NIMODIPINE- nimodipine solution
Nimodipine by
Drug Labeling and Warnings
Nimodipine by is a Prescription medication manufactured, distributed, or labeled by Camber Pharmaceuticals, Inc., Annora Pharma Private Limited. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use NIMODIPINE ORAL SOLUTION safely and effectively. See full prescribing information for NIMODIPINE ORAL SOLUTION.
NIMODIPINE oral solution
Initial U.S. Approval: 1988INDICATIONS AND USAGE
Nimodipine oral solution is a dihydropyridine calcium channel blocker indicated for the improvement of neurological outcome by reducing the incidence and severity of ischemic deficits in adult patients with subarachnoid hemorrhage (SAH) from ruptured intracranial berry aneurysms regardless of their post-ictus neurological condition (i.e., Hunt and Hess Grades I to V). (1)
DOSAGE AND ADMINISTRATION
Administer only enterally (e.g., oral, nasogastric tube, or gastric tube route). Do not administer intravenously or by other parenteral routes. (2.1)
Give one hour before a meal ortwo hours after a meal. (2.1)
Start dosing within 96 hours of the SAH. (2.1)
Recommended dose is 20 mL (60 mg) every 4 hours for 21 consecutive days. (2.2)
Nasogastric or Gastric Tube Administration:Administer 20 mL (60 mg) every 4 hours with an oral syringe. Refill syringe with 20 mL of 0.9% saline water solution; flush remaining contents from nasogastric or gastric tube into stomach. (2.3)
Patients with Cirrhosis:Reduce dosage to 10 mL (30 mg) every 4 hours. (2.4)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
Hypotension:Monitor blood pressure. (5.1)
Patients with Cirrhosis:Higher risk of adverse reactions. Monitor blood pressure and pulse. (5.2)
CYP3A4 Strong Inhibitors:May significantly increase risk of hypotension. Concomitant use with nimodipine should generally be avoided. (5.3)
CYP3A4 Strong Inducers:May significantly reduce efficacy of nimodipine. Concomitant use with nimodipine should generally be avoided. (5.4)ADVERSE REACTIONS
Most common adverse reactions (incidence ≥1% and ≥1% placebo) were hypotension, headache, nausea, and bradycardia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Annora Pharma Private Limited at 1-866-495-1995 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Anti-Hypertensives:
May increase risk of hypotension. Monitor blood pressure. (7.1)
CYP3A4 Moderate and Weak Inhibitors:May increase risk of hypotension. Monitor blood pressure. Dose reduction of nimodipine may be needed. Avoid grapefruit juice. (7.2)
CYP3A4 Moderate and Weak Inducers:May reduce efficacy of nimodipine. Dose increase may be needed. (7.3)
USE IN SPECIFIC POPULATIONS
Pregnancy:Based on animal data may cause fetal harm. (8.1)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 3/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USE
2 DOSAGE AND ADMINISTRATION
2.1 Administration Instructions
2.2 Administration by Oral Route
2.3 Administration Via Nasogastric or Gastric Tube
2.4 Dosage Adjustments in Patients with Cirrhosis
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypotension
5.2 Possible Increased Risk of Adverse Reactions in Patients with Cirrhosis
5.3 Possible Increased Risk of Hypotension with Strong CYP3A4 Inhibitors
5.4 Possible Reduced Efficacy with Strong CYP3A4 Inducers
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Blood Pressure Lowering Drugs
7.2 CYP3A4 Inhibitors
7.3 CYP3A4 Inducers
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USE
Nimodipine oral solution is indicated for the improvement of neurological outcome by reducing the incidence and severity of ischemic deficits in adult patients with subarachnoid hemorrhage (SAH) from ruptured intracranial berry aneurysms regardless of their post-ictus neurological condition (i.e., Hunt and Hess Grades I to V).
-
2 DOSAGE AND ADMINISTRATION
2.1 Administration Instructions
Administer only enterally (e.g., oral, nasogastric tube, or gastric tube route). Do not administer intravenously or by other parenteral routes. For all routes of administration, begin nimodipine oral solution within 96 hours of the onset of SAH. Administer one hour before a meal or two hours after a meal for all routes of administration [see Clinical Pharmacology (12.3)].
