IBANDRONATE SODIUM tablet
Ibandronate sodium by
Drug Labeling and Warnings
Ibandronate sodium by is a Prescription medication manufactured, distributed, or labeled by Dr. Reddy's Laboratories Limited, Dr. Reddy's Laboratories Limited - FTO III. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use IBANDRONATE SODIUM TABLETS safely and effectively.
See full prescribing information for IBANDRONATE SODIUM TABLETS.
IBANDRONATE sodium tablets, for oral use
Initial U.S. Approval: 2003INDICATIONS AND USAGE
Ibandronate sodium tablet is a bisphosphonate indicated for the treatment and prevention of postmenopausal osteoporosis. (1.1)
Limitations of Use
The optimal duration of use has not been determined. For patients at low-risk for fracture, consider drug discontinuation after 3 to 5 years of use. (1.2).
DOSAGE AND ADMINISTRATION
- Take one 150 mg tablet once monthly on the same day each month (2.1)
- Instruct patient to: (2.2)
-
- Swallow whole tablet with 6 to 8 oz of plain water only, at least 60 minutes before the first food, beverage, or medication of day.
- Avoid lying down for at least 60 minutes after taking ibandronate sodium tablets. Do not eat, drink (except for water), or take other medication for 60 minutes after taking ibandronate sodium tablets.
- Take supplemental calcium and vitamin D if dietary intake inadequate (2.3)
DOSAGE FORMS AND STRENGTHS
Tablets: 150 mg (3) (3)
CONTRAINDICATIONS
- Abnormalities of the esophagus which delay esophageal emptying such as stricture or achalasia (4, 5.1)
- Inability to stand or sit upright for at least 60 minutes (4, 5.1)
- Hypocalcemia (4)
- Hypersensitivity to ibandronate sodium tablets (4)
WARNINGS AND PRECAUTIONS
- Upper gastrointestinal Adverse Reactions can occur. Instruct patients to follow dosing instructions and discontinue use if new or worsening symptoms occur. (5.1)
- Hypocalcemia may worsen during treatment. Correct hypocalcemia before use. (5.2)
- Severe Bone, Joint, and Muscle Pain may occur. Consider discontinuing use if symptoms develop. (5.3)
- Osteonecrosis of the Jaw has been reported. (5.4)
- Atypical Femur Fractures have been reported. Patients with new thigh or groin pain should be evaluated to rule out a femoral fracture. (5.5)
ADVERSE REACTIONS
The most common adverse reactions (greater than 5%) are back pain, dyspepsia, pain in extremity, diarrhea, headache, and myalgia. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Dr. Reddy’s Laboratories Inc. at 1-888-375-3784 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 8/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Treatment and Prevention of Postmenopausal Osteoporosis
1.2 Important Limitations of Use
2 DOSAGE AND ADMINISTRATION
2.1 Dosage Information
2.2 Important Administration Instructions
2.3 Recommendations for Calcium and Vitamin D Supplementation
2.4 Administration Instructions for Missed Once-Monthly Doses
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Upper Gastrointestinal Adverse Reactions
5.2 Hypocalcemia and Mineral Metabolism
5.3 Musculoskeletal Pain
5.4 Jaw Osteonecrosis
5.5 Atypical Subtrochanteric and Diaphyseal Femoral Fractures
5.6 Severe Renal Impairment
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Calcium Supplements/Antacids
7.2 Aspirin/Nonsteroidal Anti-Inflammatory Drugs (NSAIDs)
7.3 H2 Blockers
7.4 Drug/Laboratory Test Interactions
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment Of Fertility
13.2 Animal Pharmacology
14 CLINICAL STUDIES
14.1 Treatment of Postmenopausal Osteoporosis
14.2 Prevention of Postmenopausal Osteoporosis
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
17.1 Information for Patients
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Treatment and Prevention of Postmenopausal Osteoporosis
Ibandronate sodium tablets are indicated for the treatment and prevention of osteoporosis in postmenopausal women. Ibandronate sodium tablets increases bone mineral density (BMD) and reduces the incidence of vertebral fractures.
1.2 Important Limitations of Use
The optimal duration of use has not been determined. The safety and effectiveness of ibandronate sodium tablets for the treatment of osteoporosis are based on clinical data of three years duration. All patients on bisphosphonate therapy should have the need for continued therapy re-evaluated on a periodic basis. Patients at low-risk for fracture should be considered for drug discontinuation after 3 to 5 years of use. Patients who discontinue therapy should have their risk for fracture re-evaluated periodically.
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosage Information
The dose of ibandronate sodium tablet is one 150 mg tablet taken once monthly on the same date each month.
2.2 Important Administration Instructions
Instruct Patients to do the following:
- Take ibandronate sodium tablets at least 60 minutes before the first food or drink (other than water) of the day or before taking any oral medication or supplementation, including calcium, antacids, or vitamins to maximize absorption and clinical benefit, (see Drug Interactions [7.1]). Avoid the use of water with supplements including mineral water because they may have a higher concentration of calcium.
- Swallow ibandronate sodium tablets whole with a full glass of plain water (6 to 8 oz) while standing or sitting in an upright position to reduce the potential for esophageal irritation. Avoid lying down for 60 minutes after taking ibandronate sodium tablets (see Warnings and Precautions [5.1]). Do not chew or suck the tablet because of a potential for oropharyngeal ulceration. Do not eat, drink anything except plain water, or take other medications for at least 60 minutes after taking ibandronate sodium tablets.
2.3 Recommendations for Calcium and Vitamin D Supplementation
Instruct patients to take supplemental calcium and vitamin D if their dietary intake is inadequate. Avoid the use of calcium supplements within 60 minutes of ibandronate sodium tablets administration because co-administration of ibandronate sodium tablets and calcium may interfere with the absorption of ibandronate sodium (see Drug Interactions [7.1]).
2.4 Administration Instructions for Missed Once-Monthly Doses
If the once-monthly dose is missed, instruct patients to do the following:
- If the next scheduled ibandronate sodium tablets day is more than 7 days away, take one ibandronate sodium tablet, 150 mg (acid) in the morning following the date that it is remembered.
- If the next scheduled ibandronate sodium tablets day is only 1 to 7 days away, wait until the subsequent month’s scheduled ibandronate sodium tablets day to take their tablet.
For subsequent monthly doses for both of the above scenarios, instruct patients to return to their original schedule by taking one ibandronate sodium tablet, 150 mg (acid) every month on their previous chosen day.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Ibandronate sodium tablets are contraindicated in patients with the following conditions:
- Abnormalities of the esophagus which delay esophageal emptying such as stricture or achalasia (see Warnings and Precautions[5.1])
- Inability to stand or sit upright for at least 60 minutes (see Dosage and Administration [2.2], and Warnings and Precautions [5.1])
- Hypocalcemia (see Warnings and Precautions [5.2])
- Known hypersensitivity to ibandronate sodium tablets or to any of its excipients. Cases of anaphylaxis have been reported. (see Adverse Reactions [6.2]).
-
5 WARNINGS AND PRECAUTIONS
5.1 Upper Gastrointestinal Adverse Reactions
Ibandronate sodium, like other bisphosphonates administered orally, may cause local irritation of the upper gastrointestinal mucosa. Because of these possible irritant effects and a potential for worsening of the underlying disease, caution should be used when ibandronate sodium is given to patients with active upper gastrointestinal problems (such as known Barrett’s esophagus, dysphagia, other esophageal diseases, gastritis, duodenitis or ulcers).
