats.
In a teratology study in pregnant rabbits, 0, 15, 50 or 150 mg/kg/day zileuton was administered orally to pregnant animals during organogenesis. Cleft palate was noted in three of 118 (2.5%) rabbit fetuses (or 2 of 17 litters) at 150 mg/kg/day. Additionally, two fetuses (1.7%) had domed head and two fetuses (1.7%) had hydrocephalus also at 150 mg/kg/day which was equivalent to the MRHD on a mg/m 2 basis. There were no adverse developmental outcomes at 50 mg/kg/day (approximately one-third the MRHD on a mg/m 2 basis).
Oral dose of 5 mg radiolabeled zileuton indicated that zileuton crosses the placental barrier of rats.
8.2 Lactation
Zileuton and/or its metabolites are excreted in rat milk. It is not known if zileuton is excreted in human milk, nor are there data on the effects of the drug on the breastfed infant or effects on maternal milk production. Because many drugs are excreted in human milk, and because of the potential for tumorigenicity of zileuton shown in animal studies, the developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for zileuton extended-release tablets and any potential adverse effects on the breastfed child from zileuton extended-release tablets or from the underlying maternal condition.
Data
Animal Data
Following an oral 70 mg/kg dose of radiolabeled 14C-zileuton to lactating rats, total radioactivity was distributed into the milk of dams, but the mean concentrations did not exceed those in plasma.
8.4 Pediatric Use
The safety and effectiveness of zileuton extended-release tablets in pediatric patients under 12 years of age have not been established. FDA has not required pediatric studies in patients under the age of 12 years due to risk of hepatotoxicity. Zileuton extended-release tablets are not appropriate for children less than 12 years of age.
8.5 Geriatric Use
Subgroup analysis of controlled and open-label clinical studies with zileuton immediate-release tablets suggests that females ≥65 years of age appear to be at increased risk of ALT elevations. In zileuton extended-release tablets placebo-controlled studies there were no discernable trends in ALT elevations noted in subset analyses for patients ≥65 years of age, although the database may not have been sufficiently large to detect a trend [see Pharmacokinetics (12.3)].
-
10 OVERDOSAGE
Human experience of acute overdose with zileuton is limited. A patient in a clinical study took between 6.6 and 9.0 grams of zileuton immediate-release tablets in a single dose. Vomiting was induced and the patient recovered without sequelae. Zileuton is not removed by dialysis. Should an overdose occur, the patient should be treated symptomatically and supportive measures instituted as required. If indicated, elimination of unabsorbed drug should be achieved by emesis or gastric lavage; usual precautions should be observed to maintain the airway. A Certified Poison Control Center should be consulted for up-to-date information on management of overdose with zileuton extended-release tablets.
-
11 DESCRIPTION
Zileuton is an orally active inhibitor of 5-lipoxygenase, the enzyme that catalyzes the formation of leukotrienes from arachidonic acid. Zileuton has the chemical name (±)-1-(1-Benzo[b]thien-2-ylethyl)-1-hydroxyurea and the following chemical structure:
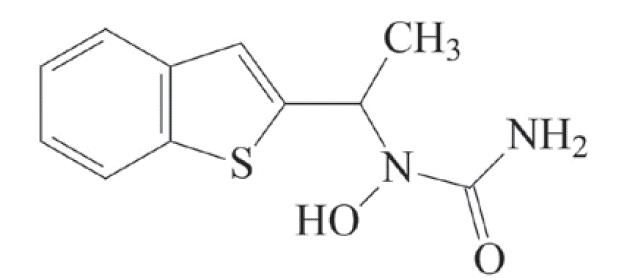
Zileuton has the molecular formula C 11H 12N 2O 2S and a molecular weight of 236.29. It is a racemic mixture (50:50) of R(+) and S(-) enantiomers. Zileuton is a practically odorless, white to off-white powder that is soluble in methanol and ethanol, slightly soluble in acetonitrile, and practically insoluble in water and hexane. The melting point ranges from 144.2° to 145.2°C.
