OLANZAPINE AND FLUOXETINE capsule
Olanzapine and Fluoxetine by
Drug Labeling and Warnings
Olanzapine and Fluoxetine by is a Prescription medication manufactured, distributed, or labeled by Sandoz Inc. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use OLANZAPINE AND FLUOXETINE CAPSULES safely and effectively. See full prescribing information for OLANZAPINE AND FLUOXETINE CAPSULES.
OLANZAPINE and FLUOXETINE capsules, for oral use.
Initial U.S. Approval: 2003WARNINGS: SUICIDAL THOUGHTS AND BEHAVIORS; AND INCREASED MORTALITY IN ELDERLY PATIENTS WITH DEMENTIA-RELATED PSYCHOSIS
See full prescribing information for complete boxed warning.
- Increased risk of suicidal thinking and behavior in children, adolescents, and young adults taking antidepressants. Olanzapine and fluoxetine hydrochloride is not approved for use in children less than 10 years of age (5.1, 8.4, 17.2).
- Monitor for worsening and emergence of suicidal thoughts and behaviors (5.1, 17.2)
- Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. Olanzapine and fluoxetine hydrochloride is not approved for the treatment of patients with dementia-related psychosis (5.2, 5.21, 17.3).
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
Olanzapine and fluoxetine capsules combines olanzapine, an atypical antipsychotic and fluoxetine, a selective serotonin reuptake inhibitor, indicated for treatment of:
- Acute Depressive Episodes Associated with Bipolar I Disorder (1)
DOSAGE AND ADMINISTRATION
- Adult Starting Dose: 6 mg olanzapine with 25 mg fluoxetine (6 mg/25 mg, once daily in the evening (2.1)
- Adult Maximum Dose: 12 mg/50 mg once daily (2.1)
- Pediatric Bipolar Depression Starting Dose: 3 mg/25 mg once daily (for ages 10 to 17 years) (2.1)
- Pediatric Bipolar Depression Maximum Dose: 12 mg/50 mg (2.1)
- Starting dose in patients predisposed to hypotensive reactions, hepatic impairment, or with potential for slowed metabolism: 3 mg/25 mg to 6 mg/25 mg . Escalate dose cautiously (2.3)
DOSAGE FORMS AND STRENGTHS
Capsules: 3 mg olanzapine/25 mg fluoxetine, 6 mg/25 mg, 6 mg/50 mg, 12 mg/25 mg, and 12 mg/50 mg (mg equivalent olanzapine/mg equivalent fluoxetine) (3)
CONTRAINDICATIONS
- Monoamine Oxidase Inhibitors (MAOI): Because of the risk of serotonin syndrome, do not use MAOIs intended to treat psychiatric disorders with olanzapine and fluoxetine hydrochloride or within 5 weeks of stopping treatment with olanzapine and fluoxetine hydrochloride. Do not use olanzapine and fluoxetine hydrochloride within 14 days of stopping an MAOI intended to treat psychiatric disorders. In addition, do not start olanzapine and fluoxetine hydrochloride in a patient who is being treated with linezolid or intravenous methylene blue. (4.1)
- Pimozide: Do not use. Risk of QT interval prolongation (4.2, 5.20, 7.7, 7.8)
- Thioridazine: Do not use. Risk of QT interval prolongation. Do not use thioridazine within 5 weeks of discontinuing olanzapine and fluoxetine hydrochloride (4.2, 5.20, 7.7, 7.8)
WARNINGS AND PRECAUTIONS
- Neuroleptic Malignant Syndrome: Manage with immediate discontinuation and close monitoring (5.3)
- Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS): Discontinue if DRESS is suspected (5.4)
- Metabolic Changes: Atypical antipsychotic drugs have been associated with metabolic changes including hyperglycemia, dyslipidemia, and weight gain (5.5)
- Hyperglycemia and Diabetes Mellitus: In some cases extreme and associated with ketoacidosis or hyperosmolar coma or death. Monitor for symptoms of hyperglycemia. Perform fasting blood glucose testing before beginning, and periodically during treatment (5.5)
- Dyslipidemia: Appropriate clinical monitoring is recommended, including fasting blood lipid testing before beginning, and periodically during, treatment (5.5)
- Weight gain: Consider potential consequences of weight gain. Monitor weight regularly (5.5)
- Serotonin Syndrome: Serotonin syndrome has been reported with SSRIs and SNRIs, including olanzapine and fluoxetine hydrochloride, both when taken alone, but especially when co-administered with other serotonergic agents (including triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, tryptophan, buspirone, amphetamines, and St. John's Wort). If such symptoms occur, discontinue olanzapine and fluoxetine hydrochloride and initiate supportive treatment. If concomitant use of olanzapine and fluoxetine hydrochloride with other serotonergic drugs is clinically warranted, patients should be made aware of a potential increased risk for serotonin syndrome, particularly during treatment initiation and dose increases (5.6)
- Angle-Closure Glaucoma: Angle-closure glaucoma has occurred in patients with untreated anatomically narrow angles treated with antidepressants (5.7)
- Allergic Reactions and Rash: Discontinue upon appearance of rash or allergic phenomena (5.8)
- Activation of Mania/Hypomania: Screen for Bipolar Disorder and monitor for activation of mania/hypomania (5.9)
- Tardive Dyskinesia: Discontinue if clinically appropriate (5.10)
- Orthostatic Hypotension: Can be associated with bradycardia and syncope. Risk is increased during initial dose titration. Use caution in patients with cardiovascular disease or cerebrovascular disease, and those conditions that could affect hemodynamic responses (5.11)
- Leukopenia, Neutropenia, and Agranulocytosis: Has been reported with antipsychotics, including olanzapine and fluoxetine hydrochloride. Patients with a history of a clinically significant low white blood cell count (WBC) or drug induced leukopenia/neutropenia should have their complete blood count (CBC) monitored frequently during the first few months of therapy. Consider discontinuing olanzapine and fluoxetine hydrochloride at the first sign of a clinically significant decline in WBC in the absence of other causative factors (5.13)
- Seizures: Use cautiously in patients with a history of seizures or with conditions that lower the seizure threshold (5.15)
- Abnormal Bleeding: SSRIs increase the risk of bleeding. Use with NSAIDs, aspirin, warfarin, or other drugs that affect coagulation may potentiate the risk of gastrointestinal or other bleeding (5.16)
- Hyponatremia: Can occur in association with syndrome of inappropriate antidiuretic hormone (SIADH). Consider discontinuing olanzapine and fluoxetine hydrochloride if symptomatic hyponatremia occurs (SIADH) (5.17)
- Potential for Cognitive and Motor Impairment: Has potential to impair judgment, thinking, and motor skills. Caution patients about operating machinery (5.18)
- QT Prolongation: QT prolongation and ventricular arrhythmia including Torsade de Pointes have been reported with fluoxetine. Use with caution in conditions that predispose to arrhythmias or increased fluoxetine exposure. Use cautiously in patients with risk factors for QT prolongation (4.2, 5.20)
- Hyperprolactinemia: May elevate prolactin levels (5.22)
- Long Elimination Half-Life of Fluoxetine: Changes in dose will not be fully reflected in plasma for several weeks (5.24)
ADVERSE REACTIONS
Most common adverse reactions (≥5% and at least twice that for placebo) in adults: sedation, weight increased, appetite increased, dry mouth, fatigue, edema, tremor, disturbance in attention, blurred vision. Children and adolescents: sedation, weight increased, appetite increased, tremor, triglyceride increased, hepatic enzymes increased (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Sandoz Inc. at 1-800-525-8747 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
DRUG INTERACTIONS
- Monoamine Oxidase Inhibitor (MAOI): (2.4, 2.5, 4.1, 5.6, 7.1)
- Drugs Metabolized by CYP2D6: Fluoxetine is a potent inhibitor of CYP2D6 enzyme pathway (7.7)
- Tricyclic Antidepressants (TCAs): Monitor TCA levels during coadministration with olanzapine and fluoxetine hydrochloride or when olanzapine and fluoxetine hydrochloride has been recently discontinued (5.6, 7.7)
- CNS Acting Drugs: Caution is advised if the concomitant administration of olanzapine and fluoxetine hydrochloride and other CNS-active drugs is required (7.2)
- Antihypertensive Agent: Enhanced antihypertensive effect (7.7)
- Levodopa and Dopamine Agonists: May antagonize levodopa/dopamine agonists (7.7)
- Benzodiazepines: May potentiate orthostatic hypotension and sedation (7.6, 7.7)
- Clozapine: May elevate clozapine levels (7.7)
- Haloperidol: Elevated haloperidol levels have been observed (7.7)
- Carbamazepine: Potential for elevated carbamazepine levels and clinical anticonvulsant toxicity (7.7)
- Phenytoin: Potential for elevated phenytoin levels and clinical anticonvulsant toxicity (7.7)
- Alcohol: May potentiate sedation and orthostatic hypotension (7.7)
- Serotonergic Drugs: (2.4, 2.5, 4.1, 5.6, 7.3)
- Fluvoxamine: May increase olanzapine levels; a lower dose of the olanzapine component of olanzapine and fluoxetine hydrochloride should be considered (7.6)
- Drugs that Interfere with Hemostasis; (e.g., NSAIDs, Aspirin, Warfarin, etc.): May potentiate the risk of bleeding (7.4)
- Drugs Tightly Bound to Plasma Proteins: Fluoxetine may cause shift in plasma concentrations (7.7)
- Drugs that Prolong the QT Interval: Do not use olanzapine and fluoxetine hydrochloride in combination with thioridazine or pimozide. Use olanzapine and fluoxetine hydrochloride with caution in combination with other drugs that prolong the QT interval (4.2, 5.20, 7.7, 7.8)
USE IN SPECIFIC POPULATIONS
- Pregnancy: Olanzapine and fluoxetine hydrochloride should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus (8.1)
- Nursing Mothers: Breast feeding is not recommended (8.3)
- Pediatric Use: Safety and efficacy of olanzapine and fluoxetine hydrochloride for the treatment of bipolar I depression in patients under 10 years of age have not been established. (8.4)
- Hepatic Impairment: Use a lower or less frequent dose in patients with cirrhosis (8.6)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 4/2017
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNINGS: SUICIDAL THOUGHTS AND BEHAVIORS; AND INCREASED MORTALITY IN ELDERLY PATIENTS WITH DEMENTIA-RELATED PSYCHOSIS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Depressive Episodes Associated with Bipolar I Disorder
2.3 Specific Populations
2.4 Switching a Patient To or From a Monoamine Oxidase Inhibitor (MAOI) Intended to Treat Psychiatric Disorders
2.5 Use of Olanzapine and Fluoxetine Capsules with Other MAOIs such as Linezolid or Methylene Blue
2.6 Discontinuation of Treatment with Olanzapine and Fluoxetine Capsules
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Monoamine Oxidase Inhibitors (MAOIs)
4.2 Other Contraindications
5 WARNINGS AND PRECAUTIONS
5.1 Suicidal Thoughts and Behaviors in Children, Adolescents, and Young Adults
5.2 Increased Mortality in Elderly Patients with Dementia-Related Psychosis
5.3 Neuroleptic Malignant Syndrome (NMS)
5.4 Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)
5.5 Metabolic Changes
5.6 Serotonin Syndrome
5.7 Angle-Closure Glaucoma
5.8 Allergic Reactions and Rash
5.9 Activation of Mania/Hypomania
5.10 Tardive Dyskinesia
5.11 Orthostatic Hypotension
5.12 Falls
5.13 Leukopenia, Neutropenia, and Agranulocytosis
5.14 Dysphagia
5.15 Seizures
5.16 Abnormal Bleeding
5.17 Hyponatremia
5.18 Potential for Cognitive and Motor Impairment
5.19 Body Temperature Dysregulation
5.20 QT Prolongation
5.21 Use in Patients with Concomitant Illness
5.22 Hyperprolactinemia
5.23 Concomitant Use of Olanzapine and Fluoxetine Products
5.24 Long Elimination Half-Life of Fluoxetine
5.25 Discontinuation Adverse Reactions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Vital Signs and Laboratory Studies
6.3 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Monoamine Oxidase Inhibitors (MAOIs)
7.2 CNS Acting Drugs
7.3 Serotonergic Drugs
7.4 Drugs that Interfere with Hemostasis (e.g., NSAIDs, Aspirin, Warfarin)
7.5 Electroconvulsive Therapy (ECT)
7.6 Potential for Other Drugs to Affect Olanzapine and Fluoxetine Hydrochloride
7.7 Potential for Olanzapine and Fluoxetine Hydrochloride to Affect Other Drugs
7.8 Drugs that Prolong the QT Interval
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Labor and Delivery
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
9 DRUG ABUSE AND DEPENDENCE
9.3 Dependence
10 OVERDOSAGE
10.1 Management of Overdose
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Specific Populations
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Depressive Episodes Associated with Bipolar I Disorder
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
17.1 Information on Medication Guide
17.2 Suicidal Thoughts and Behaviors in Children, Adolescents, and Young Adults
17.3 Elderly Patients with Dementia-Related Psychosis: Increased Mortality and Cerebrovascular Adverse Events (CVAE), Including Stroke
17.4 Neuroleptic Malignant Syndrome (NMS)
17.5 Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)
17.6 Hyperglycemia and Diabetes Mellitus
17.7 Dyslipidemia
17.8 Weight Gain
17.9 Serotonin Syndrome
17.10 Angle-Closure Glaucoma
17.11 Allergic Reactions and Rash
17.12 Orthostatic Hypotension
17.13 Abnormal Bleeding
17.14 Hyponatremia
17.15 Potential for Cognitive and Motor Impairment
17.16 Body Temperature Dysregulation
17.17 Concomitant Medication
17.18 Discontinuation of Treatment with Olanzapine and Fluoxetine Capsules
17.19 Alcohol
17.20 Use in Specific Populations
17.21 QT Prolongation
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNINGS: SUICIDAL THOUGHTS AND BEHAVIORS; AND INCREASED MORTALITY IN ELDERLY PATIENTS WITH DEMENTIA-RELATED PSYCHOSIS
Suicidal Thoughts and Behaviors
Antidepressants increased the risk of suicidal thoughts and behavior in children, adolescents, and young adults in short-term studies. These studies did not show an increase in the risk of suicidal thoughts and behavior with antidepressant use in patients over age 24; there was a reduction in risk with antidepressant use in patients aged 65 and older.
In patients of all ages who are started on antidepressant therapy, monitor closely for worsening and emergence of suicidal thoughts and behaviors. Advise families and caregivers of the need for close observation and communication with the prescriber. Olanzapine and fluoxetine hydrochloride is not approved for use in children less than 10 years of age [see WARNINGS AND PRECAUTIONS (5.1), USE IN SPECIFIC POPULATIONS (8.4), and PATIENT COUNSELING INFORMATION (17.2)].
Increased Mortality in Elderly Patients with Dementia-Related Psychosis
Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. Olanzapine and fluoxetine hydrochloride is not approved for the treatment of patients with dementia-related psychosis [see WARNINGS AND PRECAUTIONS (5.2)].
-
1 INDICATIONS AND USAGE
Olanzapine and fluoxetine capsules are indicated for the treatment of:
- Acute depressive episodes in Bipolar I Disorder [see CLINICAL STUDIES (14.1)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Depressive Episodes Associated with Bipolar I Disorder
Adults
Olanzapine and fluoxetine capsules should be administered once daily in the evening, generally beginning with the 6 mg/25 mg (mg equivalent olanzapine/mg equivalent fluoxetine) capsule. While food has no appreciable effect on the absorption of olanzapine and fluoxetine given individually, the effect of food on the absorption of olanzapine and fluoxetine capsules has not been studied. Make dosage adjustments, if indicated, according to efficacy and tolerability. Antidepressant efficacy was demonstrated with olanzapine and fluoxetine capsules in a dose range of olanzapine 6 mg to 12 mg and fluoxetine 25 mg to 50 mg [see CLINICAL STUDIES (14.1)]. The safety of doses above 18 mg of olanzapine and 75 mg of fluoxetine has not been evaluated in adult clinical studies. Periodically reexamine the need for continued pharmacotherapy.
Children and Adolescents (10 to 17 years of age)
Administer olanzapine and fluoxetine capsules once daily in the evening, generally beginning with the 3 mg/25 mg capsule, without regard to meals, with a recommended target dose within the approved dosing range (6/25; 6/50; 12/25; 12/50 mg) [see CLINICAL STUDIES (14.1)]. The safety of doses above 12 mg of olanzapine and 50 mg of fluoxetine has not been evaluated in pediatric clinical studies. Periodically reexamine the need for continued pharmacotherapy.
2.3 Specific Populations
Start olanzapine and fluoxetine capsules at 3 mg/25 mg or 6 mg/25 mg in patients with a predisposition to hypotensive reactions, patients with hepatic impairment, or patients who exhibit a combination of factors that may slow the metabolism of olanzapine and fluoxetine capsules (female gender, geriatric age, nonsmoking status) or those patients who may be pharmacodynamically sensitive to olanzapine. Titrate slowly and adjust dosage as needed in patients who exhibit a combination of factors that may slow metabolism. Olanzapine and fluoxetine capsules has not been systematically studied in patients >65 years of age or in patients <10 years of age [see WARNINGS AND PRECAUTIONS (5.21), USE IN SPECIFIC POPULATIONS (8.5), and CLINICAL PHARMACOLOGY (12.3, 12.4)].
Treatment of Pregnant Women
When treating pregnant women with fluoxetine, a component of olanzapine and fluoxetine capsules, the physician should carefully consider the potential risks and potential benefits of treatment. Neonates exposed to SSRIs or SNRIs late in the third trimester have developed complications requiring prolonged hospitalizations, respiratory support, and tube feeding [see USE IN SPECIFIC POPULATIONS (8.1)].
2.4 Switching a Patient To or From a Monoamine Oxidase Inhibitor (MAOI) Intended to Treat Psychiatric Disorders
At least 14 days should elapse between discontinuation of an MAOI intended to treat psychiatric disorders and initiation of therapy with olanzapine and fluoxetine capsules. Conversely, at least 5 weeks should be allowed after stopping olanzapine and fluoxetine capsules before starting an MAOI intended to treat psychiatric disorders [see CONTRAINDICATIONS (4.1)].
2.5 Use of Olanzapine and Fluoxetine Capsules with Other MAOIs such as Linezolid or Methylene Blue
Do not start olanzapine and fluoxetine capsules in a patient who is being treated with linezolid or intravenous methylene blue because there is an increased risk of serotonin syndrome. In a patient who requires more urgent treatment of a psychiatric condition, other interventions, including hospitalization, should be considered [see CONTRAINDICATIONS (4.1)].