2.2 Administration by Oral Route
The recommended oral dosage is 60 mg/20 mL (3 mg/mL) every 4 hours for 21 consecutive days.
2.3 Administration Via Nasogastric or Gastric Tube
Using the oral syringe, administer 60 mg/20 mL (3 mg/mL) every 4 hours into a nasogastric or gastric tube for 21 consecutive days. For each dose, refill the syringe with 20 mL of 0.9% saline solution and then flush any remaining contents from nasogastric or gastric tube into the stomach.
2.4 Dosage Adjustments in Patients with Cirrhosis
In patients with cirrhosis, reduce the dosage to 10 mL (30 mg) every 4 hours [see Warnings and Precautions(5.2), Clinical Pharmacology (12.3)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypotension
Blood pressure should be carefully monitored during treatment with nimodipine. In clinical studies of patients with subarachnoid hemorrhage, about 5% of nimodipine-treated patients compared to 1% of placebo-treated patients had hypotension and about 1% of nimodipine-treated patients left the study because of this [see Adverse Reactions (6)].
5.2 Possible Increased Risk of Adverse Reactions in Patients with Cirrhosis
Given that the plasma levels of nimodipine are increased in patients with cirrhosis, these patients are at higher risk of adverse reactions. Therefore, monitor blood pressure and pulse rate closely and administer a lower dosage [see Dosage and Administration (2.4), Clinical Pharmacology (12.3)].
5.3 Possible Increased Risk of Hypotension with Strong CYP3A4 Inhibitors
Concomitant use of strong inhibitors of CYP3A4, such as some macrolide antibiotics (e.g., clarithromycin, telithromycin), some HIV protease inhibitors (e.g., indinavir, nelfinavir, ritonavir, saquinavir), some HCV protease inhibitors (e.g., boceprevir, telaprevir), some azole antimycotics (e.g., ketoconazole, itraconazole, posaconazole, voriconazole), conivaptan, delavirdine, and nefazodone with nimodipine should generally be avoided because of a risk of significant hypotension [see Drug Interactions (7.2)].
5.4 Possible Reduced Efficacy with Strong CYP3A4 Inducers
Concomitant use of strong CYP3A4 inducers (e.g., carbamazepine, phenobarbital, phenytoin, rifampin, St. John’s wort) and nimodipine should generally be avoided, as nimodipine plasma concentration and efficacy may be significantly reduced [see Drug Interactions (7.3)].
-
6 ADVERSE REACTIONS
The safety and efficacy of nimodipine oral solution in the treatment of patients with SAH is based on adequate and well-controlled studies of nimodipine oral capsules in patients with SAH. Nimodipine oral solution has comparable bioavailability to nimodipine oral capsules.
The following clinically significant adverse reaction appears in other sections of the labeling:
Hypotension [see Warnings and Precautions (5.1)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
In clinical trials of nimodipine oral capsules in patients with SAH, eleven percent (92 of 823) of nimodipine-treated patients reported adverse events compared to six percent (29 of 479) of placebo-treated patients. The most common adverse event was decreased blood pressure in 4.4% of nimodipine-treated patients. The events reported with a frequency greater than 1% are displayed in Table 1 by dose.
