EYLEA- aflibercept injection, solution
EYLEA by
Drug Labeling and Warnings
EYLEA by is a Prescription medication manufactured, distributed, or labeled by Regeneron Pharmaceuticals, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use EYLEA safely and effectively. See full prescribing information for EYLEA.
EYLEA® (aflibercept) Injection, for Intravitreal Use
Initial U.S. Approval: 2011INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
-
Neovascular (Wet) Age-Related Macular Degeneration (AMD)
- The recommended dose for EYLEA is 2 mg (0.05 mL) administered by intravitreal injection every 4 weeks (approximately every 28 days, monthly) for the first 3 months, followed by 2 mg (0.05 mL) via intravitreal injection once every 8 weeks (2 months). (2.2)
- Although EYLEA may be dosed as frequently as 2 mg every 4 weeks (approximately every 25 days, monthly), additional efficacy was not demonstrated in most patients when EYLEA was dosed every 4 weeks compared to every 8 weeks. Some patients may need every 4 week (monthly) dosing after the first 12 weeks (3 months). (2.2)
- Although not as effective as the recommended every 8 week dosing regimen, patients may also be treated with one dose every 12 weeks after one year of effective therapy. Patients should be assessed regularly. (2.2)
-
Macular Edema Following Retinal Vein Occlusion (RVO)
- The recommended dose for EYLEA is 2 mg (0.05 mL) administered by intravitreal injection once every 4 weeks (approximately every 25 days, monthly). (2.3)
-
Diabetic Macular Edema (DME) and Diabetic Retinopathy (DR)
- The recommended dose for EYLEA is 2 mg (0.05 mL) administered by intravitreal injection every 4 weeks (approximately every 28 days, monthly) for the first 5 injections followed by 2 mg (0.05 mL) via intravitreal injection once every 8 weeks (2 months). (2.4, 2.5)
- Although EYLEA may be dosed as frequently as 2 mg every 4 weeks (approximately every 25 days, monthly), additional efficacy was not demonstrated in most patients when EYLEA was dosed every 4 weeks compared to every 8 weeks. Some patients may need every 4 week (monthly) dosing after the first 20 weeks (5 months). (2.4, 2.5)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Endophthalmitis and retinal detachments may occur following intravitreal injections. Patients should be instructed to report any symptoms suggestive of endophthalmitis or retinal detachment without delay and should be managed appropriately. (5.1)
- Increases in intraocular pressure have been seen within 60 minutes of an intravitreal injection. (5.2)
- There is a potential risk of arterial thromboembolic events following intravitreal use of VEGF inhibitors. (5.3)
ADVERSE REACTIONS
The most common adverse reactions (≥5%) reported in patients receiving EYLEA were conjunctival hemorrhage, eye pain, cataract, vitreous detachment, vitreous floaters, and intraocular pressure increased. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Regeneron at 1-855-395-3248 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 8/2019
-
Neovascular (Wet) Age-Related Macular Degeneration (AMD)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Neovascular (Wet) Age-Related Macular Degeneration (AMD)
1.2 Macular Edema Following Retinal Vein Occlusion (RVO)
1.3 Diabetic Macular Edema (DME)
1.4 Diabetic Retinopathy (DR)
2 DOSAGE AND ADMINISTRATION
2.1 Important Injection Instructions
2.2 Neovascular (Wet) Age-Related Macular Degeneration (AMD)
2.3 Macular Edema Following Retinal Vein Occlusion (RVO)
2.4 Diabetic Macular Edema (DME)
2.5 Diabetic Retinopathy (DR)
2.6 Preparation for Administration - Pre-filled Syringe
2.7 Preparation for Administration - Vial
2.8 Injection Procedure
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Ocular or Periocular Infections
4.2 Active Intraocular Inflammation
4.3 Hypersensitivity
5 WARNINGS AND PRECAUTIONS
5.1 Endophthalmitis and Retinal Detachments
5.2 Increase in Intraocular Pressure
5.3 Thromboembolic Events
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Neovascular (Wet) Age-Related Macular Degeneration (AMD)
14.2 Macular Edema Following Central Retinal Vein Occlusion (CRVO)
14.3 Macular Edema Following Branch Retinal Vein Occlusion (BRVO)
14.4 Diabetic Macular Edema (DME)
14.5 Diabetic Retinopathy (DR)
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Injection Instructions
For ophthalmic intravitreal injection. EYLEA must only be administered by a qualified physician.
Pre-filled Syringe: A 30-gauge × ½-inch sterile injection needle is needed but not provided.
2.2 Neovascular (Wet) Age-Related Macular Degeneration (AMD)
The recommended dose for EYLEA is 2 mg (0.05 mL or 50 microliters) administered by intravitreal injection every 4 weeks (approximately every 28 days, monthly) for the first 12 weeks (3 months), followed by 2 mg (0.05 mL) via intravitreal injection once every 8 weeks (2 months). Although EYLEA may be dosed as frequently as 2 mg every 4 weeks (approximately every 25 days, monthly), additional efficacy was not demonstrated in most patients when EYLEA was dosed every 4 weeks compared to every 8 weeks [see Clinical Studies (14.1)]. Some patients may need every 4 week (monthly) dosing after the first 12 weeks (3 months). Although not as effective as the recommended every 8 week dosing regimen, patients may also be treated with one dose every 12 weeks after one year of effective therapy. Patients should be assessed regularly.
2.3 Macular Edema Following Retinal Vein Occlusion (RVO)
The recommended dose for EYLEA is 2 mg (0.05 mL or 50 microliters) administered by intravitreal injection once every 4 weeks (approximately every 25 days, monthly) [see Clinical Studies (14.2), (14.3)].
2.4 Diabetic Macular Edema (DME)
The recommended dose for EYLEA is 2 mg (0.05 mL or 50 microliters) administered by intravitreal injection every 4 weeks (approximately every 28 days, monthly) for the first 5 injections, followed by 2 mg (0.05 mL) via intravitreal injection once every 8 weeks (2 months). Although EYLEA may be dosed as frequently as 2 mg every 4 weeks (approximately every 25 days, monthly), additional efficacy was not demonstrated in most patients when EYLEA was dosed every 4 weeks compared to every 8 weeks [see Clinical Studies (14.4)]. Some patients may need every 4 week (monthly) dosing after the first 20 weeks (5 months).
2.5 Diabetic Retinopathy (DR)
The recommended dose for EYLEA is 2 mg (0.05 mL or 50 microliters) administered by intravitreal injection every 4 weeks (approximately every 28 days, monthly) for the first 5 injections, followed by 2 mg (0.05 mL) via intravitreal injection once every 8 weeks (2 months). Although EYLEA may be dosed as frequently as 2 mg every 4 weeks (approximately every 25 days, monthly), additional efficacy was not demonstrated in most patients when EYLEA was dosed every 4 weeks compared to every 8 weeks [see Clinical Studies (14.5)]. Some patients may need every 4 week (monthly) dosing after the first 20 weeks (5 months).
2.6 Preparation for Administration - Pre-filled Syringe
The EYLEA pre-filled glass syringe is sterile and for single use only. It should be inspected visually prior to administration. Do not use if particulates, cloudiness, or discoloration are visible, or if the package is open or damaged.
The intravitreal injection should be performed with a 30-gauge × ½-inch injection needle (not provided).
PRE-FILLED SYRINGE DESCRIPTION – Figure 1:
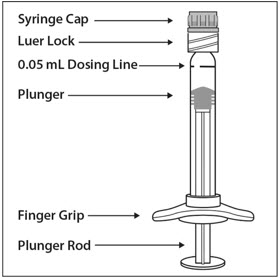
Use aseptic technique to carry out the following steps:
1. PREPARE
When ready to administer EYLEA, open the carton and remove sterilized blister pack. Carefully peel open the sterilized blister pack ensuring the sterility of its contents. Keep the syringe in the sterile tray until you are ready for assembly.