In some cases, a patient already receiving olanzapine and fluoxetine capsules therapy may require urgent treatment with linezolid or intravenous methylene blue. If acceptable alternatives to linezolid or intravenous methylene blue treatment are not available and the potential benefits of linezolid or intravenous methylene blue are judged to outweigh the risks of serotonin syndrome in a particular patient, olanzapine and fluoxetine capsules should be stopped promptly, and linezolid or intravenous methylene blue can be administered. The patient should be monitored for symptoms of serotonin syndrome for five weeks or until 24 hours after the last dose of linezolid or intravenous methylene blue, whichever comes first. Therapy with olanzapine and fluoxetine capsules may be resumed 24 hours after the last dose of linezolid or intravenous methylene blue [see WARNINGS AND PRECAUTIONS (5.6)].
The risk of administering methylene blue by non-intravenous routes (such as oral tablets or by local injection) or in intravenous doses much lower than 1 mg/kg with olanzapine and fluoxetine capsules is unclear. The clinician should, nevertheless, be aware of the possibility of emergent symptoms of serotonin syndrome with such use [see WARNINGS AND PRECAUTIONS (5.6)].
2.6 Discontinuation of Treatment with Olanzapine and Fluoxetine Capsules
Symptoms associated with discontinuation of fluoxetine, a component of olanzapine and fluoxetine capsules, SNRIs, and SSRIs, have been reported [see WARNINGS AND PRECAUTIONS (5.25)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
4.1 Monoamine Oxidase Inhibitors (MAOIs)
The use of MAOIs intended to treat psychiatric disorders with olanzapine and fluoxetine hydrochloride or within 5 weeks of stopping treatment with olanzapine and fluoxetine hydrochloride is contraindicated because of an increased risk of serotonin syndrome. The use of olanzapine and fluoxetine hydrochloride within 14 days of stopping an MAOI intended to treat psychiatric disorders is also contraindicated. [see DOSAGE AND ADMINISTRATION (2.4) and WARNINGS AND PRECAUTIONS (5.6)].
Starting olanzapine and fluoxetine hydrochloride in a patient who is being treated with MAOIs such as linezolid or intravenous methylene blue is also contraindicated because of an increased risk of serotonin syndrome. [see DOSAGE AND ADMINISTRATION (2.5) and WARNINGS AND PRECAUTIONS (5.6)].
4.2 Other Contraindications
- Pimozide [see WARNINGS AND PRECAUTIONS (5.20) and DRUG INTERACTIONS (7.7, 7.8)]
- Thioridazine [see WARNINGS AND PRECAUTIONS (5.20) and DRUG INTERACTIONS (7.7, 7.8)]
Pimozide and thioridazine prolong the QT interval. Olanzapine and fluoxetine hydrochloride can increase the levels of pimozide and thioridazine through inhibition of CYP2D6. Olanzapine and fluoxetine hydrochloride can also prolong the QT interval.
-
5 WARNINGS AND PRECAUTIONS
5.1 Suicidal Thoughts and Behaviors in Children, Adolescents, and Young Adults
Patients with Major Depressive Disorder (MDD), both adult and pediatric, may experience worsening of their depression and/or the emergence of suicidal ideation and behavior (suicidality) or unusual changes in behavior, whether or not they are taking antidepressant medications, and this risk may persist until significant remission occurs. Suicide is a known risk of depression and certain other psychiatric disorders, and these disorders themselves are the strongest predictors of suicide. There has been a long-standing concern, however, that antidepressants may have a role in inducing worsening of depression and the emergence of suicidality in certain patients during the early phases of treatment. Pooled analyses of short-term placebo-controlled trials of antidepressant drugs (SSRIs and others) showed that these drugs increase the risk of suicidal thinking and behavior (suicidality) in children, adolescents, and young adults (ages 18 to 24) with Major Depressive Disorder (MDD) and other psychiatric disorders. Short-term studies did not show an increase in the risk of suicidality with antidepressants compared to placebo in adults beyond age 24; there was a reduction with antidepressants compared to placebo in adults aged 65 and older.
The pooled analyses of placebo-controlled trials in children and adolescents with MDD, Obsessive Compulsive Disorder (OCD), or other psychiatric disorders included a total of 24 short-term trials of 9 antidepressant drugs in over 4,400 patients. The pooled analyses of placebo-controlled trials in adults with MDD or other psychiatric disorders included a total of 295 short-term trials (median duration of 2 months) of 11 antidepressant drugs in over 77,000 patients. There was considerable variation in risk of suicidality among drugs, but a tendency toward an increase in the younger patients for almost all drugs studied. There were differences in absolute risk of suicidality across the different indications, with the highest incidence in MDD. The risk differences (drug versus placebo), however, were relatively stable within age strata and across indications. These risk differences (drug-placebo difference in the number of cases of suicidality per 1,000 patients treated) are provided in Table 1.
Table 1: Suicidality per 1000 Patients Treated
Age Range
Drug-Placebo Difference in Number of Cases of Suicidality per 1000 Patients Treated
Increases Compared to Placebo
<18
14 additional cases
18-24
5 additional cases
Decreases Compared to Placebo
25-64
1 fewer case
≥65
6 fewer cases
No suicides occurred in any of the pediatric trials. There were suicides in the adult trials, but the number was not sufficient to reach any conclusion about drug effect on suicide.
It is unknown whether the suicidality risk extends to longer-term use, i.e., beyond several months. However, there is substantial evidence from placebo-controlled maintenance trials in adults with depression that the use of antidepressants can delay the recurrence of depression.
All patients being treated with antidepressants for any indication should be monitored appropriately and observed closely for clinical worsening, suicidality, and unusual changes in behavior, especially during the initial few months of a course of drug therapy, or at times of dose changes, either increases or decreases.
The following symptoms, anxiety, agitation, panic attacks, insomnia, irritability, hostility, aggressiveness, impulsivity, akathisia (psychomotor restlessness), hypomania, and mania, have been reported in adult and pediatric patients being treated with antidepressants for Major Depressive Disorder as well as for other indications, both psychiatric and nonpsychiatric. Although a causal link between the emergence of such symptoms and either the worsening of depression and/or the emergence of suicidal impulses has not been established, there is concern that such symptoms may represent precursors to emerging suicidality.
Consideration should be given to changing the therapeutic regimen, including possibly discontinuing the medication, in patients whose depression is persistently worse, or who are experiencing emergent suicidality or symptoms that might be precursors to worsening depression or suicidality, especially if these symptoms are severe, abrupt in onset, or were not part of the patient's presenting symptoms.
If the decision has been made to discontinue treatment, medication should be tapered, as rapidly as is feasible, but with recognition that abrupt discontinuation can be associated with certain symptoms [see WARNINGS AND PRECAUTIONS (5.25)].
Families and caregivers of patients being treated with antidepressants for Major Depressive Disorder or other indications, both psychiatric and nonpsychiatric, should be alerted about the need to monitor patients for the emergence of agitation, irritability, unusual changes in behavior, and the other symptoms described above, as well as the emergence of suicidality, and to report such symptoms immediately to health care providers. Such monitoring should include daily observation by families and caregivers. Prescriptions for olanzapine and fluoxetine capsules should be written for the smallest quantity of capsules consistent with good patient management, in order to reduce the risk of overdose.
It should be noted that olanzapine and fluoxetine hydrochloride is not approved for use in treating any indications in patients less than 10 years of age [see USE IN SPECIFIC POPULATIONS (8.4)].
5.2 Increased Mortality in Elderly Patients with Dementia-Related Psychosis
Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. Olanzapine and fluoxetine hydrochloride is not approved for the treatment of patients with dementia-related psychosis[see BOXED WARNING, WARNINGS AND PRECAUTIONS (5.21), and PATIENT COUNSELING INFORMATION (17.3)].
In olanzapine placebo-controlled clinical trials of elderly patients with dementia-related psychosis, the incidence of death in olanzapine-treated patients was significantly greater than placebo-treated patients (3.5% vs. 1.5%, respectively).
Meta-Analysis of Antipsychotic Use in Dementia-Related Psychosis
Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. Analyses of seventeen placebo-controlled trials (modal duration of 10 weeks), largely in patients taking atypical antipsychotic drugs, revealed a risk of death in drug-treated patients of between 1.6 to 1.7 times the risk of death in placebo-treated patients. Over the course of a typical 10-week controlled trial, the rate of death in drug-treated patients was about 4.5%, compared to a rate of about 2.6% in the placebo group. Although the causes of death were varied, most of the deaths appeared to be either cardiovascular (e.g., heart failure, sudden death) or infectious (e.g., pneumonia) in nature. Observational studies suggest that, similar to atypical antipsychotic drugs, treatment with conventional antipsychotic drugs may increase mortality. The extent to which the findings of increased mortality in observational studies may be attributed to the antipsychotic drug as opposed to some characteristic(s) of the patients is not clear. Olanzapine and fluoxetine hydrochloride is not approved for the treatment of patients with dementia-related psychosis [see WARNINGS AND PRECAUTIONS (5.21) and PATIENT COUNSELING INFORMATION (17.3)].
Cerebrovascular Adverse Events (CVAE), Including Stroke
Cerebrovascular adverse events (e.g., stroke, transient ischemic attack), including fatalities, were reported in patients in trials of olanzapine in elderly patients with dementia-related psychosis. In placebo-controlled trials, there was a significantly higher incidence of cerebrovascular adverse events in patients treated with olanzapine compared to patients treated with placebo. Olanzapine and olanzapine and fluoxetine hydrochloride are not approved for the treatment of patients with dementia-related psychosis [see BOXED WARNING and PATIENT COUNSELING INFORMATION (17.3)].
5.3 Neuroleptic Malignant Syndrome (NMS)
A potentially fatal symptom complex sometimes referred to as NMS has been reported in association with administration of antipsychotic drugs, including olanzapine. Clinical manifestations of NMS are hyperpyrexia, muscle rigidity, altered mental status, and evidence of autonomic instability (irregular pulse or blood pressure, tachycardia, diaphoresis, and cardiac dysrhythmia). Additional signs may include elevated creatinine phosphokinase, myoglobinuria (rhabdomyolysis), and acute renal failure.
The diagnostic evaluation of patients with this syndrome is complicated. In arriving at a diagnosis, it is important to exclude cases where the clinical presentation includes both serious medical illness (e.g., pneumonia, systemic infection, etc.) and untreated or inadequately treated extrapyramidal signs and symptoms (EPS). Other important considerations in the differential diagnosis include central anticholinergic toxicity, heat stroke, drug fever, and primary central nervous system pathology.
The management of NMS should include: 1) immediate discontinuation of antipsychotic drugs and other drugs not essential to concurrent therapy, 2) intensive symptomatic treatment and medical monitoring, and 3) treatment of any concomitant serious medical problems for which specific treatments are available. There is no general agreement about specific pharmacological treatment regimens for NMS.
If after recovering from NMS, a patient requires treatment with an antipsychotic, the patient should be carefully monitored, since recurrences of NMS have been reported [see WARNINGS AND PRECAUTIONS (5.5) and PATIENT COUNSELING INFORMATION (17.4), 17.9)].
5.4 Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) has been reported with olanzapine exposure. DRESS may present with a cutaneous reaction (such as rash or exfoliative dermatitis), eosinophilia, fever, and/or lymphadenopathy with systemic complications such as hepatitis, nephritis, pneumonitis, myocarditis, and/or pericarditis. DRESS is sometimes fatal. Discontinue olanzapine and fluoxetine hydrochloride if DRESS is suspected [see PATIENT COUNSELING INFORMATION (17.5)].
5.5 Metabolic Changes
Atypical antipsychotic drugs have been associated with metabolic changes including hyperglycemia, dyslipidemia, and weight gain. Metabolic changes may be associated with increased cardiovascular/cerebrovascular risk. Olanzapine's specific metabolic profile is presented below.
Hyperglycemia and Diabetes Mellitus
Adults
Physicians should consider the risks and benefits when prescribing olanzapine and fluoxetine hydrochloride to patients with an established diagnosis of diabetes mellitus, or having borderline increased blood glucose level (fasting 100 to 126 mg/dL, nonfasting 140 to 200 mg/dL). Patients taking olanzapine and fluoxetine hydrochloride should be monitored regularly for worsening of glucose control. Patients starting treatment with olanzapine and fluoxetine hydrochloride should undergo fasting blood glucose testing at the beginning of treatment and periodically during treatment. Any patient treated with atypical antipsychotics should be monitored for symptoms of hyperglycemia including polydipsia, polyuria, polyphagia, and weakness. Patients who develop symptoms of hyperglycemia during treatment with atypical antipsychotics should undergo fasting blood glucose testing. In some cases, hyperglycemia has resolved when the atypical antipsychotic was discontinued; however, some patients required continuation of anti-diabetic treatment despite discontinuation of the suspect drug [see PATIENT COUNSELING INFORMATION (17.6)].
Hyperglycemia, in some cases extreme and associated with ketoacidosis or hyperosmolar coma or death, has been reported in patients treated with atypical antipsychotics, including olanzapine alone, as well as olanzapine taken concomitantly with fluoxetine. Assessment of the relationship between atypical antipsychotic use and glucose abnormalities is complicated by the possibility of an increased background risk of diabetes mellitus in patients with schizophrenia and the increasing incidence of diabetes mellitus in the general population. Epidemiological studies suggest an increased risk of treatment-emergent hyperglycemia-related adverse reactions in patients treated with the atypical antipsychotics. While relative risk estimates are inconsistent, the association between atypical antipsychotics and increases in glucose levels appears to fall on a continuum and olanzapine appears to have a greater association than some other atypical antipsychotics.
Mean increases in blood glucose have been observed in patients treated (median exposure of 9.2 months) with olanzapine in phase 1 of the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE). The mean increase of serum glucose (fasting and nonfasting samples) from baseline to the average of the 2 highest serum concentrations was 15 mg/dL.
In a study of healthy volunteers, subjects who received olanzapine (N=22) for 3 weeks had a mean increase compared to baseline in fasting blood glucose of 2.3 mg/dL. Placebo-treated subjects (N=19) had a mean increase in fasting blood glucose compared to baseline of 0.34 mg/dL.
In an analysis of 7 controlled clinical studies, 2 of which were placebo-controlled, with treatment duration up to 12 weeks, olanzapine and fluoxetine hydrochloride was associated with a greater mean change in random glucose compared to placebo (+8.65 mg/dL vs. -3.86 mg/dL). The difference in mean changes between olanzapine and fluoxetine hydrochloride and placebo was greater in patients with evidence of glucose dysregulation at baseline (including those patients diagnosed with diabetes mellitus or related adverse reactions, patients treated with anti-diabetic agents, patients with a baseline random glucose level ≥200 mg/dL, or a baseline fasting glucose level ≥126 mg/dL). Olanzapine and fluoxetine hydrochloride-treated patients had a greater mean HbA1c increase from baseline of 0.15% (median exposure 63 days), compared to a mean HbA1c decrease of 0.04% in fluoxetine-treated subjects (median exposure 57 days) and a mean HbA1c increase of 0.12% in olanzapine-treated patients (median exposure 56 days).
In an analysis of 6 controlled clinical studies, a larger proportion of olanzapine and fluoxetine hydrochloride-treated subjects had glycosuria (4.4%) compared to placebo-treated subjects (1.4%).
The mean change in nonfasting glucose in patients exposed at least 48 weeks was +5.9 mg/dL (N=425).
Table 2 shows short-term and long-term changes in random glucose levels from adult olanzapine and fluoxetine hydrochloride studies.
Table 2: Changes in Random Glucose Levels from Adult Olanzapine and Fluoxetine Hydrochloride Studies
Laboratory Analyte
Category Change (at least once) from Baseline
Treatment Arm
Up to 12 weeks exposure
At least 48 weeks exposure
N
Patients
N
Patients
Random Glucose
Normal to High
(<140 mg/dL to ≥200 mg/dL)
Olanzapine and fluoxetine hydrochloride
609
2.3%
382
3.1%
Placebo
346
0.3%
NA*
NA*
Borderline to High
(≥140 mg/dL and <200 mg/dL to ≥200 mg/dL)
Olanzapine and fluoxetine hydrochloride
44
34.1%
27
37%
Placebo
28
3.6%
NA*
NA*
* Not Applicable.
Controlled fasting glucose data is limited for olanzapine and fluoxetine hydrochloride; however, in an analysis of 5 placebo-controlled olanzapine monotherapy studies with treatment duration up to 12 weeks, olanzapine was associated with a greater mean change in fasting glucose levels compared to placebo (+2.76 mg/dL vs. +0.17 mg/dL).
The mean change in fasting glucose for olanzapine-treated patients exposed at least 48 weeks was +4.2 mg/dL (N=487). In analyses of patients who completed 9 to 12 months of olanzapine therapy, mean change in fasting and nonfasting glucose levels continued to increase over time.
Children and Adolescents
In a single, 8-week, randomized, placebo-controlled clinical trial investigating olanzapine and fluoxetine hydrochloride for treatment of bipolar I depression in patients 10 to 17 years of age, there were no clinically meaningful differences observed between olanzapine and fluoxetine hydrochloride and placebo for mean change in fasting glucose levels. Table 4 shows categorical changes in fasting blood glucose from the pediatric olanzapine and fluoxetine hydrochloride study.
Table 4: Changes in Fasting Glucose Levels from a Single Pediatric Olanzapine and Fluoxetine Hydrochloride Study in Bipolar Depression
Laboratory Analyte
Category Change (at least once) from Baseline
Treatment Arm
Up to 8 weeks exposure
N
Patients
Fasting Glucose
Normal to High
(<100 mg/dL to ≥126 mg/dL)Olanzapine and fluoxetine hydrochloride
125
4.8%
Placebo
65
1.5%
Normal/IGT* to High
(<126 mg/dL to ≥126 mg/dL)Olanzapine and fluoxetine hydrochloride
156
5.8%
Placebo
78
1.3%
Normal/IGT* (<126 mg/dL) to ≥140 mg/dL)
Olanzapine and fluoxetine hydrochloride
156
1.9%
Placebo
78
0%
* Impaired Glucose Tolerance.
Olanzapine Monotherapy in Adolescents
In an analysis of 3 placebo-controlled olanzapine monotherapy studies of adolescent patients, including those with Schizophrenia (6 weeks) or Bipolar I Disorder (manic or mixed episodes) (3 weeks), olanzapine was associated with a greater mean change from baseline in fasting glucose levels compared to placebo (+2.68 mg/dL vs. -2.59 mg/dL). The mean change in fasting glucose for adolescents exposed at least 24 weeks was +3.1 mg/dL (N=121). Table 5 shows short-term and long-term changes in fasting blood glucose from adolescent olanzapine monotherapy studies.