Zileuton extended-release tablets for oral administration are triple-layer tablets comprised of an immediate-release layer, a middle (barrier) layer, and an extended-release layer. Zileuton extended-release tablets are oblong, film-coated tablets with one red layer between two white layers, debossed on one side with "P723" and plain on other side. Each tablet contains 600 mg of zileuton and the following inactive ingredients: colloidal silicon dioxide, glyceryl behenate, hydroxypropyl cellulose, hydroxypropyl methylcellulose, hypromellose, iron oxide red, magnesium stearate, mannitol, microcrystalline cellulose, polyethylene glycol, pregelatinized starch and sodium starch glycolate.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Zileuton is an inhibitor of 5-lipoxygenase and thus inhibits leukotriene (LTB 4, LTC 4, LTD 4 and LTE 4) formation. Both the R(+) and S(-) enantiomers are pharmacologically active as 5-lipoxygenase inhibitors in in vitro and in vivo systems. Leukotrienes are substances that induce numerous biological effects including augmentation of neutrophil and eosinophil migration, neutrophil and monocyte aggregation, leukocyte adhesion, increased capillary permeability, and smooth muscle contraction. These effects contribute to inflammation, edema, mucus secretion, and bronchoconstriction in the airways of asthmatic patients. LTB 4, a chemoattractant for neutrophils and eosinophils, and cysteinyl leukotrienes (LTC 4, LTD 4, LTE 4) can be measured in a number of biological fluids including bronchoalveolar lavage fluid (BALF), blood, urine and sputum from asthmatic patients.
Zileuton is an orally active inhibitor of ex vivo LTB 4 formation in several species, including mice, rats, rabbits, dogs, sheep, and monkeys. Zileuton inhibits arachidonic acid-induced ear edema in mice, neutrophil migration in mice in response to polyacrylamide gel, and eosinophil migration into the lungs of antigen-challenged sheep. In a mouse model of allergic inflammation, zileuton inhibited neutrophil and eosinophil influx, reduced the levels of multiple cytokines in the BALF, and reduced serum IgE levels. Zileuton inhibits leukotriene-dependent smooth muscle contractions in vitro in guinea pig and human airways. The compound inhibits leukotriene-dependent bronchospasm in antigen and arachidonic acid-challenged guinea pigs. In antigen-challenged sheep, zileuton inhibits late-phase bronchoconstriction and airway hyperreactivity. The clinical relevance of these findings is unknown.
12.2 Pharmacodynamics
Zileuton is an orally active inhibitor of ex vivo LTB 4 formation in humans. The inhibition of LTB 4 formation in whole blood is directly related to zileuton plasma levels. In patients with asthma, the IC 50 is estimated to be 0.46 μg/mL, and maximum inhibition ≥80% is reached at a zileuton concentration of 2 μg/mL. In patients with asthma receiving zileuton immediate-release tablets 600 mg four times daily, peak plasma levels averaging 5.9 μg/mL were associated with a mean LTB 4 inhibition of 98%. Zileuton inhibits the synthesis of cysteinyl leukotrienes as demonstrated by reduced urinary LTE 4 levels.
12.3 Pharmacokinetics
Information on the pharmacokinetics of zileuton following the administration of zileuton immediate-release tablets is available in healthy subjects. The results of two clinical pharmacology studies using zileuton extended-release tablets are described below.
Absorption
A three-way crossover study was conducted in healthy male and female subjects (n=23) with a mean age of 33 (range 20 to 55) following single dose of 1200 mg (2 x 600 mg) zileuton extended-release tablets under fasted and fed conditions, and two doses of 600 mg zileuton immediate-release tablets every 6 hours under fasted conditions. Food increased the peak mean plasma concentrations (C max) and the mean extent of absorption (AUC) of zileuton extended-release tablets by 18 and 34%, respectively, and prolonged T max from 2.1 hours to 4.3 hours. The relative bioavailability of zileuton extended-release tablets to zileuton immediate-release tablets with respect to C max and AUC under fasted conditions were 0.39 (90% CI: 0.36, 0.43) and 0.57 (90% CI: 0.52, 0.62), respectively. Similarly, relative bioavailability of zileuton extended-release tablets to zileuton immediate-release tablets with respect to C max and AUC under fed conditions were 0.45 (90% CI: 0.41, 0.49) and 0.76 (90% CI: 0.70, 0.83), respectively.
