Etidronate Disodium by Mylan Pharmaceuticals Inc.
Etidronate Disodium by
Drug Labeling and Warnings
Etidronate Disodium by is a Prescription medication manufactured, distributed, or labeled by Mylan Pharmaceuticals Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
ETIDRONATE DISODIUM- etidronate disodium tablet
Mylan Pharmaceuticals Inc.
----------
DESCRIPTION
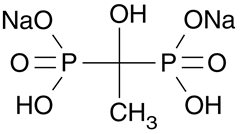
Etidronate disodium tablets, USP contain either 200 mg or 400 mg of etidronate disodium, the disodium salt of (1-hydroxyethylidene) diphosphonic acid, for oral administration. This compound, also known as EHDP, regulates bone metabolism. Etidronate disodium, USP is a white powder, highly soluble in water, with a molecular weight of 250 and the following structural formula:
Inactive ingredients: Each tablet contains magnesium stearate, microcrystalline cellulose, pregelatinized starch and starch (corn).
CLINICAL PHARMACOLOGY
Etidronate disodium acts primarily on bone. It can inhibit the formation, growth, and dissolution of hydroxyapatite crystals and their amorphous precursors by chemisorption to calcium phosphate surfaces. Inhibition of crystal resorption occurs at lower doses than are required to inhibit crystal growth. Both effects increase as the dose increases.
Etidronate disodium is not metabolized. The amount of drug absorbed after an oral dose is approximately 3%. In normal subjects, plasma half-life (t1/2) of etidronate, based on non-compartmental pharmacokinetics is 1 to 6 hours. Within 24 hours, approximately half the absorbed dose is excreted in urine; the remainder is distributed to bone compartments from which it is slowly eliminated. Animal studies have yielded bone clearance estimates up to 165 days. In humans, the residence time on bone may vary due to such factors as specific metabolic condition and bone type. Unabsorbed drug is excreted intact in the feces. Preclinical studies indicate etidronate disodium does not cross the blood-brain barrier.
Etidronate disodium therapy does not adversely affect serum levels of parathyroid hormone or calcium.
Paget’s Disease
Paget’s disease of bone (osteitis deformans) is an idiopathic, progressive disease characterized by abnormal and accelerated bone metabolism in one or more bones. Signs and symptoms may include bone pain and/or deformity, neurologic disorders, elevated cardiac output and other vascular disorders, and increased serum alkaline phosphatase and/or urinary hydroxyproline levels. Bone fractures are common in patients with Paget’s disease.
Etidronate disodium slows accelerated bone turnover (resorption and accretion) in pagetic lesions and, to a lesser extent, in normal bone. This has been demonstrated histologically, scintigraphically, biochemically, and through calcium kinetic and balance studies. Reduced bone turnover is often accompanied by symptomatic improvement, including reduced bone pain. Also, the incidence of pagetic fractures may be reduced and elevated cardiac output and other vascular disorders may be improved by etidronate disodium therapy.
Heterotopic Ossification
Heterotopic ossification, also referred to as myositis ossificans (circumscripta, progressiva or traumatica), ectopic calcification, periarticular ossification, or paraosteoarthropathy, is characterized by metaplastic osteogenesis. It usually presents with signs of localized inflammation or pain, elevated skin temperature, and redness. When tissues near joints are involved, functional loss may also be present.
Heterotopic ossification may occur for no known reason as in myositis ossificans progressiva or may follow a wide variety of surgical, occupational, and sports trauma (e.g., hip arthroplasty, spinal cord injury, head injury, burns, and severe thigh bruises). Heterotopic ossification has also been observed in non-traumatic conditions (e.g., infections of the central nervous system, peripheral neuropathy, tetanus, biliary cirrhosis, Peyronie’s disease, as well as in association with a variety of benign and malignant neoplasms).
Clinical trials have demonstrated the efficacy of etidronate disodium in heterotopic ossification following total hip replacement or due to spinal cord injury.
- Heterotopic ossification complicating total hip replacement typically develops radiographically 3 to 8 weeks postoperatively in the pericapsular area of the affected hip joint. The overall incidence is about 50%; about one-third of these cases are clinically significant.
- Heterotopic ossification due to spinal cord injury typically develops radiographically 1 to 4 months after injury. It occurs below the level of injury, usually at major joints. The overall incidence is about 40%; about one-half of these cases are clinically significant.
Etidronate disodium chemisorbs to calcium hydroxyapatite crystals and their amorphous precursors, blocking the aggregation, growth, and mineralization of these crystals. This is thought to be the mechanism by which etidronate disodium prevents or retards heterotopic ossification. There is no evidence etidronate disodium affects mature heterotopic bone.