2. REMOVE SYRINGE
Using aseptic technique, remove the syringe from the sterilized blister pack.
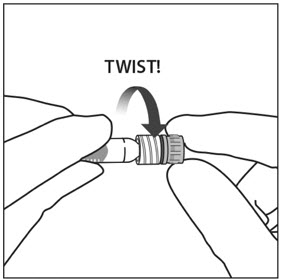
3. TWIST OFF SYRINGE CAP
Twist off the syringe cap by holding the syringe in one hand and the syringe cap with the thumb and forefinger of the other hand (see Figure 2).
Note: To avoid compromising the sterility of the product, do not pull back on the plunger.
4. ATTACH NEEDLE
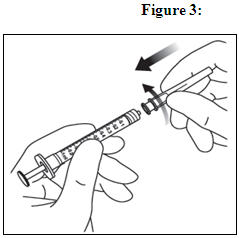
Using aseptic technique, firmly twist a 30-gauge × ½-inch injection needle onto the Luer lock syringe tip (see Figure 3).
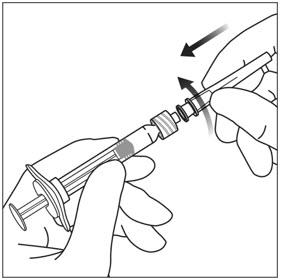
Note: When ready to administer EYLEA, remove the plastic needle shield from the needle.
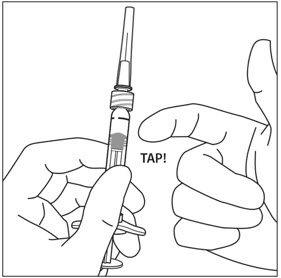
5. DISLODGE AIR BUBBLES
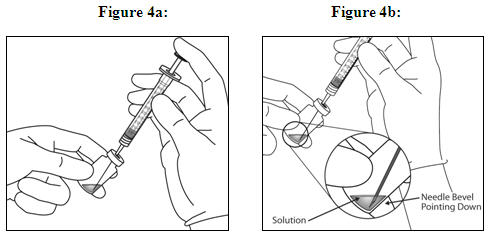
Holding the syringe with the needle pointing up, check the syringe for bubbles. If there are bubbles, gently tap the syringe with your finger until the bubbles rise to the top (see Figure 4).
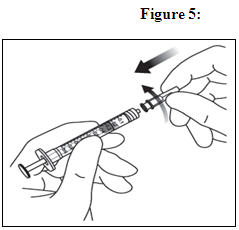
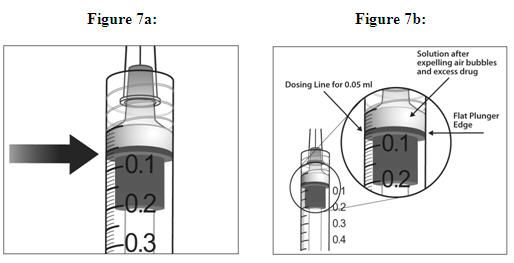
6. EXPEL AIR AND SET THE DOSE
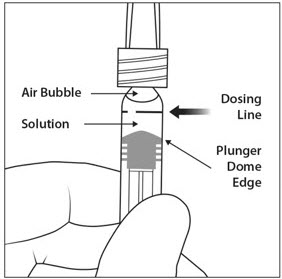
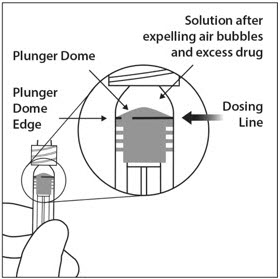
To eliminate all bubbles and to expel excess drug, slowly depress the plunger rod to align the plunger dome edge (see Figure 5a) with the black dosing line on the syringe (equivalent to 50 microliters) (see Figure 5b).
7. The pre-filled syringe is for single use only. After injection any unused product must be discarded.
2.7 Preparation for Administration - Vial
EYLEA should be inspected visually prior to administration. If particulates, cloudiness, or discoloration are visible, the vial must not be used.
The glass vial is for single use only.
Use aseptic technique to carry out the following preparation steps:
Prepare for intravitreal injection with the following medical devices for single use:
- a 5-micron sterile filter needle (19-gauge × 1½-inch)
- a 1-mL sterile Luer lock syringe (with marking to measure 0.05 mL)
- a sterile injection needle (30-gauge × ½-inch)
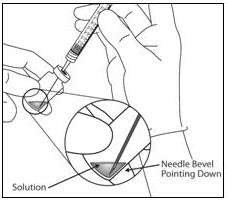
- Remove the protective plastic cap from the vial (see Figure 6).
Figure 6:
- Clean the top of the vial with an alcohol wipe (see Figure 7).
Figure 7:
-
Remove the 19-gauge × 1½-inch, 5-micron, filter needle and the 1-mL syringe from their packaging. Attach the filter needle to the syringe by twisting it onto the Luer lock syringe tip (see Figure 8).
- Push the filter needle into the center of the vial stopper until the needle is completely inserted into the vial and the tip touches the bottom or bottom edge of the vial.
- Using aseptic technique withdraw all of the EYLEA vial contents into the syringe, keeping the vial in an upright position, slightly inclined to ease complete withdrawal. To deter the introduction of air, ensure the bevel of the filter needle is submerged into the liquid. Continue to tilt the vial during withdrawal keeping the bevel of the filter needle submerged in the liquid (see Figure 9a and Figure 9b).
- Ensure that the plunger rod is drawn sufficiently back when emptying the vial in order to completely empty the filter needle.
- Remove the filter needle from the syringe and properly dispose of the filter needle. Note: Filter needle is not to be used for intravitreal injection.
- Remove the 30-gauge × ½-inch injection needle from its packaging and attach the injection needle to the syringe by firmly twisting the injection needle onto the Luer lock syringe tip (see Figure 10).
- When ready to administer EYLEA, remove the plastic needle shield from the needle.
- Holding the syringe with the needle pointing up, check the syringe for bubbles. If there are bubbles, gently tap the syringe with your finger until the bubbles rise to the top (see Figure 11).
Figure 11:
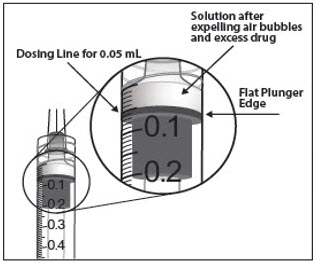
- To eliminate all of the bubbles and to expel excess drug, SLOWLY depress the plunger so that the plunger tip aligns with the line that marks 0.05 mL on the syringe (see Figure 12a and Figure 12b).
2.8 Injection Procedure
The intravitreal injection procedure should be carried out under controlled aseptic conditions, which include surgical hand disinfection and the use of sterile gloves, a sterile drape, and a sterile eyelid speculum (or equivalent). Adequate anesthesia and a topical broad–spectrum microbicide should be given prior to the injection.
Immediately following the intravitreal injection, patients should be monitored for elevation in intraocular pressure. Appropriate monitoring may consist of a check for perfusion of the optic nerve head or tonometry. If required, a sterile paracentesis needle should be available.
Following intravitreal injection, patients should be instructed to report any symptoms suggestive of endophthalmitis or retinal detachment (e.g., eye pain, redness of the eye, photophobia, blurring of vision) without delay [see Patient Counseling Information (17)].
Each sterile, pre-filled syringe or vial should only be used for the treatment of a single eye. If the contralateral eye requires treatment, a new sterile, pre-filled syringe or vial should be used and the sterile field, syringe, gloves, drapes, eyelid speculum, filter, and injection needles should be changed before EYLEA is administered to the other eye.
After injection, any unused product must be discarded.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Endophthalmitis and Retinal Detachments
Intravitreal injections, including those with EYLEA, have been associated with endophthalmitis and retinal detachments [see Adverse Reactions (6.1)]. Proper aseptic injection technique must always be used when administering EYLEA. Patients should be instructed to report any symptoms suggestive of endophthalmitis or retinal detachment without delay and should be managed appropriately [see Dosage and Administration (2.8) and Patient Counseling Information (17)].
