YARTEMLEA- narsoplimab injection
YARTEMLEA by
Drug Labeling and Warnings
YARTEMLEA by is a Prescription medication manufactured, distributed, or labeled by Omeros Corporation. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use YARTEMLEA safely and effectively. See full prescribing information for YARTEMLEA.
YARTEMLEA (narsoplimab-wuug) injection, for intravenous use
Initial U.S. Approval: 2025INDICATIONS AND USAGE
YARTEMLEA is a MASP-2 inhibitor indicated for the treatment of adult and pediatric patients 2 years of age and older with hematopoietic stem cell transplant-associated thrombotic microangiopathy (TA-TMA). ( 1)
DOSAGE AND ADMINISTRATION
Weight (kg) Recommended Dosage Greater than or equal to 50 kg 370 mg given as an intravenous infusion over 30 minutes once weekly. Increase frequency to twice weekly if there is inadequate improvement in TA-TMA signs and symptoms. ( 2.1) Less than 50 kg 4 mg/kg given as an intravenous infusion over 30 minutes once weekly. Increase frequency to twice weekly if there is inadequate improvement in TA-TMA signs and symptoms. ( 2.1) DOSAGE FORMS AND STRENGTHS
Injection: 370 mg/2 mL (185 mg/mL) in a single-dose vial. ( 3)
CONTRAINDICATIONS
None ( 4)
WARNINGS AND PRECAUTIONS
Serious infections: Monitor patients for signs/symptoms and treat appropriately. ( 5.1)
ADVERSE REACTIONS
Most common adverse reactions (incidence > 20% and independent of causality) are viral infections, sepsis, hemorrhage, diarrhea, vomiting, nausea, neutropenia, pyrexia, fatigue and hypokalemia. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Omeros Corporation at 1-844-YARTEM1 or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Important Preparation and Administration Instructions
2.3 Preparation Instructions into an Intravenous Bag
2.4 Preparation Instructions into an Intravenous Syringe
2.5 Storage and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infections
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage of YARTEMLEA is provided in Table 1.
Table 1: Recommended Dosage of YARTEMLEA in Adult and Pediatric Patients Two Years of Age and Older with TA-TMA Weight (kg) Recommended Dosage Greater than or equal to 50 kg 370 mg given as an intravenous infusion over 30 minutes once weekly. Increase frequency to twice weekly if there is inadequate improvement in TA-TMA signs and symptoms. Less than 50 kg 4 mg/kg given as an intravenous infusion over 30 minutes once weekly. Increase frequency to twice weekly if there is inadequate improvement in TA-TMA signs and symptoms. If a dose is missed, administer the dose as soon as possible. Thereafter, resume dosing at the regular scheduled time.
2.2 Important Preparation and Administration Instructions
- Administer diluted YARTEMLEA as an intravenous infusion through a polyvinyl chloride (PVC) or PVC-lined infusion line with a 0.2-micron polyethersulfone (PES) in-line filter and a polyurethane catheter.
- For adults and pediatric patients weighing 10 kg or more, prepare YARTEMLEA in an intravenous bag, diluted to final concentration of 0.8 mg/mL to 8 mg/mLand administer by gravity infusion or via an infusion pump [see Dosage and Administration ( 2.3and 2.5)] .
- For pediatric patients weighing less than 10 kg, prepare YARTEMLEA in a polypropylene syringe, diluted to final concentration of 0.8 mg/mLand administer via a syringe pump [see Dosage and Administration ( 2.4and 2.5)] .
2.3 Preparation Instructions into an Intravenous Bag
This section describes preparation of YARTEMLEA for adults and pediatric patients weighing 10 kg or more in an intravenous bag.
1. Preparation
- Use aseptic technique to prepare YARTEMLEA.
- Parenteral drug-product vial should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. YARTEMLEA is a clear to slightly opalescent, slightly yellow to yellow-brown solution. If discoloration or particles are observed in the vial, discard it.