Table 1: Adverse Events [n (%)] reported with a frequency > 1% in four clinical trials (Study 1, Study 2, Study 3, and Study 4)
Placebo
(n=479)
Nimodipine dose every 4 hours
0.35 mg/kg
(n=82)
30 mg
(n=71)
60 mg
(n=494)
90 mg
(n=172)
120 mg
(n=4)
Decreased Blood Pressure
6 (1.2)
1 (1.2)
0
19 (3.8)
14 (8.1)
2 (50.0)
Edema
3 (0.6)
0
0
2 (0.4)
2 (1.2)
0
Diarrhea
3 (0.6)
0
3 (4.2)
0
3 (1.7)
0
Rash
3 (0.6)
2 (2.4)
0
3 (0.6)
2 (1.2)
0
Headache
1 (0.2)
0
1 (1.4)
6 (1.2)
0
0
Gastrointestinal Symptoms
0
2 (2.4)
0
0
2 (1.2)
0
Nausea
0
1 (1.2)
1 (1.4)
6 (1.2)
1 (0.6)
0
Dyspnea
0
1 (1.2)
0
0
0
0
EKG Abnormalities
0
0
1 (1.4)
0
1 (0.6)
0
Tachycardia
0
0
1 (1.4)
0
0
0
Bradycardia
0
0
0
5 (1.0)
1 (0.6)
0
Muscle Pain/Cramp
0
0
1 (1.4)
1 (0.2)
1 (0.6)
0
Acne
0
0
1 (1.4)
0
0
0
Depression
0
0
1 (1.4)
0
0
0
SAH is frequently accompanied by alterations in consciousness that may lead to an under-reporting of adverse experiences. As a calcium channel blocker, nimodipine may have the potential to exacerbate heart failure in susceptible patients or to interfere with A-V conduction, but these events were not observed in SAH trials.
-
7 DRUG INTERACTIONS
7.1 Blood Pressure Lowering Drugs
Nimodipine may increase the blood pressure lowering effect of concomitantly administered anti-hypertensives such as diuretics, beta-blockers, ACE inhibitors, angiotensin receptor blockers, other calcium channel blockers, α-adrenergic blockers, PDE5 inhibitors, and α-methyldopa. In Europe, nimodipinewas observed to occasionally intensify the effect of antihypertensive drugs taken concomitantly by hypertensive patients; this phenomenon was not observed in North American clinical trials. Blood pressure should be carefully monitored, and dose adjustment of the blood pressure lowering drug(s) may be necessary.
7.2 CYP3A4 Inhibitors
Nimodipine plasma concentration can be significantly increased when concomitantly administered with strong CYP3A4 inhibitors. As a consequence, the blood pressure lowering effect may be increased. Therefore, the concomitant administration of nimodipine and strong CYP3A4 inhibitors should generally be avoided [see Warnings and Precautions (5.3)]. Strong CYP3A4 inhibitors include some members of the following classes:
- macrolide antibiotics (e.g., clarithromycin, telithromycin),
- HIV protease inhibitors (e.g., indinavir, nelfinavir, ritonavir, saquinavir),
- HCV protease inhibitors (e.g., boceprevir, telaprevir),
- azole antimycotics (e.g., ketoconazole, itraconazole, posaconazole, voriconazole),
- conivaptan, delavirdine, nefazodone
Nimodipine plasma concentration can also be increased in the presence of moderate and weak inhibitors of CYP3A4. If nimodipine is concomitantly administered with these drugs, blood pressure should be monitored, and a reduction of the nimodipine dose may be necessary. Moderate and weak CYP3A4 inhibitors include alprozalam, ameprenavir, amiodarone, aprepitant, atazanavir, cimetidine, cyclosporine, diltiazem, erythromycin, fluconazole, fluoxetine, isoniazid, oral contraceptives, quinuprestin/dalfopristin, valproic acid, and verapamil.
A study in eight healthy volunteers has shown a 50% increase in mean peak nimodipine plasma concentrations and a 90% increase in mean area under the curve, after a one-week course of cimetidine at 1,000 mg/day and nimodipine at 90 mg/day. This effect may be mediated by the known inhibition of hepatic cytochrome P-450 (CYP) by cimetidine, which could decrease first-pass metabolism of nimodipine. Grapefruit juice inhibits CYP3A4. Ingestion of grapefruit/grapefruit juice is not recommended while taking nimodipine.7.3 CYP3A4 Inducers
Nimodipine plasma concentration and efficacy may be significantly reduced when concomitantly administered with strong CYP3A4 inducers. Therefore, concomitant use of nimodipine with strong CYP3A4 inducers (e.g., carbamazepine, phenobarbital, phenytoin, rifampin, St. John’s wort) should generally be avoided [see Warnings and Precautions (5.4)].