Esophageal adverse experiences, such as esophagitis, esophageal ulcers and esophageal erosions, occasionally with bleeding and rarely followed by esophageal stricture or perforation, have been reported in patients receiving treatment with oral bisphosphonates. In some cases, these have been severe and required hospitalization. Physicians should therefore be alert to any signs or symptoms signaling a possible esophageal reaction and patients should be instructed to discontinue ibandronate sodium and seek medical attention if they develop dysphagia, odynophagia, retrosternal pain or new or worsening heartburn.
The risk of severe esophageal adverse experiences appears to be greater in patients who lie down after taking oral bisphosphonates and/or who fail to swallow it with the recommended full glass (6 to 8 oz) of water, and/or who continue to take oral bisphosphonates after developing symptoms suggestive of esophageal irritation. Therefore, it is very important that the full dosing instructions are provided to, and understood by, the patient (see Dosage and Administration [2.2]). In patients who cannot comply with dosing instructions due to mental disability, therapy with ibandronate sodium should be used under appropriate supervision.
There have been post-marketing reports of gastric and duodenal ulcers with oral bisphosphonate use, some severe and with complications, although no increased risk was observed in controlled clinical trials.
5.2 Hypocalcemia and Mineral Metabolism
Hypocalcemia has been reported in patients taking ibandronate sodium. Treat hypocalcemia and other disturbances of bone and mineral metabolism before starting ibandronate sodium therapy. Instruct patients to take supplemental calcium and vitamin D if their dietary intake is inadequate. (see Dosage and Administration [2.3]).
5.3 Musculoskeletal Pain
Severe and occasionally incapacitating bone, joint, and/or muscle pain has been reported in patients taking ibandronate sodium and other bisphosphonates (see Adverse Reactions [6]). The time to onset of symptoms varied from one day to several months after starting the drug. Most patients had relief of symptoms after stopping. A subset had recurrence of symptoms when rechallenged with the same drug or another bisphosphonate. Consider discontinuing use if severe symptoms develop.
5.4 Jaw Osteonecrosis
Osteonecrosis of the jaw (ONJ), which can occur spontaneously, is generally associated with tooth extraction and/or local infection with delayed healing, and has been reported in patients taking bisphosphonates, including ibandronate sodium. Known risk factors for osteonecrosis of the jaw include invasive dental procedures (e.g., tooth extraction, dental implants, boney surgery), diagnosis of cancer, concomitant therapies (e.g., chemotherapy, corticosteroids, angiogenesis inhibitors), poor oral hygiene, and co-morbid disorders (e.g., periodontal and/or other pre-existing dental disease, anemia, coagulopathy, infection, ill-fitting dentures). The risk of ONJ may increase with duration of exposure to bisphosphonates.
For patients requiring invasive dental procedures, discontinuation of bisphosphonate treatment may reduce the risk for ONJ. Clinical judgment of the treating physician and/or oral surgeon should guide the management plan of each patient based on individual benefit/risk assessment.
Patients who develop osteonecrosis of the jaw while on bisphosphonate therapy should receive care by an oral surgeon. In these patients, extensive dental surgery to treat ONJ may exacerbate the condition. Discontinuation of bisphosphonate therapy should be considered based on individual benefit/risk assessment.
5.5 Atypical Subtrochanteric and Diaphyseal Femoral Fractures
Atypical, low-energy, or low-trauma fractures of the femoral shaft have been reported in bisphosphonate-treated patients. These fractures can occur anywhere in the femoral shaft from just below the lesser trochanter to above the supracondylar flare and are transverse or short oblique in orientation without evidence of comminution. Causality has not been established as these fractures also occur in osteoporotic patients who have not been treated with bisphosphonates.
Atypical femur fractures most commonly occur with minimal or no trauma to the affected area. They may be bilateral and many patients report prodromal pain in the affected area, usually presenting as dull, aching thigh pain, weeks to months before a complete fracture occurs. A number of reports note that patients were also receiving treatment with glucocorticoids (e.g., prednisone) at the time of fracture.
Any patient with a history of bisphosphonate exposure who presents with thigh or groin pain should be suspected of having an atypical fracture and should be evaluated to rule out an incomplete femur fracture. Patients presenting with an atypical fracture should also be assessed for symptoms and signs of fracture in the contralateral limb. Interruption of bisphosphonate therapy should be considered, pending a risk/benefit assessment, on an individual basis.
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Treatment and Prevention of Postmenopausal Osteoporosis
Daily DosingThe safety of ibandronate sodium 2.5 mg once daily in the treatment and prevention of postmenopausal osteoporosis was assessed in 3577 patients aged 41 to 82 years. The duration of the trials was 2 to 3 years, with 1134 patients exposed to placebo and 1140 exposed to ibandronate sodium 2.5 mg. Patients with pre-existing gastrointestinal diseaseand concomitant use of non-steroidal anti-inflammatory drugs, proton pump inhibitors and H2 antagonists were included in these clinical trials. All patients received 500 mg calcium plus 400 international units vitamin D supplementation daily.
The incidence of all-cause mortality was 1% in the placebo group and 1.2% in the ibandronate sodium 2.5 mg daily group. The incidence of serious adverse reactions was 20% in the placebo group and 23% in the ibandronate sodium2.5 mg daily group. The percentage of patients who withdrew from treatment due to adverse reactions was approximately 17% in both the ibandronate sodium 2.5 mg daily group and the placebo group. Table 1 lists adverse reactions from the treatment and prevention studies reported in greater than or equal to 2% of patients and more frequently in patients treated daily with ibandronate than patients treated with placebo.
Table 1 Adverse Reactions Occurring at an Incidence Greater Than or Equal to 2% and in More Patients Treated with Ibandronate Sodium Than in Patients Treated with Placebo Daily in the Osteoporosis Treatment and Prevention Studies
Body System Placebo % (n=1134) Ibandronate Sodium 2.5 mg % (n=1140) Body as a Whole Back Pain 12 14 Pain in Extremity 6 8 Asthenia 2 4 Allergic Reaction 2 3 Digestive System Dyspepsia 10 12 Diarrhea 5 7 Tooth Disorder 2 4 Vomiting 2 3 Gastritis 2 2 Musculoskeletal System Myalgia 5 6 Joint Disorder 3 4 Arthritis 3 3 Nervous System Headache 6 7 Dizziness 3 4 Vertigo 3 3 Respiratory System Upper Respiratory Infection 33 34 Bronchitis 7 10 Pneumonia 4 6 Pharyngitis 2 3 Urogenital System Urinary Tract Infection 4 6 Gastrointestinal Adverse Reactions
The incidence of selected gastrointestinal adverse reactions in the placebo and ibandronate sodium 2.5 mg daily groups were: dyspepsia (10% vs. 12%), diarrhea (5% vs. 7%), and abdominal pain (5% vs. 6%).
Musculoskeletal Adverse Reactions
The incidence of selected musculoskeletal adverse reactions in the placebo and ibandronate sodium 2.5 mg daily groups were: back pain (12% vs. 14%), arthralgia (14% vs. 14%) and myalgia (5% vs. 6%).
Ocular Adverse Events
Reports in the medical literature indicate that bisphosphonates may be associated with ocular inflammation such as iritis and scleritis. In some cases, these events did not resolve until the bisphosphonate was discontinued.
There were no reports of ocular inflammation in studies with ibandronate 2.5 mg daily.