Table 5: Changes in Fasting Glucose Levels from Adolescent Olanzapine Monotherapy Studies
Laboratory Analyte
Category Change (at least once) from Baseline
Treatment Arm
Up to 12 weeks exposure
At least 24 weeks exposure
N
Patients
N
Patients
Fasting Glucose
Normal to High
(<100 mg/dL to ≥126 mg/dL)
Olanzapine
124
0%
108
0.9%
Placebo
53
1.9%
NA*
NA*
Borderline to High
(≥100 mg/dL and <126 mg/dL to ≥126 mg/dL)
Olanzapine
14
14.3%
13
23.1%
Placebo
13
0%
NA*
NA*
* Not Applicable.
Dyslipidemia
Undesirable alterations in lipids have been observed with olanzapine and fluoxetine hydrochloride use. Clinical monitoring, including baseline and periodic follow-up lipid evaluations in patients using olanzapine and fluoxetine hydrochloride, is recommended [see PATIENT COUNSELING INFORMATION (17.7)].
Adults
Clinically meaningful, and sometimes very high (>500 mg/dL), elevations in triglyceride levels have been observed with olanzapine and fluoxetine hydrochloride use. Clinically meaningful increases in total cholesterol have also been seen with olanzapine and fluoxetine hydrochloride use.
In an analysis of 7 controlled clinical studies, 2 of which were placebo-controlled, with treatment duration up to 12 weeks, olanzapine and fluoxetine hydrochloride-treated patients had an increase from baseline in mean random total cholesterol of 12.1 mg/dL compared to an increase from baseline in mean random total cholesterol of 4.8 mg/dL for olanzapine-treated patients and a decrease in mean random total cholesterol of 5.5 mg/dL for placebo-treated patients. Table 6 shows categorical changes in nonfasting lipid values.
In long-term olanzapine and fluoxetine in combination studies (at least 48 weeks), changes (at least once) in nonfasting total cholesterol from normal at baseline to high occurred in 12% (N=150) and changes from borderline to high occurred in 56.6% (N=143) of patients. The mean change in nonfasting total cholesterol was 11.3 mg/dL (N= 426).
Table 6: Changes in Nonfasting Lipids Values from Controlled Clinical Studies with Treatment Duration up to 12 Weeks
Laboratory Analyte
Category Change (at least once) from Baseline
Treatment Arm
N
Patients
Nonfasting
TriglyceridesIncrease by ≥50 mg/dL
Olanzapine and fluoxetine hydrochloride
174
67.8%
Olanzapine
172
72.7%
Normal to High
(<150 mg/dL to
≥500 mg/dL)Olanzapine and fluoxetine hydrochloride
57
0%
Olanzapine
58
0%
Borderline to High (≥150 mg/dL and <500 mg/dL to
≥500 mg/dL)Olanzapine and fluoxetine hydrochloride
106
15.1%
Olanzapine
103
8.7%
Nonfasting
Total CholesterolIncrease by ≥40 mg/dL
Olanzapine and fluoxetine hydrochloride
685
35%
Olanzapine
749
22.7%
Placebo
390
9%
Normal to High (<200 mg/dL to
≥240 mg/dL)Olanzapine and fluoxetine hydrochloride
256
8.2%
Olanzapine
279
2.9%
Placebo
175
1.7%
Borderline to High (≥200 mg/dL and <240 mg/dL to ≥240 mg/dL)
Olanzapine and fluoxetine hydrochloride
213
36.2%
Olanzapine
261
27.6%
Placebo
111
9.9%
Fasting lipid data is limited for olanzapine and fluoxetine hydrochloride; however, in an analysis of 5 placebo-controlled olanzapine monotherapy studies with treatment duration up to 12 weeks, olanzapine-treated patients had increases from baseline in mean fasting total cholesterol, LDL cholesterol, and triglycerides of 5.3 mg/dL, 3 mg/dL, and 20.8 mg/dL respectively compared to decreases from baseline in mean fasting total cholesterol, LDL cholesterol, and triglycerides of 6.1 mg/dL, 4.3 mg/dL, and 10.7 mg/dL for placebo-treated patients. For fasting HDL cholesterol, no clinically meaningful differences were observed between olanzapine-treated patients and placebo-treated patients. Mean increases in fasting lipid values (total cholesterol, LDL cholesterol, and triglycerides) were greater in patients without evidence of lipid dysregulation at baseline, where lipid dysregulation was defined as patients diagnosed with dyslipidemia or related adverse reactions, patients treated with lipid lowering agents, patients with high baseline lipid levels.
In long-term olanzapine studies (at least 48 weeks), patients had increases from baseline in mean fasting total cholesterol, LDL cholesterol, and triglycerides of 5.6 mg/dL, 2.5 mg/dL, and 18.7 mg/dL, respectively, and a mean decrease in fasting HDL cholesterol of 0.16 mg/dL. In an analysis of patients who completed 12 months of therapy, the mean nonfasting total cholesterol did not increase further after approximately 4 to 6 months.
The proportion of olanzapine-treated patients who had changes (at least once) in total cholesterol, LDL cholesterol or triglycerides from normal or borderline to high, or changes in HDL cholesterol from normal or borderline to low, was greater in long-term studies (at least 48 weeks) as compared with short-term studies. Table 8 shows categorical changes in fasting lipids values.
Table 8: Changes in Fasting Lipids Values from Adult Olanzapine Monotherapy Studies
Laboratory Analyte
Category Change (at least once) from Baseline
Treatment Arm
Up to 12 weeks exposure
At least 48 weeks exposure
N
Patients
N
Patients
Fasting
TriglyceridesIncrease by
≥50 mg/dLOlanzapine
745
39.6%
487
61.4%
Placebo
402
26.1%
NA*
NA*
Normal to High
(<150 mg/dL to ≥200 mg/dL)Olanzapine
457
9.2%
293
32.4%
Placebo
251
4.4%
NA*
NA*
Borderline to High
(≥150 mg/dL and <200 mg/dL to
≥200 mg/dL)Olanzapine
135
39.3%
75
70.7%
Placebo
65
20%
NA*
NA*
Fasting
Total CholesterolIncrease by
≥40 mg/dLOlanzapine
745
21.6%
489
32.9%
Placebo
402
9.5%
NA*
NA*
Normal to High
(<200 mg/dL to ≥240 mg/dL)Olanzapine
392
2.8%
283
14.8%
Placebo
207
2.4%
NA*
NA*
Borderline to High
(≥200 mg/dL and <240 mg/dL to ≥240 mg/dL)Olanzapine
222
23%
125
55.2%
Placebo
112
12.5%
NA*
NA*
Fasting
LDL CholesterolIncrease by
≥30 mg/dLOlanzapine
536
23.7%
483
39.8%
Placebo
304
14.1%
NA*
NA*
Normal to High
(<100 mg/dL to ≥160 mg/dL)Olanzapine
154
0%
123
7.3%
Placebo
82
1.2%
NA*
NA*
Borderline to High
(≥100 mg/dL and <160 mg/dL to ≥160 mg/dL)Olanzapine
302
10.6%
284
31%
Placebo
173
8.1%
NA*
NA*
*Not Applicable.
In phase 1 of the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE), over a median exposure of 9.2 months, the mean increase in triglycerides in patients taking olanzapine was 40.5 mg/dL. In phase 1 of CATIE, the median increase in total cholesterol was 9.4 mg/dL.
Children and Adolescents
In a single, 8-week, randomized, placebo-controlled clinical trial investigating olanzapine and fluoxetine hydrochloride for treatment of bipolar I depression in patients 10 to 17 years of age, there were clinically meaningful and statistically significant differences observed between olanzapine and fluoxetine hydrochloride and placebo for mean change in fasting total cholesterol (+16.3 mg/dL vs. -4.3 mg/dL, respectively), LDL cholesterol (+9.7 mg/dL vs. -3.5 mg/dL, respectively), and triglycerides (+35.4 mg/dL vs. -3.5 mg/dL, respectively).
The magnitude and frequency of changes in lipids were greater in children and adolescents than previously observed in adults. Table 9 shows categorical changes in fasting lipids values from the pediatric olanzapine and fluoxetine hydrochloride study.
Table 9: Changes in Fasting Lipids Values from a Single Pediatric Olanzapine and Fluoxetine Hydrochloride Study in Bipolar Depression
Laboratory Analyte
Category Change (at least once) from Baseline
Treatment Arm
Up to 8 weeks exposure
N
Patients
Fasting
TriglyceridesIncrease by ≥50 mg/dL
Olanzapine and fluoxetine hydrochloride
158
70.3%
Placebo
81
38.3%
Normal to High
(<90 mg/dL to
≥130 mg/dL)Olanzapine and fluoxetine hydrochloride
71
39.4%
Placebo
31
19.4%
Borderline to High
(≥90 mg/dL and
<130 mg/dL to
≥130 mg/dL)Olanzapine and fluoxetine hydrochloride
13
84.6%
Placebo
12
33.3%
Normal/Borderline to High
(<130 mg/dL to
≥130 mg/dL)Olanzapine and fluoxetine hydrochloride
106
52.8%
Placebo
56
25%
Normal to Borderline/High
(<90 mg/dL to
≥90 mg/dL)Olanzapine and fluoxetine hydrochloride
71
73.2%
Placebo
31
41.9%
Normal/Borderline/High to very high (<500 mg/dL to
≥500 mg/dL)Olanzapine and fluoxetine hydrochloride
158
2.5%
Placebo
81
1.2%
Fasting
Total CholesterolIncrease by ≥40 mg/dL
Olanzapine and fluoxetine hydrochloride
158
52.5%
Placebo
81
8.6%
Normal to High
(<170 mg/dL to
≥200 mg/dL)Olanzapine and fluoxetine hydrochloride
81
12.3%
Placebo
44
4.5%
Borderline to High
(≥170 mg/dL and
<200 mg/dL to
≥200 mg/dL)Olanzapine and fluoxetine hydrochloride
22
72.7%
Placebo
11
24.3%
Normal/Borderline to High
(<200 mg/dL to
≥200 mg/dL)Olanzapine and fluoxetine hydrochloride
126
32.5%
Placebo
67
10.4%
Normal to Borderline/High
(<170 mg/dL to
≥170 mg/dL)Olanzapine and fluoxetine hydrochloride
81
58%
Placebo
44
31.8%
Fasting
LDL CholesterolIncrease by ≥30 mg/dL
Olanzapine and fluoxetine hydrochloride
158
53.8%
Placebo
81
23.5%
Normal to High
(<110 mg/dL to
≥130 mg/dL)Olanzapine and fluoxetine hydrochloride
112
13.4%
Placebo
62
6.5%
Borderline to High
(≥110 mg/dL and
<130 mg/dL to
≥130 mg/dL)Olanzapine and fluoxetine hydrochloride
12
75%
Placebo
3
0%
Normal/Borderline to High
(<130 mg/dL to
≥130 mg/dL)Olanzapine and fluoxetine hydrochloride
138
21.7%
Placebo
77
7.8%
Normal to Borderline/high
(<110 mg/dL to
≥110 mg/dL)Olanzapine and fluoxetine hydrochloride
112
30.4%
Placebo
62
14.5%
Olanzapine Monotherapy in Adolescents
In an analysis of 3 placebo-controlled olanzapine monotherapy studies of adolescents, including those with Schizophrenia (6 weeks) or Bipolar I Disorder (manic or mixed episodes) (3 weeks), olanzapine-treated adolescents had increases from baseline in mean fasting total cholesterol, LDL cholesterol, and triglycerides of 12.9 mg/dL, 6.5 mg/dL, and 28.4 mg/dL, respectively, compared to increases from baseline in mean fasting total cholesterol and LDL cholesterol of 1.3 mg/dL and 1 mg/dL, and a decrease in triglycerides of 1.1 mg/dL for placebo-treated adolescents. For fasting HDL cholesterol, no clinically meaningful differences were observed between olanzapine-treated adolescents and placebo-treated adolescents.
In long-term olanzapine studies (at least 24 weeks), adolescents had increases from baseline in mean fasting total cholesterol, LDL cholesterol, and triglycerides of 5.5 mg/dL, 5.4 mg/dL, and 20.5 mg/dL, respectively, and a mean decrease in fasting HDL cholesterol of 4.5 mg/dL. Table 10 shows categorical changes in fasting lipids values in adolescents.
Table 10: Changes in Fasting Lipids Values from Adolescent Olanzapine Monotherapy Studies
Laboratory Analyte
Category Change (at least once) from Baseline
Treatment Arm
Up to 6 weeks exposure
At least 24 weeks exposure
N
Patients
N
Patients
Fasting
TriglyceridesIncrease by
≥50 mg/dLOlanzapine
138
37%
122
45.9%
Placebo
66
15.2%
NA*
NA*
Normal to High
(<90 mg/dL to
>130 mg/dL)Olanzapine
67
26.9%
66
36.4%
Placebo
28
10.7%
NA*
NA*
Borderline to High
(≥90 mg/dL and
≤130 mg/dL to
>130 mg/dL)Olanzapine
37
59.5%
31
64.5%
Placebo
17
35.3%
NA*
NA*
Fasting
Total CholesterolIncrease by
≥40 mg/dLOlanzapine
138
14.5%
122
14.8%
Placebo
66
4.5%
NA*
NA*
Normal to High
(<170 mg/dL to
≥200 mg/dL)Olanzapine
87
6.9%
78
7.7%
Placebo
43
2.3%
NA*
NA*
Borderline to High
(≥170 mg/dL and
<200 mg/dL to
≥200 mg/dL)Olanzapine
36
38.9%
33
57.6%
Placebo
13
7.7%
NA*
NA*
Fasting
LDL CholesterolIncrease by
≥30 mg/dLOlanzapine
137
17.5%
121
22.3%
Placebo
63
11.1%
NA*
NA*
Normal to High
(<110 mg/dL to
≥130 mg/dL)Olanzapine
98
5.1%
92
10.9%
Placebo
44
4.5%
NA*
NA*
Borderline to High
(≥110 mg/dL and
<130 mg/dL to
≥130 mg/dL)Olanzapine
29
48.3%
21
47.6%
Placebo
9
0%
NA*
NA*
*Not Applicable.
Weight Gain
Potential consequences of weight gain should be considered prior to starting olanzapine and fluoxetine hydrochloride. Patients receiving olanzapine and fluoxetine hydrochloride should receive regular monitoring of weight [see PATIENT COUNSELING INFORMATION (17.8)].
Adults
In an analysis of 7 controlled clinical studies, 2 of which were placebo-controlled, the mean weight increase for olanzapine and fluoxetine hydrochloride-treated patients was greater than placebo-treated patients [4 kg (8.8 lb) vs. -0.3 kg (-0.7 lb)]. Twenty-two percent of olanzapine and fluoxetine hydrochloride-treated patients gained at least 7% of their baseline weight, with a median exposure to event of 6 weeks. This was greater than in placebo-treated patients (1.8%). Approximately 3% of olanzapine and fluoxetine hydrochloride-treated patients gained at least 15% of their baseline weight, with a median exposure to event of 8 weeks. This was greater than in placebo-treated patients (0%). Clinically significant weight gain was observed across all baseline Body Mass Index (BMI) categories. Discontinuation due to weight gain occurred in 2.5% of olanzapine and fluoxetine hydrochloride-treated patients and 0% of placebo-treated patients.
In long-term olanzapine and fluoxetine in combination studies (at least 48 weeks), the mean weight gain was 6.7 kg (14.7 lb) (median exposure of 448 days, N=431). The percentages of patients who gained at least 7%, 15% or 25% of their baseline body weight with long-term exposure were 66%, 33%, and 10%, respectively. Discontinuation due to weight gain occurred in 1.2% of patients treated with olanzapine and fluoxetine in combination following at least 48 weeks of exposure.
In long-term olanzapine studies (at least 48 weeks), the mean weight gain was 5.6 kg (12.3 lb) (median exposure of 573 days, N=2021). The percentages of patients who gained at least 7%, 15%, or 25% of their baseline body weight with long-term exposure were 64%, 32%, and 12%, respectively. Discontinuation due to weight gain occurred in 0.4% of olanzapine-treated patients following at least 48 weeks of exposure.
Table 12 includes data on adult weight gain with olanzapine pooled from 86 clinical trials. The data in each column represent data for those patients who completed treatment periods of the durations specified.
Table 12: Weight Gain with Olanzapine Use in Adults
Amount Gained
kg (lb)6 Weeks
(N=7465)
(%)6 Months
(N=4162)
(%)12 Months
(N=1345)
(%)24 Months
(N=474)
(%)36 Months
(N=147)
(%)≤0
26.2
24.3
20.8
23.2
17
0 to ≤5 (0-11 lb)
57
36
26
23.4
25.2
>5 to ≤10 (11-22 lb)
14.9
24.6
24.2
24.1
18.4
>10 to ≤15 (22-33 lb)
1.8
10.9
14.9
11.4
17
>15 to ≤20 (33-44 lb)
0.1
3.1
8.6
9.3
11.6
>20 to ≤25 (44-55 lb)
0
0.9
3.3
5.1
4.1
>25 to ≤30 (55-66 lb)
0
0.2
1.4
2.3
4.8
>30 (>66 lb)
0
0.1
0.8
1.2
2
Dose group differences with respect to weight gain have been observed. In a single 8-week randomized, double-blind, fixed-dose study comparing 10 (N=199), 20 (N=200) and 40 (N=200) mg/day of oral olanzapine in adult patients with schizophrenia or schizoaffective disorder, mean baseline to endpoint increase in weight (10 mg/day: 1.9 kg; 20 mg/day: 2.3 kg; 40 mg/day: 3 kg) was observed with significant differences between 10 vs. 40 mg/day.
Children and Adolescents
In a single, 8-week, randomized, placebo-controlled clinical trial investigating olanzapine and fluoxetine hydrochloride for the treatment of bipolar I depression in patients 10 to 17 years of age, olanzapine and fluoxetine hydrochloride was associated with greater mean change in weight compared to placebo (+4.4 kg vs +0.5 kg, respectively). The percentages of children and adolescents who gained at least 7%, 15%, or 25% of their baseline body weight with 8-week exposure were 52%, 14%, and 1%, respectively. The proportion of patients who had clinically significant weight gain was greater in children and adolescent patients compared to short-term data in adults. Discontinuation due to weight gain occurred in 2.9% of olanzapine and fluoxetine hydrochloride-treated patients and 0% of placebo-treated patients. Table 13 depicts weight gain observed in the pediatric olanzapine and fluoxetine hydrochloride study.
