MULPLETA- lusutrombopag tablet, film coated
Mulpleta by
Drug Labeling and Warnings
Mulpleta by is a Prescription medication manufactured, distributed, or labeled by SHIONOGI INC.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use MULPLETA safely and effectively. See full prescribing information for MULPLETA.
MULPLETA ® (lusutrombopag tablets) for oral use
Initial U.S. Approval: 2018INDICATIONS AND USAGE
MULPLETA is a thrombopoietin receptor agonist indicated for the treatment of thrombocytopenia in adult patients with chronic liver disease who are scheduled to undergo a procedure. ( 1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Tablet: 3 mg. ( 3)
CONTRAINDICATIONS
- None.
WARNINGS AND PRECAUTIONS
- Thrombotic/Thromboembolic Complications: MULPLETA is a thrombopoietin (TPO) receptor agonist, and TPO receptor agonists have been associated with thrombotic and thromboembolic complications in patients with chronic liver disease. Monitor platelet counts and for thromboembolic events and institute treatment promptly. ( 5.1)
ADVERSE REACTIONS
The most common adverse reaction (≥3%): headache. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Shionogi Inc. at 1-800-849-9707 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 5/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Monitoring
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Thrombotic/Thromboembolic Complications
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Begin MULPLETA dosing 8-14 days prior to a scheduled procedure.
Patients should undergo their procedure 2-8 days after the last dose.
The recommended dosage of MULPLETA is 3 mg taken orally once daily with or without food for 7 days. In the case of a missed dose of MULPLETA, patients should take the missed dose as soon as possible on the same day and return to the normal schedule the following day.
MULPLETA has been investigated only as a single 7-day once daily dosing regimen in clinical trials in patients with chronic liver disease [see Clinical Studies (14)] . MULPLETA should not be administered to patients with chronic liver disease in an attempt to normalize platelet counts.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Thrombotic/Thromboembolic Complications
MULPLETA is a thrombopoietin (TPO) receptor agonist, and TPO receptor agonists have been associated with thrombotic and thromboembolic complications in patients with chronic liver disease. Portal vein thrombosis has been reported in patients with chronic liver disease treated with TPO receptor agonists. Portal vein thrombosis was reported in 1% (2 of 171) of MULPLETA-treated patients and 1% (2 of 170) of placebo-treated patients in 3 randomized, double-blind trials and was identified post-procedure in protocol-specified imaging. The thromboses were not associated with a marked increase in platelet count.
Consider the potential increased thrombotic risk when administering MULPLETA to patients with known risk factors for thromboembolism, including genetic pro-thrombotic conditions (Factor V Leiden, Prothrombin 20210A, Antithrombin deficiency, or Protein C or S deficiency). In patients with ongoing or prior thrombosis or absence of hepatopetal blood flow, MULPLETA should only be used if the potential benefit to the patient justifies the potential risk.
MULPLETA should not be administered to patients with chronic liver disease in an attempt to normalize platelet counts.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed in detail in other sections of the labeling:
- Thrombotic/Thromboembolic Complications [see Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of MULPLETA was evaluated in 3 randomized, double-blind, placebo-controlled trials, L-PLUS 1, L-PLUS 2, and M0626, in which patients with chronic liver disease and thrombocytopenia were treated with MULPLETA (N=171) or placebo (N=170) at a dose of 3 mg daily for up to 7 days prior to a scheduled procedure.
The majority of patients were males (59%), and median age was 61 years (range 19-88). The racial and ethnic distribution was White (50%), Asian (47%), Black (<1%), and Other (3%).
The most common adverse reactions (those occurring in at least 3%) in the MULPLETA-treated group across the pooled data from the three trials are summarized in table 1.
Table 1. Adverse Reactions with a Frequency ≥3% in Patients Treated with MULPLETA (Pooled Data (L-PLUS 1, L-PLUS 2, and M0626)) Adverse Reaction * MULPLETA 3 mg
(N=171)
%Placebo
(N=170)
%- * Includes treatment-emergent adverse reactions occurring at a rate higher than placebo.