5.2 Increase in Intraocular Pressure
Acute increases in intraocular pressure have been seen within 60 minutes of intravitreal injection, including with EYLEA [see Adverse Reactions (6.1)]. Sustained increases in intraocular pressure have also been reported after repeated intravitreal dosing with vascular endothelial growth factor (VEGF) inhibitors. Intraocular pressure and the perfusion of the optic nerve head should be monitored and managed appropriately [see Dosage and Administration (2.8)].
5.3 Thromboembolic Events
There is a potential risk of arterial thromboembolic events (ATEs) following intravitreal use of VEGF inhibitors, including EYLEA. ATEs are defined as nonfatal stroke, nonfatal myocardial infarction, or vascular death (including deaths of unknown cause). The incidence of reported thromboembolic events in wet AMD studies during the first year was 1.8% (32 out of 1824) in the combined group of patients treated with EYLEA compared with 1.5% (9 out of 595) in patients treated with ranibizumab; through 96 weeks, the incidence was 3.3% (60 out of 1824) in the EYLEA group compared with 3.2% (19 out of 595) in the ranibizumab group. The incidence in the DME studies from baseline to week 52 was 3.3% (19 out of 578) in the combined group of patients treated with EYLEA compared with 2.8% (8 out of 287) in the control group; from baseline to week 100, the incidence was 6.4% (37 out of 578) in the combined group of patients treated with EYLEA compared with 4.2% (12 out of 287) in the control group. There were no reported thromboembolic events in the patients treated with EYLEA in the first six months of the RVO studies.
-
6 ADVERSE REACTIONS
The following potentially serious adverse reactions are described elsewhere in the labeling:
- Hypersensitivity [see Contraindications (4.3)]
- Endophthalmitis and retinal detachments [see Warnings and Precautions (5.1)]
- Increase in intraocular pressure [see Warnings and Precautions (5.2)]
- Thromboembolic events [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in other clinical trials of the same or another drug and may not reflect the rates observed in practice.
A total of 2980 patients treated with EYLEA constituted the safety population in eight phase 3 studies. Among those, 2379 patients were treated with the recommended dose of 2 mg. Serious adverse reactions related to the injection procedure have occurred in <0.1% of intravitreal injections with EYLEA including endophthalmitis and retinal detachment. The most common adverse reactions (≥5%) reported in patients receiving EYLEA were conjunctival hemorrhage, eye pain, cataract, vitreous detachment, vitreous floaters, and intraocular pressure increased.
Neovascular (Wet) Age-Related Macular Degeneration (AMD)
The data described below reflect exposure to EYLEA in 1824 patients with wet AMD, including 1223 patients treated with the 2-mg dose, in 2 double-masked, controlled clinical studies (VIEW1 and VIEW2) for 24 months (with active control in year 1) [see Clinical Studies (14.1)].
Safety data observed in the EYLEA group in a 52-week, double-masked, Phase 2 study were consistent with these results.
Table 1: Most Common Adverse Reactions (≥1%) in Wet AMD Studies Adverse Reactions Baseline to Week 52 Baseline to Week 96 EYLEA
(N=1824)Active Control (ranibizumab)
(N=595)EYLEA
(N=1824)Control (ranibizumab)
(N=595)Conjunctival hemorrhage 25% 28% 27% 30% Eye pain 9% 9% 10% 10% Cataract 7% 7% 13% 10% Vitreous detachment 6% 6% 8% 8% Vitreous floaters 6% 7% 8% 10% Intraocular pressure increased 5% 7% 7% 11% Ocular hyperemia 4% 8% 5% 10% Corneal epithelium defect 4% 5% 5% 6% Detachment of the retinal pigment epithelium 3% 3% 5% 5% Injection site pain 3% 3% 3% 4% Foreign body sensation in eyes 3% 4% 4% 4% Lacrimation increased 3% 1% 4% 2% Vision blurred 2% 2% 4% 3% Intraocular inflammation 2% 3% 3% 4% Retinal pigment epithelium tear 2% 1% 2% 2% Injection site hemorrhage 1% 2% 2% 2% Eyelid edema 1% 2% 2% 3% Corneal edema 1% 1% 1% 1% Retinal detachment <1% <1% 1% 1% Less common serious adverse reactions reported in <1% of the patients treated with EYLEA were hypersensitivity, retinal tear, and endophthalmitis.
Macular Edema Following Retinal Vein Occlusion (RVO)
The data described below reflect 6 months exposure to EYLEA with a monthly 2 mg dose in 218 patients following central retinal vein occlusion (CRVO) in 2 clinical studies (COPERNICUS and GALILEO) and 91 patients following branch retinal vein occlusion (BRVO) in one clinical study (VIBRANT) [see Clinical Studies (14.2), (14.3)].
Table 2: Most Common Adverse Reactions (≥1%) in RVO Studies Adverse Reactions CRVO BRVO EYLEA
(N=218)Control
(N=142)EYLEA
(N=91)Control
(N=92)Eye pain 13% 5% 4% 5% Conjunctival hemorrhage 12% 11% 20% 4% Intraocular pressure increased 8% 6% 2% 0% Corneal epithelium defect 5% 4% 2% 0% Vitreous floaters 5% 1% 1% 0% Ocular hyperemia 5% 3% 2% 2% Foreign body sensation in eyes 3% 5% 3% 0% Vitreous detachment 3% 4% 2% 0% Lacrimation increased 3% 4% 3% 0% Injection site pain 3% 1% 1% 0% Vision blurred 1% <1% 1% 1% Intraocular inflammation 1% 1% 0% 0% Cataract <1% 1% 5% 0% Eyelid edema <1% 1% 1% 0% Less common adverse reactions reported in <1% of the patients treated with EYLEA in the CRVO studies were corneal edema, retinal tear, hypersensitivity, and endophthalmitis.
Diabetic Macular Edema (DME) and Diabetic Retinopathy (DR)
The data described below reflect exposure to EYLEA in 578 patients with DME treated with the 2-mg dose in 2 double-masked, controlled clinical studies (VIVID and VISTA) from baseline to week 52 and from baseline to week 100 [see Clinical Studies (14.4)].
Table 3: Most Common Adverse Reactions (≥1%) in DME Studies Adverse Reactions Baseline to Week 52 Baseline to Week 100 EYLEA
(N=578)Control
(N=287)EYLEA
(N=578)Control
(N=287)Conjunctival hemorrhage 28% 17% 31% 21% Eye pain 9% 6% 11% 9% Cataract 8% 9% 19% 17% Vitreous floaters 6% 3% 8% 6% Corneal epithelium defect 5% 3% 7% 5% Intraocular pressure increased 5% 3% 9% 5% Ocular hyperemia 5% 6% 5% 6% Vitreous detachment 3% 3% 8% 6% Foreign body sensation in eyes 3% 3% 3% 3% Lacrimation increased 3% 2% 4% 2% Vision blurred 2% 2% 3% 4% Intraocular inflammation 2% <1% 3% 1% Injection site pain 2% <1% 2% <1% Eyelid edema <1% 1% 2% 1% Less common adverse reactions reported in <1% of the patients treated with EYLEA were hypersensitivity, retinal detachment, retinal tear, corneal edema, and injection site hemorrhage.
Safety data observed in 269 patients with nonproliferative diabetic retinopathy (NPDR) through week 52 in the PANORAMA trial were consistent with those seen in the phase 3 VIVID and VISTA trials (see Table 3 above).
6.2 Immunogenicity
As with all therapeutic proteins, there is a potential for an immune response in patients treated with EYLEA. The immunogenicity of EYLEA was evaluated in serum samples. The immunogenicity data reflect the percentage of patients whose test results were considered positive for antibodies to EYLEA in immunoassays. The detection of an immune response is highly dependent on the sensitivity and specificity of the assays used, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to EYLEA with the incidence of antibodies to other products may be misleading.