Monthly Dosing
The safety of ibandronate sodium 150 mg (acid) once monthly in the treatment of postmenopausal osteoporosis was assessed in a two year trial which enrolled 1583 patients aged 54 to 81 years, with 395 patients exposed to ibandronate sodium 2.5 mg (acid) daily and 396 exposed to ibandronate sodium 150 mg (acid) monthly. Patients with active or significant pre-existing gastrointestinal disease were excluded from this trial. Patients with dyspepsia or concomitant use of nonsteroidal anti-inflammatory drugs, proton pump inhibitors and H2 antagonists were included in this study. All patients received 500 mg calcium plus 400 international units vitamin D supplementation daily.
After one year, the incidence of all-cause mortality was 0.3% in both the ibandronate sodium 2.5 mg (acid) daily group and the ibandronate sodium 150 mg (acid) monthly group. The incidence of serious adverse events was 5% in the ibandronate sodium 2.5 mg (acid) daily group and 7% in the ibandronate sodium 150 mg (acid) monthly group. The percentage of patients who withdrew from treatment due to adverse events was 9% in the ibandronate sodium 2.5 mg (acid) daily group and 8% in the ibandronate sodium 150 mg (acid) monthly group. Table 2 lists the adverse events reported in greater than or equal to 2% of patients.
Table 2 Adverse Events with an Incidence of at Least 2% in Patients Treated with the Ibandronate Sodium 2.5 mg (acid) Daily or Ibandronate Sodium 150 mg (acid) Once-Monthly for Treatment of Postmenopausal Osteoporosis
Body System/Adverse Event Ibandronate
Sodium 2.5 mg (acid) Daily
%
(n=395)Ibandronate Sodium 150 mg (acid)
Monthly
%
(n=396)Vascular Disorders
Hypertension
7.3
6.3Gastrointestinal Disorders
Dyspepsia
Nausea
Diarrhea
Constipation
Abdominal Paina
7.1
4.8
4.1
2.5
5.3
5.6
5.1
5.1
4.0
7.8Musculoskeletal and ConnectiveTissue Disorders
Arthralgia
Back Pain
Pain in Extremity
Localized Osteoarthritis
Myalgia
Muscle Cramp
3.5
4.3
1.3
1.3
0.8
2.0
5.6
4.5
4.0
3.0
2.0
1.8Infections and Infestations
Influenza
Nasopharyngitis
Bronchitis
Urinary Tract Infection
Upper Respiratory Tract Infection
3.8
4.3
3.5
1.8
2.0
4.0
3.5
2.5
2.3
2.0Nervous System Disorders
Headache
Dizziness
4.1
1.0
3.3
2.3General Disorders andAdministration Site Conditions
Influenza-like Illnessb
0.8
3.3Skin and Subcutaneous TissueDisorders
Rashc
1.3
2.3Psychiatric Disorders
Insomnia
0.8
2.0a Combination of abdominal pain and abdominal pain upper
b Combination of influenza-like illness and acute phase reaction
c Combination of rash pruritic, rash macular, rash papular, rash generalized, rash erythematous, dermatitis, dermatitis allergic, dermatitis medicamentosa, erythema and exanthema
Gastrointestinal Adverse Events
The incidence of adverse events in the ibandronate sodium 2.5 mg (acid) daily and ibandronate sodium 150 mg (acid) monthly groups were: dyspepsia (7% vs. 6%), diarrhea (4% vs. 5%), and abdominal pain (5% vs. 8%).
Musculoskeletal Adverse Events
The incidence of adverse events in the ibandronate sodium 2.5 mg (acid) daily and ibandronate sodium 150 mg (acid) monthly groups were: back pain (4% vs. 5%), arthralgia (4% vs. 6%) and myalgia (1% vs. 2%).
Acute Phase Reactions
Symptoms consistent with acute phase reactions have been reported with bisphosphonate use. Over the two years of the study, the overall incidence of acute phase reaction symptoms was 3% in the ibandronate sodium 2.5 mg (acid) daily group and 9% in the ibandronate sodium 150 mg (acid) monthly group. These incidence rates are based on the reporting of any of 33 acute-phase reaction like symptoms within 3 days of the monthly dosing and lasting 7 days or less. Influenza like illness was reported in no patients in the ibandronate sodium 2.5 mg (acid) daily group and 2% in the ibandronate sodium 150 mg (acid) monthly group.
Ocular Adverse Events
Two patients who received ibandronate sodium 150 mg (acid) once-monthly experienced ocular inflammation, one was a case of uveitis and the other scleritis.
One hundred sixty (160) postmenopausal women without osteoporosis participated in a 1-year, double-blind, placebo-controlled study of ibandronate sodium 150 mg (acid) once-monthly for prevention of bone loss. Seventy-seven subjects received ibandronate sodium and 83 subjects received placebo. The overall pattern of adverse events was similar to that previously observed.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of ibandronate sodium. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hypersensitivity
Allergic reactions including anaphylactic reaction/shock; with fatalities, angioedema, bronchospasm, asthma exacerbations,rash, Stevens-Johnson syndrome, erythema muliforme, and dermatitis bullous have been reported (see Contraindications [4])
Hypocalcemia
Hypocalcemia has been reported in patients treated with ibandronate sodium (see Warnings and Precautions [5.2]).
Musculoskeletal Pain
Bone, joint, or muscle pain (musculoskeletal pain), described as severe or incapacitating, has been reported (see Warnings and Precautions [5.3]).
Jaw Osteonecrosis
Osteonecrosis of the jaw has been reported in patients treated with ibandronate sodium (see Warnings and Precautions [5.4]).
Atypical Femoral Shaft Fracture
Atypical, low-energy, or low-trauma fractures of the femoral shaft (see Warnings and Precautions [5.5]).
-
7 DRUG INTERACTIONS
7.1 Calcium Supplements/Antacids
Products containing calcium and other multivalent cations (such as aluminum, magnesium, iron) are likely to interfere with absorption of ibandronate sodium. Therefore, instruct patients to take ibandronate sodium at least 60 minutes before any oral medications, including medications containing multivalent cations (such as antacids, supplements or vitamins). Also, patients should wait at least 60 minutes after dosing before taking any other oral medications (see Dosage and Administration [2.3]).
7.2 Aspirin/Nonsteroidal Anti-Inflammatory Drugs (NSAIDs)
Because aspirin, NSAIDs, and bisphosphonates are all associated with gastrointestinal irritation, caution should be exercised in the concomitant use of aspirin or NSAIDs with ibandronate.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Ibandronate is not indicated for use in women of reproductive potential. There are no data with ibandronate use in pregnant women to inform any drug-associated risks.
In reproductive toxicity studies in the rat, ibandronate caused post-implantation loss and obstruction of labor with maternal and fetal periparturient mortality at greater than or equal to 3 times human exposure at the recommended 2.5 mg daily oral dose, or at greater than or equal to 1 times human exposure at the recommended 150 mg once-monthly oral dose. In pregnant rats, kidney developmental toxicity occurred in offspring at greater than or equal to 30 times the daily 2.5 mg human dose or at greater than or equal to 9 times the once-monthly 150 mg human dose. In rat reproductive studies, impaired pup neuromuscular development was observed at 45 times the daily 2.5 mg dose and 13 times the once-monthly 150 mg dose. In reproductive studies in the rabbit, ibandronate caused maternal mortality at greater than or equal to 8 times the daily 2.5 mg dose and greater than or equal to 4 times the once-monthly 150 mg dose (see Data).