INDICATIONS AND USAGE
Etidronate disodium tablets, USP are indicated for the treatment of symptomatic Paget’s disease of bone and in the prevention and treatment of heterotopic ossification following total hip replacement or due to spinal cord injury. Etidronate disodium tablets are not approved for the treatment of osteoporosis.
Paget’s Disease
Etidronate disodium tablets are indicated for the treatment of symptomatic Paget’s disease of bone. Etidronate disodium therapy usually arrests or significantly impedes the disease process as evidenced by:
- Symptomatic relief, including decreased pain and/or increased mobility (experienced by 3 out of 5 patients).
- Reductions in serum alkaline phosphatase and urinary hydroxyproline levels (30% or more in 4 out of 5 patients).
- Histomorphometry showing reduced numbers of osteoclasts and osteoblasts, and more lamellar bone formation.
- Bone scans showing reduced radionuclide uptake at pagetic lesions.
In addition, reductions in pagetically elevated cardiac output and skin temperature have been observed in some patients.
In many patients, the disease process will be suppressed for a period of at least one year following cessation of therapy. The upper limit of this period has not been determined.
The effects of the etidronate disodium treatment in patients with asymptomatic Paget’s disease have not been studied. However, etidronate disodium treatment of such patients may be warranted if extensive involvement threatens irreversible neurologic damage, major joints, or major weight-bearing bones.
Heterotopic Ossification
Etidronate disodium tablets are indicated in the prevention and treatment of heterotopic ossification following total hip replacement or due to spinal cord injury.
Etidronate disodium tablets reduce the incidence of clinically important heterotopic bone by about two-thirds. Among those patients who form heterotopic bone, etidronate disodium tablets retard the progression of immature lesions and reduces the severity by at least half. Follow-up data (at least 9 months post-therapy) suggests these benefits persist.
In total hip replacement patients, etidronate disodium tablets do not promote loosening of the prosthesis or impede trochanteric reattachment.
In spinal cord injury patients, etidronate disodium tablets do not inhibit fracture healing or stabilization of the spine.
CONTRAINDICATIONS
- Abnormalities of the esophagus which delay esophageal emptying such as stricture or achalasia.
- Known hypersensitivity to etidronate disodium or in patients with clinically overt osteomalacia.
WARNINGS
General
Upper Gastrointestinal Adverse Reactions
Etidronate disodium, like other bisphosphonates administered orally, may cause local irritation of the upper gastrointestinal mucosa. Because of these possible irritant effects and a potential for worsening of the underlying disease, caution should be used when etidronate disodium is given to patients with active upper gastrointestinal problems (such as known Barrett’s esophagus, dysphagia, other esophageal diseases, gastritis, duodenitis or ulcers).
Esophageal adverse experiences, such as esophagitis, esophageal ulcers and esophageal erosions, occasionally with bleeding and rarely followed by esophageal stricture or perforation, have been reported in patients receiving treatment with oral bisphosphonates. In some cases, these have been severe and required hospitalization. Physicians should therefore be alert to any signs or symptoms signaling a possible esophageal reaction and patients should be instructed to discontinue etidronate disodium and seek medical attention if they develop dysphagia, odynophagia, retrosternal pain or new or worsening heartburn.
The risk of severe esophageal adverse experiences appears to be greater in patients who lie down after taking oral bisphosphonates and/or who fail to swallow it with the recommended full glass (6 to 8 oz) of water, and/or who continue to take oral bisphosphonates after developing symptoms suggestive of esophageal irritation. Therefore, it is very important that the full dosing instructions are provided to, and understood by, the patient (see DOSAGE AND ADMINISTRATION). In patients who cannot comply with dosing instructions due to mental disability, therapy with etidronate disodium should be used under appropriate supervision.
There have been post-marketing reports of gastric and duodenal ulcers with oral bisphosphonate use, some severe and with complications, although no increased risk was observed in controlled clinical trials.
PRECAUTIONS
General
Patients should maintain an adequate nutritional status, particularly an adequate intake of calcium and vitamin D.
Therapy has been withheld from some patients with enterocolitis since diarrhea may be experienced, particularly at higher doses.