- Calculate the dose (mg) based on the patient's body weight and the total volume (mL) of YARTEMLEA solution required.
- Remove the YARTEMLEA vial from the refrigerator and allow the vial to come to room temperature (18°C to 25°C [64°F to 77°F]) for 30 minutes. Vial must be used to prepare the appropriate dosing solution within 4 hours following removal from refrigerated storage.
2. Dilution for Intravenous Bag Infusion
- Withdraw the required volume of YARTEMLEA solution from the vial using a polypropylene syringe and dilute in a polyvinyl chloride (PVC) infusion bag of 5% Dextrose Injection, USP to make a final concentration of 0.8 mg/mL to 8 mg/mLwith a total volume not to exceed 50 mL.
- Discard any unused portion left in the vial.
- Gently invert infusion bag 10 times to mix the diluted solution. Do not shake.
- Following dilution of YARTEMLEA in the infusion bag, the solution may become opalescent, and small translucent-to-white particles may appear. Discard prepared solution if particulate matter, other than translucent-to-white particles, is observed.
2.4 Preparation Instructions into an Intravenous Syringe
This section describes preparation of YARTEMLEA for pediatric patients weighing less than 10 kg in a syringe.
1. Preparation
- Use aseptic technique to prepare YARTEMLEA.
- Parenteral drug-product vial should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. YARTEMLEA is a clear to slightly opalescent, slightly yellow to yellow-brown solution. If discoloration or particles are observed in the vial, discard it.
- Calculate the dose (mg) based on the patient's body weight and the total volume (mL) of YARTEMLEA solution required.
- Remove the YARTEMLEA vial from the refrigerator and allow the vial to come to room temperature (18°C to 25°C [64°F to 77°F]) for 30 minutes. Vial must be used to prepare the appropriate dosing solution within 4 hours following removal from refrigerated storage.
2. Dilution for Intravenous Syringe Infusion
- Withdraw the required volume of YARTEMLEA solution from the vial using a polypropylene syringe. Discard any unused portion left in the vial.
- Dilute in 5% Dextrose Injection, USP to make a final concentration of 0.8 mg/mLwith a total volume not to exceed 50 mL.
- Gently invert syringe 10 times to mix the diluted solution. Do not shake.
- Following dilution of YARTEMLEA in the syringe, the solution may become opalescent, and small translucent-to-white particles may appear. Discard prepared solution if particulate matter, other than translucent-to-white particles, is observed.
- Remove air bubbles from the syringe before administration.
2.5 Storage and Administration
Storage of Diluted Product
- If the prepared diluted solution is not used immediately, store the diluted YARTEMLEA solution at room temperature at 18°C to 25°C (64°F to 77°F) for up to 4 additional hours.
- Discard unused YARTEMLEA solution if not used within 4 hours from the time of dilution to the end of the infusion.
Administration
- Administer YARTEMLEA diluted solution intravenously by gravity infusion or via infusion pump (for solution prepared in an intravenous bag) or via syringe pump (for solution prepared in a syringe) over 30 minutes through a PVC or PVC-lined infusion line with a 0.2-micron polyethersulfone (PES) in-line filter and a polyurethane catheter.
- Flush the intravenous line at the end of the infusion with sufficient volume of 5% Dextrose Injection, USP to clear the line of YARTEMLEA infusion solution.
- Do not co-administer other drugs through the same intravenous line.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infections
Serious and life-threatening infections have occurred in patients treated with YARTEMLEA.
Serious infections, independent of causality, were reported in 36% (10/28) of patients with TA-TMA receiving YARTEMLEA in clinical trials. These infections included sepsis, viral infections, pneumonia, bacteremia, fungal infection, gastroenteritis, respiratory tract infection and urosepsis.