Data
Animal Data
In female rats given ibandronate at oral doses greater than or equal to 3 times human exposure at therecommended daily oral dose of 2.5 mg or greater than or equal to 1 times human exposure at the recommended once-monthly oral dose of 150 mg beginning 14 days before mating and continuing through lactation, maternal deaths were observed at the time of delivery in all dose groups. Perinatal pup loss in dams given doses producing 45 times human exposure at the recommended daily dose and 13 times human exposure at the recommended once-monthly dose was likely related to maternal dystocia. Calcium supplementation did not completely prevent dystocia and periparturient mortality in any of the treated groups at greater than or equal to 16 times the recommended daily dose and greater than or equal to 4.6 times the recommended once-monthly dose. A low incidence of postimplantation loss was observed in rats treated from 14 days before mating throughout lactation or during gestation, only at doses causing maternal dystocia and periparturient mortality. In pregnant rats dosed orally from gestation day 17 through lactation day 21 (following closure of the hard palate through weaning), maternal toxicity, including dystocia and mortality, fetal perinatal and postnatal mortality, were observed at doses equivalent to human exposure at the recommended daily dose and greater than or equal to 4 times the recommended once-monthly dose. Periparturient mortality has also been observed with other bisphosphonates and appears to be a class effect related to inhibition of skeletal calcium mobilization resulting in hypocalcemia and dystocia.
Exposure of pregnant rats during the period of organogenesis resulted in an increased fetal incidence of RPU (renal pelvis ureter) syndrome at oral doses producing 30 times human exposure at the recommended daily oral dose of 2.5 mg and greater than or equal to 9 times human exposure at the recommended once-monthly oral dose of 150 mg. Impaired pup neuromuscular development (cliff avoidance test) was observed at 45 times human exposure at the daily dose and 13 times the once-monthly dose.
In pregnant rabbits treated orally with ibandronate during gestation at doses greater than or equal to 8 times the recommended human daily oral dose of 2.5 mg and greater than or equal to 4 times the recommended human once-monthly oral dose of 150 mg, dose-related maternal mortality was observed in all treatment groups. The deaths occurred prior to parturition and were associated with lung edema and hemorrhage. No significant fetal anomalies were observed.
Exposure multiples for the rat studies were calculated for the recommended daily oral dose of 2.5 mg or once monthly dose of 150 mg based on area under the curve (AUC) comparison. Exposure multiples for the rabbit study were calculated for the recommended human daily oral dose of 2.5 mg or once-monthly dose of 150 mg based on dose/body surface area comparison. Doses used in pregnant animals were 1, 4, 5, 6, 16, 10, 20, 30, 60 or 100 mg/kg/day in rats, and 1, 4 or 20 mg/kg/day in rabbits.
8.2 Lactation
Risk Summary
Ibandronate sodium is not indicated for use in women of reproductive potential. There is no information on the presence of ibandronate in human milk, the effects of ibandronate on the breastfed infant, or the effects of ibandronate on milk production. Ibandronate is present in rat milk (see Data).The clinical relevance of these data is unclear.DataAnimal DataIn lactating rats treated with intravenous doses of 0.08 mg/kg, ibandronate was present in breast milk from 2 to 24 hours after dose administration. Concentrations in milk averaged 1.5 times plasma concentrations.
8.5 Geriatric Use
Of the patients receiving ibandronate 2.5 mg daily in postmenopausal osteoporosis studies, 52% were over65 years of age, and 10% were over 75 years of age. Of the patients receiving ibandronate sodium 150 mg (acid) once-monthly in the postmenopausal osteoporosis 1-year study, 52% were over 65 years of age, and 9% were over 75 years of age. No overall differences in effectiveness or safety were observed between these patients and younger patients but greater sensitivity in some older individuals cannot be ruled out.
-
10 OVERDOSAGE
No specific information is available on the treatment of overdosage of ibandronate sodium. However, based on knowledge of this class of compounds, oral overdosage may result in hypocalcemia, hypophosphatemia, and upper gastrointestinal adverse events, such as upset stomach, dyspepsia, esophagitis, gastritis, or ulcer. Milk or antacids should be given to bind ibandronate sodium. Due to the risk of esophageal irritation, vomiting should not be induced, and the patient should remain fully upright. Dialysis would not be beneficial.
-
11 DESCRIPTION
Ibandronate sodium is a nitrogen-containing bisphosphonate that inhibits osteoclast-mediated bone resorption. The chemical name for ibandronate sodium is 3-(N-methyl-N-pentyl) amino-1-hydroxypropane-1,1diphosphonic acid, monosodium salt, with the molecular formula C9H22NO7P2Na and a molecular weight of 341. Ibandronate sodium is an off-white to white colored powder. It is sparingly soluble in water. Ibandronate sodium has the following structural formula:
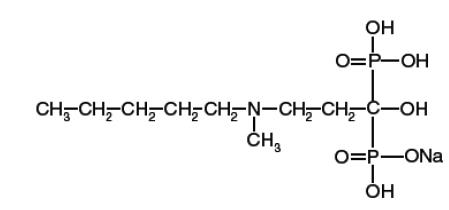
Ibandronate sodium tablets are available as a white, capsule shaped 150-mg coated tablets for once-monthly oral administration. One 150 mg coated tablet contains 160.33 mg ibandronate sodium, equivalent to 150 mg of ibandronic acid. Ibandronate sodium tablets also contains the following inactive ingredients: colloidal silicon dioxide, crospovidone, croscarmellose sodium, lactose monohydrate, microcrystalline cellulose, povidone, and sodium stearyl fumarate. The tablet coating contains hypromellose, polyethylene glycol 400, and titanium dioxide. Imprinting ink contains: ammonium hydroxide, black iron oxide, propylene glycol and shellac.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The action of ibandronate on bone tissue is based on its affinity for hydroxyapatite, which is part of the mineral matrix of bone. Ibandronate inhibits osteoclast activity and reduces bone resorption and turnover. In postmenopausal women, it reduces the elevated rate of bone turnover, leading to, on average, a net gain in bone mass.
12.2 Pharmacodynamics
Osteoporosis is characterized by decreased bone mass and increased fracture risk, most commonly at the spine, hip, and wrist. The diagnosis can be confirmed by a finding of low bone mass, evidence of fracture on x-ray, a history of osteoporotic fracture, or height loss or kyphosis indicative of vertebral fracture. While osteoporosis occurs in both men and women, it is most common among women following menopause. In healthy humans, bone formation and resorption are closely linked; old bone is resorbed and replaced by newly formed bone. In postmenopausal osteoporosis, bone resorption exceeds bone formation, leading to bone loss and increased risk of fracture. After menopause, the risk of fractures of the spine and hip increases; approximately 40% of 50-year-old women will experience an osteoporosis-related fracture during their remaining lifetimes.
Ibandronate produced biochemical changes indicative of dose-dependent inhibition of bone resorption, including decreases of biochemical markers of bone collagen degradation (such as deoxypyridinoline, and cross-linked C-telopeptide of Type I collagen) in the once-monthly doses from 100 mg to 150 mg in postmenopausal women.
Treatment with 2.5 mg daily ibandronate sodium resulted in decreases in biochemical markers of bone turnover, including urinary C-terminal telopeptide of Type I collagen (uCTX) and serum osteocalcin, to levels similar to those in premenopausal women. Changes in markers of bone formation were observed later than changes in resorption markers, as expected, due to the coupled nature of bone resorption and formation. Treatment with 2.5 mg daily ibandronate sodium decreased levels of uCTX within 1 month of starting treatment and decreased levels of osteocalcin within 3 months. Bone turnover markers reached a nadir of approximately 64% below baseline values by6 months of treatment and remained stable with continued treatment for up to 3 years. Following treatment discontinuation, there is a return to pretreatment baseline rates of elevated bone resorption associated with postmenopausal osteoporosis.