Etidronate disodium is not metabolized and is excreted intact via the kidney. Hyperphosphatemia may occur at doses of 10 to 20 mg/kg/day, apparently as a result of drug-related increases in tubular reabsorption of phosphate. Serum phosphate levels generally return to normal 2 to 4 weeks post therapy. There is no experience to specifically guide treatment in patients with impaired renal function. Etidronate disodium dosage should be reduced when reductions in glomerular filtration rates are present. Patients with renal impairment should be closely monitored. In approximately 10% of patients in clinical trials of etidronate disodium I.V. infusion, for hypercalcemia of malignancy, occasional, mild-to-moderate abnormalities in renal function (increases of > 0.5 mg/dL serum creatinine) were observed during or immediately after treatment.
Etidronate disodium suppresses bone turnover, and may retard mineralization of osteoid laid down during the bone accretion process. These effects are dose and time dependent. Osteoid, which may accumulate noticeably at doses of 10 to 20 mg/kg/day, mineralizes normally post therapy. In patients with fractures, especially of long bones, it may be advisable to delay or interrupt treatment until callus is evident.
Osteonecrosis of the Jaw (ONJ)
ONJ, which can occur spontaneously, is generally associated with tooth extraction and/or local infection with delayed healing, and has been reported in patients taking bisphosphonates, including etidronate sodium. Known risk factors for osteonecrosis of the jaw include invasive dental procedures (e.g., tooth extraction, dental implants, boney surgery), diagnosis of cancer, concomitant therapies (e.g., chemotherapy, corticosteroids, angiogenesis inhibitors), poor oral hygiene, and co-morbid disorders (e.g., periodontal and/or other pre-existing dental disease, anemia, coagulopathy, infection, ill-fitting dentures). The risk of ONJ may increase with duration of exposure to bisphosphonates.
For patients requiring invasive dental procedures, discontinuation of bisphosphonate treatment may reduce the risk for ONJ. Clinical judgment of the treating physician and/or oral surgeon should guide the management plan of each patient based on individual benefit/risk assessment.
Patients who develop osteonecrosis of the jaw while on bisphosphonate therapy should receive care by an oral surgeon. In these patients, extensive dental surgery to treat ONJ may exacerbate the condition. Discontinuation of bisphosphonate therapy should be considered based on individual benefit/risk assessment.
Musculoskeletal Pain
In post-marketing experience, there have been infrequent reports of severe and occasionally incapacitating bone, joint, and/or muscle pain in patients taking bisphosphonates (see ADVERSE REACTIONS). The time to onset of symptoms varied from one day to several months after starting the drug. Most patients had relief of symptoms after stopping medication. A subset had recurrence of symptoms when rechallenged with the same drug or another bisphosphonate.
Paget’s Disease
In Paget’s patients, treatment regimens exceeding the recommended (see DOSAGE AND ADMINISTRATION) daily maximum dose of 20 mg/kg or continuous administration of medication for periods greater than 6 months may be associated with osteomalacia and an increased risk of fracture.
Long bones predominantly affected by lytic lesions, particularly in those patients unresponsive to etidronate disodium therapy, may be especially prone to fracture.
Patients with predominantly lytic lesions should be monitored radiographically and biochemically to permit termination of etidronate disodium in those patients unresponsive to treatment.
Drug Interactions
There have been isolated reports of patients experiencing increases in their prothrombin times when etidronate was added to warfarin therapy. The majority of these reports concerned variable elevations in prothrombin times without clinically significant sequelae. Although the relevance of these reports and any mechanism of coagulation alterations is unclear, patients on warfarin should have their prothrombin time monitored.
Carcinogenesis
Long-term studies in rats have indicated that etidronate disodium is not carcinogenic.
Pregnancy
Teratogenic Effects. Pregnancy Category C
In teratology and developmental toxicity studies conducted in rats and rabbits treated with dosages of up to 100 mg/kg (5 to 20 times the clinical dose), no adverse or teratogenic effects have been observed in the offspring. Etidronate disodium has been shown to cause skeletal abnormalities in rats when given at oral dose levels of 300 mg/kg (15 to 60 times the human dose). Other effects on the offspring (including decreased live births) are at dosages that cause significant toxicity in the parent generation and are 25 to 200 times the human dose. The skeletal effects are thought to be the result of the pharmacological effects of the drug on bone.
Bisphosphonates are incorporated into the bone matrix, from which they are gradually released over periods of weeks to years. The amount of bisphosphonate incorporation into adult bone, and hence, the amount available for release back into the systemic circulation, is directly related to the dose and duration of bisphosphonate use. There are no data on fetal risk in humans. However, there is a theoretical risk of fetal harm, predominantly skeletal, if a woman becomes pregnant after completing a course of bisphosphonate therapy. The impact of variables such as time between cessation of bisphosphonate therapy to conception, the particular bisphosphonate used, and the route of administration (intravenous vs. oral) on this risk has not been studied.