In the wet AMD, RVO, and DME studies, the pre-treatment incidence of immunoreactivity to EYLEA was approximately 1% to 3% across treatment groups. After dosing with EYLEA for 24-100 weeks, antibodies to EYLEA were detected in a similar percentage range of patients. There were no differences in efficacy or safety between patients with or without immunoreactivity.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Adequate and well-controlled studies with EYLEA have not been conducted in pregnant women. Aflibercept produced adverse embryofetal effects in rabbits, including external, visceral, and skeletal malformations. A fetal No Observed Adverse Effect Level (NOAEL) was not identified. At the lowest dose shown to produce adverse embryofetal effects, systemic exposures (based on AUC for free aflibercept) were approximately 6 times higher than AUC values observed in humans after a single intravitreal treatment at the recommended clinical dose [see Animal Data].
Animal reproduction studies are not always predictive of human response, and it is not known whether EYLEA can cause fetal harm when administered to a pregnant woman. Based on the anti-VEGF mechanism of action for aflibercept [see Clinical Pharmacology (12.1)], treatment with EYLEA may pose a risk to human embryofetal development. EYLEA should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
In two embryofetal development studies, aflibercept produced adverse embryofetal effects when administered every three days during organogenesis to pregnant rabbits at intravenous doses ≥3 mg per kg, or every six days during organogenesis at subcutaneous doses ≥0.1 mg per kg.
Adverse embryofetal effects included increased incidences of postimplantation loss and fetal malformations, including anasarca, umbilical hernia, diaphragmatic hernia, gastroschisis, cleft palate, ectrodactyly, intestinal atresia, spina bifida, encephalomeningocele, heart and major vessel defects, and skeletal malformations (fused vertebrae, sternebrae, and ribs; supernumerary vertebral arches and ribs; and incomplete ossification). The maternal No Observed Adverse Effect Level (NOAEL) in these studies was 3 mg per kg. Aflibercept produced fetal malformations at all doses assessed in rabbits and the fetal NOAEL was not identified. At the lowest dose shown to produce adverse embryofetal effects in rabbits (0.1 mg per kg), systemic exposure (AUC) of free aflibercept was approximately 6 times higher than systemic exposure (AUC) observed in humans after a single intravitreal dose of 2 mg.
8.2 Lactation
Risk Summary
There is no information regarding the presence of aflibercept in human milk, the effects of the drug on the breastfed infant, or the effects of the drug on milk production/excretion. Because many drugs are excreted in human milk, and because the potential for absorption and harm to infant growth and development exists, EYLEA is not recommended during breastfeeding.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for EYLEA and any potential adverse effects on the breastfed child from EYLEA.
8.3 Females and Males of Reproductive Potential
Contraception
Females of reproductive potential are advised to use effective contraception prior to the initial dose, during treatment, and for at least 3 months after the last intravitreal injection of EYLEA.
Infertility
There are no data regarding the effects of EYLEA on human fertility. Aflibercept adversely affected female and male reproductive systems in cynomolgus monkeys when administered by intravenous injection at a dose approximately 1500 times higher than the systemic level observed humans with an intravitreal dose of 2 mg. A No Observed Adverse Effect Level (NOAEL) was not identified. These findings were reversible within 20 weeks after cessation of treatment [see Nonclinical Toxicology (13.1)].
-
11 DESCRIPTION
Aflibercept is a recombinant fusion protein consisting of portions of human VEGF receptors 1 and 2 extracellular domains fused to the Fc portion of human IgG1 formulated as an iso-osmotic solution for intravitreal administration. Aflibercept is a dimeric glycoprotein with a protein molecular weight of 97 kilodaltons (kDa) and contains glycosylation, constituting an additional 15% of the total molecular mass, resulting in a total molecular weight of 115 kDa. Aflibercept is produced in recombinant Chinese hamster ovary (CHO) cells.
EYLEA (aflibercept) Injection is a sterile, clear, and colorless to pale yellow solution. EYLEA is supplied as a preservative-free, sterile, aqueous solution for intravitreal injection in a single-dose pre-filled glass syringe or a single-dose glass vial designed to deliver 0.05 mL (50 microliters) of solution containing 2 mg of aflibercept in 10 mM sodium phosphate, 40 mM sodium chloride, 0.03% polysorbate 20, and 5% sucrose, with a pH of 6.2.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Vascular endothelial growth factor-A (VEGF-A) and placental growth factor (PlGF) are members of the VEGF family of angiogenic factors that can act as mitogenic, chemotactic, and vascular permeability factors for endothelial cells. VEGF acts via two receptor tyrosine kinases, VEGFR-1 and VEGFR-2, present on the surface of endothelial cells. PlGF binds only to VEGFR-1, which is also present on the surface of leucocytes. Activation of these receptors by VEGF-A can result in neovascularization and vascular permeability.
Aflibercept acts as a soluble decoy receptor that binds VEGF-A and PlGF, and thereby can inhibit the binding and activation of these cognate VEGF receptors.
12.2 Pharmacodynamics
Neovascular (Wet) Age-Related Macular Degeneration (AMD)
In the clinical studies anatomic measures of disease activity improved similarly in all treatment groups from baseline to week 52. Anatomic data were not used to influence treatment decisions during the first year.
Macular Edema Following Retinal Vein Occlusion (RVO)
Reductions in mean retinal thickness were observed in COPERNICUS, GALILEO, and VIBRANT at week 24 compared to baseline. Anatomic data were not used to influence treatment decisions [see Clinical Studies (14.2), (14.3)].
Diabetic Macular Edema (DME)
Reductions in mean retinal thickness were observed in VIVID and VISTA at weeks 52 and 100 compared to baseline. Anatomic data were not used to influence EYLEA treatment decisions [see Clinical Studies (14.4)].
12.3 Pharmacokinetics
EYLEA is administered intravitreally to exert local effects in the eye. In patients with wet AMD, RVO, or DME, following intravitreal administration of EYLEA, a fraction of the administered dose is expected to bind with endogenous VEGF in the eye to form an inactive aflibercept: VEGF complex. Once absorbed into the systemic circulation, aflibercept presents in the plasma as free aflibercept (unbound to VEGF) and a more predominant stable inactive form with circulating endogenous VEGF (i.e., aflibercept: VEGF complex).
Absorption/Distribution
Following intravitreal administration of 2 mg per eye of EYLEA to patients with wet AMD, RVO, and DME, the mean Cmax of free aflibercept in the plasma was 0.02 mcg/mL (range: 0 to 0.054 mcg/mL), 0.05 mcg/mL (range: 0 to 0.081 mcg/mL), and 0.03 mcg/mL (range: 0 to 0.076 mcg/mL), respectively and was attained in 1 to 3 days. The free aflibercept plasma concentrations were undetectable two weeks post-dosing in all patients. Aflibercept did not accumulate in plasma when administered as repeated doses intravitreally every 4 weeks. It is estimated that after intravitreal administration of 2 mg to patients, the mean maximum plasma concentration of free aflibercept is more than 100 fold lower than the concentration of aflibercept required to half-maximally bind systemic VEGF.
The volume of distribution of free aflibercept following intravenous (I.V.) administration of aflibercept has been determined to be approximately 6L.
Metabolism/Elimination
Aflibercept is a therapeutic protein and no drug metabolism studies have been conducted. Aflibercept is expected to undergo elimination through both target-mediated disposition via binding to free endogenous VEGF and metabolism via proteolysis. The terminal elimination half-life (t1/2) of free aflibercept in plasma was approximately 5 to 6 days after I.V. administration of doses of 2 to 4 mg/kg aflibercept.