Table 13: Weight Gain with Olanzapine and Fluoxetine Hydrochloride Use Seen in a Single Pediatric Study in Bipolar Depression
Amount Gained
kg (lb)Up to 8 Weeks
(N=170)
(%)≤0
7.1
0 to ≤5 (0-11 lb)
54.7
>5 to ≤10 (11-22 lb)
31.2
>10 to ≤15 (22-33 lb)
7.1
>15 to ≤20 (33-44 lb)
0
>20 to ≤25 (44-55 lb)
0
>25 to ≤30 (55-66 lb)
0
>30 (>66 lb)
0
Olanzapine Monotherapy in Adolescents
Mean increase in weight in adolescents was greater than in adults. In 4 placebo-controlled trials, discontinuation due to weight gain occurred in 1% of olanzapine-treated patients, compared to 0% of placebo-treated patients.
Table 14: Weight Gain with Olanzapine Use in Adolescents from 4 Placebo-Controlled Trials
Olanzapine-treated patients
Placebo-treated
patientsMean change in body weight from baseline (median exposure = 3 weeks)
4.6 kg (10.1 lb)
0.3 kg (0.7 lb)
Percentage of patients who gained at least 7% of baseline body weight
40.6%
(median exposure to 7% = 4 weeks)9.8%
(median exposure to
7% = 8 weeks)Percentage of patients who gained at least 15% of baseline body weight
7.1%
(median exposure to
15% = 19 weeks)2.7%
(median exposure to
15% = 8 weeks)In long-term olanzapine studies (at least 24 weeks), the mean weight gain was 11.2 kg (24.6 lb) (median exposure of 201 days, N=179). The percentages of adolescents who gained at least 7%, 15%, or 25% of their baseline body weight with long-term exposure were 89%, 55%, and 29%, respectively. Among adolescent patients, mean weight gain by baseline BMI category was 11.5 kg (25.3 lb), 12.1 kg (26.6 lb), and 12.7 kg (27.9 lb), respectively, for normal (N=106), overweight (N=26) and obese (N=17). Discontinuation due to weight gain occurred in 2.2% of olanzapine-treated patients following at least 24 weeks of exposure.
Table 15 shows data on adolescent weight gain with olanzapine pooled from 6 clinical trials. The data in each column represent data for those patients who completed treatment periods of the durations specified. Little clinical trial data is available on weight gain in adolescents with olanzapine beyond 6 months of treatment.
Table 15: Weight Gain with Olanzapine Use in Adolescents
Amount Gained
kg (lb)6 Weeks
(N=243)
(%)6 Months
(N=191)
(%)≤0
2.9
2.1
0 to ≤5 (0-11 lb)
47.3
24.6
>5 to ≤10 (11-22 lb)
42.4
26.7
>10 to ≤15 (22-33 lb)
5.8
22
>15 to ≤20 (33-44 lb)
0.8
12.6
>20 to ≤25 (44-55 lb)
0.8
9.4
>25 to ≤30 (55-66 lb)
0
2.1
>30 to ≤35 (66-77 lb)
0
0
>35 to ≤40 (77-88 lb)
0
0
>40 (>88 lb)
0
0.5
5.6 Serotonin Syndrome
The development of a potentially life-threatening serotonin syndrome has been reported with SNRIs and SSRIs, including olanzapine and fluoxetine hydrochloride, alone, but particularly with concomitant use of other serotonergic drugs (including triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, tryptophan, buspirone, amphetamines, and St. John's Wort) and with drugs that impair metabolism of serotonin (in particular, MAOIs, both those intended to treat psychiatric disorders and also others, such as linezolid and intravenous methylene blue).
Serotonin syndrome symptoms may include mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). Patients should be monitored for the emergence of serotonin syndrome.
The concomitant use of olanzapine and fluoxetine hydrochloride with MAOIs intended to treat psychiatric disorders is contraindicated. Olanzapine and fluoxetine hydrochloride should also not be started in a patient who is being treated with MAOIs such as linezolid or intravenous methylene blue. All reports with methylene blue that provided information on the route of administration involved intravenous administration in the dose range of 1 mg/kg to 8 mg/kg. No reports involved the administration of methylene blue by other routes (such as oral tablets or local tissue injection) or at lower doses. There may be circumstances when it is necessary to initiate treatment with an MAOI such as linezolid or intravenous methylene blue in a patient taking olanzapine and fluoxetine hydrochloride. Olanzapine and fluoxetine hydrochloride should be discontinued before initiating treatment with the MAOI [see DOSAGE AND ADMINISTRATION (2.4, 2.5) and CONTRAINDICATIONS (4.1)].
If concomitant use of olanzapine and fluoxetine hydrochloride with other serotonergic drugs including triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, buspirone, amphetamines, tryptophan and St. John's Wort is clinically warranted, patients should be made aware of a potential increased risk for serotonin syndrome, particularly during treatment initiation and dose increases.
Treatment with olanzapine and fluoxetine hydrochloride and any concomitant serotonergic agents should be discontinued immediately if the above events occur and supportive symptomatic treatment should be initiated.
5.7 Angle-Closure Glaucoma
Angle Closure Glaucoma
The pupillary dilation that occurs following use of many antidepressant drugs including olanzapine and fluoxetine hydrochloride may trigger an angle-closure attack in a patient with anatomically narrow angles who does not have a patent iridectomy.
5.8 Allergic Reactions and Rash
In olanzapine and fluoxetine hydrochloride premarketing controlled clinical studies, the overall incidence of rash or allergic reactions in olanzapine and fluoxetine hydrochloride-treated patients [4.6% (26/571)] was similar to that of placebo [5.2% (25/477)]. The majority of the cases of rash and/or urticaria were mild; however, 3 patients discontinued (1 due to rash, which was moderate in severity and 2 due to allergic reactions, 1 of which included face edema).
In fluoxetine US clinical studies, 7% of 10,782 fluoxetine-treated patients developed various types of rashes and/or urticaria. Among the cases of rash and/or urticaria reported in premarketing clinical studies, almost a third were withdrawn from treatment because of the rash and/or systemic signs or symptoms associated with the rash. Clinical findings reported in association with rash include fever, leukocytosis, arthralgias, edema, carpal tunnel syndrome, respiratory distress, lymphadenopathy, proteinuria, and mild transaminase elevation. Most patients improved promptly with discontinuation of fluoxetine and/or adjunctive treatment with antihistamines or steroids, and all patients experiencing these reactions were reported to recover completely.
In fluoxetine premarketing clinical studies, 2 patients are known to have developed a serious cutaneous systemic illness. In neither patient was there an unequivocal diagnosis, but 1 was considered to have a leukocytoclastic vasculitis, and the other, a severe desquamating syndrome that was considered variously to be a vasculitis or erythema multiforme. Other patients have had systemic syndromes suggestive of serum sickness.
Since the introduction of fluoxetine, systemic reactions, possibly related to vasculitis, have developed in patients with rash. Although these reactions are rare, they may be serious, involving the lung, kidney, or liver. Death has been reported to occur in association with these systemic reactions.
Anaphylactoid reactions, including bronchospasm, angioedema, and urticaria alone and in combination, have been reported.
Pulmonary reactions, including inflammatory processes of varying histopathology and/or fibrosis, have been reported rarely. These reactions have occurred with dyspnea as the only preceding symptom.
Whether these systemic reactions and rash have a common underlying cause or are due to different etiologies or pathogenic processes is not known. Furthermore, a specific underlying immunologic basis for these reactions has not been identified. Upon the appearance of rash or of other possible allergic phenomena for which an alternative etiology cannot be identified, olanzapine and fluoxetine hydrochloride should be discontinued.
5.9 Activation of Mania/Hypomania
A major depressive episode may be the initial presentation of Bipolar Disorder. It is generally believed (though not established in controlled trials) that treating such an episode with an antidepressant alone may increase the likelihood of precipitation of a manic episode in patients at risk for Bipolar Disorder. Whether any of the symptoms described for clinical worsening and suicide risk represent such a conversion is unknown. However, prior to initiating treatment with an antidepressant, patients with depressive symptoms should be adequately screened to determine if they are at risk for Bipolar Disorder; such screening should include a detailed psychiatric history, including a family history of suicide, Bipolar Disorder, and depression. It should be noted that olanzapine and fluoxetine hydrochloride is approved for the acute treatment of depressive episodes associated with Bipolar I Disorder.
In the 3 controlled bipolar depression studies (2 in adults and 1 in children and adolescents [10 to 17 years of age]) there was no statistically significant difference in the incidence of manic reactions (manic reaction or manic depressive reaction) between olanzapine and fluoxetine hydrochloride - and placebo-treated patients. In 1 adult study, the incidence of manic reactions was (7% [3/43]) in olanzapine and fluoxetine hydrochloride -treated patients compared to (3% [5/184]) in placebo-treated patients. In the other adult study, the incidence of manic reactions was (2% [1/43]) in olanzapine and fluoxetine hydrochloride -treated patients compared to (8% [15/193]) in placebo-treated patients. In a single, 8-week, randomized, placebo-controlled clinical trial investigating olanzapine and fluoxetine hydrochloride for the treatment of bipolar I depression in patients 10 to 17 years of age, the incidence of manic reactions was (1% [2/170]) in olanzapine and fluoxetine hydrochloride -treated patients compared to (0% [0/84]) in placebo-treated patients. Because of the cyclical nature of Bipolar I Disorder, patients should be monitored closely for the development of symptoms of mania/hypomania during treatment with olanzapine and fluoxetine hydrochloride.
5.10 Tardive Dyskinesia
A syndrome of potentially irreversible, involuntary, dyskinetic movements may develop in patients treated with antipsychotic drugs. Although the prevalence of the syndrome appears to be highest among the elderly, especially elderly women, it is impossible to rely upon prevalence estimates to predict, at the inception of antipsychotic treatment, which patients are likely to develop the syndrome. Whether antipsychotic drug products differ in their potential to cause tardive dyskinesia is unknown.
The risk of developing tardive dyskinesia and the likelihood that it will become irreversible are believed to increase as the duration of treatment and the total cumulative dose of antipsychotic drugs administered to the patient increase. However, the syndrome can develop, although much less commonly, after relatively brief treatment periods at low doses or may even arise after discontinuation of treatment.
There is no known treatment for established cases of tardive dyskinesia, although the syndrome may remit, partially or completely, if antipsychotic treatment is withdrawn. Antipsychotic treatment itself, however, may suppress (or partially suppress) the signs and symptoms of the syndrome and thereby may possibly mask the underlying process. The effect that symptomatic suppression has upon the long-term course of the syndrome is unknown.
The incidence of dyskinetic movement in olanzapine and fluoxetine hydrochloride-treated patients was infrequent. The mean score on the Abnormal Involuntary Movement Scale (AIMS) in the olanzapine and fluoxetine hydrochloride-controlled database across clinical studies involving olanzapine and fluoxetine hydrochloride-treated patients decreased from baseline. Nonetheless, olanzapine and fluoxetine hydrochloride should be prescribed in a manner that is most likely to minimize the risk of tardive dyskinesia. If signs and symptoms of tardive dyskinesia appear in a patient on olanzapine and fluoxetine hydrochloride, drug discontinuation should be considered. However, some patients may require treatment with olanzapine and fluoxetine hydrochloride despite the presence of the syndrome. The need for continued treatment should be reassessed periodically.
5.11 Orthostatic Hypotension
Olanzapine and fluoxetine hydrochloride may induce orthostatic hypotension associated with dizziness, tachycardia, bradycardia and, in some patients, syncope, especially during the initial dose-titration period [see PATIENT COUNSELING INFORMATION (17.12)].
In the olanzapine and fluoxetine hydrochloride-controlled clinical trials across all indications, there were no significant differences between olanzapine and fluoxetine hydrochloride-treated patients and olanzapine, fluoxetine- or placebo-treated patients in exposure-adjusted rates of orthostatic systolic blood pressure decreases of at least 30 mm Hg. Orthostatic systolic blood pressure decreases of at least 30 mm Hg occurred in 4% (28/705), 2.3% (19/831), 4.5% (18/399), and 1.8% (8/442) of the olanzapine and fluoxetine hydrochloride, olanzapine, fluoxetine, and placebo groups, respectively. In this group of studies, the incidence of syncope-related adverse reactions (i.e., syncope and/or loss of consciousness) in olanzapine and fluoxetine hydrochloride-treated patients was 0.4% (3/771) compared to placebo 0.2% (1/477).
In a clinical pharmacology study of olanzapine and fluoxetine hydrochloride, 3 healthy subjects were discontinued from the trial after experiencing severe, but self-limited, hypotension and bradycardia that occurred 2 to 9 hours following a single 12 mg/50 mg dose of olanzapine and fluoxetine hydrochloride. Reactions consisting of this combination of hypotension and bradycardia (and also accompanied by sinus pause) have been observed in at least 3 other healthy subjects treated with various formulations of olanzapine (1 oral, 2 intramuscular). In controlled clinical studies, the incidence of patients with a ≥20 bpm decrease in orthostatic pulse concomitantly with a ≥20 mm Hg decrease in orthostatic systolic blood pressure was 0.3% (2/706) in the olanzapine and fluoxetine hydrochloride group, 0.2% (1/445) in the placebo group, 0.7% (6/837) in the olanzapine group, and 0% (0/404) in the fluoxetine group.
Olanzapine and fluoxetine hydrochloride should be used with particular caution in patients with known cardiovascular disease (history of myocardial infarction or ischemia, heart failure, or conduction abnormalities), cerebrovascular disease, or conditions that would predispose patients to hypotension (dehydration, hypovolemia, and treatment with antihypertensive medications).
5.12 Falls
Olanzapine and fluoxetine hydrochloride may cause somnolence, postural hypotension, motor and sensory instability, which may lead to falls and, consequently, fractures or other injuries. For patients with diseases, conditions, or medications that could exacerbate these effects, complete fall risk assessments when initiating antipsychotic treatment and recurrently for patients on long-term antipsychotic therapy.
5.13 Leukopenia, Neutropenia, and Agranulocytosis
Class Effect
In clinical trial and/or postmarketing experience, events of leukopenia/neutropenia have been reported temporally related to antipsychotic agents, including olanzapine and fluoxetine hydrochloride. Agranulocytosis has also been reported.
Possible risk factors for leukopenia/neutropenia include preexisting low white blood cell count (WBC) and history of drug induced leukopenia/neutropenia. Patients with a history of a clinically significant low WBC or drug induced leukopenia/neutropenia should have their complete blood count (CBC) monitored frequently during the first few months of therapy and discontinuation of olanzapine and fluoxetine hydrochloride should be considered at the first sign of a clinically significant decline in WBC in the absence of other causative factors.
Patients with clinically significant neutropenia should be carefully monitored for fever or other symptoms or signs of infection and treated promptly if such symptoms or signs occur. Patients with severe neutropenia (absolute neutrophil count <1000/mm3) should discontinue olanzapine and fluoxetine hydrochloride and have their WBC followed until recovery.
5.14 Dysphagia
Esophageal dysmotility and aspiration have been associated with antipsychotic drug use. Aspiration pneumonia is a common cause of morbidity and mortality in patients with advanced Alzheimer's disease. Olanzapine and fluoxetine hydrochloride is not approved for the treatment of patients with Alzheimer's disease.
5.15 Seizures
Seizures occurred in 0.2% (4/2,547) of olanzapine and fluoxetine hydrochloride-treated patients during open-label clinical studies. No seizures occurred in the controlled olanzapine and fluoxetine hydrochloride studies. Seizures have also been reported with both olanzapine and fluoxetine monotherapy. Olanzapine and fluoxetine hydrochloride should be used cautiously in patients with a history of seizures or with conditions that potentially lower the seizure threshold, e.g., Alzheimer's dementia. Olanzapine and fluoxetine hydrochloride is not approved for the treatment of patients with Alzheimer's disease. Conditions that lower the seizure threshold may be more prevalent in a population of ≥65 years of age.
5.16 Abnormal Bleeding
SNRIs and SSRIs , including fluoxetine, may increase the risk of bleeding reactions. Concomitant use of aspirin, nonsteroidal anti-inflammatory drugs, warfarin, and other anti-coagulants may add to this risk. Case reports and epidemiological studies (case-control and cohort design) have demonstrated an association between use of drugs that interfere with serotonin reuptake and the occurrence of gastrointestinal bleeding. Bleeding reactions related to SNRIs and SSRIs use have ranged from ecchymoses, hematomas, epistaxis, and petechiae to life-threatening hemorrhages.
Patients should be cautioned about the risk of bleeding associated with the concomitant use of olanzapine and fluoxetine hydrochloride and NSAIDs, aspirin, or other drugs that affect coagulation [see DRUG INTERACTIONS (7.4) and PATIENT COUNSELING INFORMATION (17.13)].
5.17 Hyponatremia
Hyponatremia has been reported during treatment with SNRIs and SSRIs, including fluoxetine and olanzapine and fluoxetine hydrochloride. In many cases, this hyponatremia appears to be the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH). Cases with serum sodium lower than 110 mmol/L have been reported and appeared to be reversible when olanzapine and fluoxetine hydrochloride was discontinued [see USE IN SPECIFIC POPULATIONS (8.5)]. Elderly patients may be at greater risk of developing hyponatremia with SNRIs and SSRIs. Also, patients taking diuretics or who are otherwise volume depleted may be at greater risk. Discontinuation of olanzapine and fluoxetine hydrochloride should be considered in patients with symptomatic hyponatremia and appropriate medical intervention should be instituted.
Signs and symptoms of hyponatremia include headache, difficulty concentrating, memory impairment, confusion, weakness, and unsteadiness, which may lead to falls. More severe and/or acute cases have been associated with hallucination, syncope, seizure, coma, respiratory arrest, and death. [See PATIENT COUNSELING INFORMATION (17.14)].
5.18 Potential for Cognitive and Motor Impairment
Olanzapine and fluoxetine hydrochloride has the potential to impair judgment, thinking, or motor skills. Patients should be cautioned about operating hazardous machinery, including automobiles, until they are reasonably certain that olanzapine and fluoxetine hydrochloride therapy does not affect them adversely [see PATIENT COUNSELING INFORMATION (17.15)].
Adults
Sedation-related adverse reactions were commonly reported with olanzapine and fluoxetine hydrochloride treatment occurring at an incidence of 26.6% in olanzapine and fluoxetine hydrochloride-treated patients compared with 10.9% in placebo-treated patients. Sedation-related adverse reactions (sedation, somnolence, hypersomnia, and lethargy) led to discontinuation in 2% (15/771) of patients in the controlled clinical studies.