In a 1-year, study comparing once-monthly vs. once-daily oral dosing regimens, the median decrease from baseline in serum CTX values was -76% for patients treated with the 150 mg once-monthly regimen and -67%for patients treated with the 2.5 mg daily regimen. In a 1-year, prevention study comparing ibandronate sodium 150 mg once-monthly to placebo, the median placebo-subtracted decrease in sCTX was -49.8%.
12.3 Pharmacokinetics
Absorption
The absorption of oral ibandronate occurs in the upper gastrointestinal tract. Plasma concentrations increase in a dose-linear manner up to 50 mg oral intake and increases nonlinearly above this dose.
Following oral dosing, the time to maximum observed plasma ibandronate concentrations ranged from 0.5 to 2 hours (median 1 hour) in fasted healthy postmenopausal women. The mean oral bioavailability of 2.5 mgibandronate was about 0.6% compared to intravenous dosing.The extent of absorption is impaired by food or beverages (other than plain water). The oral bioavailability of ibandronate is reduced by about 90% when ibandronate sodium tablet is administered concomitantly with a standard breakfast in comparison with bioavailability observed in fasted subjects. There is no meaningful reduction in bioavailability when ibandronate is taken at least 60 minutes before a meal. However, both bioavailability and the effect on bone mineral density (BMD) are reduced when food or beverages are taken less than 60 minutes following an ibandronate dose.
Distribution
After absorption, ibandronate either rapidly binds to bone or is excreted into urine. In humans, the apparent terminal volume of distribution is at least 90 L, and the amount of dose removed from the circulation via the bone is estimated to be 40% to 50% of the circulating dose. In vitro protein binding in human serum was 99.5% to 90.9% over an ibandronate concentration range of 2 to 10 ng/mL in one study and approximately 85.7% over a concentration range of 0.5 to 10 ng/mL in another study.
Metabolism
Ibandronate does not undergo hepatic metabolism and does not inhibit the hepatic cytochrome P450 system. Ibandronate is eliminated by renal excretion. Based on a rat study, the ibandronate secretory pathway does not appear to include known acidic or basic transport systems involved in the excretion of other drugs. There is no evidence that ibandronate is metabolized in humans.
Elimination
The portion of ibandronate that is not removed from the circulation via bone absorption is eliminated unchanged by the kidney (approximately 50% to 60% of the absorbed dose). Unabsorbed ibandronate is eliminated unchanged in the feces.
The plasma elimination of ibandronate is multiphasic. Its renal clearance and distribution into bone accounts for a rapid and early decline in plasma concentrations, reaching 10% of the Cmax within 3 or 8 hours after intravenous or oral administration, respectively. This is followed by a slower clearance phase as ibandronate redistributes back into the blood from bone. The observed apparent terminal half-life for ibandronate is generally dependent on the dose studied and on assay sensitivity. The observed apparent terminal half-life for the 150 mg ibandronate tablet upon oral administration to healthy postmenopausal women ranges from 37 to 157 hours.
Total clearance of ibandronate is low, with average values in the range 84 to 160 mL/min. Renal clearance (about 60 mL/min in healthy postmenopausal females) accounts for 50% to 60% of total clearance and is related to creatinine clearance. The difference between the apparent total and renal clearances likely reflects bone uptake of the drug.
Specific Populations
Pediatrics
The pharmacokinetics of ibandronate has not been studied in patients less than 18 years of age.
Geriatric
Because ibandronate is not known to be metabolized, the only difference in ibandronate elimination for geriatric patients versus younger patients is expected to relate to progressive age-related changes in renal function.
Gender
The bioavailability and pharmacokinetics of ibandronate are similar in both men and women.
Race
Pharmacokinetic differences due to race have not been studied.
Renal Impairment
Renal clearance of ibandronate in patients with various degrees of renal impairment is linearly related to creatinine clearance (CLcr).
Following a single dose of 0.5 mg ibandronate by intravenous administration, patients with CLcr 40 to 70 mL/min had 55% higher exposure (AUC∞) than the exposure observed in subjects with CLcr greater than 90 mL/min. Patients with CLcr less than 30 mL/min had more than a two-fold increase in exposure compared to the exposure for healthy subjects (see Dosageand Administration [2.4]).
Hepatic Impairment
No studies have been performed to assess the pharmacokinetics of ibandronate in patients with hepatic impairment because ibandronate is not metabolized in the human liver.
Drug Interaction Studies
Products containing calcium and other multivalent cations (such as aluminum, magnesium, iron), including milk, food, and antacids are likely to interfere with absorption of ibandronate, which is consistent with findings in animal studies.
H2 Blockers
A pharmacokinetic interaction study in healthy volunteers demonstrated that 75 mg ranitidine (25 mg injected intravenously 90 and 15 minutes before and 30 minutes after ibandronate administration) increased the oral bioavailability of 10 mg ibandronate by about 20%. This degree of increase is not considered to be clinically relevant.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment Of Fertility
In a 104-week carcinogenicity study, doses of 3, 7, or 15 mg/kg/day were administered by oral gavage to male and female Wistar rats (systemic exposures up to 12 and 7 times, respectively, human exposure at the recommended daily oral dose of 2.5 mg, and cumulative exposures up to 3.5 and 2 times, respectively, human exposure at the recommended once-monthly oral dose of 150 mg, based on AUC comparison). There were no significant drug-related tumor findings in male or female rats. In a 78-week carcinogenicity study, doses of 5, 20, or 40 mg/kg/day were administered by oral gavage to male and female NMRI mice (exposures up to 475 and 70 times, respectively, human exposure at the recommended daily oral dose of 2.5 mg and cumulative exposures up to 135 and 20 times, respectively, human exposure at the recommended once-monthly oral dose of 150 mg, based on AUC comparison). There were no significant drug-related tumor findings in male or female mice. In a 90-week carcinogenicity study, doses of 5, 20, or 80 mg/kg/day were administered in the drinking water to NMRI mice (cumulative monthly exposures in males and females up to 70 and 115 times, respectively, human exposure at the recommended dose of 150 mg, based on AUC comparison). A dose-related increased incidence of adrenal subcapsular adenoma/carcinoma was observed in female mice, which was statistically significant at 80 mg/kg/day (220 to 400 times human exposure at the recommended daily oral dose of 2.5 mg and115 times human exposure at the recommended once-monthly oral dose of 150 mg, based on AUC comparison). The relevance of these findings to humans is unknown.
Mutagenesis There was no evidence for a mutagenic or clastogenic potential of ibandronate in the following assays: in vitro bacterial mutagenesis assay in Salmonella typhimurium and Escherichia coli (Ames test), mammalian cell mutagenesis assay in Chinese hamster V79 cells, and chromosomal aberration test in human peripheral lymphocytes, each with and without metabolic activation. Ibandronate was not genotoxic in the in vivo mouse micronucleus tests for chromosomal damage.
Impairment of Fertility In female rats treated from 14 days prior to mating through gestation, decreases in fertility, corpora lutea, and implantation sites were observed at an oral dose of 16 mg/kg/day (45 times human exposure at the recommended daily oral dose of 2.5 mg and 13 times human exposure at the recommended once-monthly oral dose of 150 mg, based on AUC comparison).