Moderate and weak inducers of CYP3A4 may also reduce the efficacy of nimodipine. Patients on these should be closely monitored for lack of effectiveness, and a nimodipine dosage increase may be required. Moderate and weak CYP3A4 inducers include, for example: amprenavir, aprepitant, armodafinil, bosentan, efavirnenz, etravirine, Echinacea, modafinil, nafcillin, pioglitazone, prednisone, rufinamide, and vemurafenib.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate data on the developmental risk associated with the use of nimodipine in pregnant women. In animal studies, oral administration of nimodipine during pregnancy resulted in adverse effects on development (increased embryofetal mortality, increased incidences of fetal structural abnormalities, decreased fetal growth) at doses equivalent to (rat) or less than (rabbit) those used clinically [see Data].
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
Nimodipine has been shown to have a teratogenic effect in two studies in rabbit. In one study, incidences of malformations and stunted fetuses were increased at oral doses of 1 mg/kg/day and 10 mg/kg/day administered throughout organogenesis but not at 3 mg/kg/day. In the second study, an increased incidence of stunted fetuses was seen at 1 mg/kg/day but not at higher doses (3 mg/kg/day and 10 mg/kg/day). The lowest effect dose in rabbits (1 mg/kg/day) is less than the recommended human dose (RHD) of 360 mg/day on a body surface area (mg/m 2) basis.
Nimodipine was embryotoxic, causing resorption and stunted growth of fetuses in rats at 100 mg/kg/day administered orally throughout organogenesis; this dose is approximately 3 times the RHD on a mg/m 2basis. In two other studies in rats, nimodipine administered orally at 30 mg/kg/day throughout organogenesis and continued until sacrifice (day 20 of pregnancy or day 21 postpartum) was associated with higher incidences of skeletal variation, stunted fetuses, and stillbirths but no malformations; this dose is similar to the RHD on a mg/m 2basis.8.2 Lactation
Risk Summary
Nimodipine has been detected in human milk. There are no data on the effects of nimodipine on the breastfed infant or on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for nimodipine and any potential adverse effects on the breastfed infant from nimodipine or from the underlying maternal condition.
Data
Animal Data
[ 14C]Nimodipine and its radiolabeled metabolites were secreted in milk of orally dosed lactating rats. The milk concentration of nimodipine and/or metabolites was higher than that in plasma, with a milk/plasma ratio of 0.65 to 4.7.
8.5 Geriatric Use
Clinical studies of nimodipine did not include sufficient numbers of subjects aged 65 and over to determine whether they had a different clinical response than younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dosing in elderly patients should be cautious, reflecting the greater frequency of decreased hepatic, renal or cardiac function, and of concomitant disease or other drug therapy [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
There have been no reports of overdosage from the oral administration of nimodipine. Symptoms of overdosage would be expected to be related to cardiovascular effects such as excessive peripheral vasodilation with marked systemic hypotension. Clinically significant hypotension due to nimodipine overdosage may require active cardiovascular support with pressor agents and specific treatments for calcium channel blocker overdose. Since nimodipine is highly protein-bound, dialysis is not likely to be of benefit.
-
11 DESCRIPTION
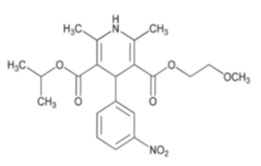
Nimodipine oral solution contains nimodipine, a dihydropyridine calcium channel blocker. Nimodipine is isopropyl 2-methoxyethyl 1,4-dihydro-2,6-dimethyl-4-( m-nitrophenyl)-3,5-pyridinedicarboxylate. It has a molecular weight of 418.45 and a molecular formula of C 21H 26N 2O 7. The structural formula is:
Nimodipine USP is a light yellow or yellow, crystalline powder, freely soluble in ethyl acetate, sparingly soluble in ethanol, practically insoluble in water.