Children and Adolescents
In a single, 8-week, randomized, placebo-controlled clinical trial investigating olanzapine and fluoxetine hydrochloride for the treatment of bipolar I depression in patients 10 to 17 years of age, somnolence-related adverse events were commonly reported with olanzapine and fluoxetine hydrochloride treatment occurring at an incidence of 23.5% in olanzapine and fluoxetine hydrochloride-treated patients compared with 2.4% in placebo-treated patients. Somnolence-related adverse events led to discontinuation in 1.2% (2/170) of patients.
5.19 Body Temperature Dysregulation
Disruption of the body's ability to reduce core body temperature has been attributed to antipsychotic drugs. Appropriate care is advised when prescribing olanzapine and fluoxetine hydrochloride for patients who will be experiencing conditions which may contribute to an elevation in core body temperature (e.g., exercising strenuously, exposure to extreme heat, receiving concomitant medication with anticholinergic activity, or being subject to dehydration). [see PATIENT COUNSELING INFORMATION (17.16)].
5.20 QT Prolongation
Post-marketing cases of QT interval prolongation and ventricular arrhythmia including Torsade de Pointes have been reported in patients treated with fluoxetine. Olanzapine and fluoxetine hydrochloride should be used with caution in patients with congenital long QT syndrome; a previous history of QT prolongation; a family history of long QT syndrome or sudden cardiac death; and other conditions that predispose to QT prolongation and ventricular arrhythmia. Such conditions include concomitant use of drugs that prolong the QT interval; hypokalemia or hypomagnesemia; recent myocardial infarction, uncompensated heart failure, bradyarrhythmias, and other significant arrhythmias; and conditions that predispose to increased fluoxetine exposure (overdose, hepatic impairment, use of CYP2D6 inhibitors, CYP2D6 poor metabolizer status, or use of other highly protein-bound drugs). Fluoxetine is primarily metabolized by CYP2D6 [see CONTRAINDICATIONS (4.2), ADVERSE REACTIONS (6), DRUG INTERACTIONS (7.7, 7.8), OVERDOSE (10.1), and CLINICAL PHARMACOLOGY (12.3)].
Pimozide and thioridazine are contraindicated for use with olanzapine and fluoxetine hydrochloride. Avoid the concomitant use of drugs known to prolong the QT interval. These include specific antipsychotics (e.g., ziprasidone, iloperidone, chlorpromazine, mesoridazine, droperidol); specific antibiotics (e.g.,erythromycin, gatifloxacin, moxifloxacin, sparfloxacin); Class 1A antiarrhythmic medications (e.g., quinidine, procainamide); Class III antiarrhythmics (e.g., amiodarone, sotalol); and others (e.g., pentamidine, levomethadyl acetate, methadone, halofantrine, mefloquine, dolasetron mesylate, probucol or tacrolimus) [see DRUG INTERACTIONS (7.7, 7.8) and CLINICAL PHARMACOLOGY (12.3)].
Consider ECG assessment and periodic ECG monitoring if initiating treatment with olanzapine and fluoxetine hydrochloride in patients with risk factors for QT prolongation and ventricular arrhythmia. Consider discontinuing olanzapine and fluoxetine hydrochloride and obtaining a cardiac evaluation if patients develop signs or symptoms consistent with ventricular arrhythmia.
In a single, 8-week, randomized, placebo-controlled clinical trial investigating olanzapine and fluoxetine hydrochloride for the treatment of bipolar I depression in patients 10 to 17 years of age, there was a statistically significant difference in QTc interval for patients treated with olanzapine and fluoxetine hydrochloride compared with patients on placebo: mean change in QTcF (Fridericia correction factor) from baseline to endpoint in patients treated with olanzapine and fluoxetine hydrochloride was 8.2 msec (95% CI 6.2, 10.2). No patient developed QTc increases ≥60 msec or QTc ≥480 msec. Clinicians should use olanzapine and fluoxetine hydrochloride with caution in those children or adolescents who are known to be particularly at risk for QT prolongation [see ADVERSE REACTIONS (6.2)].
5.21 Use in Patients with Concomitant Illness
Clinical experience with olanzapine and fluoxetine hydrochloride in patients with concomitant systemic illnesses is limited [see CLINICAL PHARMACOLOGY (12.4)]. The following precautions for the individual components may be applicable to olanzapine and fluoxetine hydrochloride.
Anticholinergic Adverse Reactions
Olanzapine exhibits in vitro muscarinic receptor affinity. In premarketing clinical studies, olanzapine and fluoxetine hydrochloride was associated with constipation, dry mouth, and tachycardia, all adverse reactions possibly related to cholinergic antagonism. Such adverse reactions were not often the basis for study discontinuations; olanzapine and fluoxetine hydrochloride should be used with caution in patients with clinically significant prostatic hypertrophy, angle-closure glaucoma, a history of paralytic ileus, or related conditions.
Elderly Patients with Dementia-related Psychosis
In 5 placebo-controlled studies of olanzapine in elderly patients with dementia-related psychosis (n=1184), the following treatment-emergent adverse reactions were reported in olanzapine-treated patients at an incidence of at least 2% and significantly greater than placebo-treated patients: falls, somnolence, peripheral edema, abnormal gait, urinary incontinence, lethargy, increased weight, asthenia, pyrexia, pneumonia, dry mouth, and visual hallucinations. The rate of discontinuation due to adverse reactions was significantly greater with olanzapine than placebo (13% vs 7%). Elderly patients with dementia-related psychosis treated with olanzapine are at an increased risk of death compared to placebo. Olanzapine is not approved for the treatment of patients with dementia-related psychosis [see BOXED WARNING, WARNINGS AND PRECAUTIONS (5.2), and PATIENT COUNSELING INFORMATION (17.3)].
As with other CNS-active drugs, olanzapine and fluoxetine hydrochloride should be used with caution in elderly patients with dementia. Olanzapine and fluoxetine hydrochloride is not approved for the treatment of patients with dementia-related psychosis. If the prescriber elects to treat elderly patients with dementia-related psychosis, vigilance should be exercised [see BOXED WARNING and WARNINGS AND PRECAUTIONS (5.2), and PATIENT COUNSELING INFORMATION (17.3)].
Olanzapine and fluoxetine hydrochloride has not been evaluated or used to any appreciable extent in patients with a recent history of myocardial infarction or unstable heart disease. Patients with these diagnoses were excluded from clinical studies during the premarket testing.
Patients with Cardiovascular Disease
Caution is advised when using olanzapine and fluoxetine hydrochloride in cardiac patients and in patients with diseases or conditions that could affect hemodynamic responses [see WARNINGS AND PRECAUTIONS (5.9)].
5.22 Hyperprolactinemia
As with other drugs that antagonize dopamine D2 receptors, olanzapine and fluoxetine hydrochloride elevates prolactin levels, and the elevation persists during administration. Hyperprolactinemia may suppress hypothalamic GnRH, resulting in reduced pituitary gonadotropin secretion. This, in turn, may inhibit reproductive function by impairing gonadal steroidogenesis in both female and male patients. Galactorrhea, amenorrhea, gynecomastia, and erectile dysfunction have been reported in patients receiving prolactin-elevating compounds. Long-standing hyperprolactinemia when associated with hypogonadism may lead to decreased bone density in both female and male subjects.
Tissue culture experiments indicate that approximately one-third of human breast cancers are prolactin dependent in vitro, a factor of potential importance if the prescription of these drugs is contemplated in a patient with previously detected breast cancer. As is common with compounds that increase prolactin release, an increase in mammary gland neoplasia was observed in the olanzapine carcinogenicity studies conducted in mice and rats [see NONCLINICAL TOXICOLOGY (13.1)]. Neither clinical studies nor epidemiologic studies conducted to date have shown an association between chronic administration of this class of drugs and tumorigenesis in humans; the available evidence is considered too limited to be conclusive at this time.
Adults
In controlled clinical studies of olanzapine and fluoxetine hydrochloride (up to 12 weeks), changes from normal to high in prolactin concentrations were observed in 28% of adults treated with olanzapine and fluoxetine hydrochloride as compared to 5% of placebo-treated patients. The elevations persisted throughout administration of olanzapine and fluoxetine hydrochloride. In a pooled analysis from clinical studies including 2,929 adults treated with olanzapine and fluoxetine hydrochloride, potentially associated clinical manifestations included menstrual-related events1 (1% [20/1,946] of females), sexual function-related events2 (7% [192/2,929] of females and males), and breast-related events3 (0.8% [16/1,946] of females, 0.2% [2/983] of males).
Children and Adolescents
In a single, 8-week, randomized, placebo-controlled clinical trial investigating olanzapine and fluoxetine hydrochloride for the treatment of bipolar I depression in patients 10 to 17 years of age, olanzapine and fluoxetine hydrochloride was associated with a statistically significant greater mean change from baseline in prolactin levels compared to placebo (8.7 mcg/L vs. 0.7 mcg/L, respectively). Although prolactin concentrations were very commonly (>10%) elevated above normal in both the olanzapine and fluoxetine hydrochloride and placebo groups, more than twice as many olanzapine and fluoxetine hydrochloride-treated patients were seen with these elevations compared to placebo-treated patients. Five patients experienced an adverse event potentially associated with elevated prolactin; these events included dysmenorrhoea, galactorrhoea, and ovulation disorder.
The magnitude and frequency of change in prolactin in children and adolescents was larger than observed in adult patients treated with olanzapine and fluoxetine hydrochloride, but was similar to that observed in adolescents treated with olanzapine monotherapy.
Olanzapine Monotherapy
In placebo-controlled olanzapine clinical studies (up to 12 weeks), changes from normal to high in prolactin concentrations were observed in 30% of adults treated with olanzapine as compared to 10.5% of adults treated with placebo. In a pooled analysis from clinical studies including 8,136 adults treated with olanzapine, potentially associated clinical manifestations included menstrual-related events1 (2% [49/3,240] of females), sexual function-related events2 (2% [150/8,136] of females and males), and breast-related events3 (0.7% [23/3,240] of females, 0.2% [9/4,896] of males).
In placebo-controlled olanzapine monotherapy studies in adolescent patients (up to 6 weeks) with schizophrenia or bipolar I disorder (manic or mixed episodes), changes from normal to high in prolactin concentrations were observed in 47% of olanzapine-treated patients compared to 7% of placebo-treated patients. In a pooled analysis from clinical trials including 454 adolescents treated with olanzapine, potentially associated clinical manifestations included menstrual-related events1 (1% [2/168] of females), sexual function-related events2 (0.7% [3/454] of females and males), and breast-related events3 (2% [3/168] of females, 2% [7/286] of males), [see USE IN SPECIFIC POPULATIONS (8.4)].
1Based on a search of the following terms: amenorrhea, hypomenorrhea, menstruation delayed, and oligomenorrhea.
2Based on a search of the following terms: anorgasmia, delayed ejaculation, erectile dysfunction, decreased libido, loss of libido, abnormal orgasm, and sexual dysfunction.
3Based on a search of the following terms: breast discharge, enlargement or swelling, galactorrhea, gynecomastia, and lactation disorder.
Dose group differences with respect to prolactin elevation have been observed. In a single 8-week randomized, double-blind, fixed-dose study comparing 10 (n=199), 20 (n=200) and 40 (n=200) mg/day of oral olanzapine in adult patients with schizophrenia or schizoaffective disorder, incidence of prolactin elevation >24.2 ng/mL (female) or >18.77 ng/mL (male) at any time during the trial (10 mg/day: 31.2%; 20 mg/day: 42.7%; 40 mg/day: 61.1%) indicated significant differences between 10 vs. 40 mg/day and 20 vs. 40 mg/day.
5.23 Concomitant Use of Olanzapine and Fluoxetine Products
Olanzapine and fluoxetine hydrochloride contains the same active ingredients that are in Zyprexa®, Zyprexa® Zydis®, Zyprexa® RelprevvTM (olanzapine) and in Prozac®, Prozac® Weekly™, and Sarafem® (fluoxetine HCl). Caution should be exercised when prescribing these medications concomitantly with olanzapine and fluoxetine hydrochloride [see OVERDOSAGE (10)].
5.24 Long Elimination Half-Life of Fluoxetine
Because of the long elimination half-lives of fluoxetine and its major active metabolite, changes in dose will not be fully reflected in plasma for several weeks, affecting both strategies for titration to final dose and withdrawal from treatment. This is of potential consequence when drug discontinuation is required or when drugs are prescribed that might interact with fluoxetine and norfluoxetine following the discontinuation of fluoxetine [see CLINICAL PHARMACOLOGY (12.3)].
5.25 Discontinuation Adverse Reactions
During marketing of fluoxetine, a component of olanzapine and fluoxetine hydrochloride, SNRIs, and SSRIs, there have been spontaneous reports of adverse reactions occurring upon discontinuation of these drugs, particularly when abrupt, including the following: dysphoric mood, irritability, agitation, dizziness, sensory disturbances (e.g., paresthesias such as electric shock sensations), anxiety, confusion, headache, lethargy, emotional lability, insomnia, and hypomania. While these reactions are generally self-limiting, there have been reports of serious discontinuation symptoms. Patients should be monitored for these symptoms when discontinuing treatment with fluoxetine. A gradual reduction in the dose rather than abrupt cessation is recommended whenever possible. If intolerable symptoms occur following a decrease in the dose or upon discontinuation of treatment, then resuming the previously prescribed dose may be considered. Subsequently, the physician may continue decreasing the dose but at a more gradual rate. Plasma fluoxetine and norfluoxetine concentration decrease gradually at the conclusion of therapy, which may minimize the risk of discontinuation symptoms with this drug [see DOSAGE AND ADMINISTRATION (2.4) and PATIENT COUNSELING INFORMATION (17.18)].
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in more detail in other sections of the labeling
- Suicidal Thoughts and Behaviors in Children, Adolescents, and Young Adults [see BOXED WARNING and WARNINGS AND PRECAUTIONS (5.1)]
- Increased Mortality in Elderly Patients with Dementia-Related Psychosis [see WARNINGS AND PRECAUTIONS (5.2)]
- Neuroleptic Malignant syndrome (NMS) [see WARNINGS AND PRECAUTIONS (5.3)]
- Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) [see WARNINGS AND PRECAUTIONS (5.4)]
- Hyperglycemia [see WARNINGS AND PRECAUTIONS (5.5)]
- Dyslipidemia [see WARNINGS AND PRECAUTIONS (5.5)]
- Weight Gain [see WARNINGS AND PRECAUTIONS (5.5)]
- Serotonin Syndrome [see WARNINGS AND PRECAUTIONS (5.6)]
- Angle-Closure Glaucoma [see WARNINGS AND PRECAUTIONS (5.7)]
- Allergic Reactions and Rash [see WARNINGS AND PRECAUTIONS (5.8)]
- Activation of Mania/Hypomania [see WARNINGS AND PRECAUTIONS (5.9)]
- Tardive Dyskinesia [see WARNINGS AND PRECAUTIONS (5.10)]
- Orthostatic Hypotension [see WARNINGS AND PRECAUTIONS (5.11)]
- Falls [see WARNINGS AND PRECAUTIONS (5.12)]
- Leukopenia, Neutropenia, and Agranulocytosis [see WARNINGS AND PRECAUTIONS (5.13)]
- Dysphagia [see WARNINGS AND PRECAUTIONS (5.14)]
- Seizures [see WARNINGS AND PRECAUTIONS (5.15)]
- Abnormal Bleeding [see WARNINGS AND PRECAUTIONS (5.16)]
- Hyponatremia [see WARNINGS AND PRECAUTIONS (5.17)]
- Potential for Cognitive and Motor Impairment [see WARNINGS AND PRECAUTIONS (5.18)]
- Body Temperature Dysregulation [see WARNINGS AND PRECAUTIONS (5.19)]
- QT Prolongation [see WARNINGS AND PRECAUTIONS (5.20)]
- Hyperprolactinemia [see WARNINGS AND PRECAUTIONS (5.22)]
- Discontinuation Adverse Reactions [see WARNINGS AND PRECAUTIONS (5.25)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect or predict the rates observed in practice.
The data in the tables represent the proportion of individuals who experienced, at least once, a treatment-emergent adverse reaction of the type listed. A reaction was considered treatment-emergent if it occurred for the first time or worsened while receiving therapy following baseline evaluation.
Adults
The information below is derived from a clinical study database for olanzapine and fluoxetine hydrochloride consisting of 2,547 patients. The conditions and duration of treatment with olanzapine and fluoxetine hydrochloride varied greatly and included (in overlapping categories) open-label and double-blind phases of studies, inpatients and outpatients, fixed-dose and dose-titration studies, and short-term or long-term exposure.
Adverse Reactions Associated with Discontinuation of Treatment in Short-Term, Controlled Studies
Overall, 11.3% of the 771 patients in the olanzapine and fluoxetine hydrochloride group discontinued due to adverse reactions compared with 4.4% of the 477 patients for placebo. Adverse reactions leading to discontinuation associated with the use of olanzapine and fluoxetine hydrochloride (incidence of at least 1% for olanzapine and fluoxetine hydrochloride and greater than that for placebo) using MedDRA Dictionary coding were weight increased (2%) and sedation (1%) versus placebo patients which had 0% incidence of weight increased and sedation.
Commonly Observed Adverse Reactions in Controlled Studies
In short term studies, the most commonly observed adverse reactions associated with the use of olanzapine and fluoxetine hydrochloride (incidence ≥5% and at least twice that for placebo in the olanzapine and fluoxetine hydrochloride-controlled database) using MedDRA Dictionary coding were: disturbance in attention, dry mouth, fatigue, hypersomnia, increased appetite, peripheral edema, sedation, somnolence, tremor, vision blurred, and weight increased. Adverse reactions reported in clinical trials of olanzapine and fluoxetine in combination are generally consistent with treatment-emergent adverse reactions during olanzapine or fluoxetine monotherapy.
In a 47-week maintenance study in adults with adverse reactions associated with olanzapine and fluoxetine hydrochloride use were generally similar to those seen in short-term studies. Weight gain, hyperlipidemia, and hyperglycemia were observed in olanzapine and fluoxetine hydrochloride-treated patients throughout the study.
Adverse Reactions Occurring at an Incidence of 2% or More in Short-Term Controlled Studies
Table 16 enumerates the treatment-emergent adverse reactions associated with the use of olanzapine and fluoxetine hydrochloride (incidence of at least 2% for olanzapine and fluoxetine hydrochloride and twice or more than for placebo). The olanzapine and fluoxetine hydrochloride-controlled column includes patients with various diagnoses while the placebo column includes only patients with bipolar depression and major depression with psychotic features.