There are no adequate and well controlled studies in pregnant women. Etidronate disodium should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when etidronate disodium is administered to a nursing woman.
Pediatric Use
Safety and effectiveness in pediatric patients have not been established. Pediatric patients have been treated with etidronate disodium, at doses recommended for adults, to prevent heterotopic ossifications or soft tissue calcifications. A rachitic syndrome has been reported infrequently at doses of 10 mg/kg/day and more for prolonged periods approaching or exceeding a year. The epiphyseal radiologic changes associated with retarded mineralization of new osteoid and cartilage, and occasional symptoms reported, have been reversible when medication is discontinued.
Geriatric Use
Clinical studies of etidronate disodium did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between elderly and younger patients. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy. This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken when prescribing this drug therapy. As stated in PRECAUTIONS, etidronate disodium dosage should be reduced when reductions in glomerular filtration rates are present. In addition, patients with renal impairment should be closely monitored.
ADVERSE REACTIONS
The incidence of gastrointestinal complaints (diarrhea, nausea) is the same for etidronate disodium at 5 mg/kg/day as for placebo, about 1 patient in 15. At 10 to 20 mg/kg/day the incidence may increase to 2 or 3 in 10. These complaints are often alleviated by dividing the total daily dose.
Paget’s Disease
In Paget’s patients, increased or recurrent bone pain at pagetic sites, and/or the onset of pain at previously asymptomatic sites has been reported. At 5 mg/kg/day about 1 patient in 10 (vs. 1 in 15 in the placebo group) report these phenomena. At higher doses the incidence rises to about 2 in 10. When therapy continues, pain resolves in some patients but persists in others.
Worldwide Post-Marketing Experience
The worldwide post-marketing experience for etidronate disodium reflects its use in the following approved indications: Paget’s disease, heterotopic ossification, and hypercalcemia of malignancy. It also reflects the use of etidronate disodium for osteoporosis where approved in countries outside the US. Other adverse events that have been reported and were thought to be possibly related to etidronate disodium include the following: alopecia; arthropathies, including arthralgia and arthritis; bone fracture; esophagitis; glossitis; hypersensitivity reactions, including angioedema, follicular eruption, macular rash, maculopapular rash, pruritus, Stevens-Johnson syndrome, toxic epidermal necrolysis, and urticaria; osteomalacia; neuropsychiatric events, including amnesia, confusion, depression, and hallucination; and paresthesias.
In patients receiving etidronate disodium, there have been rare reports of agranulocytosis, pancytopenia, and a report of leukopenia with recurrence on rechallenge. In addition, there have been rare reports of exacerbation of asthma. Exacerbation of existing peptic ulcer disease including perforation has been reported rarely.
In osteoporosis clinical trials, headache, gastritis, leg cramps, and arthralgia occurred at a significantly greater incidence in patients who received etidronate as compared with those who received placebo.
OVERDOSAGE
Clinical experience with acute etidronate disodium overdosage is extremely limited. Decreases in serum calcium following substantial overdosage may be expected in some patients. Signs and symptoms of hypocalcemia also may occur in some of these patients. Some patients may develop vomiting. In one event, an 18 year old female who ingested an estimated single dose of 4000 mg to 6000 mg (67 to 100 mg/kg) of etidronate disodium was reported to be mildly hypocalcemic (7.52 mg/dL) and experienced paresthesia of the fingers. Hypocalcemia resolved 6 hours after lavage and treatment with intravenous calcium gluconate. A 92 year old female who accidentally received 1600 mg of etidronate disodium per day for 3.5 days experienced marked diarrhea and required treatment for electrolyte imbalance. Orally administered etidronate disodium may cause hematologic abnormalities in some patients (see ADVERSE REACTIONS).
Etidronate disodium suppresses bone turnover and may retard mineralization of osteoid laid down during the bone accretion process. These effects are dose and time dependent. Osteoid which may accumulate noticeably at doses of 10 to 20 mg/kg/day of chronic, continuous dosing mineralizes normally post therapy.
Prolonged continuous treatment (chronic overdosage) has been reported to cause nephrotic syndrome and fracture.
Gastric lavage may remove unabsorbed drug. Standard procedures for treating hypocalcemia, including the administration of Ca++ intravenously, would be expected to restore physiologic amounts of ionized calcium and relieve signs and symptoms of hypocalcemia. Such treatment has been effective.