Nimodipine oral solution contains 60 mg of nimodipine per 20 mL. In addition, the oral solution contains the following inactive ingredients: dibasic sodium phosphate anhydrous, ethanol 0.63% v/v, glycerin, methylparaben, monobasic sodium phosphate monohydrate, polyethylene glycol 400 and purified water. -
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Nimodipine is a dihydropyridine calcium channel blocker. The contractile processes of smooth muscle cells are dependent upon calcium ions, which enter these cells during depolarization as slow ionic transmembrane currents. Nimodipine inhibits calcium ion transfer into these cells and thus inhibits contractions of vascular smooth muscle. In animal experiments, nimodipine had a greater effect on cerebral arteries than on arteries elsewhere in the body perhaps because it is highly lipophilic, allowing it to cross the blood-brain barrier; concentrations of nimodipine as high as 12.5 ng/mL have been detected in the cerebrospinal fluid of nimodipine-treated SAH patients.
The precise mechanism of action of nimodipine in reducing the incidence and severity of ischemic deficits in adult patients with SAH from ruptured intracranial berry aneurysms is unknown. Although the clinical studies demonstrate a favorable effect of nimodipine on the severity of neurological deficits caused by cerebral vasospasm following SAH, there is no arteriographic evidence that nimodipine either prevents or relieves the spasm of these arteries. However, whether or not the arteriographic methodology utilized was adequate to detect a clinically meaningful effect, if any, on vasospasm is unknown.
12.3 Pharmacokinetics
After a single 60 mg oral dose of nimodipine, mean (CV%) C maxwas 69.9 ng/mL (36.1%), AUC infwas 151 h.ng/mL (36.0%) and within subject variability (CV%) was 21.7% and 12.4%, respectively. There were no signs of accumulation when nimodipine was given three times a day for seven days.
Absorption
In humans, nimodipine was absorbed with a time to maximum concentration (T max) ranging from 0.25 to 1.05 hours following oral administration. Because of a high first-pass metabolism, the bioavailability of nimodipine averages 13% after oral administration.
Effect of Food
In a study of 24 healthy male volunteers, administration of nimodipine capsules following a standard breakfast resulted in a 68% lower peak plasma concentration and 38% lower bioavailability relative to dosing under fasted conditions [see Dosage and Administration (2.1)].
Distribution
Nimodipine is over 95% bound to plasma proteins. The binding was concentration independent over the range of 10 ng/mL to 10 mcg/mL.
Elimination
The terminal elimination half-life is approximately 8 to 9 hours but earlier elimination rates are much more rapid, equivalent to a half-life of 1 to 2 hours; a consequence is the need for frequent (every 4 hours) dosing.
Metabolism
Numerous metabolites, all of which are either inactive or considerably less active than the parent compound, have been identified. The metabolism of nimodipine is mediated by CYP3A4 [see Drug Interactions (7.2, 7.3)].
Excretion
Nimodipine is eliminated almost exclusively in the form of metabolites and less than 1% is recovered in the urine as unchanged drug.
Specific Populations
Patients with Cirrhosis
The bioavailability of nimodipine is significantly increased in patients with cirrhosis, with C maxapproximately double that in normals, which necessitates lowering the dose in this group of patients [see Dosage and Administration (2.4), Warnings and Precautions (5.2)].
Geriatric Patients
In a single parallel-group study involving 24 elderly subjects (aged 59 to 79 years) and 24 younger subjects (aged 22 to 40 years), the observed AUC and C maxof nimodipine was approximately 2-fold higher in the elderly population compared to the younger study subjects following oral administration (given as a single dose of 30 mg and dosed to steady-state with 30 mg three times daily [less than the recommended dosing regimen] for 6 days). The clinical response to these age-related pharmacokinetic differences, however, was not considered significant [see Use in Specific Populations (8.5)]. -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In a two-year study in rats, the incidences of adenocarcinoma of the uterus and Leydig cell adenoma of the testes were increased at 1800 ppm nimodipine in the diet (approximately 90 to 120 mg/kg/day). The increases were not statistically significant, however, and the higher rates were within the historical control range for these tumors. Nimodipine was found not to be carcinogenic in a 91-week mouse study, but the high dose of 1800 ppm nimodipine in the diet (approximately 550 to 775 mg/kg/day) was associated with an increased mortality rate.