Headache 5 4 The incidence of serious adverse events was 5% (9 of 171 patients) in the MULPLETA group and 7% (12 of 170 patients) in the placebo group. The most common serious adverse reaction reported with MULPLETA was portal vein thrombosis [see Warnings and Precautions (5.1)] . No adverse reactions resulted in discontinuation of MULPLETA.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on MULPLETA in pregnant women to inform the drug-associated risk. In animal reproduction studies, oral administration of lusutrombopag to pregnant rats during organogenesis and the lactation period resulted in adverse developmental outcomes. These findings were observed at exposures based on AUC that were substantially higher than the AUC observed in patients (approximately 89 times) at the recommended clinical dose of 3 mg once daily . Advise pregnant women of the potential risk to a fetus (see Data) .
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the general population in the United States, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryo-fetal development study in rats, lusutrombopag was orally administered during organogenesis at doses of 4, 12.5, 40, and 80 mg/kg/day. Low body weight and a decrease in the number of ossified sternebrae were noted in fetuses at 80 mg/kg/day (approximately 251 times the AUC observed in patients at the recommended clinical dose of 3 mg once daily). Minor skeletal variations (supernumerary ribs) were observed at doses of 4 mg/kg/day (approximately 23 times the AUC observed in patients at the recommended clinical dose of 3 mg once daily).
In an embryo-fetal development study in rabbits following oral administration of lusutrombopag at doses up to 1000 mg/kg/day, no effect of lusutrombopag was observed on any parameter of embryo-fetal development.
In a pre- and postnatal development study in rats at oral doses of 1, 4, 12.5, and 40 mg/kg/day, there were adverse effects of lusutrombopag on postnatal development at 40 mg/kg/day (approximately 230 times the AUC observed in patients at the recommended clinical dose of 3 mg once daily). The effects included prolongation of the gestation period in dams, low viability before weaning, delayed postnatal growth (delayed negative geotaxis, delayed eyelid opening, or low pup body weight), abnormal clinical signs (prominent annular rings on the tail after weaning), low fertility index, a low number of corpora lutea or implantations, and increased pre-implantation loss. The incidence of short thoracolumbar supernumerary ribs on postnatal Day 4 of F1 pups was high at doses of 12.5 mg/kg/day or more (approximately 89 times the AUC observed in patients at the recommended clinical dose of 3 mg once daily).
8.2 Lactation
Risk Summary
There is no information regarding the presence of lusutrombopag in human milk, the effects on the breastfed child, and the effects on milk production. Lusutrombopag was present in the milk of lactating rats. Due to the potential for serious adverse reactions in a breastfed child, breastfeeding is not recommended during treatment with MULPLETA and for at least 28 days after the last dose (see Clinical Considerations) .
8.5 Geriatric Use
Clinical studies of MULPLETA did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients.
-
10 OVERDOSAGE
No antidote for MULPLETA overdose is known.
In the event of overdose, platelet count may increase excessively and result in thrombotic or thromboembolic complications. Closely monitor the patient and platelet count. Treat thrombotic complications in accordance with standard of care.
Hemodialysis is not expected to enhance the elimination of MULPLETA because lusutrombopag is highly bound to protein in plasma [see Clinical Pharmacology (12.3)] .
-
11 DESCRIPTION
MULPLETA (lusutrombopag), a thrombopoietin (TPO) receptor agonist, contains lusutrombopag as the active ingredient.
The chemical name for lusutrombopag is (2 E)-3-{2,6-Dichloro-4-[(4-{3-[(1 S)-1-(hexyloxy) ethyl]-2-methoxyphenyl}-1,3-thiazol-2-yl) carbamoyl]phenyl}-2-methylprop-2-enoic acid.
The structural formula is:
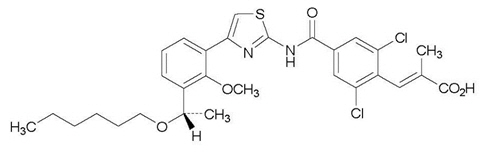
The empirical formula for lusutrombopag is C 29H 32Cl 2N 2O 5S and the molecular weight is 591.54.
Lusutrombopag is a white to slightly yellowish white powder, and is freely soluble in N,N-dimethylformamide, slightly soluble in ethanol (99.5%) and methanol, very slightly soluble in acetonitrile, and practically insoluble in water. Lusutrombopag is slightly soluble in the buffer solution at pH 11 and practically insoluble in buffer solutions with pH ranges of 1 to 9.