If YARTEMLEA is administered to patients with active infections, monitor closely for signs and symptoms of worsening infection and treat promptly.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Serious Infections [see Warnings and Precautions ( 5.1)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety data described in this section reflect exposure to YARTEMLEA in the TA-TMA Study in which 28 adult patients received YARTEMLEA. In total, 24 patients received YARTEMLEA at a dose of 4 mg/kg intravenously once weekly for 4 or 8 weeks and 4 patients received 370 mg intravenously weekly for 8 weeks [see Clinical Studies ( 14)] . The median duration of treatment with YARTEMLEA was 8 weeks (range: 2 to 16.4 weeks).
Serious adverse reactions were reported in 61% of patients receiving YARTEMLEA. Serious adverse reactions in > 5% of patients who received YARTEMLEA included acute kidney injury, confusional state, acute respiratory failure, neutropenic sepsis, septic shock, pulmonary edema, and vomiting. Fatal adverse reactions occurred in 7% of patients, including neutropenic sepsis and septic shock.
Adverse reactions leading to dosage interruptions occurred in 7% of patients who received YARTEMLEA and included Escherichiasepsis, pyrexia, pulmonary alveolar hemorrhage, and acute myocardial infarction.
The most common adverse reactions (≥ 20%) were viral infections, sepsis, hemorrhage, diarrhea, vomiting, nausea, neutropenia, pyrexia, fatigue, and hypokalemia.
Table 2summarizes the adverse reactions, without regard to causality or relatedness to YARTEMLEA, in the TA-TMA Study.
Table 2: Adverse Reactions (≥ 15%) in Patients Receiving YARTEMLEA in the TA-TMA Study * Grouped terms
Adverse Reaction All Grades
n (%)
N = 28Grade ≥ 3
n (%)
N = 28Hemorrhage* 12 (43) 2 (7) Diarrhea 10 (36) 2 (7) Infection, viral* 10 (36) 2 (7) Neutropenia* 10 (36) 10 (36) Pyrexia 10 (36) 1 (4) Vomiting 9 (32) 2 (7) Fatigue* 8 (29) 1 (4) Hypokalemia* 7 (25) 3 (11) Nausea 7 (25) 1 (4) Sepsis* 7 (25) 6 (21) Pneumonia* 5 (18) 4 (14) Hypotension* 5 (18) 3 (11) Abdominal pain* 5 (18) 1 (4) Anemia* 5 (18) 3 (11) Back pain 5 (18) 0 (0) An additional 221 adult and pediatric patients with TA-TMA were treated with YARTEMLEA in a global expanded access program (EAP) that included patients for whom YARTEMLEA was their initial treatment following diagnosis of TA-TMA as well as patients who had previously failed or stopped other treatments. The median number of YARTEMLEA doses received by the 221 patients in the EAP was 8 and the median duration of therapy was 5.5 weeks. No new clinically significant safety signals were identified in patients treated in the EAP.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The available data on the use of YARTEMLEA during pregnancy are insufficient to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. In animal reproduction studies, narsoplimab-wuug was administered subcutaneously and intravenously twice weekly to pregnant mice and rabbits during organogenesis at dose exposures up to 22 and 91-fold, respectively, the human exposure at the maximum recommended human dose (MRHD) based on area under the concentration-time curve (AUC). There were no adverse effects observed in the absence of maternal toxicity (see Data) .
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Transport of endogenous IgG antibodies across the placenta increases as pregnancy progresses and peaks during the third trimester. Therefore, it is expected that YARTEMLEA, following administration, will be present in infants exposed in utero during the third trimester. The potential clinical impact of narsoplimab-wuug exposure on infants exposed in utero should be considered.