Table 16: Adverse Reactions: Incidence in the Short-Term Controlled Clinical Studies in Adults
System Organ Class
Adverse Reaction
Percentage of Patients Reporting Event
Olanzapine and Fluoxetine Hydrochloride-Controlled
(N=771)
Placebo
(N=477)
Eye disorders
Vision blurred
5
2
Gastrointestinal disorders
Dry mouth
15
6
Flatulence
3
1
Abdominal distension
2
0
General disorders and administration site conditions
Fatigue
12
2
Edema*
15
2
Asthenia
3
1
Pain
2
1
Pyrexia
2
1
Infections and infestations
Sinusitis
2
1
Investigations
Weight increased
25
3
Metabolism and nutrition disorders
Increased appetite
20
4
Musculoskeletal and connective tissue disorders
Arthralgia
4
1
Pain in extremity
3
1
Musculoskeletal stiffness
2
1
Nervous system disorders
Somnolence†
27
11
Tremor
9
3
Disturbance in attention
5
1
Psychiatric disorders
Restlessness
4
1
Thinking abnormal
2
1
Nervousness
2
1
Reproductive system and breast disorders
Erectile dysfunction
2
1
*Includes edema, edema peripheral, pitting edema, generalized edema, eyelid edema, face edema, gravitational edema, localized edema, periorbital edema, swelling, joint swelling, swelling face, and eye swelling.
†Includes somnolence, sedation, hypersomnia, and lethargy.
Extrapyramidal Symptoms
Dystonia, Class Effect for Antipsychotics
Symptoms of dystonia, prolonged abnormal contractions of muscle groups, may occur in susceptible individuals during the first few days of treatment. Dystonic symptoms include: spasm of the neck muscles, sometimes progressing to tightness of the throat, swallowing difficulty, difficulty breathing, and/or protrusion of the tongue. While these symptoms can occur at low doses, the frequency and severity are greater with high potency and at higher doses of first generation antipsychotic drugs. In general, an elevated risk of acute dystonia may be observed in males and younger age groups receiving antipsychotics; however, events of dystonia have been reported infrequently (<1%) with the olanzapine and fluoxetine combination.
Additional Findings Observed in Clinical Studies
Sexual Dysfunction
In the pool of controlled olanzapine and fluoxetine hydrochloride studies in patients with bipolar depression, there were higher rates of the treatment-emergent adverse reactions decreased libido, anorgasmia, erectile dysfunction and abnormal ejaculation in the olanzapine and fluoxetine hydrochloride group than in the placebo group. One case of decreased libido led to discontinuation in the olanzapine and fluoxetine hydrochloride group. In the controlled studies that contained a fluoxetine arm, the rates of decreased libido and abnormal ejaculation in the olanzapine and fluoxetine hydrochloride group were less than the rates in the fluoxetine group. None of the differences were statistically significant.
Sexual dysfunction, including priapism, has been reported with all SSRIs. While it is difficult to know the precise risk of sexual dysfunction associated with the use of SSRIs, physicians should routinely inquire about such possible side effects.
There are no adequate and well-controlled studies examining sexual dysfunction with olanzapine and fluoxetine hydrochloride or fluoxetine treatment. Symptoms of sexual dysfunction occasionally persist after discontinuation of fluoxetine treatment.
Difference Among Dose Levels Observed in Other Olanzapine Clinical Trials
In a single 8-week randomized, double-blind, fixed-dose study comparing 10 (N=199), 20 (N=200), and 40 (N=200) mg/day of olanzapine in patients with Schizophrenia or Schizoaffective Disorder, statistically significant differences among 3 dose groups were observed for the following safety outcomes: weight gain, prolactin elevation, fatigue, and dizziness. Mean baseline to endpoint increase in weight (10 mg/day: 1.9 kg; 20 mg/day: 2.3 kg; 40 mg/day: 3 kg) was observed with significant differences between 10 vs. 40 mg/day. Incidence of treatment-emergent prolactin elevation >24.2 ng/mL (female) or >18.77 ng/mL (male) at any time during the trial (10 mg/day: 31.2%; 20 mg/day: 42.7%; 40 mg/day: 61.1%) with significant differences between 10 vs. 40 mg/day and 20 vs. 40 mg/day; fatigue (10 mg/day: 1.5%; 20 mg/day: 2.1%; 40 mg/day: 6.6%) with significant differences between 10 vs. 40 and 20 vs. 40 mg/day; and dizziness (10 mg/day: 2.6%; 20 mg/day: 1.6%; 40 mg/day: 6.6%) with significant differences between 20 vs. 40 mg, was observed.
Other Adverse Reactions Observed in Clinical Studies
Following is a list of treatment-emergent adverse reactions reported by patients treated with olanzapine and fluoxetine hydrochloride in clinical trials. This listing is not intended to include reactions (1) already listed in previous tables or elsewhere in labeling, (2) for which a drug cause was remote, (3) which were so general as to be uninformative, (4) which were not considered to have significant clinical implications, or (5) which occurred at a rate equal to or less than placebo.
Reactions are classified by body system using the following definitions: frequent adverse reactions are those occurring in at least 1/100 patients; infrequent adverse reactions are those occurring in 1/100 to 1/1000 patients; and rare reactions are those occurring in fewer than 1/1000 patients.
Body as a Whole — Frequent:chills, neck rigidity, photosensitivity reaction; Rare: death1.
Cardiovascular System — Frequent: vasodilatation.
Digestive System — Frequent:diarrhea; Infrequent: gastritis, gastroenteritis, nausea and vomiting, peptic ulcer; Rare: gastrointestinal hemorrhage, intestinal obstruction, liver fatty deposit, pancreatitis.
Hemic and Lymphatic System — Frequent: ecchymosis; Infrequent: anemia, thrombocytopenia; Rare: leukopenia, purpura.
Metabolic and Nutritional — Frequent: generalized edema, weight loss; Rare:bilirubinemia, creatinine increased, gout.
Musculoskeletal System — Rare: osteoporosis.
Nervous System — Frequent: amnesia; Infrequent: ataxia, buccoglossal syndrome, coma, depersonalization, dysarthria, emotional lability, euphoria, hypokinesia, movement disorder, myoclonus; Rare: hyperkinesia, libido increased, withdrawal syndrome.
Respiratory System — Infrequent: epistaxis, yawn; Rare: laryngismus.
Skin and Appendages — Infrequent: alopecia, dry skin, pruritus; Rare: exfoliative dermatitis.
Special Senses — Frequent:taste perversion; Infrequent: abnormality of accommodation, dry eyes.
Urogenital System — Frequent: breast pain, menorrhagia2, urinary frequency, urinary incontinence; Infrequent: amenorrhea2, female lactation2, hypomenorrhea2, metrorrhagia2, urinary retention, urinary urgency, urination impaired; Rare: breast engorgement2.
1 This term represents a serious adverse event but does not meet the definition for adverse drug reactions. It is included here because of its seriousness.
2 Adjusted for gender.
Other Adverse Reactions Observed with Olanzapine or Fluoxetine Monotherapy
The following adverse reactions were not observed in olanzapine and fluoxetine hydrochloride-treated patients during premarketing clinical studies but have been reported with olanzapine or fluoxetine monotherapy: aplastic anemia, bruxism, cholestatic jaundice, diabetic coma, dysuria, eosinophilic pneumonia3, erythema multiforme, esophageal ulcer, gynecological bleeding, headache, hypotension, jaundice, neutropenia, restless legs syndrome, sudden unexpected death3, sweating, and violent behaviors3. Random triglyceride levels of ≥1000 mg/dL have been reported.
3 These terms represent serious adverse events but do not meet the definition for adverse drug reactions. They are included here because of their seriousness.
Children and Adolescent Patients (aged 10 to 17 years) with a Diagnosis of Bipolar Depression
The information below is derived from a single, 8-week, randomized, placebo-controlled clinical trial investigating olanzapine and fluoxetine hydrochloride for the treatment of bipolar I depression in patients 10 to 17 years of age.
Adverse Reactions Associated with Discontinuation of Treatment in the Single Pediatric Study
Overall, 14.1% of the 170 patients in the olanzapine and fluoxetine hydrochloride group discontinued due to adverse reactions compared with 5.9% of the 85 patients for placebo. Adverse reactions leading to discontinuation associated with the use of olanzapine and fluoxetine hydrochloride (incidence of at least 1% for olanzapine and fluoxetine hydrochloride and greater than that for placebo) using MedDRA Dictionary coding were weight increased (2.9%), suicidal ideation (1.8%), bipolar disorder (1.2%), and somnolence (1.2%) versus placebo patients which had 0% incidence of weight increased, bipolar disorder, and somnolence, and a 1.2% incidence of suicidal ideation.
Adverse Reactions Occurring at an Incidence of 2% or More and Greater than Placebo
Table 17 enumerates the treatment-emergent adverse reactions associated with the use of olanzapine and fluoxetine hydrochloride (incidence of at least 2% for olanzapine and fluoxetine hydrochloride and twice or more than for placebo).
Table 17: Treatment-Emergent Adverse Reactions: Incidence in a 8-week Randomized, Double-blind, Placebo-Controlled Clinical Trial in Pediatric Bipolar I Depression.
System Organ Class
Adverse Reaction
Percentage of Patients
Reporting EventOlanzapine and Fluoxetine Hydrochloride
(N=170)Placebo
(N=85)Nervous system disorders
Somnolence*
24
2
Tremor
9
1
Investigations
Weight increased
20
1
Blood triglycerides increased
7
2
Blood cholesterol increased
4
0
Hepatic enzyme increased†
9
1
Gastrointestinal disorders
Dyspepsia
3
1
Metabolism and nutrition disorders
Increased appetite
17
1
Psychiatric disorders
Anxiety
3
1
Restlessness
3
1
Suicidal ideation
2
1
Musculoskeletal and connective tissue disorders
Back pain
2
1
Injury, poisoning and procedural complications
Accidental overdose
3
1
Reproductive system and breast disorders
Dysmenorrhea
2
0
*Includes somnolence, sedation, and hypersomnia. No lethargy was reported.
†Includes alanine aminotransferase increased, aspartate aminotransferase increased, hepatic enzyme increased, liver function test abnormal, gamma-glutamyltransferase increased, and transaminases increased.
6.2 Vital Signs and Laboratory Studies
Adults
Vital Signs
Tachycardia, bradycardia, and orthostatic hypotension have occurred in olanzapine and fluoxetine hydrochloride-treated patients [see WARNINGS AND PRECAUTIONS (5.11)]. The mean standing pulse rate of olanzapine and fluoxetine hydrochloride-treated patients was reduced by 0.7 beats/min.
Laboratory Changes
In olanzapine and fluoxetine hydrochloride clinical studies, olanzapine and fluoxetine hydrochloride was associated with statistically significantly greater frequencies for the following treatment-emergent findings in laboratory analytes (normal at baseline to abnormal at any time during the trial) compared to placebo: elevated prolactin ( 28% vs. 5%); elevated urea nitrogen (3% vs. 0.8%); elevated uric acid (3% vs. 0.5%); low albumin (3% vs. 0.3%); low bicarbonate (14% vs. 9%); low hemoglobin (3% vs. 0%); low inorganic phosphorus (2% vs. 0.3%); low lymphocytes (2% vs. 0%); and low total bilirubin (15% vs. 4%).
As with olanzapine, asymptomatic elevations of hepatic aminotransferases [ALT, AST, and GGT] and alkaline phosphatase have been observed with olanzapine and fluoxetine hydrochloride. In the olanzapine and fluoxetine hydrochloride-controlled database, clinically significant ALT elevations (change from <3 times the upper limit of normal [ULN] at baseline to ≥3 times ULN) were observed in 5% (38/698) of patients exposed to olanzapine and fluoxetine hydrochloride compared with 0.5% (2/378) of placebo-treated patients and 4% (33/751) of olanzapine-treated patients. ALT elevations ≥5 times ULN were observed in 2% (11/701) of olanzapine and fluoxetine hydrochloride-treated patients, compared to 0.3% (1/379) of placebo-treated patients and 1% (11/760) of olanzapine-treated patients. No patient with elevated ALT values experienced jaundice or liver failure, or met the criteria for Hy’s Rule. ALT values returned to normal, or were decreasing, at last follow-up in the majority of patients who either continued treatment with olanzapine and fluoxetine hydrochloride or discontinued olanzapine and fluoxetine hydrochloride.
Rare postmarketing reports of hepatitis have been received in patients treated with olanzapine. Very rare cases of cholestatic or mixed liver injury have also been reported in the postmarketing period in patients treated with olanzapine.
Caution should be exercised in patients with signs and symptoms of hepatic impairment, in patients with pre-existing conditions associated with limited hepatic functional reserve, and in patients who are being treated with potentially hepatotoxic drugs.
An increase in creatine phosphokinase has been reported very rarely in olanzapine and fluoxetine hydrochloride-treated patients and infrequently in clinical trials of olanzapine-treated patients.
QT Interval Prolongation
In patients treated with olanzapine and fluoxetine hydrochloride QTcF≥450 msec for males and QTcF≥470 msec for females has been reported frequently (≥1%). The incidence of QTcF>500 msec associated with olanzapine and fluoxetine hydrochloride treatment in clinical trials has been rare and was not significantly different from the incidence associated with placebo. The mean increase in QTc interval for olanzapine and fluoxetine hydrochloride-treated patients (5.17 msec) in the one clinical study directly comparing olanzapine and fluoxetine hydrochloride to placebo in adult patients was significantly greater than that for placebo-treated patients (-1.66 msec).
Children and Adolescents (aged 10 to 17 years)
In a single 8-week randomized, placebo-controlled clinical trial investigating olanzapine and fluoxetine hydrochloride for treatment of bipolar I depression in patients 10 to 17 years of age, the following was observed:
Vital Signs
In the olanzapine and fluoxetine hydrochloride-treated patients compared with placebo-treated patients, the mean orthostatic blood pressure and standing pulse rate were not significantly different between treatment groups.
Body Weight
An increase in weight greater than or equal to 7% occurred in 52.4% of the olanzapine and fluoxetine hydrochloride group and 3.6% of the placebo group. Weight gain greater than or equal to 15% occurred in 14.1% of the olanzapine and fluoxetine hydrochloride group and none of the placebo group.
Laboratory Changes
Olanzapine and fluoxetine hydrochloride was associated with statistically significantly greater frequencies for the following treatment-emergent findings in laboratory analytes (normal or low at baseline to abnormal at any time during the trial) compared to placebo: elevated ALT (45.9% vs. 2.5%); elevated AST (33.7% vs. 7.6%); high fasting total cholesterol (28.9% vs. 8.2%); high fasting LDL cholesterol (19.7% vs. 6.5%); high fasting triglycerides (52.3% vs. 27.3%), and elevated prolactin (85% vs 36%). No patient with elevated hepatic enzyme values experienced jaundice or liver failure, or met the criteria for Hy's Rule. Five patients experienced an adverse event potentially associated with elevated prolactin; these events included dysmenorrhoea, galactorrhoea, and ovulation disorder.
QT Interval Prolongation
Olanzapine and fluoxetine hydrochloride was associated with a statistically significantly greater mean increase in QTcF interval (8.2 msec [95% CI 6.2, 10.2]) compared with placebo. No patients developed QTc increases ≥60 msec or QTc ≥480 msec [see WARNINGS AND PRECAUTIONS (5.20)].
6.3 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of olanzapine and fluoxetine hydrochloride. Because these reactions are reported voluntarily from a population of uncertain size, it is difficult to reliably estimate their frequency or evaluate a causal relationship to drug exposure.
Adverse reactions reported since market introduction that were temporally (but not necessarily causally) related to olanzapine and fluoxetine hydrochloride therapy include the following: rhabdomyolysis and venous thromboembolic events (including pulmonary embolism and deep venous thrombosis).
-
7 DRUG INTERACTIONS
The risks of using olanzapine and fluoxetine hydrochloride in combination with other drugs have not been extensively evaluated in systematic studies. The drug-drug interactions sections of fluoxetine and olanzapine are applicable to olanzapine and fluoxetine hydrochloride. As with all drugs, the potential for interaction by a variety of mechanisms (e.g., pharmacodynamic, pharmacokinetic drug inhibition or enhancement, etc.) is a possibility. In evaluating individual cases, consideration should be given to using lower initial doses of the concomitantly administered drugs, using conservative titration schedules, and monitoring of clinical status [see CLINICAL PHARMACOLOGY (12.3)].
7.1 Monoamine Oxidase Inhibitors (MAOIs)
[See DOSAGE AND ADMINISTRATION (2.4, 2.5), CONTRAINDICATIONS (4.1), and WARNINGS AND PRECAUTIONS (5.6)]
7.2 CNS Acting Drugs
Caution is advised if the concomitant administration of olanzapine and fluoxetine hydrochloride and other CNS-active drugs is required. In evaluating individual cases, consideration should be given to using lower initial doses of the concomitantly administered drugs, using conservative titration schedules, and monitoring of clinical status [see CLINICAL PHARMACOLOGY (12.3 )].
7.3 Serotonergic Drugs
[See DOSAGE AND ADMINISTRATION (2.4, 2.5), CONTRAINDICATIONS (4.1), and WARNINGS AND PRECAUTIONS(5.6)]
7.4 Drugs that Interfere with Hemostasis (e.g., NSAIDs, Aspirin, Warfarin)
Serotonin release by platelets plays an important role in hemostasis. Epidemiological studies of the case-control and cohort design that have demonstrated an association between use of psychotropic drugs that interfere with serotonin reuptake and the occurrence of upper gastrointestinal bleeding have also shown that concurrent use of an NSAID or aspirin may potentiate this risk of bleeding. Altered anticoagulant effects, including increased bleeding, have been reported when SNRIs or SSRIs are coadministered with warfarin [see WARNINGS AND PRECAUTIONS (5.16)]. Warfarin (20 mg single dose) did not affect olanzapine pharmacokinetics. Single doses of olanzapine did not affect the pharmacokinetics of warfarin. Patients receiving warfarin therapy should be carefully monitored when olanzapine and fluoxetine hydrochloride is initiated or discontinued.
7.5 Electroconvulsive Therapy (ECT)
There are no clinical studies establishing the benefit of the combined use of ECT and fluoxetine. There have been rare reports of prolonged seizures in patients on fluoxetine receiving ECT treatment [see WARNINGS AND PRECAUTIONS (5.15)].
7.6 Potential for Other Drugs to Affect Olanzapine and Fluoxetine Hydrochloride
Benzodiazepines
Co-administration of diazepam with olanzapine potentiated the orthostatic hypotension observed with olanzapine [see DRUG INTERACTIONS (7.7)].