13.2 Animal Pharmacology
Animal studies have shown that ibandronate is an inhibitor of osteoclast-mediated bone resorption. In the Schenk assay in growing rats, ibandronate inhibited bone resorption and increased bone volume, based on histologic examination of the tibial metaphyses. There was no evidence of impaired mineralization at the highest dose of 5 mg/kg/day (subcutaneously), which is 1000 times the lowest antiresorptive dose of 0.005 mg/kg/day in this model, and 5000 times the optimal antiresorptive dose of 0.001 mg/kg/day in the aged ovariectomized rat. This indicates that ibandronate sodium tablet administered at therapeutic doses is unlikely to induce osteomalacia.
Long-term daily or once-monthly intermittent administration of ibandronate to ovariectomized rats or monkeys was associated with suppression of bone turnover and increases in bone mass. In both rats and monkeys, vertebral BMD, trabecular density, and biomechanical strength were increased dose-dependently at doses up to 15 times the recommended human daily oral dose of 2.5 mg, or cumulative monthly doses up to 8 times (rat) or 6 times (monkey) the recommended human once-monthly oral dose of 150 mg, based on body surface area (mg/m2) or area under curve (AUC) comparison. In monkeys, ibandronate maintained the positive correlation between bone mass and strength at the ulna and femoral neck. New bone formed in the presence of ibandronate had normal histologic structure and did not show mineralization defects.
-
14 CLINICAL STUDIES
14.1 Treatment of Postmenopausal Osteoporosis
Daily Dosing
The effectiveness and safety of ibandronate were demonstrated in a randomized, double-blind, placebo-controlled, multinational study (Treatment Study) of 2946 women aged 55 to 80 years, who were on average 21 years postmenopause, who had lumbar spine BMD 2 to 5 SD below the premenopausal mean (T-score) in at least one vertebra [L1 to L4], and who had 1 to 4 prevalent vertebral fractures. Ibandronate sodium was evaluated at oral doses of 2.5 mg daily and 20 mg intermittently. The main outcome measure was the occurrence of new radiographically diagnosed vertebral fractures after 3 years of treatment. The diagnosis of an incident vertebral fracture was based on both qualitative diagnosis by the radiologist and quantitative morphometric criterion. The morphometric criterion required the dual occurrence of 2 events: a relative height ratio or relative height reduction in a vertebral body of at least 20%, together with at least a 4 mm absolute decrease in height. All women received 400 international units vitamin D and 500 mg calcium supplementation per day.
Effect on Fracture Incidence
Ibandronate sodium 2.5 mg (acid) daily significantly reduced the incidence of new vertebral (primary efficacy measure) and of new and worsening vertebral fractures. Over the course of the 3-year study, the risk for vertebral fracture was 9.6% in the placebo-treated women and 4.7% in the women treated with ibandronate sodium 2.5 mg (acid) (p<0.001) (see Table 3).
Table 3 Effect of Ibandronate Sodium on the Incidence of Vertebral Fracture in the 3-Year Osteoporosis Treatment Study*.
Proportion of Patients with Fracture (%) Placebo n=975 Ibandronate 2.5 mg (acid)
Daily
n=977Absolute Risk Reduction
(%)
95% CIRelative Risk Reduction
(%)
95% CINew Vertebral Fracture
0 to 3 Year9.6 4.7 4.9
(2.3, 7.4)52 **
(29, 68)New and Worsening Vertebral Fracture
0 to 3 Year10.4 5.1 5.3
(2.6, 7.9)52
(30, 67)Clinical (Symptomatic) Vertebral Fracture
0 to 3 Year5.3 2.8 2.5
(0.6, 4.5)49
(14, 69)*The endpoint value is the value at the study's last time point, 3 years, for all patients who had a fracture identified at that time; otherwise, the last postbaseline value prior to the study's last time point is used. **p=0.0003 vs. placebo
Ibandronate sodium 2.5 mg (acid) daily did not reduce the incidence of nonvertebral fractures (secondary efficacy measure). There was a similar number of nonvertebral osteoporotic fractures at 3 years reported in women treated with Ibandronate sodium 2.5 mg (acid) daily [9.1%, (95% CI: 7.1%, 11.1%)] and placebo [8.2%, (95% CI: 6.3%, 10.2%)]. The two treatment groups were also similar with regard to the number of fractures reported at the individual nonvertebral sites: pelvis, femur, wrist, forearm, rib, and hip.
Bone Mineral Density (BMD)
Ibandronate significantly increased BMD at the lumbar spine and hip relative to treatment with placebo. In the 3-year osteoporosis treatment study, Ibandronate sodium 2.5 mg (acid) daily produced increases in lumbar spine BMD that were progressive over 3 years of treatment and were statistically significant relative to placebo at 6 months and at all later time points. Lumbar spine BMD increased by 6.4% after 3 years of treatment with 2.5 mg daily ibandronate compared with 1.4% in the placebo group. Table 4 displays the significant increases in BMD seen at the lumbar spine, total hip, femoral neck, and trochanter compared to placebo. Thus, overall ibandronate sodium reverses the loss of BMD, a central factor in the progression of osteoporosis
Table 4 Mean Percent Change in BMD from Baseline to Endpoint in Patients Treated Daily with Ibandronate Sodium2.5 mg (acid) or Placebo in the 3-Year Osteoporosis Treatment Study*
Placebo Ibandronate Sodium 2.5 mg (acid)Daily Lumbar Spine 1.4
(n=693)6.4
(n=712)Total Hip -0.7
(n=638)3.1
(n=654)Femoral Neck -0.7
(n=683)2.6
(n=699)Trochanter 0.2
(n=683)5.3
(n=699)*The endpoint value is the value at the study's last time point, 3 years, for all patients who had BMD measured at that time; otherwise, the last postbaseline value prior to the study's last time point is used.
Bone Histology
The effects of Ibandronate sodium 2.5 mg (acid) daily on bone histology were evaluated in iliac crest biopsies from 16 women after 22 months of treatment and 20 women after 34 months of treatment.
The histological analysis of bone biopsies showed bone of normal quality and no indication of osteomalacia or a mineralization defect.
Once-Monthly Dosing
The effectiveness and safety of ibandronate sodium once-monthly were demonstrated in a randomized, double-blind, multinational, noninferiority trial in 1602 women aged 54 to 81 years, who were on average 18 years postmenopause, and had L2-L4 lumbar spine BMD T-score below -2.5 SD at baseline. The main outcome measure was the comparison of the percentage change from baseline in lumbar spine BMD after 1 year of treatment with once-monthly ibandronate (100 mg, 150 mg) to daily ibandronate (2.5 mg). All patients received 400 international units vitamin D and 500 mg calcium supplementation per day.
Ibandronate sodium 150 mg once-monthly (n=327) was shown to be noninferior to ibandronate sodium 2.5 mg daily (n=318) in lumbar spine BMD in a 1-year, double-blind, multicenter study of women with postmenopausal osteoporosis. In the primary efficacy analysis (per-protocol population), the mean increases from baseline in lumbar spine BMD at 1 year were 3.86% (95% CI: 3.40%, 4.32%) in the 2.5 mg daily group and 4.85% (95% CI: 4.41%, 5.29%) in the 150 mg once-monthly group; the mean difference between 2.5 mg daily and 150 mg once-monthly was 0.99% (95% CI: 0.38%, 1.60%), which was statistically significant (p=0.002). The results of the intent-to-treat analysis were consistent with the primary efficacy analysis. The 150 mg once-monthly group also had consistently higher BMD increases at the other skeletal sites compared to the 2.5 mg daily group.