Specific Populations
Renal Impairment
Pharmacokinetic analysis of a subgroup of patients (n=492) in one wet AMD study, of which 43% had renal impairment (mild n=120, moderate n=74, and severe n=16), revealed no differences with respect to plasma concentrations of free aflibercept after intravitreal administration every 4 or 8 weeks. Similar results were seen in patients in a RVO study and in patients in a DME study. No dose adjustment based on renal impairment status is needed for either wet AMD, RVO, or DME patients.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No studies have been conducted on the mutagenic or carcinogenic potential of aflibercept. Effects on male and female fertility were assessed as part of a 6-month study in monkeys with intravenous administration of aflibercept at weekly doses ranging from 3 to 30 mg per kg. Absent or irregular menses associated with alterations in female reproductive hormone levels and changes in sperm morphology and motility were observed at all dose levels. In addition, females showed decreased ovarian and uterine weight accompanied by compromised luteal development and reduction of maturing follicles. These changes correlated with uterine and vaginal atrophy. A No Observed Adverse Effect Level (NOAEL) was not identified. Intravenous administration of the lowest dose of aflibercept assessed in monkeys (3 mg per kg) resulted in systemic exposure (AUC) for free aflibercept that was approximately 1500 times higher than the systemic exposure observed in humans after an intravitreal dose of 2 mg. All changes were reversible within 20 weeks after cessation of treatment.
13.2 Animal Toxicology and/or Pharmacology
Erosions and ulcerations of the respiratory epithelium in nasal turbinates in monkeys treated with aflibercept intravitreally were observed at intravitreal doses of 2 or 4 mg per eye. At the NOAEL of 0.5 mg per eye in monkeys, the systemic exposure (AUC) was 56 times higher than the exposure observed in humans after an intravitreal dose of 2 mg. Similar effects were not seen in clinical studies [see Clinical Studies (14)].
-
14 CLINICAL STUDIES
14.1 Neovascular (Wet) Age-Related Macular Degeneration (AMD)
The safety and efficacy of EYLEA were assessed in two randomized, multi-center, double-masked, active-controlled studies in patients with wet AMD. A total of 2412 patients were treated and evaluable for efficacy (1817 with EYLEA) in the two studies (VIEW1 and VIEW2). In each study, up to week 52, patients were randomly assigned in a 1:1:1:1 ratio to 1 of 4 dosing regimens: 1) EYLEA administered 2 mg every 8 weeks following 3 initial monthly doses (EYLEA 2Q8); 2) EYLEA administered 2 mg every 4 weeks (EYLEA 2Q4); 3) EYLEA 0.5 mg administered every 4 weeks (EYLEA 0.5Q4); and 4) ranibizumab administered 0.5 mg every 4 weeks (ranibizumab 0.5 mg Q4). Protocol-specified visits occurred every 28±3 days. Patient ages ranged from 49 to 99 years with a mean of 76 years.
In both studies, the primary efficacy endpoint was the proportion of patients who maintained vision, defined as losing fewer than 15 letters of visual acuity at week 52 compared to baseline. Both EYLEA 2Q8 and EYLEA 2Q4 groups were shown to have efficacy that was clinically equivalent to the ranibizumab 0.5 mg Q4 group in year 1.
Detailed results from the analysis of the VIEW1 and VIEW2 studies are shown in Table 4 and Figure 13 below.
Table 4: Efficacy Outcomes at Week 52 (Full Analysis Set with LOCF) in VIEW1 and VIEW2 Studies VIEW1 VIEW2 EYLEA
2 mg Q8 weeks *EYLEA
2 mg Q4 weeksranibizumab
0.5 mg Q4 weeksEYLEA
2 mg Q8 weeks *EYLEA
2 mg Q4 weeksranibizumab
0.5 mg Q4 weeksFull Analysis Set N=301 N=304 N=304 N=306 N=309 N=291 BCVA = Best Corrected Visual Acuity; CI = Confidence Interval; ETDRS = Early Treatment Diabetic Retinopathy Study; LOCF = Last Observation Carried Forward (baseline values are not carried forward); 95.1% confidence intervals were presented to adjust for safety assessment conducted during the study - * After treatment initiation with 3 monthly doses
- † EYLEA group minus the ranibizumab group
Efficacy Outcomes Proportion of patients who maintained visual acuity (%)
(<15 letters of BCVA loss)94% 95% 94% 95% 95% 95% Difference† (%)
(95.1% CI)0.6
(-3.2, 4.4)1.3
(-2.4, 5.0)0.6
(-2.9, 4.0)-0.3
(-4.0, 3.3)Mean change in BCVA as measured by ETDRS letter score from Baseline 7.9 10.9 8.1 8.9 7.6 9.4 Difference† in LS mean
(95.1% CI)0.3
(-2.0, 2.5)3.2
(0.9, 5.4)-0.9
(-3.1, 1.3)-2.0
(-4.1, 0.2)Number of patients who gained at least 15 letters of vision from Baseline (%) 92
(31%)114
(38%)94
(31%)96
(31%)91
(29%)99
(34%)Difference† (%)
(95.1% CI)-0.4
(-7.7, 7.0)6.6
(-1.0, 14.1)-2.6
(-10.2, 4.9)-4.6
(-12.1, 2.9)Treatment effects in evaluable subgroups (e.g., age, gender, race, baseline visual acuity) in each study were in general consistent with the results in the overall populations.
- * Patient dosing schedules were individualized from weeks 52 to 96 using a modified 12-week dosing regimen.
Figure 13: Mean Change in Visual Acuity from Baseline to Week 96* in VIEW1 and VIEW2 Studies 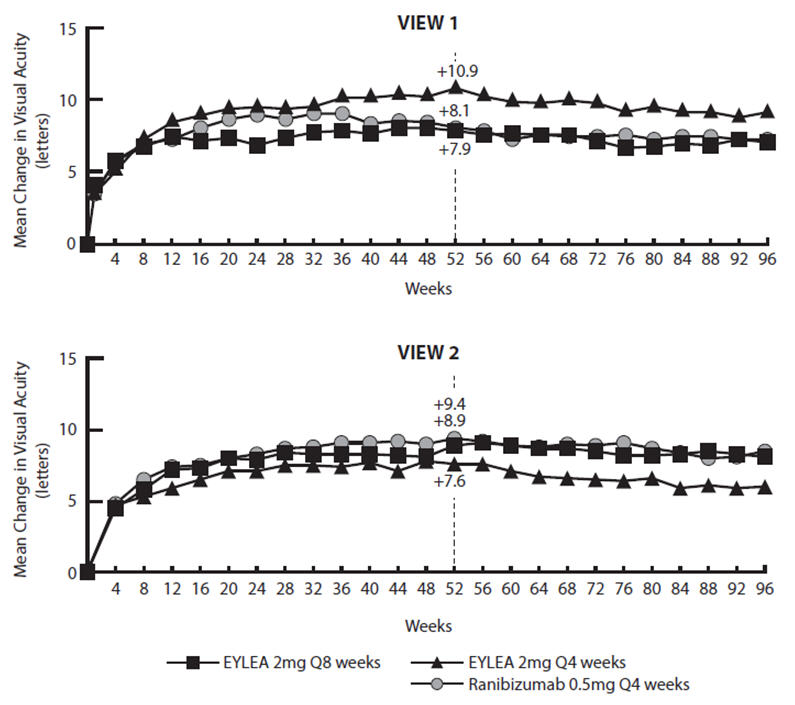
VIEW1 and VIEW2 studies were both 96 weeks in duration. However after 52 weeks patients no longer followed a fixed dosing schedule. Between week 52 and week 96, patients continued to receive the drug and dosage strength to which they were initially randomized on a modified 12 week dosing schedule (doses at least every 12 weeks and additional doses as needed). Therefore, during the second year of these studies there was no active control comparison arm.
14.2 Macular Edema Following Central Retinal Vein Occlusion (CRVO)
The safety and efficacy of EYLEA were assessed in two randomized, multi-center, double-masked, sham-controlled studies in patients with macular edema following CRVO. A total of 358 patients were treated and evaluable for efficacy (217 with EYLEA) in the two studies (COPERNICUS and GALILEO). In both studies, patients were randomly assigned in a 3:2 ratio to either 2 mg EYLEA administered every 4 weeks (2Q4), or sham injections (control group) administered every 4 weeks for a total of 6 injections. Protocol-specified visits occurred every 28±7 days. Patient ages ranged from 22 to 89 years with a mean of 64 years.