MULPLETA (lusutrombopag) tablets for oral use contain lusutrombopag 3 mg.
Excipients are D-mannitol, microcrystalline cellulose, magnesium oxide, sodium lauryl sulfate, hydroxypropyl cellulose, carboxymethylcellulose calcium, magnesium stearate, hypromellose, triethyl citrate, titanium dioxide, red ferric oxide, and talc.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Lusutrombopag is an orally bioavailable, small molecule TPO receptor agonist that interacts with the transmembrane domain of human TPO receptors expressed on megakaryocytes to induce the proliferation and differentiation of megakaryocytic progenitor cells from hematopoietic stem cells and megakaryocyte maturation.
12.2 Pharmacodynamics
Platelet Response
Lusutrombopag upregulates the production of platelets through its agonistic effect on human TPO receptors.
The effect of lusutrombopag on platelet count increase was correlated with the AUC across the studied dose range of 0.25 mg to 4 mg in thrombocytopenic patients with chronic liver disease. With the 3 mg daily dose, the mean (standard deviation) maximum platelet count in patients (N=74) without platelet transfusion was 86.9 (27.2) × 10 9/L, and the median time to reach the maximum platelet count was 12.0 (5 to 35) days.
12.3 Pharmacokinetics
Lusutrombopag demonstrated dose-proportional pharmacokinetics after single doses ranging from 1 mg (0.33 times the lowest approved dosage) to 50 mg (16.7 times the highest recommended dosage). Healthy subjects administered 3 mg of lusutrombopag had a geometric mean (%CV) maximal concentration (C max) of 111 (20.4) ng/mL and area under the time-concentration curve extrapolated to infinity (AUC 0-inf) of 2931 (23.4) ng.hr/mL. The pharmacokinetics of lusutrombopag were similar in both healthy subjects and the chronic liver disease population.
The accumulation ratios of C max and AUC were approximately 2 with once-daily multiple-dose administration, and steady-state plasma lusutrombopag concentrations were achieved after Day 5.
Absorption
In patients with chronic liver disease, the time to peak lusutrombopag concentration (T max) was observed 6 to 8 hours after oral administration.
Distribution
The mean (%CV) lusutrombopag apparent volume of distribution in healthy adult subjects was 39.5 (23.5) L. The plasma protein binding of lusutrombopag is more than 99.9%.
Elimination
The terminal elimination half-life (t 1/2) in healthy adult subjects was approximately 27 hours. The mean (%CV) clearance of lusutrombopag in patients with chronic liver disease is estimated to be 1.1 (36.1) L/hr.
Specific Populations
No clinically significant differences in the pharmacokinetics of lusutrombopag were observed based on age or race/ethnicity. Though lusutrombopag exposure tends to decrease with increasing body weight, differences in exposure are not considered clinically relevant.
Patients with Renal Impairment
A population pharmacokinetic analysis did not find a clinically meaningful effect of mild (creatinine clearance (CLcr) 60 to less than 90 mL/min) and moderate (CLcr 30 to less than 60 mL/min) renal impairment on the pharmacokinetics of lusutrombopag. Data in patients with severe renal impairment (CLcr less than 30 mL/min) are limited.
Patients with Hepatic Impairment
No clinically significant differences in the pharmacokinetics of lusutrombopag were observed based on mild to moderate (Child-Pugh class A and B) hepatic impairment.
The mean observed lusutrombopag C max and AUC 0-τ decreased by 20% to 30% in patients (N=5) with severe (Child-Pugh class C) hepatic impairment compared to patients with Child-Pugh class A and class B liver disease. However, the ranges for C max and AUC 0-τ overlapped among patients with Child-Pugh class A, B, and C liver disease.
Drug Interaction Studies
Clinical Studies
No clinically significant changes in lusutrombopag exposure were observed when co-administered with cyclosporine (an inhibitor of P-gp and BCRP) or an antacid containing a multivalent cation (calcium carbonate).
No clinically significant changes in midazolam (a CYP3A substrate) exposure were observed when co-administered with lusutrombopag.