14.2 Prevention of Postmenopausal Osteoporosis
Daily Dosing
The safety and effectiveness of ibandronate sodium 2.5 mg (acid) daily for the prevention of postmenopausal osteoporosis were demonstrated in a randomized, double-blind, placebo-controlled 2-year study (Prevention Study) of 653 postmenopausal women without osteoporosis at baseline. Women were aged 41 to 82 years, were on average 8.5 years postmenopause, and had lumbar spine BMD T-scores greater than-2.5. Women were stratified according to time since menopause (1 to 3 years, greater than 3 years) and baseline lumbar spine BMD (T-score: greater than-1, -1 to -2.5). The study compared daily ibandronate sodium at three dose levels (0.5 mg, 1 mg, 2.5 mg) with placebo. All women received 500 mg of supplemental calcium per day.
The primary efficacy measure was the change in BMD of lumbar spine after 2 years of treatment. Ibandronate sodium 2.5 mg (acid) daily resulted in a mean increase in lumbar spine BMD of 3.1% compared with placebo following 2 years of treatment. Increases in BMD were seen at 6 months and at all later time points. Irrespective of the time since menopause or the degree of pre-existing bone loss, treatment with ibandronate resulted in a higher BMD response at the lumbar spine compared with placebo across all four baseline strata [time since menopause (1 to 3 years, greater than 3 years) and baseline lumbar spine BMD (T-score: greater than-1, -1 to -2.5)].
Compared with placebo, treatment with ibandronate sodium 2.5 mg (acid) daily increased BMD of the total hip by 1.8%, the femoral neck by 2%, and the trochanter by 2.1%.
Once-Monthly Dosing
The safety and effectiveness of ibandronate sodium 150 mg (acid) once-monthly for the prevention of postmenopausal osteoporosis were demonstrated in a randomized, double-blind, placebo-controlled 1-year study (Monthly Prevention Study) of 160 postmenopausal women with low bone mass at baseline (T-score of -1 to -2.5). Women, aged 46 to 60 years, were on average 5.4 years postmenopause. All women received 400 international units of vitamin D and 500 mg calcium supplementation daily.
The primary efficacy measure was the relative change in BMD at the lumbar spine after 1 year of treatment. Ibandronate sodium 150 mg (acid) once-monthly resulted in a mean increase in lumbar spine BMD of 4.12% (95% confidence interval 2.96 to 5.28) compared with placebo following 1 year of treatment (p<0.0001), based on a 3.73% and -0.39% mean change in BMD from baseline in the 150 mg once-monthly ibandronate sodium and placebo treatment groups, respectively. BMD at other skeletal sites was also increased relative to baseline values.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Ibandronate sodium tablets 150 mg (acid) are white, film coated and capsule shaped tablets printed with R575 on one side and plain on other side. They are supplied in carton of 3 packs containing 1 tablet each.
Carton of 3 packs (NDC: 55111-575-03), each pack containing 1 tablet (NDC: 55111-575-11)
Carton of 1 blister pack containing 3 tablets (NDC: 55111-575-43).
-
17 PATIENT COUNSELING INFORMATION
“See FDA-approved patient labeling (Medication Guide)”
17.1 Information for Patients
Instruct patients should be instructed to read the Medication Guide carefully before taking ibandronate sodium tablets and to re-read it each time the prescription is renewed because it contains important information the patient should know about ibandronate sodium tablets. The Medication Guide also includes to the dosing instructions in order to maximize absorption and clinical benefit.
- Ibandronate sodium tablets should be taken at least 60 minutes before the first food or drink (other than water) of the day and before taking any oral medication or supplementation including calcium, antacids or vitamins (see Drug Interactions [7.1]).
- To facilitate delivery to the stomach, and thus reduce the potential for esophageal irritation, ibandronate sodium tablets should be swallowed whole with a full glass of plain water (6 to 8 oz) while the patient is standing or sitting in an upright position. Patients should not lie down for 60 minutes after taking ibandronate sodium tablets.
- Patients should not eat, drink anything except for water, or take other medications for 60 minutes after taking ibandronate sodium tablets.
- Plain water is the only drink that should be taken with ibandronate sodium tablets. Note that some mineral waters may have a higher concentration of calcium and therefore should not be used.
- Patients should not chew or suck the tablet because of a potential for oropharyngeal ulceration.
- The ibandronate sodium tablet, 150 mg (acid) should be taken on the same date each month (i.e., the patient’s ibandronate sodium day).
- The patient must not take two 150 mg tablets within the same week.
- If the once-monthly dose is missed, and the patient’s next scheduled ibandronate sodium tablets day is more than 7 days away, the patient should be instructed to take one ibandronate sodium tablet, 150 mg (acid) in the morning following the date that it is remembered (see Dosage and Administration [2.3]). The patient should then return to taking one ibandronate sodium tablet, 150 mg (acid) every month in the morning of their chosen day, according to their original schedule.
- If the once-monthly dose is missed, and the patient’s next scheduled ibandronate sodium tablets day is only 1 to 7 days away, the patient must wait until the subsequent month’s scheduled ibandronate sodium tablets day to take their tablet. The patient should then return to taking one ibandronate sodium tablet, 150 mg (acid) every month in the morning of their chosen day, according to their original schedule.
Patients should receive supplemental calcium and vitamin D if dietary intake is inadequate. Intake of supplemental calcium and vitamin D should be delayed for at least 60 minutes following oral administration of ibandronate sodium tablets in order to maximize absorption of ibandronate sodium tablets.
Physicians should be alert to signs or symptoms signaling a possible esophageal reaction during therapy, and patients should be instructed to discontinue ibandronate sodium tablets and seek medical attention if they develop symptoms of esophageal irritation such as new or worsening dysphagia, pain on swallowing, retrosternal pain, or heartburn.
-
MEDICATION GUIDE
Medication Guide
Ibandronate Sodium Tablets
Read the Medication Guide that comes with ibandronate sodium tablets before you start taking it and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking with your doctor about your medical condition or your treatment. Talk to your doctor if you have any questions about ibandronate sodium tablets.
What is the most important information I should know about ibandronate sodium tablets?
Ibandronate sodium tablets may cause serious side effects including:
1. Esophagus problems
2. Low calcium levels in your blood (hypocalcemia)
3. Bone, joint or muscle pain
4. Severe jaw bone problems (osteonecrosis)
5. Unusual thigh bone fractures
1. Esophagus problems.
Some people who take ibandronate sodium tablets may develop problems in the esophagus (the tube that connects the mouth and the stomach). These problems include irritation, inflammation, or ulcers of the esophagus which may sometimes bleed.
It is important that you take ibandronate sodium tablets exactly as prescribed to help lower your chance of getting esophagus problems (see the section “How should I take ibandronate sodium tablets?”).
Stop taking ibandronate sodium tablets and call your doctor right away if you get chest pain, new or worsening heartburn, or have trouble or pain when you swallow.
2. Low calcium levels in your blood (hypocalcemia).
Ibandronate sodium tablets may lower the calcium levels in your blood. If you have low blood calcium before you start taking ibandronate sodium tablets, it may get worse during treatment. Your low blood calcium must be treated before you take ibandronate sodium tablets. Most people with low blood calcium levels do not have symptoms, but some people may have symptoms. Call your doctor right away if you have symptoms of low blood calcium such as:
- Spasms, twitches, or cramps in your muscles
- Numbness or tingling in your fingers, toes, or around your mouth
Your doctor may prescribe calcium and vitamin D to help prevent low calcium levels in your blood, while you take ibandronate sodium tablets. Take calcium and vitamin D as your doctor tells you to. 3. Bone, joint, or muscle pain.