A three-way crossover study was conducted in healthy male and female subjects (n=24) with a mean age of 35 (range 19 to 56) following multiple doses of 1200 mg (2 × 600 mg) zileuton extended-release tablets administered every 12 hours under fasted and fed conditions, and 600 mg zileuton immediate-release tablets every 6 hours under fed conditions until steady state zileuton levels were achieved. Food increased AUC and C min of zileuton extended-release tablets by 43% and 170%, respectively, but had no effect on C max. Therefore, zileuton extended-release tablets are recommended to be administered with food [see Dosage and Administration (2)]. At steady state, relative bioavailability of zileuton extended-release tablets to zileuton immediate-release tablets with respect to C max, C min, and AUC were 0.65 (90% CI: 0.60, 0.71), 1.05 (90% CI: 0.88, 1.25) and 0.85 (90% CI: 0.78, 0.92) respectively. These data indicate that at steady state under fed conditions the C max of zileuton extended-release tablets is about 35% lower than that of zileuton immediate-release tablets but the C min and AUC are similar for both formulations.
Distribution
The apparent volume of distribution (V/F) of zileuton is approximately 1.2 L/kg. Zileuton is 93% bound to plasma proteins, primarily to albumin, with minor binding to α1–acid glycoprotein.
Elimination
Elimination of zileuton is predominantly via metabolism with a mean terminal half-life of 3.2 hours. Apparent oral clearance (CL/F) of zileuton is 669 mL/min. Zileuton activity is primarily due to the parent drug. Studies with radiolabeled drug have demonstrated that orally administered zileuton is well absorbed into the systemic circulation with 94.5% and 2.2% of the radiolabeled dose recovered in urine and feces, respectively.
Metabolism
In vitro studies utilizing human liver microsomes have shown that zileuton and its N-dehydroxylated metabolite can be oxidatively metabolized by CYP1A2, CYP2C9 and CYP3A4.
Several zileuton metabolites have been identified in human plasma and urine. These include two diastereomeric O-glucuronide conjugates (major metabolites) and an N-dehydroxylated metabolite (A-66193) of zileuton. The urinary excretion of the inactive A-66193 metabolite and unchanged zileuton each accounted for less than 0.5% of the single radiolabeled dose. Multiple doses of 1200 mg zileuton extended-release tablets twice daily resulted in peak plasma levels of 4.9 μg/mL of the inactive metabolite A-66193 with an AUC of 93 μg .hr/mL, showing large inter-subject variability. This inactive metabolite has been shown to be formed by the gastrointestinal microflora prior to the absorption of zileuton and its formation increases with delayed absorption of zileuton.
Renal Impairment
The pharmacokinetics of zileuton immediate-release tablets were similar in healthy subjects and in subjects with mild, moderate, and severe renal insufficiency. In subjects with renal failure requiring hemodialysis, zileuton pharmacokinetics were not altered by hemodialysis and a very small percentage of the administered zileuton dose (<0.5%) was removed by hemodialysis. Hence, dosing adjustment in patients with renal dysfunction or undergoing hemodialysis is not necessary.
Hepatic Impairment
The pharmacokinetics of zileuton immediate-release tablets were compared between subjects with mild and moderate chronic hepatic insufficiency. The mean apparent plasma clearance of total zileuton in subjects with hepatic impairment was approximately half the value of the healthy subjects. The percent binding of zileuton to plasma proteins after multiple dosing was significantly reduced in patients with moderate hepatic impairment. Zileuton extended-release tablets are contraindicated in patients with active liver disease or persistent ALT elevations ≥3×ULN [see Warnings and Precautions (5)].
Geriatric Use
The pharmacokinetics of zileuton immediate-release tablets were investigated in healthy elderly subjects (ages 65 to 81 years, 9 males, 9 females) and healthy young subjects (ages 20 to 40 years, 5 males, 4 females) after single and multiple oral doses of 600 mg zileuton every 6 hours. Zileuton pharmacokinetics were similar in healthy elderly subjects (≥65 years) compared to healthy younger adults (20 to 40 years).
-
13 NONCLINICAL TOXICOLOGY
13.3 Carcinogenesis, Mutagenesis, Impairment of Fertility
In 2-year carcinogenicity studies, increases in the incidence of liver, kidney, and vascular tumors in female mice and a trend toward an increase in the incidence of liver tumors in male mice were observed at 450 mg/kg/day (providing approximately 5 times [females] or 8 times [males] the systemic exposure [AUC=64 μg .hr/mL] achieved at the MRHD). No increase in the incidence of tumors was observed at 150 mg/kg/day (providing approximately 2 to 3 times the systemic exposure [AUC] achieved at the MRHD). In rats, an increase in the incidence of kidney tumors was observed in both sexes at 170 mg/kg/day (providing approximately 8 times [males] or 16 times [females] the systemic exposure [AUC] achieved at the MRHD). No increased incidence of kidney tumors was seen at 80 mg/kg/day (providing approximately 4 times [males] or 7 times [females] the systemic exposure [AUC] achieved at the MRHD). Although a dose-related increased incidence of benign Leydig cell tumors was observed, Leydig cell tumorigenesis was prevented by supplementing male rats with testosterone.