In both studies, the primary efficacy endpoint was the proportion of patients who gained at least 15 letters in BCVA compared to baseline. At week 24, the EYLEA 2 mg Q4 group was superior to the control group for the primary endpoint.
Results from the analysis of the COPERNICUS and GALILEO studies are shown in Table 5 and Figure 14 below.
Table 5: Efficacy Outcomes at Week 24 (Full Analysis Set with LOCF) in COPERNICUS and GALILEO Studies COPERNICUS GALILEO Control EYLEA
2 mg Q4 weeksControl EYLEA
2 mg Q4 weeksN=73 N=114 N=68 N=103 - * Difference is EYLEA 2 mg Q4 weeks minus Control
- † Difference and CI are calculated using Cochran-Mantel-Haenszel (CMH) test adjusted for baseline factors; 95.1% confidence intervals were presented to adjust for the multiple assessments conducted during the study
- ‡ p<0.01 compared with Control
- § LS mean and CI based on an ANCOVA model
Efficacy Outcomes Proportion of patients who gained at least 15 letters in BCVA from Baseline (%) 12% 56% 22% 60% Weighted Difference *, † (%)
(95.1% CI)44.8%‡
(32.9, 56.6)38.3%‡
(24.4, 52.1)Mean change in BCVA as measured by ETDRS letter score from Baseline (SD) -4.0
(18.0)17.3
(12.8)3.3
(14.1)18.0
(12.2)Difference in LS mean *, §
(95.1% CI)21.7‡
(17.3, 26.1)14.7‡
(10.7, 18.7)Figure 14: Mean Change in BCVA as Measured by ETDRS Letter Score from Baseline to Week 24 in COPERNICUS and GALILEO Studies
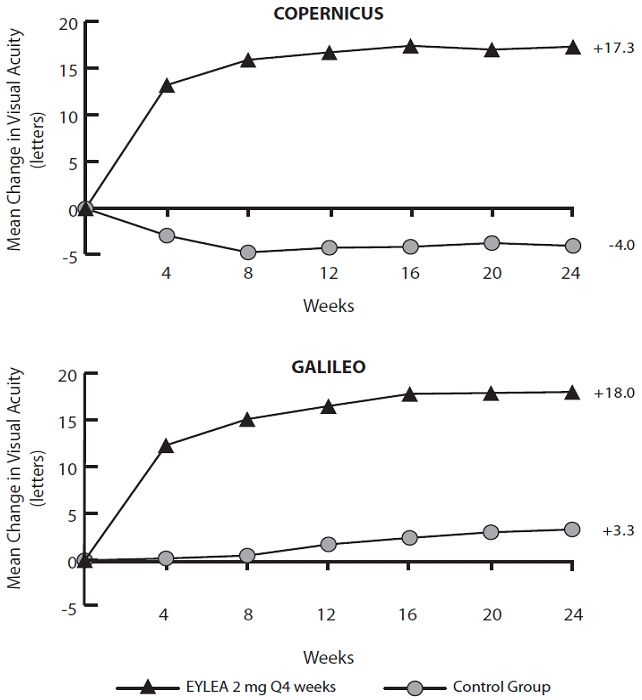
Treatment effects in evaluable subgroups (e.g., age, gender, race, baseline visual acuity, retinal perfusion status, and CRVO duration) in each study and in the combined analysis were in general consistent with the results in the overall populations.
14.3 Macular Edema Following Branch Retinal Vein Occlusion (BRVO)
The safety and efficacy of EYLEA were assessed in a 24-week, randomized, multi-center, double-masked, controlled study in patients with macular edema following BRVO. A total of 181 patients were treated and evaluable for efficacy (91 with EYLEA) in the VIBRANT study. In the study, patients were randomly assigned in a 1:1 ratio to either 2 mg EYLEA administered every 4 weeks (2Q4) or laser photocoagulation administered at baseline and subsequently as needed (control group). Protocol-specified visits occurred every 28±7 days. Patient ages ranged from 42 to 94 years with a mean of 65 years.
In the VIBRANT study, the primary efficacy endpoint was the proportion of patients who gained at least 15 letters in BCVA at week 24 compared to baseline. At week 24, the EYLEA 2 mg Q4 group was superior to the control group for the primary endpoint.
Detailed results from the analysis of the VIBRANT study are shown in Table 6 and Figure 15 below.
Table 6: Efficacy Outcomes at Week 24 (Full Analysis Set with LOCF) in VIBRANT Study VIBRANT Control EYLEA
2 mg Q4 weeksN=90 N=91 - * Difference is EYLEA 2 mg Q4 weeks minus Control
- † Difference and CI are calculated using Mantel-Haenszel weighting scheme adjusted for region (North America vs. Japan) and baseline BCVA category (> 20/200 and ≤ 20/200)
- ‡ p<0.01 compared with Control
- § LS mean and CI based on an ANCOVA model
Efficacy Outcomes Proportion of patients who gained at least 15 letters in BCVA from Baseline (%) 26.7% 52.7% Weighted Difference *, † (%)
(95% CI)26.6%‡
(13.0, 40.1)Mean change in BCVA as measured by ETDRS letter score from Baseline (SD) 6.9
(12.9)17.0
(11.9)Difference in LS mean *, §
(95% CI)10.5‡
(7.1, 14.0)Figure 15: Mean Change in BCVA as Measured by ETDRS Letter Score from Baseline to Week 24 in VIBRANT Study
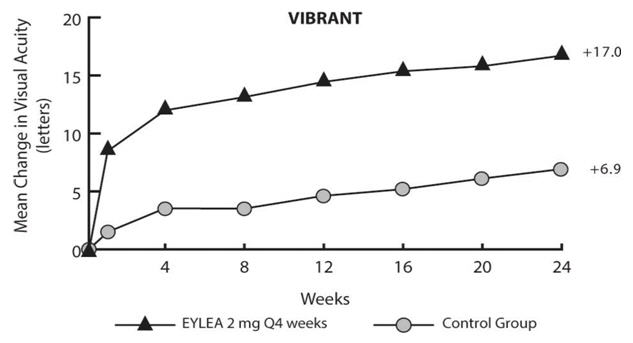
Treatment effects in evaluable subgroups (e.g., age, gender, and baseline retinal perfusion status) in the study were in general consistent with the results in the overall populations.
14.4 Diabetic Macular Edema (DME)
The safety and efficacy of EYLEA were assessed in two randomized, multi-center, double-masked, controlled studies in patients with DME. A total of 862 randomized and treated patients were evaluable for efficacy. Protocol-specified visits occurred every 28±7 days. Patient ages ranged from 23 to 87 years with a mean of 63 years.
Of those, 576 were randomized to EYLEA groups in the two studies (VIVID and VISTA). In each study, patients were randomly assigned in a 1:1:1 ratio to 1 of 3 dosing regimens: 1) EYLEA administered 2 mg every 8 weeks following 5 initial monthly injections (EYLEA 2Q8); 2) EYLEA administered 2 mg every 4 weeks (EYLEA 2Q4); and 3) macular laser photocoagulation (at baseline and then as needed). Beginning at week 24, patients meeting a pre-specified threshold of vision loss were eligible to receive additional treatment: patients in the EYLEA groups could receive laser and patients in the laser group could receive EYLEA.
In both studies, the primary efficacy endpoint was the mean change from baseline in BCVA at week 52 as measured by ETDRS letter score. Efficacy of both EYLEA 2Q8 and EYLEA 2Q4 groups was statistically superior to the control group. This statistically superior improvement in BCVA was maintained at week 100 in both studies.
Results from the analysis of the VIVID and VISTA studies are shown in Table 7 and Figure 16 below.