In Vitro Studies
CYP Enzymes: lusutrombopag has low potential to inhibit CYP enzymes (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5). Lusutrombopag did not induce CYP1A2, CYP2C9, or CYP3A4.
UGT Enzymes: lusutrombopag did not induce UGT1A2, UGT1A6, or UGT2B7.
Transporter Systems: lusutrombopag is a substrate of P-gp and BCRP. Lusutrombopag has low potential to inhibit P-gp, BCRP, OATP1B1, OATP1B3, OCT1, OCT2, OAT1, OAT3, MATE1, MATE2-K, and BSEP.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In 2-year studies, lusutrombopag was not carcinogenic to rats at oral doses up to 20 mg/kg/day in males and 2 mg/kg/day in females (a dose 49 times and 30 times, respectively, the human exposure (AUC) at the recommended clinical dose of 3 mg/day for 7 days) and to mice at oral doses up to 20 mg/kg/day in males and females (a dose approximately 45 times the human exposure (AUC) at the recommended clinical dose of 3 mg/day for 7 days).
Lusutrombopag was not genotoxic based on an in vitro bacterial reverse mutation (Ames) assay, a chromosomal aberration assay with cultured Chinese hamster lung cells, and an in vivo micronucleus assay with mouse bone marrow cells.
In a fertility and early embryonic development study, lusutrombopag did not affect fertility in male and female rats at oral doses up to 100 mg/kg/day (a dose in males and females approximately 176 and 252 times, respectively, the human exposure (AUC) at the recommended clinical dose of 3 mg/day for 7 days).
-
14 CLINICAL STUDIES
The efficacy of MULPLETA for the treatment of thrombocytopenia in patients with chronic liver disease who are scheduled to undergo a procedure was evaluated in 2 randomized, double-blind, placebo-controlled trials (L-PLUS 1 (N=97) and L-PLUS 2 (N=215; NCT02389621)). Patients with chronic liver disease who were undergoing an invasive procedure and had a platelet count less than 50 × 10 9/L were eligible to participate. Patients undergoing laparotomy, thoracotomy, open-heart surgery, craniotomy, or organ resection were excluded. Patients with a history of splenectomy, partial splenic embolization, or thrombosis and those with Child-Pugh class C liver disease, absence of hepatopetal blood flow, or a prothrombotic condition other than chronic liver disease were not allowed to participate.
The patient populations were similar between the MULPLETA and placebo arms and consisted of 60% male and 40% female; median age was 60 years (range 19-88). The racial and ethnic distribution was White (55%), Asian (41%), and Other (4%).
Patients were randomized 1:1 to receive 3 mg of MULPLETA or placebo once daily for up to 7 days. Randomization was stratified by liver ablation/coagulation or other procedures and the platelet count at screening/baseline. In L-PLUS 1, 57% of patients underwent procedures other than liver ablation/coagulation and 43% underwent liver ablation/coagulation (RFA/MCT). In L-PLUS 2, 98% of patients underwent procedures other than liver ablation/coagulation and 2% underwent liver ablation/coagulation (RFA/MCT). Procedures other than liver ablation/coagulation (RFA/MCT) included liver-related procedures (transcatheter arterial chemoembolization, liver biopsy, and others), upper and lower gastrointestinal endoscopy-related procedures (endoscopic variceal ligation, endoscopic injection sclerotherapy, polypectomy, and biopsy), and other procedures (dental extraction, diagnostic paracentesis or laparocentesis, septoplasty, embolization of splenic artery aneurysm, bone marrow biopsy, removal of cervical polyp, and inguinal hernia repair (non-laparotomy based)).
In L-PLUS 1, the major efficacy outcome was the proportion of patients who require no platelet transfusion prior to the primary invasive procedure. In L-PLUS 2, the major efficacy outcome was the proportion of patients who require no platelet transfusion prior to the primary invasive procedure and no rescue therapy for bleeding (i.e., platelet preparations, other blood preparations, including red blood cells and plasma, volume expanders) from randomization through 7 days after the primary invasive procedure. In both trials, additional efficacy outcomes included the proportion of patients who require no platelet transfusion during the study, proportion of responders, duration of the increase in platelet count defined as the number of days during which the platelet count was maintained as ≥50 × 10 9/L, and the time course of platelet counts.