Zileuton was negative in genotoxicity studies including bacterial reverse mutation (Ames) using S. typhimurium and E. coli, chromosome aberration in human lymphocytes, in vitro unscheduled DNA synthesis (UDS), in rat hepatocytes with or without zileuton pretreatment and in mouse and rat kidney cells with zileuton pretreatment, and mouse micronucleus assays. However, a dose-related increase in DNA adduct formation was reported in kidneys and livers of female mice treated with zileuton. Although some evidence of DNA damage was observed in a UDS assay in hepatocytes isolated from Aroclor-1254-treated rats, no such finding was noticed in hepatocytes isolated from monkeys, where the metabolic profile of zileuton is more similar to that of humans.
In reproductive performance/fertility studies, zileuton produced no effects on fertility in rats at oral doses up to 300 mg/kg/day (providing at least 10 times [male rats] and greater than 20 times [female rats] the systemic exposure [AUC] achieved at the MRHD). However, reduction in fetal implants was observed at oral doses of 150 mg/kg/day and higher (providing approximately 20 times the systemic exposure [AUC] achieved at the MRHD). Comparative systemic exposure (AUC) is based on measurements in male rats or nonpregnant female rats obtained from the comparable doses of 3-month or 1-year general toxicity study at similar dosages. Increases in gestation length, prolongation of estrus cycle, and increases in stillbirths were observed at oral doses of 75 mg/kg/day and higher (providing approximately 7 times the systemic exposure [AUC] achieved at the MRHD on an AUC basis with data obtained from the comparable doses of 2-year dietary carcinogenicity study). No adverse effects were observed at 15 mg/kg/day in the study at estimated exposure similar to that at the MRHD.
-
14 CLINICAL STUDIES
The efficacy of zileuton extended-release tablets was evaluated in a randomized, double-blind, parallel-group, placebo-controlled, multicenter trial of 12 weeks duration in patients 12 years of age and older with asthma. The 12-week trial included 199 patients randomized to zileuton extended-release tablets (two 600 mg tablets twice daily) and 198 to placebo. Eighty-three percent of patients were white, 48% were male, and the mean age was 34 years. The mean baseline FEV 1 percent predicted was 58.5%.
Assessment of efficacy was based upon forced expiratory volume in one second (FEV 1) at 12 weeks. Zileuton extended-release tablets demonstrated a significantly greater improvement in mean change from baseline trough FEV 1 at 12 weeks compared to placebo (0.39 L vs. 0.27 L; p=0.021). The mean change from baseline FEV 1 over the course of the 12-week study is shown in Figure 1. Secondary endpoints (PEFR and rescue beta-agonist use) were supportive of efficacy.
Examination of gender subgroups did not identify differences in response between men and women. The database was not large enough to assess whether there were differences in response in age or racial subgroups.
Figure 1. Mean Change from Baseline in Trough FEV 1 in 12-Week Clinical Trial in Patients with Asthma.
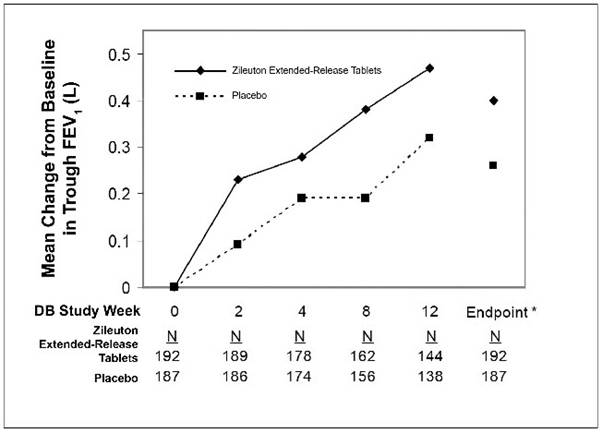
*p ≤0.050. Endpoint analysis based on last-observation-carried-forward (LOCF) methodology.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Zileuton Extended-Release Tablets, 600 mg are oblong, film-coated tablets with one red layer between two white layers, debossed on one side with "P723" and plain on other side; they are available in bottles of 120 tablets (NDC: 51407-741-12).