Table 7: Efficacy Outcomes at Weeks 52 and 100 (Full Analysis Set with LOCF) in VIVID and VISTA Studies VIVID VISTA EYLEA
2 mg Q8 weeks *EYLEA
2 mg Q4 weeksControl EYLEA
2 mg Q8 weeks *EYLEA
2 mg Q4 weeksControl Full Analysis Set N=135 N=136 N=132 N=151 N=154 N=154 - * After treatment initiation with 5 monthly injections
- † LS mean and CI based on an ANCOVA model with baseline BCVA measurement as a covariate and a factor for treatment group. Additionally, protocol specified stratification factors were included in the model
- ‡ Difference is EYLEA group minus Control group
- § p<0.01 compared with Control
- ¶ Difference with confidence interval (CI) and statistical test is calculated using Mantel-Haenszel weighting scheme adjusted by protocol specified stratification factors
Efficacy Outcomes at Week 52 Mean change in BCVA as measured by ETDRS letter score from Baseline (SD) 10.7
(9.3)10.5
(9.6)1.2
(10.6)10.7
(8.2)12.5
(9.5)0.2
(12.5)Difference†, ‡ in LS mean
(97.5% CI)9.1§
(6.3, 11.8)9.3§
(6.5, 12.0)10.5§
(7.7, 13.2)12.2§
(9.4, 15.0)Proportion of patients who gained at least 15 letters in BCVA from Baseline (%) 33.3% 32.4% 9.1% 31.1% 41.6% 7.8% Adjusted Difference‡, ¶ (%)
(97.5% CI)24.2%§
(13.5, 34.9)23.3%§
(12.6, 33.9)23.3%§
(13.5, 33.1)34.2%§
(24.1, 44.4)Efficacy Outcomes at Week 100 Mean change in BCVA as measured by ETDRS letter score from Baseline (SD) 9.4
(10.5)11.4
(11.2)0.7
(11.8)11.1
(10.7)11.5
(13.8)0.9
(13.9)Difference†, ‡ in LS mean
(97.5% CI)8.2§
(5.2, 11.3)10.7§
(7.6, 13.8)10.1§
(7.0, 13.3)10.6§
(7.1, 14.2)Proportion of patients who gained at least 15 letters in BCVA from Baseline (%) 31.1% 38.2% 12.1% 33.1% 38.3% 13.0% Adjusted Difference‡, ¶ (%)
(97.5% CI)19.0%§
(8.0, 29.9)26.1%§
(14.8, 37.5)20.1%§
(9.6, 30.6)25.8%§
(15.1, 36.6)Figure 16: Mean Change in BCVA as Measured by ETDRS Letter Score from Baseline to Week 100 in VIVID and VISTA Studies
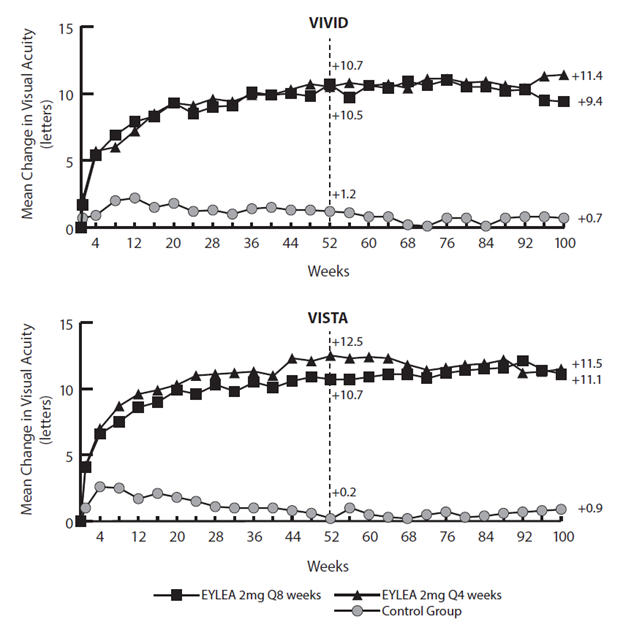
Treatment effects in the subgroup of patients who had previously been treated with a VEGF inhibitor prior to study participation were similar to those seen in patients who were VEGF inhibitor naïve prior to study participation.
Treatment effects in evaluable subgroups (e.g., age, gender, race, baseline HbA1c, baseline visual acuity, prior anti-VEGF therapy) in each study were in general consistent with the results in the overall populations.
14.5 Diabetic Retinopathy (DR)
Efficacy and safety data of EYLEA in diabetic retinopathy (DR) are derived from the VIVID, VISTA, and PANORAMA studies.
VIVID AND VISTA
In the VIVID and VISTA studies, an efficacy outcome was the change in the Early Treatment Diabetic Retinopathy Study (ETDRS) Diabetic Retinopathy Severity Scale (ETDRS-DRSS). The ETDRS-DRSS score was assessed at baseline and approximately every 6 months thereafter for the duration of the studies [see Clinical Studies (14.4)].
All enrolled patients had DR and DME at baseline. The majority of patients enrolled in these studies (77%) had moderate-to-severe nonproliferative diabetic retinopathy (NPDR) based on the ETDRS-DRSS. At week 100, the proportion of patients improving by at least 2 steps on the ETDRS-DRSS was significantly greater in both EYLEA treatment groups (2Q4 and 2Q8) when compared to the control group.
Results from the analysis of ETDRS-DRSS at week 100 in the VIVID and VISTA studies are shown in Table 8 below.
Table 8: Proportion of Patients Who Achieved a ≥2-Step Improvement from Baseline in the ETDRS-DRSS Score at Week 100 in VIVID and VISTA Studies VIVID VISTA EYLEA
2 mg Q8 weeks *EYLEA
2 mg Q4 weeksControl EYLEA
2 mg Q8 weeks *EYLEA
2 mg Q4 weeksControl Evaluable Patients† N=101 N=97 N=99 N=148 N=153 N=150 Non-gradable post-baseline ETDRS-DRSS values were treated as missing and were imputed using the last gradable ETDRS-DRSS values (including baseline values if all post-baseline values were missing or non-gradable) - * After treatment initiation with 5 monthly injections
- † The number of evaluable patients included all patients who had valid ETDRS-DRSS data at baseline
- ‡ Difference with confidence interval (CI) was calculated using Mantel-Haenszel weighting scheme adjusted by protocol specified stratification factors
- § Difference is EYLEA minus Control group
- ¶ p<0.01 compared with Control
Number of patients with a ≥2-step improvement on ETDRS-DRSS from Baseline (%) 32
(32%)27
(28%)7
(7%)56
(38%)58
(38%)24
(16%)Difference ‡, §(%)
(97.5% CI)24%¶
(12, 36)21%¶
(9, 33)22%¶
(11, 33)22%¶
(11, 33)Results of the evaluable subgroups (e.g., age, gender, race, baseline HbA1c, baseline visual acuity) on the proportion of patients who achieved a ≥2-step improvement on the ETDRS-DRSS from baseline to week 100 were, in general, consistent with those in the overall population.
PANORAMA
The PANORAMA study assessed the safety and efficacy of EYLEA in a randomized, multi-center, double-masked, controlled study in patients with moderately severe to severe nonproliferative diabetic retinopathy (NPDR) (ETDRS-DRSS of 47 or 53), without central-involved DME (CI-DME). A total of 402 randomized patients were evaluable for efficacy. Protocol-specified visits occurred every 28±7 days for the first 5 visits, then every 8 weeks (56±7 days). Patient ages ranged from 25 to 85 years with a mean of 55.7 years.
Patients were randomly assigned in a 1:1:1 ratio to 1 of 3 dosing regimens: 1) 3 initial monthly EYLEA 2 mg injections followed by one injection after 8 weeks and then one injection every 16 weeks (EYLEA 2Q16); 2) 5 monthly EYLEA 2 mg injections followed by one injection every 8 weeks (EYLEA 2Q8); and 3) sham treatment.