Data
Animal Data
Narsoplimab-wuug was administered to pregnant mice at doses of 50, 150, or 300 mg/kg by subcutaneous injection and 300 mg/kg by intravenous injection approximately twice weekly during the major period of organogenesis. Intravenous administration caused reduced body weight gain in dams, reduced fetal body weight up to 4.8%, and an increase of approximately 33% in post-implantation loss at 22-fold the exposure expected at the MRHD (based on AUC) of 4 mg/kg intravenously once weekly in humans. Narsoplimab-wuug was administered to pregnant rabbits at doses of 50 or 150 mg/kg by subcutaneous injection and 150 mg/kg by intravenous injection approximately twice weekly during the major period of organogenesis. Intravenous administration increased post-implantation loss during early gestation days and reduced fetal body weights up to 8.5% below controls at 91-fold the exposure expected at the MRHD (based on AUC) of 4 mg/kg intravenously once weekly in humans.
In an enhanced pre- and postnatal development study, pregnant mice were dosed with narsoplimab-wuug at doses up to 300 mg/kg intravenously twice weekly from day 6 of gestation through lactation. There was a transiently reduced maternal body weight gain but no adverse effects of narsoplimab-wuug on pregnancy nor on the viability, growth, or development of the infants up to 22-fold the exposure expected at the MRHD (based on AUC).
8.2 Lactation
Risk Summary
There are no data on the presence of narsoplimab-wuug in human milk, the effects on the breastfed child, or the effects on milk production. Narsoplimab-wuug is present in the milk of lactating mice (see Data) . When a drug is present in animal milk, it is likely that the drug will be present in human milk. Endogenous maternal IgG and monoclonal antibodies are transferred into human milk. The effects of local gastrointestinal exposure and the extent of systemic exposure in the breastfed child to narsoplimab-wuug are unknown. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for YARTEMLEA and any potential adverse effects on the breastfed child from YARTEMLEA or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of YARTEMLEA for treatment of TA-TMA have been established in pediatric patients aged 2 years and older. Use of YARTEMLEA for this indication is supported by evidence from studies in adults and in 6 pediatric patients, and additional safety data from 83 more pediatric patients aged 2 years and older [see Adverse Reactions ( 6.1) and Clinical Studies ( 14.1)] .
The safety and effectiveness of YARTEMLEA have not been established in pediatric patients younger than 2 years old.
8.5 Geriatric Use
Of the total number of YARTEMLEA-treated patients in the clinical trial and EAP for TA-TMA, 21 (8.4%) were 65 years of age and older, while 1 (0.4%) was at least 75 years of age [see Clinical Studies ( 14)]. Clinical studies of YARTEMLEA did not include sufficient numbers of subjects at least 65 years of age to determine whether they respond differently than younger subjects. Other reported clinical experience has not identified differences in responses between elderly and younger patients.
- 10 OVERDOSAGE
-
11 DESCRIPTION
Narsoplimab-wuug, a mannan-binding lectin-associated serine protease 2 (MASP-2) inhibitor, is a recombinant human immunoglobulin G4 (IgG4) monoclonal antibody produced in Chinese Hamster Ovary cells. The approximate molecular weight is 143 kDa.
YARTEMLEA (narsoplimab-wuug) injection is a sterile, preservative-free, clear to slightly opalescent, slightly yellow to yellow-brown solution for intravenous infusion. Each 2-mL single-dose glass vial contains 370 mg of narsoplimab-wuug, arginine hydrochloride (84.3 mg), citric acid monohydrate (1.42 mg), polysorbate 80 (0.2 mg), sodium citrate (8.6 mg), and Water for Injection, USP. Sodium hydroxide and hydrochloric acid were added to adjust the pH to 5.8.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Narsoplimab-wuug inhibits MASP-2, the effector enzyme of the lectin pathway of the complement system, blocking lectin-dependent activation of complement component 3 (C3) and C4 without affecting the classical and alternative pathways of complement.
In hematopoietic stem cell transplant-associated thrombotic microangiopathy (TA-TMA), MASP-2 inhibition is thought to prevent lectin pathway-mediated cellular injury, including endothelial cell injury in small blood vessels.