DOSAGE AND ADMINISTRATION
Etidronate disodium tablets should be taken as a single, oral dose. As with other bisphosphonates, it is recommended that etidronate disodium tablets should be swallowed with a full glass of water (6 to 8 oz). Patients should not lie down after taking the medication. However, should gastrointestinal discomfort occur, the dose may be divided. To maximize absorption, patients should avoid taking the following items within 2 hours of dosing:
- Food, especially food high in calcium, such as milk or milk products.
- Vitamins with mineral supplements or antacids which are high in metals such as calcium, iron, magnesium, or aluminum.
Paget’s Disease
Initial Treatment Regimens
5 to 10 mg/kg/day, not to exceed 6 months or 11 to 20 mg/kg/day, not to exceed 3 months.
The recommended initial dose is 5 mg/kg/day for a period not to exceed 6 months. Doses above 10 mg/kg/day should be reserved for when 1) lower doses are ineffective or 2) there is an overriding need to suppress rapid bone turnover (especially when irreversible neurologic damage is possible) or reduce elevated cardiac output. Doses in excess of 20 mg/kg/day are not recommended.
Retreatment Guidelines
Retreatment should be initiated only after 1) an etidronate disodium-free period of at least 90 days and 2) there is biochemical, symptomatic or other evidence of active disease process. It is advisable to monitor patients every 3 to 6 months although some patients may go drug free for extended periods. Retreatment regimens are the same as for initial treatment. For most patients the original dose will be adequate for retreatment. If not, consideration should be given to increasing the dose within the recommended guidelines.
Heterotopic Ossification
The following treatment regimens have been shown to be effective:
- Total Hip Replacement Patients: 20 mg/kg/day for 1 month before and 3 months after surgery (4 months total).
- Spinal Cord Injured Patients: 20 mg/kg/day for 2 weeks followed by 10 mg/kg/day for 10 weeks (12 weeks total). Etidronate disodium therapy should begin as soon as medically feasible following the injury, preferably prior to evidence of heterotopic ossification.
Retreatment has not been studied.
HOW SUPPLIED
Etidronate Disodium Tablets, USP are available containing 200 mg or 400 mg of etidronate disodium, USP.
The 200 mg tablets are white, rectangular-shaped tablets with ED 200 on one side and G on the other side. They are available as follows:
NDC: 0378-3286-91
bottles of 60
The 400 mg tablets are white, capsule-shaped tablets with ED 400 on one side and G on the other side. They are available as follows:
NDC: 0378-3288-91
bottles of 60
Store at 20° to 25°C (68° to 77°F). [See USP Controlled Room Temperature.]
Avoid excessive heat (over 104°F or 40°C).
Dispense in a tight, light-resistant container as defined in the USP using a child-resistant closure.
Manufactured by:
ALPHAPHARM PTY LTD
15 Garnet Street
Carole Park QLD 4300
Australia
ALP:ETDN:R8
Revised: 3/2017
PRINCIPAL DISPLAY PANEL - 200 mg
NDC: 0378-3286-91
Etidronate
Disodium
Tablets, USP
200 mg
Rx only 60 Tablets
Each tablet contains:
Etidronate disodium, USP 200 mg
Dispense in a tight, light-resistant
container as defined in the USP
using a child-resistant closure.
Keep container tightly closed.
Keep this and all medication
out of the reach of children.
Store at 20° to 25°C (68° to 77°F).
[See USP Controlled Room
Temperature.]
Avoid excessive heat (over 104°F
or 40°C).
Usual Dosage: See accompanying
prescribing information.
Manufactured for:
Mylan Pharmaceuticals Inc.
Morgantown, WV 26505 U.S.A.
Made in Australia
3290/0
RM3286D4

PRINCIPAL DISPLAY PANEL - 400 mg
NDC: 0378-3288-91
Etidronate
Disodium
Tablets, USP
400 mg
Rx only 60 Tablets
Each tablet contains:
Etidronate disodium, USP 400 mg
Dispense in a tight, light-resistant
container as defined in the USP
using a child-resistant closure.
Keep container tightly closed.
Keep this and all medication
out of the reach of children.
Store at 20° to 25°C (68° to 77°F).
[See USP Controlled Room
Temperature.]
Avoid excessive heat (over 104°F
or 40°C).
Usual Dosage: See accompanying
prescribing information.
Manufactured for:
Mylan Pharmaceuticals Inc.
Morgantown, WV 26505 U.S.A.
Made in Australia
3291/0
RM3288D4

| ETIDRONATE DISODIUM
etidronate disodium tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| ETIDRONATE DISODIUM
etidronate disodium tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - Mylan Pharmaceuticals Inc. (059295980) |