Some people who take ibandronate sodium tablets develop severe bone, joint, or muscle pain.
4. Severe jaw bone problems (osteonecrosis).
Severe jaw bone problems may happen when you take ibandronate sodium tablets. Your doctor may examine your mouth before you start ibandronate sodium tablets. Your doctor may tell you to see your dentist before you start ibandronate sodium tablets. It is important for you to practice good mouth care during treatment with ibandronate sodium tablets.
5. Unusual thigh bone fractures.
Some people have developed unusual fractures in their thigh bone. Symptoms of a fracture may include new or unusual pain in your hip, groin, or thigh.
Call your doctor right away if you have any of these side effects.
What is ibandronate sodium tablet?
Ibandronate sodium tablet is a prescription medicine used to treat or prevent osteoporosis in women after menopause. Ibandronate sodium tablets helps increase bone mass and helps reduce the chance of having a spinal fracture (break).
It is not known how long ibandronate sodium tablets works for the treatment and prevention of osteoporosis. You should see your doctor regularly to determine if ibandronate sodium tablet are still right for you.
It is not known if ibandronate sodium tablets are safe and effective in children.
Who should not take ibandronate sodium tablets?
Do not take ibandronate sodium tablets if you:
- Have certain problems with your esophagus, the tube that connects your mouth with your stomach
- Cannot stand or sit upright for at least 60 minutes
- Have low levels of calcium in your blood
- Are allergic to ibandronate sodium tablets or any of its ingredients. A list of ingredients is at the end of this leaflet
What should I tell my doctor before taking ibandronate sodium tablets?
Before you start ibandronate sodium tablets, be sure to talk to your doctor if you:
- Have problems with swallowing
- Have stomach or digestive problems
- Have low blood calcium
- Plan to have dental surgery or teeth removed
- Have kidney problems
- Have been told you have trouble absorbing minerals in your stomach or intestines (malabsorption syndrome)
- Are pregnant, or plan to become pregnant. It is not known if ibandronate sodium tablets can harm your unborn baby.
- Are breast-feeding or plan to breast-feed. It is not known if ibandronate sodium passes into your milk and may harm your baby.
Tell your doctor and dentist about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements. Certain medicines may affect how ibandronate sodium tablets works.
Especially tell your doctor if you take:
- antacids
- aspirin
- Nonsteroidal Anti-Inflammatory (NSAID) medicines
Know the medicines you take. Keep a list of them and show it to your doctor and pharmacist each time you get a new medicine.
How should I take ibandronate sodium tablets?
- Take ibandronate sodium tablets exactly as your doctor tells you.
- Take 1 ibandronate sodium tablet 150 mg 1 time every month on the same day each month.
- Ibandronate sodium tablets works only if taken on an empty stomach.
- Take 1 ibandronate sodium tablet, after you get up for the day and before taking your first food, drink, or other medicine.
- Take ibandronate sodium tablets while you are sitting or standing.
- Donot chew or suck on a tablet of ibandronate sodium
- Swallow ibandronate sodium tablet with a full glass (6 to 8 oz) of plain water only.
- Do not take ibandronate sodium tablets with mineral water, coffee, tea, soda, or juice.
After swallowing ibandronate sodium tablet, wait at least 60 minutes:
- Before you lie down. You may sit, stand or walk, and do normal activities like reading.
- Before you take your first food or drink except for plain water.
- Before you take other medicines, including antacids, calcium, and other supplements and vitamins.
Do not lie down for at least 60 minutes after you take ibandronate sodium tablets and do not eat your first food of the day for at least 60 minutes after you take ibandronate sodium tablets.
If you miss a dose of ibandronate sodium tablets, do not take it later in the day. Call your doctor for instructions.
If you take too much ibandronate sodium tablets, call your doctor. Do not try to vomit. Do not lie down.
What are the possible side effects of ibandronate sodium tablets?
Ibandronate sodium tablets may cause serious side effects.
See “What is the most important information I should know about ibandronate sodium tablets?”
The most common side effects of ibandronate sodium tablets are:
- Back pain
- Heartburn
- Stomach area (abdominal) pain
- Pain in your arms and legs
- Diarrhea
- Headache
- Muscle pain
- Flu-like symptoms
You may get allergic reactions, such as hives, breathing difficulties, swelling of your face, lips, tongue or throat, or feeling faint.
Tell your doctor if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of ibandronate sodium tablets. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects to Dr. Reddy's at 1-888-375-3784.
How do I store ibandronate sodium tablets?
- Store ibandronate sodium tablets at 20°-25°C (68°-77°F).
- Keep ibandronate sodium tablets in a tightly closed container.
Keep ibandronate sodium tablets and all medicines out of the reach of children.
General information about the safe and effective use of ibandronate sodium tablets.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use ibandronate sodium tablets for a condition for which it was not prescribed. Do not give ibandronate sodium tablets to other people, even if they have the same symptoms you have. It may harm them.
This Medication Guide summarizes the most important information about ibandronate sodium tablets. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about ibandronate sodium tablets that is written for health professionals.
What are the ingredients in ibandronate sodium tablets?
Active ingredient: ibandronate sodium
Inactive ingredients: colloidal silicon dioxide, crospovidone, croscarmellose sodium, lactose monohydrate, microcrystalline cellulose, povidone, and sodium stearyl fumarate. The tablet coating contains hypromellose, polyethylene glycol 400, and titanium dioxide. Imprinting ink contains: ammonium hydroxide, black iron oxide, propylene glycol and shellac.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
RX Only
Manufactured by:
Dr. Reddy’s Laboratories Limited
Bachupally - 500 090 INDIA
Revised: 0818
Dispense with medication guide available at:
www.drreddys.com/medguide/ibandronatetabs.pdf
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL SECTION
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
IBANDRONATE SODIUM
ibandronate sodium tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 55111-575 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ibandronate sodium (UNII: J12U072QL0) (IBANDRONIC ACID - UNII:UMD7G2653W) IBANDRONIC ACID 150 mg Inactive Ingredients Ingredient Name Strength Silicon Dioxide (UNII: ETJ7Z6XBU4) Crospovidone, Unspecified (UNII: 2S7830E561) croscarmellose sodium (UNII: M28OL1HH48) lactose monohydrate (UNII: EWQ57Q8I5X) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POVIDONE (UNII: FZ989GH94E) sodium stearyl fumarate (UNII: 7CV7WJK4UI) HYPROMELLOSES (UNII: 3NXW29V3WO) polyethylene glycol 400 (UNII: B697894SGQ) titanium dioxide (UNII: 15FIX9V2JP) AMMONIA (UNII: 5138Q19F1X) FERROSOFERRIC OXIDE (UNII: XM0M87F357) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) shellac (UNII: 46N107B71O) Product Characteristics Color WHITE Score no score Shape CAPSULE Size 13mm Flavor Imprint Code R;575 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55111-575-03 3 in 1 CARTON 06/21/2012 1 NDC: 55111-575-11 1 in 1 BLISTER PACK; Type 0: Not a Combination Product 2 NDC: 55111-575-43 1 in 1 CARTON 12/30/2018 2 NDC: 55111-575-13 3 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA078997 06/21/2012 Labeler - Dr. Reddy's Laboratories Limited (650562841) Establishment Name Address ID/FEI Business Operations Dr. Reddy's Laboratories Limited - FTO III 918608162 analysis(55111-575) , manufacture(55111-575)




© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.