12.2 Pharmacodynamics
The effect of YARTEMLEA on lectin pathway activity, assessed using inhibition of C4d deposition, was investigated in healthy subjects and patients with TA-TMA (TA-TMA Study). YARTEMLEA concentration levels achieved in patients with TA-TMA resulted in > 80% inhibition of lectin pathway activity. Based on pharmacokinetic/pharmacodynamic (PK/PD) modeling, a wash-out period of 6 weeks is sufficient to reduce YARTEMLEA concentrations to below pharmacologically active levels.
12.3 Pharmacokinetics
The PK profile of narsoplimab-wuug has been characterized in healthy subjects and in patients. PK of narsoplimab-wuug is less than dose-proportional for 2 and 4 mg/kg weekly IV dosing, with an accumulation ratio ranging from 1.02 to 1.75 at 4 mg/kg IV weekly dosing. Narsoplimab-wuug steady state (measured at day 36) geometric mean C maxis 36.9 μg/mL, with geometric mean AUC 0-tauof 2314 μg·h/mL following 4 mg/kg administered intravenously in healthy subjects. Narsoplimab-wuug steady state is reached after three once-weekly IV doses (day 15) of 4 mg/kg in healthy subjects.
Absorption
After intravenous administration, peak plasma concentrations of narsoplimab-wuug occur approximately at the end of each infusion.
Distribution
Narsoplimab-wuug is distributed in the blood and hydrophilic extravascular space with an average (CV%) volume of distribution of 10.9 L (65%) in patients.
Elimination
Total clearance of narsoplimab-wuug is concentration-dependent, with an estimated mean (CV%) value of 0.12 L/hour (68%) in patients. The mean (CV%) terminal elimination half-life was estimated to be 209 hours (73%) in patients.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies (ADA) is highly dependent on the sensitivity and the specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the narsoplimab-wuug studies described below with the incidence of ADA in other studies.
Anti-Drug Antibody Effects on Pharmacokinetics and Pharmacodynamics
In the 8-week treatment period in the TA-TMA Study, treatment-emergent antibodies to narsoplimab-wuug were detected in 3 of 28 patients (11%) with TA-TMA. One of the 3 patients (33%) developed neutralizing antibodies. There is no apparent correlation of antibody development to PK or PD response, and no alteration in clinical response or adverse events was observed.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Genotoxicity and animal carcinogenicity studies of narsoplimab-wuug have not been conducted.
A fertility study of narsoplimab-wuug in male and female mice produced no adverse effects on reproductive parameters, although a transient decrease in mean body-weight gain in females (premating) and a slight increase in mean percentage of sperm with abnormal morphology in males were observed in the highest dose groups. These effects were noted at exposures corresponding to 21-fold the exposure expected at the MRHD (based on AUC). In the absence of any functional effects on mating, fertility, or reproductive organ weight, these effects on sperm morphology were not considered adverse.
-
14 CLINICAL STUDIES
The efficacy of YARTEMLEA was assessed in (i) a single-arm, open-label study (TA-TMA Study) that enrolled 28 adult patients who developed TA-TMA following hematopoietic stem-cell transplantation (HCT) and (ii) 19 adult and pediatric patients with TA-TMA with evaluable patient-level response data enrolled in an expanded access program (EAP). In the TA-TMA Study, 24 patients received YARTEMLEA 4 mg/kg intravenously once weekly and 4 patients received YARTEMLEA 370 mg intravenously once weekly. The median number of YARTEMLEA administrations received by the 28 TA-TMA Study patients plus the 19 EAP patients was 8 (range: 2-34), and the median duration of therapy was 8 weeks (range: 2-16 weeks).