Store between 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature]. Protect from light.
-
17 PATIENT COUNSELING INFORMATION
17.1 Information for Patients
- Zileuton extended-release tablets are indicated for the chronic treatment of asthma and should be taken regularly as prescribed, even during symptom-free periods.
- Zileuton extended-release tablets are a leukotriene synthesis inhibitor which works by inhibiting the formation of leukotrienes.
- Zileuton extended-release tablets should be taken within one hour after morning and evening meals.
- Zileuton extended-release tablets should not be cut, chewed or crushed.
- Zileuton extended-release tablets are not a bronchodilator and should not be used to treat acute episodes of asthma.
- When taking zileuton extended-release tablets, they should not decrease the dose or stop taking any other antiasthma medications unless instructed by a health care provider. If a dose is missed, they should take the next dose at the scheduled time and not double the dose.
- While using zileuton extended-release tablets, medical attention should be sought if short-acting bronchodilators are needed more often than usual, or if more than the maximum number of inhalations of short-acting bronchodilator treatment prescribed for a 24-hour period are needed.
- The most serious side effect of zileuton extended-release tablets is potential elevation of liver enzymes (in 2% of patients) and that, while taking zileuton extended-release tablets, they must return for liver enzyme test monitoring on a regular basis.
- If they experience signs and/or symptoms of liver dysfunction (e.g., right upper quadrant pain, nausea, fatigue, lethargy, pruritus, jaundice, or "flu-like" symptoms), they should contact their health care provider immediately.
- Patients should be instructed to notify their healthcare provider if neuropsychiatric events occur while using zileuton extended-release tablets.
- Zileuton extended-release tablets can interact with other drugs and that, while taking zileuton extended-release tablets, they should consult their health care provider before starting or stopping any prescription or non-prescription medicines.
- A patient leaflet is included with the tablets.
17.2 FDA-Approved Patient Labeling
PATIENT INFORMATION
Zileuton (zi· leu· ton) Extended-Release Tablets
Read the Patient Information that comes with zileuton extended-release tablets carefully before you start taking them and read it each time you get a refill. There may be new information. This leaflet does not take the place of talking with your health care provider about your medical condition or your treatment.
What are zileuton extended-release tablets?
Zileuton extended-release tablets are a medicine that is used to prevent asthma attacks and for long-term management of asthma in adults and children 12 years of age and older. Zileuton extended-release tablets blocks the production of leukotrienes. Leukotrienes are substances that may contribute to your asthma.
Zileuton extended-release tablets are not a rescue medicine (it is not a bronchodilator) and should not be used if you need relief right away for an asthma attack.
Who should not take zileuton extended-release tablets?
Do not take zileuton extended-release tablets if you have:
- active liver disease or repeated blood tests showing elevated liver enzymes (substances released by the liver).
- ever had an allergic reaction to zileuton extended-release tablets or any of the ingredients in zileuton extended-release tablets.
Zileuton extended-release tablets may not be right for you. Tell your health care provider if you:
- have ever had liver problems, including hepatitis, jaundice (yellow eyes or skin), or dark urine.
- drink alcohol. Tell your health care provider how much and how often you drink alcohol.
- have difficulty swallowing pills.
- are pregnant or planning to become pregnant. It is not known if zileuton extended-release tablets will harm your unborn baby. Do not take zileuton extended-release tablets during pregnancy unless you and your health care provider decide that taking the medicine is more important than the possible risk to your unborn baby.
- are breastfeeding. It is not known if zileuton passes into your breast milk. You and your health care provider should decide if you will take zileuton extended-release tablets or breastfeed. You should not do both.
Know the medicines you take. Keep a list of your medicines and show it to your health care provider and pharmacist when you get a new medicine.
How should I take zileuton extended-release tablets?
- Take zileuton extended-release tablets exactly as prescribed by your health care provider. Do not decrease the dose of zileuton extended-release tablets or stop taking the medicine without talking to your health care provider first, even if you have no asthma symptoms.
- Take two zileuton extended-release tablets two times each day within one hour after your morning and evening meals.
- Swallow zileuton extended-release tablets whole. Do not chew, cut or crush zileuton extended-release tablets . Tell your health care provider if you cannot swallow the tablets whole.
- Follow your health care provider's instructions for what to do if you get sudden symptoms of an asthma attack. You can continue taking zileuton extended-release tablets during asthma attacks.