Inducers of 1A2
Carbamazepine therapy (200 mg BID) causes an approximate 50% increase in the clearance of olanzapine. This increase is likely due to the fact that carbamazepine is a potent inducer of CYP1A2 activity. Higher daily doses of carbamazepine may cause an even greater increase in olanzapine clearance [see DRUG INTERACTIONS (7.7)].
Alcohol
Ethanol (45 mg/70 kg single dose) did not have an effect on olanzapine pharmacokinetics [see DRUG INTERACTIONS (7.7)].
Inhibitors of CYP1A2
Fluvoxamine decreases the clearance of olanzapine. This results in a mean increase in olanzapine Cmax following fluvoxamine administration of 54% in female nonsmokers and 77% in male smokers. The mean increase in olanzapine AUC is 52% and 108%, respectively. Lower doses of the olanzapine component of olanzapine and fluoxetine hydrochloride should be considered in patients receiving concomitant treatment with fluvoxamine.
The Effect of Other Drugs on Olanzapine
Fluoxetine, an inhibitor of CYP2D6, decreases olanzapine clearance a small amount [see CLINICAL PHARMACOLOGY (12.3)]. Agents that induce CYP1A2 or glucuronyl transferase enzymes, such as omeprazole and rifampin, may cause an increase in olanzapine clearance. The effect of CYP1A2 inhibitors, such as fluvoxamine and some fluoroquinolone antibiotics, on olanzapine and fluoxetine hydrochloride has not been evaluated. Although olanzapine is metabolized by multiple enzyme systems, induction or inhibition of a single enzyme may appreciably alter olanzapine clearance. Therefore, a dosage increase (for induction) or a dosage decrease (for inhibition) may need to be considered with specific drugs.
7.7 Potential for Olanzapine and Fluoxetine Hydrochloride to Affect Other Drugs
Pimozide
Concomitant use of olanzapine and fluoxetine hydrochloride and pimozide is contraindicated. Pimozide can prolong the QT interval. Olanzapine and fluoxetine hydrochloride can increase the level of pimozide through inhibition of CYP2D6. Olanzapine and fluoxetine hydrochloride can also prolong the QT interval. Clinical studies of pimozide with other antidepressants demonstrate an increase in drug interaction or QTc prolongation. While a specific study with pimozide and olanzapine and fluoxetine hydrochloride has not been conducted, the potential for drug interactions or QTc prolongation warrants restricting the concurrent use of pimozide and olanzapine and fluoxetine hydrochloride [see CONTRAINDICATIONS (4.2), WARNINGS AND PRECAUTIONS (5.20), and DRUG INTERACTIONS (7.8)].
Carbamazepine
Patients on stable doses of carbamazepine have developed elevated plasma anticonvulsant concentrations and clinical anticonvulsant toxicity following initiation of concomitant fluoxetine treatment.
Alcohol
The co-administration of ethanol with olanzapine and fluoxetine hydrochloride may potentiate sedation and orthostatic hypotension [see DRUG INTERACTIONS (7.6)].
Thioridazine
Thioridazine should not be administered with olanzapine and fluoxetine hydrochloride or administered within a minimum of 5 weeks after discontinuation of olanzapine and fluoxetine hydrochloride because of the risk of QT prolongation [see CONTRAINDICATIONS (4.2), WARNINGS AND PRECAUTIONS (5.20), and DRUG INTERACTIONS (7.8)].
In a study of 19 healthy male subjects, which included 6 slow and 13 rapid hydroxylators of debrisoquin, a single 25 mg oral dose of thioridazine produced a 2.4-fold higher Cmax and a 4.5-fold higher AUC for thioridazine in the slow hydroxylators compared with the rapid hydroxylators. The rate of debrisoquin hydroxylation is felt to depend on the level of CYP2D6 isozyme activity. Thus, this study suggests that drugs that inhibit CYP2D6, such as certain SSRIs, including fluoxetine, will produce elevated plasma levels of thioridazine [see CONTRAINDICATIONS (4.2)].
Thioridazine administration produces a dose-related prolongation of the QTc interval, which is associated with serious ventricular arrhythmias, such as torsades de pointes-type arrhythmias and sudden death. This risk is expected to increase with fluoxetine-induced inhibition of thioridazine metabolism [see CONTRAINDICATIONS (4.2)].
Due to the risk of serious ventricular arrhythmias and sudden death potentially associated with elevated thioridazine plasma levels, thioridazine should not be administered with fluoxetine or within a minimum of 5 weeks after fluoxetine has been discontinued [see CONTRAINDICATIONS (4.2)].
Tricyclic Antidepressants (TCAs)
Single doses of olanzapine did not affect the pharmacokinetics of imipramine or its active metabolite desipramine.
In 2 fluoxetine studies, previously stable plasma levels of imipramine and desipramine have increased >2 to 10-fold when fluoxetine has been administered in combination. This influence may persist for 3 weeks or longer after fluoxetine is discontinued. Thus, the dose of TCA may need to be reduced and plasma TCA concentrations may need to be monitored temporarily when olanzapine and fluoxetine hydrochloride is coadministered or has been recently discontinued [see WARNINGS AND PRECAUTIONS (5.6) and CLINICAL PHARMACOLOGY (12.3)].
Antihypertensive Agents
Because of the potential for olanzapine to induce hypotension, olanzapine and fluoxetine hydrochloride may enhance the effects of certain antihypertensive agents [see WARNINGS AND PRECAUTIONS (5.11)].
Levodopa and Dopamine Agonists
The olanzapine component of olanzapine and fluoxetine hydrochloride may antagonize the effects of levodopa and dopamine agonists.
Benzodiazepines
Multiple doses of olanzapine did not influence the pharmacokinetics of diazepam and its active metabolite N-desmethyldiazepam.
When concurrently administered with fluoxetine, the half-life of diazepam may be prolonged in some patients [see CLINICAL PHARMACOLOGY (12.3)]. Coadministration of alprazolam and fluoxetine has resulted in increased alprazolam plasma concentrations and in further psychomotor performance decrement due to increased alprazolam levels.
Clozapine
Elevation of blood levels of clozapine has been observed in patients receiving concomitant fluoxetine.
Haloperidol
Elevation of blood levels of haloperidol has been observed in patients receiving concomitant fluoxetine.
Phenytoin
Patients on stable doses of phenytoin have developed elevated plasma levels of phenytoin with clinical phenytoin toxicity following initiation of concomitant fluoxetine.
Drugs Metabolized by CYP2D6
In vitro studies utilizing human liver microsomes suggest that olanzapine has little potential to inhibit CYP2D6. Thus, olanzapine is unlikely to cause clinically important drug interactions mediated by this enzyme.
Fluoxetine inhibits the activity of CYP2D6 and may make individuals with normal CYP2D6 metabolic activity resemble a poor metabolizer. Coadministration of fluoxetine with other drugs that are metabolized by CYP2D6, including certain antidepressants (e.g., TCAs), antipsychotics (e.g., phenothiazines and most atypicals), and antiarrhythmics (e.g., propafenone, flecainide, and others) should be approached with caution. Therapy with medications that are predominantly metabolized by the CYP2D6 system and that have a relatively narrow therapeutic index should be initiated at the low end of the dose range if a patient is receiving fluoxetine concurrently or has taken it in the previous 5 weeks. If fluoxetine is added to the treatment regimen of a patient already receiving a drug metabolized by CYP2D6, the need for a decreased dose of the original medication should be considered. Drugs with a narrow therapeutic index represent the greatest concern (including but not limited to, flecainide, propafenone, vinblastine, and TCAs).
Drugs Metabolized by CYP3A
In vitro studies utilizing human liver microsomes suggest that olanzapine has little potential to inhibit CYP3A. Thus, olanzapine is unlikely to cause clinically important drug interactions mediated by these enzymes.
In an in vivo interaction study involving the coadministration of fluoxetine with single doses of terfenadine (a CYP3A substrate), no increase in plasma terfenadine concentrations occurred with concomitant fluoxetine. In addition, in vitro studies have shown ketoconazole, a potent inhibitor of CYP3A activity, to be at least 100 times more potent than fluoxetine or norfluoxetine as an inhibitor of the metabolism of several substrates for this enzyme, including astemizole, cisapride, and midazolam. These data indicate that fluoxetine's extent of inhibition of CYP3A activity is not likely to be of clinical significance.
Effect of Olanzapine on Drugs Metabolized by Other CYP Enzymes
In vitro studies utilizing human liver microsomes suggest that olanzapine has little potential to inhibit CYP1A2, CYP2C9, and CYP2C19. Thus, olanzapine is unlikely to cause clinically important drug interactions mediated by these enzymes.
Lithium
Multiple doses of olanzapine did not influence the pharmacokinetics of lithium.
There have been reports of both increased and decreased lithium levels when lithium was used concomitantly with fluoxetine. Cases of lithium toxicity and increased serotonergic effects have been reported. Lithium levels should be monitored in patients taking olanzapine and fluoxetine hydrochloride concomitantly with lithium [see WARNINGS AND PRECAUTIONS (5.5)].
Drugs Tightly Bound to Plasma Proteins
The in vitro binding of olanzapine and fluoxetine hydrochloride to human plasma proteins is similar to the individual components. The interaction between olanzapine and fluoxetine hydrochloride and other highly protein-bound drugs has not been fully evaluated. Because fluoxetine is tightly bound to plasma protein, the administration of fluoxetine to a patient taking another drug that is tightly bound to protein (e.g., Coumadin, digitoxin) may cause a shift in plasma concentrations potentially resulting in an adverse effect. Conversely, adverse effects may result from displacement of protein-bound fluoxetine by other tightly bound drugs [see CLINICAL PHARMACOLOGY (12.3)].
Valproate
In vitro studies using human liver microsomes determined that olanzapine has little potential to inhibit the major metabolic pathway, glucuronidation, of valproate. Further, valproate has little effect on the metabolism of olanzapine in vitro. Thus, a clinically significant pharmacokinetic interaction between olanzapine and valproate is unlikely.
Biperiden
Multiple doses of olanzapine did not influence the pharmacokinetics of biperiden.
Theophylline
Multiple doses of olanzapine did not affect the pharmacokinetics of theophylline or its metabolites.
7.8 Drugs that Prolong the QT Interval
Do not use olanzapine and fluoxetine hydrochloride in combination with thioridazine or pimozide. Use olanzapine and fluoxetine hydrochloride with caution in combination with other drugs that cause QT prolongation. These include: specific antipsychotics (e.g., ziprasidone, iloperidone, chlorpromazine, mesoridazine, droperidol); specific antibiotics (e.g., erythromycin, gatifloxacin, moxifloxacin, sparfloxacin); Class 1A antiarrhythmic medications (e.g., quinidine, procainamide); Class III antiarrhythmics (e.g., amiodarone, sotalol); and others (e.g., pentamidine, levomethadyl acetate, methadone, halofantrine, mefloquine, dolasetron mesylate, probucol or tacrolimus). Fluoxetine is primarily metabolized by CYP2D6. Concomitant treatment with CYP2D6 inhibitors can increase the concentration of fluoxetine. Concomitant use of other highly protein-bound drugs can increase the concentration of fluoxetine [see CONTRAINDICATIONS (4.2), WARNINGS AND PRECAUTIONS (5.20), DRUG INTERACTIONS (7.7), and CLINICAL PHARMACOLOGY (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C
Risk Summary
There are no adequate and well-controlled clinical studies with olanzapine and fluoxetine hydrochloride or its components (olanzapine and fluoxetine) in pregnant women.
A number of published epidemiological studies assessing the risk of fluoxetine exposure during the first trimester of pregnancy have demonstrated inconsistent results.
Neonates exposed to fluoxetine, a component of olanzapine and fluoxetine hydrochloride, and other SSRIs and SNRIs late in the third trimester have developed complications arising immediately upon delivery (respiratory distress, cyanosis, apnea, seizures, temperature instability, feeding difficulty, vomiting, hypoglycemia, hypotonia, hypertonia, hyperreflexia, tremor, jitteriness, irritability, and constant crying) requiring prolonged hospitalization, respiratory support, and tube feeding. These features are consistent with either a direct toxic effect of SSRIs and SNRIs or, possibly, a drug discontinuation syndrome. In some cases, the clinical picture is consistent with serotonin syndrome. Neonates exposed to olanzapine, a component of olanzapine and fluoxetine hydrochloride, during the third trimester of pregnancy are at risk for extrapyramidal and/or withdrawal symptoms following delivery including agitation, hypertonia, hypotonia, tremor, somnolence, respiratory distress and feeding disorder. These complications have varied in severity; while in some cases symptoms have been self-limited, in other cases neonates have required intensive care unit support and prolonged hospitalization.
Infants exposed to SSRIs in pregnancy may have an increased risk for persistent pulmonary hypertension of the newborn (PPHN).
Olanzapine and fluoxetine hydrochloride should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus, taking into account the risk of untreated Bipolar I Depression.
Human Data
There are no adequate and well-controlled clinical studies with olanzapine and fluoxetine hydrochloride, olanzapine or fluoxetine in pregnant women. Seven pregnancies were observed during premarketing clinical studies with olanzapine, including 2 resulting in normal births, 1 resulting in neonatal death due to a cardiovascular defect, 3 therapeutic abortions, and 1 spontaneous abortion. Results of a number of published epidemiological studies assessing the risk of fluoxetine exposure during the first trimester of pregnancy have demonstrated inconsistent results. More than 10 cohort studies and case-control studies failed to demonstrate an increased risk for congenital malformations overall. However, one prospective cohort study conducted by the European Network of Teratology Information Services reported an increased risk of cardiovascular malformations in infants born to women (N = 253) exposed to fluoxetine during the first trimester of pregnancy compared to infants of women (N = 1359) who were not exposed to fluoxetine. There was no specific pattern of cardiovascular malformations. Overall, however, a causal relationship has not been established.
Neonates exposed to fluoxetine, a component of olanzapine and fluoxetine hydrochloride, and other SSRIs or serotonin and norepinephrine reuptake inhibitors (SNRIs), late in the third trimester have developed complications requiring prolonged hospitalization, respiratory support, and tube feeding. Such complications can arise immediately upon delivery. Reported clinical findings have included respiratory distress, cyanosis, apnea, seizures, temperature instability, feeding difficulty, vomiting, hypoglycemia, hypotonia, hypertonia, hyperreflexia, tremor, jitteriness, irritability, and constant crying. These features are consistent with either a direct toxic effect of SSRIs and SNRIs or, possibly, a drug discontinuation syndrome. It should be noted that, in some cases, the clinical picture is consistent with serotonin syndrome [see DOSAGE AND ADMINISTRATION (2.3) and WARNINGS AND PRECAUTIONS (5.6)].
Infants exposed to SSRIs in pregnancy may have an increased risk for persistent pulmonary hypertension of the newborn (PPHN). PPHN occurs in 1 to 2 per 1,000 live births in the general population and is associated with substantial neonatal morbidity and mortality. Several recent epidemiologic studies suggest a positive statistical association between SSRI (including fluoxetine) use in pregnancy and PPHN. Other studies do not show a significant statistical association.
Neonates exposed to antipsychotic drugs (including olanzapine, a component of olanzapine and fluoxetine hydrochloride), during the third trimester of pregnancy are at risk for extrapyramidal and/or withdrawal symptoms following delivery. There have been reports of agitation, hypertonia, hypotonia, tremor, somnolence, respiratory distress and feeding disorder in these neonates. These complications have varied in severity; while in some cases symptoms have been self-limited, in other cases neonates have required intensive care unit support and prolonged hospitalization.
Physicians should also note the results of a prospective longitudinal study of 201 pregnant women with a history of major depression, who were either on antidepressants or had received antidepressants less than 12 weeks prior to their last menstrual period, and were in remission. Women who discontinued antidepressant medication during pregnancy showed a significant increase in relapse of their major depression compared to those women who remained on antidepressant medication throughout pregnancy.
When treating a pregnant woman with olanzapine and fluoxetine hydrochloride, the physician should carefully consider both the potential risks of taking an SSRI, along with the established benefits of treating depression with an antidepressant. This decision can only be made on a case by case basis [see DOSAGE AND ADMINISTRATION (2.3)].
Olanzapine and fluoxetine hydrochloride should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Animal Data
Olanzapine and Fluoxetine Hydrochloride
Embryo fetal development studies were conducted in rats and rabbits with olanzapine and fluoxetine in low-dose and high-dose combinations. In rats, the doses were: 2 and 4 mg/kg/day (low-dose) [1 and 0.5 times the maximum recommended human dose (MRHD) for olanzapine (20 mg) and fluoxetine (80 mg), respectively, on a mg/m2 body surface area], and 4 and 8 mg/kg/day (high-dose) [2 and 1 times the MRHD on a mg/m2 body surface area, respectively]. In rabbits, the doses were 4 and 4 mg/kg/day (low-dose) [4 and 1 times the MRHD on a mg/m2 basis, respectively], and 8 and 8 mg/kg/day (high-dose) [9 and 2 times the MRHD on a mg/m2 basis, respectively]. In these studies, olanzapine and fluoxetine were also administered alone at the high-doses (4 and 8 mg/kg/day, respectively, in the rat; 8 and 8 mg/kg/day, respectively, in the rabbit). In the rabbit, there was no evidence of teratogenicity; however, the high-dose combination produced decreases in fetal weight and retarded skeletal ossification in conjunction with maternal toxicity. Similarly, in the rat there was no evidence of teratogenicity; however, a decrease in fetal weight was observed with the high-dose combination.
In a pre- and postnatal study conducted in rats, olanzapine and fluoxetine were orally administered during pregnancy and throughout lactation in combination at dose levels up to 2 (olanzapine) plus 4 (fluoxetine) mg/kg/day (1 and 0.5 times the MRHD on a mg/m2 body surface area). An elevation of early postnatal mortality (survival through postnatal day 4 was 69% per litter) and reduced bodyweight (approximately 8% in female) occurred among offspring at the highest dose: the no-effect dose was 0.5 (olanzapine) plus 1 (fluoxetine) mg/kg/day (0.25 and 0.13 times the MRHD on a mg/m2 body surface area). Among the surviving progeny, there were no adverse effects on physical or neurobehavioral development and reproductive performance at any dose.