Baseline demographic and disease-related characteristics are shown in Table 3. The TA-TMA Study patients had a confirmed diagnosis of TA-TMA per the diagnostic criteria as follows: platelet count < 150,000/μL, evidence of microangiopathic hemolysis (presence of schistocytes, serum LDH greater than the upper limit of normal [ULN] and/or haptoglobin less than the lower limit of normal [LLN]), and renal dysfunction. Patients in the EAP were similarly thrombocytopenic with evidence of microangiopathic hemolytic anemia.
Table 3: Characteristics of TA-TMA Patients Treated with YARTEMLEA in the TA-TMA Study and Expanded Access Program Parameter TA-TMA Study Expanded Access Program
(N = 19)aAdult
(N = 28)Pediatric
(n = 6)Adult
(n = 13)aIncludes patients from EAP with available patient-level data. The entire EAP consisted of 221 patients; patient-level response data were available in 13 adult and 6 pediatric patients.
bRace and ethnicity data were not available for the 19 patients in the EAP.
Median age (years)
(range)48
(22, 68)10.5
(5, 15)62
(19, 71)Race, n (%)b
White
Asian
Black or African American
Native Hawaiian or Other Pacific Islander
Other
17 (61)
7 (25)
2 (7)
1 (4)
1 (4)
--
--Ethnicity, n (%)a
Hispanic or Latino/a
Not Hispanic or Latino/a
2 (7)
26 (93)
--
--Gender, n (%)
Male
Female
20 (71)
8 (29)
2 (33)
4 (67)
5 (38)
8 (62)Elevated LDH ≥ 2x ULN, n (%) 20 (71) 6 (100) 7 (54) Grade II-IV acute GvHD, n (%) 19 (68) 5 (83) 11 (85) Organ dysfunction, n (%) 27 (96) 6 (100) 13 (100) Renal dysfunction, n (%) 21 (75) 5 (83) 13 (100) Pulmonary dysfunction, n (%) 5 (18) 0 (0) 2 (15) Neurological dysfunction, n (%) 16 (57) 2 (33) 4 (31) Infection, n (%) 24 (86) 6 (100) 8 (62) Among patients in the TA-TMA Study, the median time from HCT to TMA diagnosis was 73.5 days (range: 21–436) and the median time from TMA diagnosis to first dose of narsoplimab was 13.5 days (range: 4–196). Among the 19 adult and pediatric patients in the EAP, the median time from HCT to TMA diagnosis was 81 days (range: 24–452) and the median time from TMA diagnosis to first dose of narsoplimab was 3 days (range: 0–52).
The primary efficacy assessment of YARTEMLEA was based on TMA response defined as improvement in both of two laboratory TMA markers (LDH and platelet counts) and either improvement in organ function or independence from transfusions. The same response criteria were applied to both the TA-TMA Study and EAP patients.
Improvement in platelet count was defined as follows:
- For baseline platelet count ≤ 20,000/μL: (i) ≥ 3-fold increase in platelet count, (ii) post-baseline platelet count ≥ 30,000/μL, and (iii) receipt of no platelet transfusions within 2 days prior to the platelet count assessment.
- For baseline platelet count > 20,000/μL: (i) ≥ 50% increase in platelet count, (ii) platelet count ≥ 75,000/μL, and (iii) receipt of no platelet transfusions within 2 days prior to the platelet count assessment.
To meet LDH improvement criteria, LDH levels were required to be < 1.5x ULN.
In the TA-TMA Study and the EAP, TA-TMA response was achieved in 17/28 (60.7%) and 13/19 (68.4%) patients, respectively ( Table 4).
Table 4: Efficacy Results for TA-TMA Study and Expanded Access Program TA-TMA Study Expanded Access Program
(N = 19)Adult
(N = 28)Pediatric
(n = 6)Adult
(n = 13)aConfidence intervals were calculated using the exact (Clopper–Pearson) method.
bThese component findings are based on the number of patients with evaluable data.
cDefined as no transfusions for at least 4 weeks from the last transfusion; only evaluated in patients who received transfusions within the 2 weeks prior to or on the first narsoplimab dose date.