In both the L-PLUS 1 and L-PLUS 2 trials, responders were defined as patients who had a platelet count of ≥50 × 10 9/L with an increase of ≥20 × 10 9/L from baseline.
Table 2. L-PLUS 1 Trial: Proportion of Patients Not Requiring Platelet Transfusion Prior to Invasive Procedure and Proportion of Responders Endpoint Proportion (n/N)
Exact 95% Confidence IntervalTreatment Difference
(95% Confidence Interval)
p valueMULPLETA
(N=49)Placebo
(N=48)- * A platelet transfusion was required if the platelet count was less than 50 × 10 9/L.
- † Cochran-Mantel-Haenszel test with baseline platelet count as stratum; p value and confidence interval calculated using Wald method.
- ‡ Platelet count reached at least 50 × 10 9/L and increased at least 20 × 10 9/L from baseline.
Not requiring platelet transfusion prior to invasive procedure * 78% (38/49)
(63, 88)13% (6/48)
(4.7, 25)64 (49, 79)
<0.0001 †Responder ‡ during study 76% (37/49)
(61, 87)6% (3/48)
(1.3, 17)68 (54, 82)
<0.0001 †Table 3. L-PLUS 2 Trial: Proportion of Patients Not Requiring Platelet Transfusion Prior to Invasive Procedure or Rescue Therapy for Bleeding Through 7 Days After Invasive Procedure and Proportion of Responders Endpoint Proportion (n/N)
Exact 95% Confidence IntervalTreatment Difference
(95% Confidence Interval)
p valueMULPLETA
(N=108)Placebo
(N=107)- * A platelet transfusion was required if the platelet count was less than 50 × 10 9/L.
- † Cochran-Mantel-Haenszel test with baseline platelet count as stratum; p value and confidence interval calculated using Wald method.
- ‡ Platelet count reached at least 50 × 10 9/L and increased at least 20 × 10 9/L from baseline.
Not requiring platelet transfusion prior to invasive procedure * or rescue therapy for bleeding from randomization through 7 days after invasive procedure 65% (70/108)
(55, 74)29% (31/107)
(21, 39)37 (25, 49)
<0.0001 †Responder ‡ during study 65% (70/108)
(55, 74)13% (14/107)
(7.3, 21)52 (41, 62)
<0.0001 †The median (Q1, Q3) duration of platelet count increase to at least 50 × 10 9/L was 22 (17, 27) days in MULPLETA-treated patients without platelet transfusion and 1.8 (0.0, 8.3) days in placebo-treated patients with platelet transfusion in L-PLUS 1 and 19 (13, 28) days in MULPLETA-treated patients without platelet transfusion and 0.0 (0.0, 5.0) days in placebo-treated patients with platelet transfusion in L-PLUS 2.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
MULPLETA is supplied as 3 mg lusutrombopag tablets in a child-resistant blister pack containing 7 tablets - NDC: 59630-551-07.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Prior to treatment, patients should fully understand and be informed of the following risks and considerations for MULPLETA.
Risks
Thrombotic/Thromboembolic Complications
MULPLETA is a thrombopoietin (TPO) receptor agonist, and TPO receptor agonists have been associated with thrombotic and thromboembolic complications in patients with chronic liver disease. Portal vein thrombosis has been reported in patients with chronic liver disease treated with TPO receptor agonists.
Pregnancy
Advise women of reproductive potential who become pregnant or are planning to become pregnant that MULPLETA should be used during pregnancy only if the potential benefit to the mother justifies the potential risk to the fetus [see Use in Specific Populations (8.1)] .
Lactation
Advise women not to breastfeed during treatment with MULPLETA and for 28 days after the last dose of MULPLETA. Advise women to pump and discard breast milk during this period [see Use in Specific Populations (8.2)] .
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
MULPLETA ® (mul ple' tah)
(lusutrombopag)
TabletsThis Patient Information has been approved by the U.S. Food and Drug Administration. Issued: 7/2018 What is MULPLETA? MULPLETA is a prescription medicine used to treat low platelet count (thrombocytopenia) in adults with chronic liver disease who are scheduled to have a procedure. MULPLETA is not used to make platelet count normal in people with chronic liver disease. It is not known if MULPLETA is safe and effective in children. Before taking MULPLETA, tell your healthcare provider about all of your medical conditions, including if you: - have a blood clot or have had a history of a blood clot.