- Get medical help right away if you need to use your rescue medicine more often than usual or if you use the highest number of "puffs" prescribed for one 24-hour period. These could be signs that your asthma is getting worse. This means that your asthma therapy may need to be changed.
- Keep taking your other asthma medicines as directed while taking zileuton extended-release tablets.
- If you miss a dose, just take your next scheduled dose when it is due. Do not double the dose.
- If you take too much zileuton extended-release tablets, call your health care provider or a Poison Control Center right away.
Zileuton extended-release tablets can cause serious side effects.
Liver problems. Liver function enzymes and bilirubin can increase while taking zileuton extended-release tablets, and severe liver injury can occur.
Sleep disorders and changes in your behavior can happen while you take zileuton extended-release tablets. Tell your healthcare provider if you have any sleep problems or changes in behavior.
- Keep all of your health care provider's appointments and be sure to follow all of your health care provider's instructions. Have your blood tests done as ordered to check your liver enzymes.
- Tell your health care provider right away if you get any of the following signs or symptoms: pain on the right side of your abdomen (stomach area), nausea, tiredness, lack of energy, itching, yellow skin or yellow color in the whites of your eyes, dark urine, or "flu-like" symptoms.
- nose and throat irritation
- sinusitis
- upper respiratory infection
- throat pain
- headache
- muscle aches
- nausea
- diarrhea
Tell your health care provider if you have any new or unusual symptoms that bother you or do not go away while taking zileuton extended-release tablets.
These are not all of the possible side effects of zileuton extended-release tablets. For more information, ask your health care provider or pharmacist.
How should I store zileuton extended-release tablets?
- Store zileuton extended-release tablets between 68°F to 77°F (20° to 25°C).
- Protect zileuton extended-release tablets from light and replace the cap each time after use.
Keep zileuton extended-release tablets and all medicines out of the reach of children.
General Information about zileuton extended-release tablets:
Medicines are sometimes prescribed for conditions that are not mentioned in the patient leaflet. Do not use zileuton extended-release tablets for a condition for which it was not prescribed. Do not give zileuton extended-release tablets to other people, even if they have the same symptoms you have. They may harm them.
This patient information leaflet summarizes the most important information about zileuton extended-release tablets. If you would like more information about zileuton extended-release tablets, talk with your health care provider or pharmacist. You can ask your health care provider or pharmacist for information about zileuton extended-release tablets that is written for health professionals.
For more information go to www.strides.com or call 1-877-244-9825.
What are the ingredients in zileuton extended-release tablets?
Active ingredient: zileuton
Inactive ingredients: colloidal silicon dioxide, glyceryl behenate, hydroxypropyl cellulose, hypromellose, iron oxide red, magnesium stearate, mannitol, microcrystalline cellulose, polyethylene glycol, pregelatinized starch and sodium starch glycolate.
Distributed by:
Strides Pharma Inc.
East Brunswick, NJ 08816
Issued: 12/2021
OS723-01-1-02Marketed/Packaged by:
GSMS, Inc.
Camarillo, CA USA 93012
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
ZILEUTON
zileuton tablet, multilayer, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 51407-741(NDC:64380-189) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ZILEUTON (UNII: V1L22WVE2S) (ZILEUTON - UNII:V1L22WVE2S) ZILEUTON 600 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) GLYCERYL BEHENATE/EICOSADIOATE (UNII: 73CJJ317SR) HYDROXYPROPYL CELLULOSE (110000 WAMW) (UNII: 5Y0974F5PW) MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) POLYETHYLENE GLYCOL 1000 (UNII: U076Q6Q621) STARCH, CORN (UNII: O8232NY3SJ) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) HYPROMELLOSE 2910 (10000 MPA.S) (UNII: 0HO1H52958) FERRIC OXIDE RED (UNII: 1K09F3G675) HYPROMELLOSE 2208 (100 MPA.S) (UNII: B1QE5P712K) Product Characteristics Color white (one red layer between two white layers) Score no score Shape OVAL Size 19mm Flavor Imprint Code P723 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 51407-741-12 120 in 1 BOTTLE; Type 0: Not a Combination Product 01/19/2023 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA212670 12/16/2019 Labeler - Golden State Medical Supply, Inc. (603184490) Establishment Name Address ID/FEI Business Operations Golden State Medical Supply, Inc. 603184490 relabel(51407-741) , repack(51407-741)

© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.