TMA response
95% CI a17/28 (61%)
(40.6, 78.5)4/6 (67%)
(22.3, 95.7)9/13 (69%)
(38.6, 90.9)Improvement in TMA
markers
Platelet count
LDH17/28 (61%)
14/23 (61%) b
21/28 (75%)4/6 (67%)
4/6 (67%)
5/6 (83%)9/13 (69%)
9/13 (69%)
11/13 (85%)Improvement in organ function 20/27 (74%) b 5/6 (83%) 11/13 (85%) Freedom from red blood cell or platelet transfusion c 12/25 (48%) b 3/5 (60%) b 9/13 (69%) In the TA-TMA Study, the 100-day survival from time of TMA diagnosis was 73.4% (95% CI: 52.2, 86.4). In the EAP cohort (N = 19), 100-day survival from time of TMA diagnosis was 73.7% (95% CI: 47.9, 88.1)
-
16 HOW SUPPLIED/STORAGE AND HANDLING
YARTEMLEA (narsoplimab-wuug) injection is a sterile, preservative-free, clear to slightly opalescent, slightly yellow to yellow-brown solution supplied as one 370 mg/2 mL (185 mg/mL) single-dose vial in a carton (NDC: 62225-300-00).
Store YARTEMLEA vials refrigerated at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light until time of use. Do not freeze. Do not shake. Do not use beyond the expiration date stamped on the carton.
-
17 PATIENT COUNSELING INFORMATION
Serious infections: Inform patients and caregivers that treatment with complement inhibitors has been associated with an increased risk of serious infections. As YARTEMLEA is a complement inhibitor, advise patients and/or caregivers to immediately report any signs or symptoms suggestive of infections [see Warnings and Precautions ( 5.1)] .
Manufactured by:
Omeros Corporation
201 Elliott Avenue West
Seattle, WA 98119
US license 2141
This product, or its use, may be covered by one or more US patents, including US Patent Nos. 9,011,860, 10,047,165, and 10,683,367, in addition to others, including patents pending.