The primary efficacy endpoint was the proportion of patients who improved by ≥2 steps on the DRSS from baseline to week 24 in the combined EYLEA groups and at week 52 in the 2Q16 and 2Q8 groups individually versus sham. A key secondary endpoint was the proportion of patients developing the composite endpoint of proliferative diabetic retinopathy or anterior segment neovascularization through week 52.
At week 52, efficacy in the 2Q16 and 2Q8 groups was superior to the sham group (see Table 9 and Table 10). The proportion of patients with a ≥2-step improvement over time is shown in Figure 17.
Table 9: Proportion of Patients Who Achieved a ≥2-Step Improvement from Baseline in the ETDRS-DRSS Score at Weeks 24 and 52 in PANORAMA PANORAMA Week 24 Week 52 EYLEA
CombinedControl
(sham)EYLEA
2Q16EYLEA
2Q8Control
(sham)Full Analysis Set N=269 N=133 N=135 N=134 N=133 Non-gradable post-baseline ETDRS-DRSS values were treated as missing and were imputed using the last gradable ETDRS-DRSS values (including baseline values if all post-baseline values were missing or non-gradable) - * Difference is EYLEA group minus sham
- † Difference with CI was calculated using the Mantel-Haenszel weighting scheme adjusted by baseline DRSS stratification variable
- ‡ p<0.01 compared with Control. p-value was calculated using a 2-sided Cochran-Mantel-Haenszel test adjusted by baseline DRSS stratification variable.
Proportion of patients with a ≥2-step improvement on ETDRS-DRSS from Baseline (%) 58% 6% 65% 80% 15% Adjusted Difference*
(%)
(95% CI)†52% ‡
(45, 60)50%‡
(40, 60)65%‡
(56, 74)Figure 17: Proportion of Patients Who Achieved a ≥2-Step Improvement from Baseline in the ETDRS-DRSS Score Through Week 52 in PANORAMA
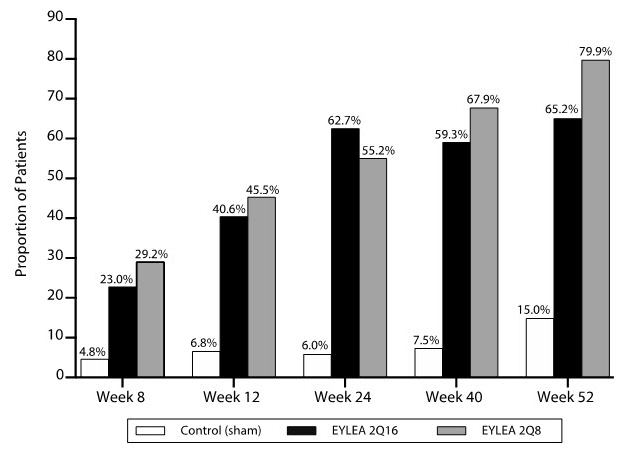
Table 10: Effect of EYLEA on Worsening of Diabetic Retinopathy in PANORAMA through Week 52 EYLEA
2Q16EYLEA
2Q8Control
(Sham)Full Analysis Set N=135 N=134 N=133 PDR = Proliferative Diabetic Retinopathy; ASNV = Anterior Segment Neovascularization - * As diagnosed by either the Reading Center or Investigator through week 52
- † Estimated using Kaplan-Meier method
- ‡ p<0.01 compared with Control
- § Defined as ≥2-step worsening on the ETDRS-DRSS score through week 52
Composite Endpoint of Developing PDR or ASNV* Event Rate† 4.0%‡ 2.4%‡ 20.1% Hazard Ratio 0.15 0.12 Development of Proliferative Diabetic Retinopathy§ Event Rate† 1.6%‡ 0.0%‡ 11.9% Hazard Ratio 0.11 0.00 -
16 HOW SUPPLIED/STORAGE AND HANDLING
Each pre-filled syringe or vial is for single eye use only. EYLEA is supplied in the following presentations [see Dosage and Administration (2.6), (2.7), and (2.8)].
NDC NUMBER CARTON TYPE CARTON CONTENTS 61755-005-01 Pre-filled Syringe one blister pack containing one EYLEA 2 mg/0.05 mL sterile, single-dose pre-filled glass syringe
one package insert61755-005-02 Vial Kit with Injection Components one EYLEA 2 mg/0.05 mL single-dose glass vial
one 19-gauge × 1½-inch, 5-micron, filter needle for withdrawal of the vial contents
one 30-gauge × ½-inch injection needle for intravitreal injection
one 1-mL syringe for administration
one package insert -
17 PATIENT COUNSELING INFORMATION
In the days following EYLEA administration, patients are at risk of developing endophthalmitis or retinal detachment. If the eye becomes red, sensitive to light, painful, or develops a change in vision, advise patients to seek immediate care from an ophthalmologist [see Warnings and Precautions (5.1)].
Patients may experience temporary visual disturbances after an intravitreal injection with EYLEA and the associated eye examinations [see Adverse Reactions (6)]. Advise patients not to drive or use machinery until visual function has recovered sufficiently.
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL - 2 mg/0.05 mL Vial Carton
NDC: 61755-005-02
EYLEA®
(aflibercept) Injection
For Intravitreal Injection2 mg/0.05 mL
Single-use VialCarton contents: Each EYLEA carton contains
- one single-use, 3-mL, glass vial of EYLEA
- one 19-gauge x 1½-inch, 5-micron, filter needle
for withdrawal of the vial contents (filter needle not
to be used for intravitreal injection) - one 30-gauge x ½-inch needle for intravitreal injection
- one 1-mL plastic syringe for administration
- one package insert
Rx ONLY

-
PRINCIPAL DISPLAY PANEL - 2 mg/0.05 mL Syringe Carton
NDC: 61755-005-01
Rx ONLYEYLEA®
(aflibercept) Injection
For Intravitreal Injection2 mg/0.05 mL
Single-dose Pre-filled Syringe
Carton contents:
- one blister pack containing
one sterile, single dose
pre-filled glass syringe - package insert
REGENERON

- one blister pack containing
-
INGREDIENTS AND APPEARANCE
EYLEA
aflibercept injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 61755-005 Route of Administration INTRAVITREAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength aflibercept (UNII: 15C2VL427D) (aflibercept - UNII:15C2VL427D) aflibercept 40 mg in 1 mL Inactive Ingredients Ingredient Name Strength sodium phosphate (UNII: SE337SVY37) sodium chloride (UNII: 451W47IQ8X) polysorbate 20 (UNII: 7T1F30V5YH) sucrose (UNII: C151H8M554) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 61755-005-02 1 in 1 CARTON 11/21/2011 1 0.05 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product 2 NDC: 61755-005-55 1 in 1 CARTON 01/16/2014 2 0.05 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product 3 NDC: 61755-005-01 1 in 1 CARTON 12/09/2019 3 0.05 mL in 1 SYRINGE, GLASS; Type 0: Not a Combination Product 4 NDC: 61755-005-54 1 in 1 CARTON 12/09/2019 4 0.05 mL in 1 SYRINGE, GLASS; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125387 11/21/2011 Labeler - Regeneron Pharmaceuticals, Inc. (194873139) Establishment Name Address ID/FEI Business Operations Regeneron Pharmaceuticals, Inc. 945589711 API MANUFACTURE(61755-005) , ANALYSIS(61755-005) , LABEL(61755-005) , MANUFACTURE(61755-005)
Trademark Results [EYLEA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 EYLEA 90701063 not registered Live/Pending |
Regeneron Pharmaceuticals, Inc. 2021-05-10 |
 EYLEA 87722615 5524177 Live/Registered |
Regeneron Pharmaceuticals, Inc. 2017-12-15 |
 EYLEA 85209921 4100104 Live/Registered |
Regeneron Pharmaceuticals, Inc. 2011-01-04 |
 EYLEA 77510978 4088223 Live/Registered |
Regeneron Pharmaceuticals, Inc. 2008-06-30 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.