Mutagenesis
Mutagenicity studies, including the Ames, micronucleus, and dominant lethal assays, were negative.
Impairment of Fertility
Nimodipine did not impair the fertility and general reproductive performance of male and female rats following oral doses of up to 30 mg/kg/day when administered prior to mating and continuing in females to day 7 of pregnancy. This dose in a rat is similar to a clinical dose of 60 mg every 4 hours in a 60 kg patient, on a body surface area (mg/m 2) basis. -
14 CLINICAL STUDIES
The safety and efficacy of nimodipine oral solution in the treatment of patients with SAH is based on adequate and well-controlled studies of nimodipine oral capsules in patients with SAH. Nimodipine oral solution has comparable bioavailability to nimodipine oral capsules.
Nimodipine has been shown in 4 randomized, double-blind, placebo-controlled trials to reduce the severity of neurological deficits resulting from vasospasm in patients who have had a recent SAH (Studies 1, 2, 3, and 4).
The trials used doses ranging from 20 to 30 mg to 90 mg every 4 hours, with drug given for 21 days in 3 studies, and for at least 18 days in the other. Three of the four trials followed patients for 3 to 6 months. Three of the trials studied relatively well patients, with all or most patients in Hunt and Hess Grades I to III (essentially free of focal deficits after the initial bleed). Study 4 studied much sicker patients with Hunt and Hess Grades III to V. Studies 1 and 2 were similar in design, with relatively unimpaired SAH patients randomized to nimodipine or placebo. In each, a judgment was made as to whether any late-developing deficit was due to spasm or other causes, and the deficits were graded. Both studies showed significantly fewer severe deficits due to spasm in the nimodipine group; Study 2 showed fewer spasm-related deficits of all severities. No effect was seen on deficits not related to spasm. See Table 2.
Table 2: Deficits in Patients with Hunt and Hess Grades I to III in Study 1 and Study 2
Study
Grade*
Treatment
Patients
Number
Analyzed
Number of
Patients with Any Deficit Due to Spasm
Numbers with Severe Deficit
Study 1
I to III
Nimodipine 20 to 30 mg every 4 hours
56
13
1
Placebo
60
16
8**
Study 2
I to III
Nimodipine 60 mg every 4 hours
31
4
2
Placebo
39
11
10**
*H *Hunt and Hess Grade
**p=0.03
Study 3 was a 554-patient trial that included SAH patients with all grades of severity (89% were in Hunt and Hess Grades I to III). In Study 3, patients were treated with placebo or 60 mg of nimodipine every 4 hours. Outcomes were not defined as spasm related or not but there was a significant reduction in the overall rate of brain infarction and severely disabling neurological outcome at 3 months (Table 3):
Table 3: Degree of Recovery or Disability in Study 3 (89% Hunt and Hess Grades I to III)
Nimodipine
Placebo
Total patients
278
276
Good recovery
199*
169
Moderate disability
24
16
Severe disability
12**
31
Death
43***
60
*p = 0.0444 – good and moderate vs severe and dead
** p = 0.001 – severe disability
***p = 0.056 – death
Study 4 enrolled much sicker patients, (Hunt and Hess Grades III to V), who had a high rate of death and
disability, and used a dose of 90 mg every 4 hours, but was otherwise similar to Study 1 and Study 2. Analysis of delayed ischemic deficits, many of which result from spasm, showed a significant reduction in spasm-related deficits. Among analyzed patients (72 nimodipine, 82 placebo), there were the following outcomes (Table 4).