YARTEMLEA is a trademark of Omeros Corporation.
© 2025 Omeros Corporation
-
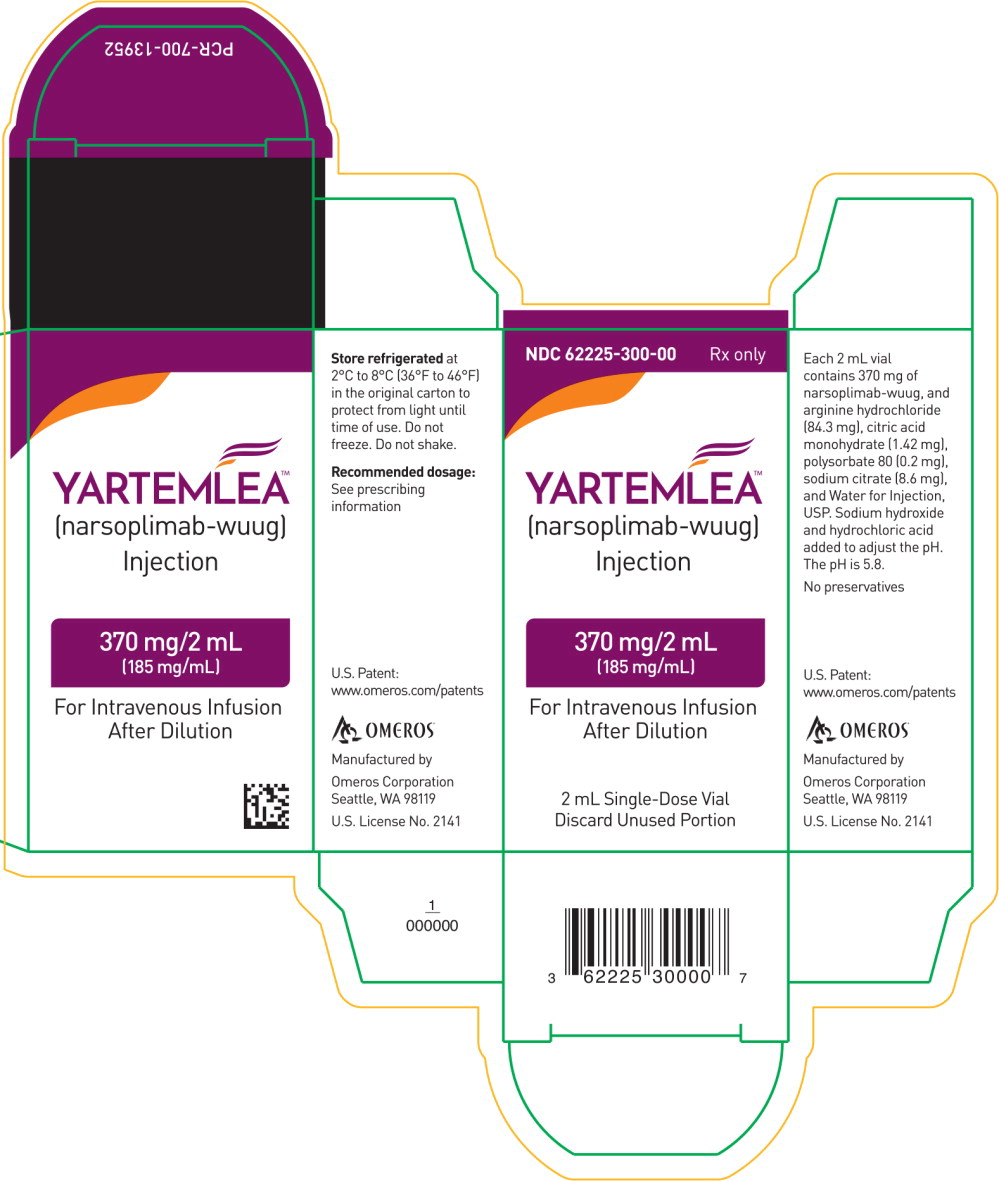
PRINCIPAL DISPLAY PANEL
Principal Display Panel - 370mg Carton Label
NDC: 62225-300-00Rx only
YARTEMLEA™
(narsoplimab-wuug)
Injection370 mg/2 mL
(185 mg/mL)For Intravenous Infusion After Dilution
2 mL Single-Dose Vial
Discard Unused Portion -
INGREDIENTS AND APPEARANCE
YARTEMLEA
narsoplimab injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 62225-300 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength NARSOPLIMAB (UNII: FT24ZQQ5RP) (NARSOPLIMAB - UNII:FT24ZQQ5RP) NARSOPLIMAB 370 mg in 2 mL Inactive Ingredients Ingredient Name Strength ARGININE HYDROCHLORIDE (UNII: F7LTH1E20Y) CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SODIUM CITRATE, UNSPECIFIED FORM (UNII: 1Q73Q2JULR) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 62225-300-00 1 in 1 CARTON 01/20/2026 1 2 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761152 01/20/2026 Labeler - Omeros Corporation (033364923)
Trademark Results [YARTEMLEA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 YARTEMLEA 98475095 not registered Live/Pending |
Omeros Corporation 2024-03-29 |
 YARTEMLEA 98171267 not registered Live/Pending |
Omeros Corporation 2023-09-08 |
 YARTEMLEA 98171265 not registered Live/Pending |
Omeros Corporation 2023-09-08 |
 YARTEMLEA 88906252 not registered Live/Pending |
Omeros Corporation 2020-05-07 |
 YARTEMLEA 88906250 not registered Live/Pending |
Omeros Corporation 2020-05-07 |
 YARTEMLEA 88453133 not registered Live/Pending |
Omeros Corporation 2019-05-30 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.