- have any blood clotting problems, other than thrombocytopenia.
- are pregnant or plan to become pregnant. MULPLETA may harm your baby.
- are breastfeeding or plan to breastfeed. It is not known if MULPLETA passes into your breast milk. Do not breastfeed during your treatment with MULPLETA and for at least 28 days after your last dose. Talk to your healthcare provider about the best way to feed your baby during your treatment with MULPLETA.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. How should I take MULPLETA? - Take MULPLETA exactly as your healthcare provider tells you to take it.
- Your healthcare provider will tell you when to start taking MULPLETA.
- Take MULPLETA 1 time each day for 7 days.
- MULPLETA may be taken with or without food.
- If you miss a dose of MULPLETA, take the missed dose as soon as possible on the same day and return to your normal schedule the following day.
- If you take too much MULPLETA, call your healthcare provider or go to the nearest hospital emergency room right away.
- Your healthcare provider will check your platelet count before you start treatment with MULPLETA and before your procedure.
What are the possible side effects of MULPLETA? MULPLETA may cause serious side effects, including: Blood clots, including blood clots in the liver, may happen in people with chronic liver disease and who take MULPLETA. You may have an increased risk of blood clots if you have certain blood clotting conditions. The most common side effect of MULPLETA is headache. These are not all of the possible side effects of MULPLETA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. How should I store MULPLETA? - Store MULPLETA at room temperature between 68°F and 77°F (20°C to 25°C).
- MULPLETA comes in a child-resistant blister pack. Keep MULPLETA in the packaging that it comes in.
Keep MULPLETA and all medicines out of the reach of children. General information about the safe and effective use of MULPLETA. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use MULPLETA for a condition for which it was not prescribed. Do not give MULPLETA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about MULPLETA that is written for healthcare professionals. What are the ingredients of MULPLETA? Active ingredient: lusutrombopag. Inactive ingredients: D-mannitol, microcrystalline cellulose, magnesium oxide, sodium lauryl sulfate, hydroxypropyl cellulose, carboxymethylcellulose calcium, magnesium stearate, hypromellose, triethyl citrate, titanium dioxide, red ferric oxide, and talc. Manufactured for: Shionogi Inc., Florham Park, NJ 07932 © Shionogi Inc. 2018 For more information, go to www.mulpleta.com or call 1-800-849-9707. -
PRINCIPAL DISPLAY PANEL - 3 mg Tablet Blister Card Carton
NDC: 59630-551-07
Rx onlyFOR ORAL USE ONLY
Mulpleta ®
(lusutrombopag) tablets
3 mg per tabletSHIONOGI INC.
Lift Here to Open
One blister card
with 7 tablets
-
INGREDIENTS AND APPEARANCE
MULPLETA
lusutrombopag tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 59630-551 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LUSUTROMBOPAG (UNII: 6LL5JFU42F) (LUSUTROMBOPAG - UNII:6LL5JFU42F) LUSUTROMBOPAG 3 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) MAGNESIUM OXIDE (UNII: 3A3U0GI71G) SODIUM LAURYL SULFATE (UNII: 368GB5141J) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) CARBOXYMETHYLCELLULOSE CALCIUM (UNII: UTY7PDF93L) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) TRIETHYL CITRATE (UNII: 8Z96QXD6UM) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) TALC (UNII: 7SEV7J4R1U) Product Characteristics Color red (light red) Score no score Shape ROUND Size 7mm Flavor Imprint Code 551;3 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 59630-551-07 1 in 1 CARTON 08/27/2018 1 7 in 1 DOSE PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA210923 08/27/2018 Labeler - SHIONOGI INC. (098241610)
 ) above the identifier code "551" on one side and with a "3" on the other side.
) above the identifier code "551" on one side and with a "3" on the other side.
Trademark Results [Mulpleta]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 MULPLETA 79163223 4890938 Live/Registered |
SHIONOGI & CO., LTD. 2014-12-22 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.