Table 4: Neurological Ischemic Deficits in Study 4 [Hunt and Hess Grades III to V]
Delayed Ischemic Deficits (DID)
Permanent Deficits
Nimodipine 90 mg every 4 hours
n (%)
Placebo
n (%)
Nimodipine 90 mg every 4 hours
n (%)
Placebo
n (%)
DID Spasm Alone
8 (11)*
25 (31)
5 (7)*
22 (27)
DID Spasm Contributing
18 (25)
21 (26)
16 (22)
17 (21)
DID Without Spasm
7 (10)
8 (10)
6 (8)
7 (9)
No DID
39 (54)
28 (34)
45 (63)
36 (44)
*p = 0.001, Nimodipine vs placebo
When data were combined for Study 3 and Study 4, the treatment difference on success rate (i.e., good recovery) on the Glasgow Outcome Scale was 25.3% (nimodipine) versus 10.9% (placebo) for Hunt and Hess Grades IV or V. Table 5 demonstrates that nimodipine tends to improve good recovery of SAH patients with poor neurological status post-ictus, while decreasing the numbers with severe disability and vegetative survival.
Table 5: Glasgow Outcome Scale in Combined Studies 3 and 4Glasgow Outcome*
Nimodipine
(n=87)
Placebo
(n=101)
Good Recovery
22 (25.3%)
11 (10.9%)
Moderate Disability
8 (9.2%)
12 (11.9%)
Severe Disability
6 (6.9%)
15 (14.9%)
Vegetative Survival
4 (4.6%)
9 (8.9%)
Death
47 (54.0%)
54 (53.5%)
*p = 0.045, nimodipine vs. placebo
A dose-ranging study comparing 30 mg, 60 mg, and 90 mg doses found a generally low rate of spasm-related neurological deficits but no dose response relationship. -
16 HOW SUPPLIED/STORAGE AND HANDLING
Nimodipine Oral Solution 3 mg/mL is a pale yellow solution and is supplied as follows:
· NDC: 31722-039-47: 16 oz. bottle (473 mL) 60 mg/20 mL (3 mg/mL).
Store between 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F). [see USP Controlled Room Temperature].
Protect from light.
Do not refrigerate.
-
17 PATIENT COUNSELING INFORMATION
Inform patients that the most frequent adverse reaction associated with nimodipine is decreased blood pressure [see Warnings and Precautions (5.1)]. Inform them that use of nimodipine oral solution with anti-hypertensives can cause increased drop in blood pressure [see Drug Interactions (7.1)] .
Patients should be aware that ingestion of grapefruit or grapefruit juice should be avoided when taking nimodipine oral solution due to its ability to increase nimodipine plasma concentrations and potential to increase the risk of hypotension [see Drug Interactions (7.2)].
Advise patients to notify their healthcare provider if they become pregnant during treatment or plan to become pregnant during therapy [see Use in Specific Populations (8.1)].
Advise female patients to notify their physicians if they intend to breastfeed or are breastfeeding an infant [see Use in Specific Populations (8.2)].
For more information, call at 1-866-495-1995.

Manufactured for:
Camber Pharmaceuticals, Inc.
Piscataway, NJ 08854.
By: Annora Pharma Pvt. Ltd.
Sangareddy - 502313, Telangana, India.
Revised: 03/2024
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
NIMODIPINE
nimodipine solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 31722-039 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength NIMODIPINE (UNII: 57WA9QZ5WH) (NIMODIPINE - UNII:57WA9QZ5WH) NIMODIPINE 60 mg in 20 mL Inactive Ingredients Ingredient Name Strength METHYLPARABEN (UNII: A2I8C7HI9T) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) ALCOHOL (UNII: 3K9958V90M) GLYCERIN (UNII: PDC6A3C0OX) SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) SODIUM PHOSPHATE, DIBASIC, ANHYDROUS (UNII: 22ADO53M6F) WATER (UNII: 059QF0KO0R) Product Characteristics Color yellow (pale yellow) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 31722-039-47 473 mL in 1 BOTTLE; Type 0: Not a Combination Product 07/09/2024 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA216937 07/09/2024 Labeler - Camber Pharmaceuticals, Inc. (826774775) Establishment Name Address ID/FEI Business Operations Annora Pharma Private Limited 650980746 manufacture(31722-039)

© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.