Olanzapine
In oral embryo-fetal studies in rats and rabbits, there was no evidence of teratogenicity following administration of olanzapine at doses up to 18 and 30 mg/kg/day, respectively (9 and 30 times the MRHD on a mg/m2 basis, respectively). In a rat teratology study, early resorptions and increased numbers of dead fetuses were observed at a dose of 18 mg/kg/day (9 times the MRHD on a mg/m2 basis). Gestation was prolonged at 10 mg/kg/day (5 times the MRHD on a mg/m2 basis). In a rabbit teratology study, fetal toxicity (manifested as increased resorptions and decreased fetal weight) occurred at a maternally toxic dose of 30 mg/kg/day (30 times the MRHD on a mg/m2 basis). Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Placental transfer of olanzapine occurs in rat pups.
Fluoxetine
In oral embryo-fetal development studies in rats and rabbits, there was no evidence of teratogenicity following administration of fluoxetine at doses up to 12.5 and 15 mg/kg/day, respectively (1.5 and 3.6 times, respectively, the (MRHD) of 80 mg/day on a mg/m2 basis,) throughout organogenesis. However, in rat reproduction studies, an increase in stillborn pups, a decrease in pup weight, and an increase in pup deaths during the first 7 days postpartum occurred following maternal exposure to 12 mg/kg/day (1.5 times the MRHD on a mg/m2 basis) during gestation or 7.5 mg/kg/day (0.9 times the MRHD on a mg/m2 basis) during gestation and lactation. There was no evidence of developmental neurotoxicity in the surviving offspring of rats treated with 12 mg/kg/day during gestation. The no-effect dose for rat pup mortality was 5 mg/kg/day (0.6 times the MRHD on a mg/m2 basis).
8.2 Labor and Delivery
Olanzapine and Fluoxetine Hydrochloride
The effect of olanzapine and fluoxetine hydrochloride on labor and delivery in humans is unknown. Parturition in rats was not affected by olanzapine and fluoxetine hydrochloride. Olanzapine and fluoxetine hydrochloride should be used during labor and delivery only if the potential benefit justifies the potential risk.
Olanzapine
The effect of olanzapine on labor and delivery in humans is unknown. Parturition in rats was not affected by olanzapine.
Fluoxetine
The effect of fluoxetine on labor and delivery in humans is unknown. Fluoxetine crosses the placenta; therefore, there is a possibility that fluoxetine may be associated with adverse effects on the newborn.
8.3 Nursing Mothers
Olanzapine and Fluoxetine Hydrochloride
Studies evaluating the individual components of olanzapine and fluoxetine hydrochloride in nursing mothers are described below. Because of the potential for serious adverse reactions in nursing infants from olanzapine and fluoxetine hydrochloride, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. It is recommended that women not breast-feed when receiving olanzapine and fluoxetine hydrochloride.
Olanzapine
In a study in lactating, healthy women, olanzapine was excreted in breast milk. Mean infant dose at steady state was estimated to be 1.8% of the maternal olanzapine dose. It is recommended that women receiving olanzapine should not breast-feed.
Fluoxetine
Fluoxetine is excreted in human breast milk. In 1 breast milk sample, the concentration of fluoxetine plus norfluoxetine was 70.4 ng/mL. The concentration in the mother's plasma was 295 ng/mL. No adverse effects on the infant were reported. In another case, an infant nursed by a mother on fluoxetine developed crying, sleep disturbance, vomiting, and watery stools. The infant's plasma drug levels were 340 ng/mL of fluoxetine and 208/mL of norfluoxetine on the 2nd day of feeding.
8.4 Pediatric Use
Olanzapine and Fluoxetine Hydrochloride
The safety and efficacy of olanzapine and fluoxetine hydrochloride in patients 10 to 17 years of age has been established for the acute treatment of Depressive Episodes Associated with Bipolar I Disorder in a single 8-week randomized, placebo-controlled clinical trial (N = 255) [see CLINICAL STUDIES (14.1)]. Patients were initiated at a dose of 3/25 mg/day and force-titrated to the maximum dose of 12/50 mg/day over two weeks. After Week 2, there was flexible dosing of olanzapine and fluoxetine hydrochloride in the range of 6/25, 6/50, 12/25, or 12/50 mg/day. The average dose was olanzapine 7.7 mg and fluoxetine 37.6 mg. The recommended starting dose for children and adolescents is 3/25 mg per day (lower than that for adults). Flexible dosing is recommended, rather than the forced titration used in the study [see DOSAGE AND ADMINISTRATION (2.1)].
The types of adverse events observed with olanzapine and fluoxetine hydrochloride in children and adolescents were generally similar to those observed in adults. However, the magnitude and frequency of some changes were greater in children and adolescents than adults. These included increases in lipids, hepatic enzymes, and prolactin, as well as increases in the QT interval. [see WARNINGS AND PRECAUTIONS (5.5, 5.20, 5.22), andVital Signs and Laboratory Studies(6.2)]. The frequency of weight gain ≥7%, and the magnitude and frequency of increases in lipids, hepatic analytes, and prolactin in children and adolescents treated with olanzapine and fluoxetine hydrochloride were similar to those observed in adolescents treated with olanzapine monotherapy.
The safety and efficacy of olanzapine and fluoxetine in combination for the treatment of bipolar I depression in patients under the age of 10 years have not been established.
Anyone considering the use of olanzapine and fluoxetine hydrochloride in a child or adolescent must balance the potential risks with the clinical need [see BOXED WARNING and WARNINGS AND PRECAUTIONS (5.1)].
Olanzapine
Safety and effectiveness of olanzapine in children <13 years of age have not been established.
Compared to patients from adult clinical trials, adolescents treated with oral olanzapine were likely to gain more weight, experience increased sedation, and have greater increases in total cholesterol, triglycerides, LDL cholesterol, prolactin and hepatic aminotransferase levels.
Animal Data
Fluoxetine
Juvenile animal toxicity studies were performed for fluoxetine alone. Significant toxicity on muscle tissue, neurobehavior, reproductive organs, and bone development has been observed following exposure of juvenile rats to fluoxetine from weaning through maturity. Oral administration of fluoxetine to rats from weaning postnatal day 21 through adulthood day 90 at 3, 10, or 30 mg/kg/day was associated with testicular degeneration and necrosis, epididymal vacuolation and hypospermia (at 30 mg/kg/day corresponding to plasma exposures [AUC] approximately 5 to 10 times the average AUC in pediatric patients at the MRHD of 20 mg/day), increased serum levels of creatine kinase (at AUC as low as 1 to 2 times the average AUC in pediatric patients at the MRHD of 20 mg/day), skeletal muscle degeneration and necrosis, decreased femur length/growth and body weight gain (at AUC 5 to 10 times the average AUC in pediatric patients at the MRHD of 20 mg/day). The high dose of 30 mg/kg/day exceeded a maximum tolerated dose. When animals were evaluated after a drug-free period (up to 11 weeks after cessation of dosing), fluoxetine was associated with neurobehavioral abnormalities (decreased reactivity at AUC as low as approximately 0.1 to 0.2 times the average AUC in pediatric patients at the MRHD and learning deficit at the high dose), and reproductive functional impairment (decreased mating at all doses and impaired fertility at the high dose). In addition, the testicular and epididymal microscopic lesions and decreased sperm concentrations found in high dose group were also observed, indicating that the drug effects on reproductive organs are irreversible. The reversibility of fluoxetine-induced muscle damage was not assessed.
These fluoxetine toxicities in juvenile rats have not been observed in adult animals. Plasma exposures (AUC) to fluoxetine in juvenile rats receiving 3, 10, or 30 mg/kg/day doses in this study are approximately 0.1 to 0.2, 1 to 2, and 5 to 10 times, respectively, the average exposure in pediatric patients receiving the MRHD of 20 mg/day. Rat exposures to the major metabolite, norfluoxetine, are approximately 0.3 to 0.8, 1 to 8, and 3 to 20 times, respectively, the pediatric exposure at the MRHD.
A specific effect on bone development was reported in juvenile mice administered fluoxetine by the intraperitoneal route to 4 week old mice for 4 weeks at doses 0.5 and 2 times the oral MRHD of 20 mg/day on mg/m2 basis. There was a decrease in bone mineralization and density at both doses, but the overall growth (body weight gain or femur length) was not affected.
8.5 Geriatric Use
Olanzapine and Fluoxetine Hydrochloride
Clinical studies of olanzapine and fluoxetine hydrochloride did not include sufficient numbers of patients ≥65 years of age to determine whether they respond differently from younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy [see DOSAGE AND ADMINISTRATION (2.3)].
Olanzapine
Of the 2,500 patients in premarketing clinical studies with olanzapine, 11% (263 patients) were ≥65 years of age. In patients with Schizophrenia, there was no indication of any different tolerability of olanzapine in the elderly compared with younger patients. Studies in patients with dementia-related psychosis have suggested that there may be a different tolerability profile in this population compared with younger patients with Schizophrenia. Elderly patients with dementia-related psychosis treated with olanzapine are at an increased risk of death compared to placebo. In placebo-controlled studies of olanzapine in elderly patients with dementia-related psychosis, there was a higher incidence of cerebrovascular adverse reactions (e.g., stroke, transient ischemic attack) in patients treated with olanzapine compared to patients treated with placebo. Olanzapine is not approved for the treatment of patients with dementia-related psychosis [see BOXED WARNING, DOSAGE AND ADMINISTRATION (2.3), and WARNINGS AND PRECAUTIONS (5.2 )].
Also, the presence of factors that might decrease pharmacokinetic clearance or increase the pharmacodynamic response to olanzapine should lead to consideration of a lower starting dose for any geriatric patient.
Fluoxetine
US fluoxetine clinical studies included 687 patients ≥65 years of age and 93 patients ≥75 years of age. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. SNRIs and SSRIs, including olanzapine and fluoxetine hydrochloride, have been associated with cases of clinically significant hyponatremia in elderly patients, who may be at greater risk for this adverse reaction [see WARNINGS AND PRECAUTIONS (5.17)].
8.6 Hepatic Impairment
In subjects with cirrhosis of the liver, the clearances of fluoxetine and its active metabolite, norfluoxetine, were decreased, thus increasing the elimination half-lives of these substances. A lower or less frequent dose of the fluoxetine-component of olanzapine and fluoxetine hydrochloride should be used in patients with cirrhosis. Caution is advised when using olanzapine and fluoxetine hydrochloride in patients with diseases or conditions that could affect its metabolism [see DOSAGE AND ADMINISTRATION (2.3) and CLINICAL PHARMACOLOGY (12.4)].
-
9 DRUG ABUSE AND DEPENDENCE
9.3 Dependence
Olanzapine and fluoxetine hydrochloride, as with fluoxetine and olanzapine, has not been systematically studied in humans for its potential for abuse, tolerance, or physical dependence. While the clinical studies did not reveal any tendency for any drug-seeking behavior, these observations were not systematic, and it is not possible to predict on the basis of this limited experience the extent to which a CNS-active drug will be misused, diverted, and/or abused once marketed. Consequently, physicians should carefully evaluate patients for history of drug abuse and follow such patients closely, observing them for signs of misuse or abuse of olanzapine and fluoxetine hydrochloride (e.g., development of tolerance, incrementation of dose, drug-seeking behavior).
In studies in rats and rhesus monkeys designed to assess abuse and dependence potential, olanzapine alone was shown to have acute depressive CNS effects but little or no potential of abuse or physical dependence at oral doses up to 15 (rat) and 8 (monkey) times the MRHD (20 mg) on a mg/m2 basis.
-
10 OVERDOSAGE
Olanzapine and Fluoxetine Hydrochloride
During premarketing clinical studies of olanzapine and fluoxetine in combination, overdose of both fluoxetine and olanzapine were reported in 5 study subjects. Four of the 5 subjects experienced loss of consciousness (3) or coma (1). No fatalities occurred.
Adverse reactions involving overdose of fluoxetine and olanzapine in combination, and olanzapine and fluoxetine hydrochloride, have been reported spontaneously. An overdose of combination therapy is defined as confirmed or suspected ingestion of a dose of >20 mg olanzapine in combination with a dose of >80 mg fluoxetine. Adverse reactions associated with these reports included somnolence (sedation), impaired consciousness (coma), impaired neurologic function (ataxia, confusion, convulsions, dysarthria), arrhythmias, lethargy, essential tremor, agitation, acute psychosis, hypotension, hypertension, and aggression. Fatalities have been confounded by exposure to additional substances including alcohol, thioridazine, oxycodone, and propoxyphene.
Olanzapine
In postmarketing reports of overdose with olanzapine alone, symptoms have been reported in the majority of cases. In symptomatic patients, symptoms with ≥10% incidence included agitation/aggressiveness, dysarthria, tachycardia, various extrapyramidal symptoms, and reduced level of consciousness ranging from sedation to coma. Among less commonly reported symptoms were the following potentially medically serious reactions: aspiration, cardiopulmonary arrest, cardiac arrhythmias (such as supraventricular tachycardia as well as a patient that experienced sinus pause with spontaneous resumption of normal rhythm), delirium, possible neuroleptic malignant syndrome, respiratory depression/arrest, convulsion, hypertension, and hypotension. Reports of fatality in association with overdose of olanzapine alone have been received. In 1 case of death, the amount of acutely ingested olanzapine was reported to be possibly as low as 450 mg of oral olanzapine; however, in another case, a patient was reported to survive an acute olanzapine ingestion of approximately 2 g of oral olanzapine.
Fluoxetine
Worldwide exposure to fluoxetine is estimated to be over 38 million patients (circa 1999). Of the 1,578 cases of overdose involving fluoxetine, alone or with other drugs, reported from this population, there were 195 deaths.
Among 633 adult patients who overdosed on fluoxetine alone, 34 resulted in a fatal outcome, 378 completely recovered, and 15 patients experienced sequelae after overdose, including abnormal accommodation, abnormal gait, confusion, unresponsiveness, nervousness, pulmonary dysfunction, vertigo, tremor, elevated blood pressure, erectile dysfunction, movement disorder, and hypomania. The remaining 206 patients had an unknown outcome. The most common signs and symptoms associated with non-fatal overdose were seizures, somnolence, nausea, tachycardia, and vomiting. The largest known ingestion of fluoxetine in adult patients was 8 grams in a patient who took fluoxetine alone and who subsequently recovered. However, in an adult patient who took fluoxetine alone, an ingestion as low as 520 mg has been associated with lethal outcome, but causality has not been established.
Among pediatric patients (ages 3 months to 17 years), there were 156 cases of overdose involving fluoxetine alone or in combination with other drugs. Six patients died, 127 patients completely recovered, 1 patient experienced renal failure, and 22 patients had an unknown outcome. One of the 6 fatalities was a 9-year-old boy who had a history of OCD, Tourette's Syndrome with tics, attention deficit disorder, and fetal alcohol syndrome. He had been receiving 100 mg of fluoxetine daily for 6 months in addition to clonidine, methylphenidate, and promethazine. Mixed-drug ingestion or other methods of suicide complicated all 6 overdoses in children that resulted in fatalities. The largest ingestion in pediatric patients was 3 grams, which was non-lethal.
Other important adverse reactions reported with fluoxetine overdose (single or multiple drugs) included coma, delirium, ECG abnormalities (such as nodal rhythm, QT-interval prolongation and ventricular arrhythmias, including torsades de pointes-type arrhythmias), hypotension, mania, neuroleptic malignant syndrome-like reactions, pyrexia, stupor, and syncope.
10.1 Management of Overdose
For current information on the management of olanzapine and fluoxetine hydrochloride overdose, contact a certified poison control center (1-800-222-1222 or www.poison.org). In managing overdose, consider the possibility of multiple drug involvement. In case of acute overdose, establish and maintain an airway and ensure adequate ventilation, which may include intubation. Induction of emesis is not recommended as the possibility of obtundation, seizures, or dystonic reactions of the head and neck following overdose may create a risk for aspiration. Commence cardiovascular monitoring immediately and include continuous electrocardiographic monitoring to detect possible arrhythmias.
A specific precaution involves patients who are taking or have recently taken olanzapine and fluoxetine hydrochloride and may have ingested excessive quantities of a TCA (tricyclic antidepressant). In such cases, accumulation of the parent TCA and/or an active metabolite increases the possibility of serious sequelae and extends the time needed for close medical observation.
Due to the large volume of distribution of olanzapine and fluoxetine, forced diuresis, dialysis, hemoperfusion, and exchange transfusion are unlikely to be of benefit. No specific antidote for either fluoxetine or olanzapine overdose is known. Treat hypotension and circulatory collapse with appropriate measures such as intravenous fluids and/or sympathomimetic agents. Do not use epinephrine, dopamine, or other sympathomimetics with β-agonist activity, since beta stimulation may worsen hypotension in the setting of olanzapine-induced alpha blockade.
-
11 DESCRIPTION
Olanzapine and fluoxetine hydrochloride capsules, USP combine an atypical antipsychotic and a selective serotonin reuptake inhibitor, olanzapine (the active ingredient in Zyprexa, and Zyprexa Zydis)and fluoxetine hydrochloride (the active ingredient in Prozac, Prozac Weekly, and Sarafem).
Olanzapine belongs to the thienobenzodiazepine class. The chemical designation is 2-methyl-4-(4-methyl-1-piperazinyl)-10H-thieno[2,3-b] [1,5]benzodiazepine. The molecular formula is C17H20N4S, which corresponds to a molecular weight of 312.44.
Fluoxetine hydrochloride is a selective serotonin reuptake inhibitor (SSRI). The chemical designation is (±)-N-methyl-3-phenyl-3-[(α,α,α-trifluoro-p-tolyl)oxy]propylamine hydrochloride. The molecular formula is C17H18F3NOHCl, which corresponds to a molecular weight of 345.79.
The chemical structures are:
Olanzapine is a yellow crystalline solid, which is practically insoluble in water.
Fluoxetine hydrochloride is a white to off-white crystalline solid with a solubility of 14 mg/mL in water.
Olanzapine and fluoxetine capsules, USP are available for oral administration in the following strength combinations:
3 mg/25 mg
6 mg/25 mg
6 mg/50 mg
12 mg/25 mg
12 mg/50 mg
olanzapine equivalent
3
6
6
12
12
fluoxetine base equivalent
25
25
50
25
50
Each capsule also contains pregelatinized starch.
The capsule shell for the 3 mg/25 mg strength consists of gelatin, FDA/E172 yellow iron oxide, FDA/E172 red iron oxide, titanium dioxide. The black ink is comprised of black iron oxide, potassium hydroxide, propylene glycol, shellac.
The capsule shell for the 6 mg/25 mg strength consists of D&C red # 28, D&C yellow # 10, FDA/E172 yellow iron oxide, gelatin, titanium dioxide. The black ink is comprised of black iron oxide, potassium hydroxide, propylene glycol, shellac.
The capsule shell for the 6 mg/50 mg strength consists of D&C red # 28, D&C yellow # 10, gelatin, FDA/E172 black iron oxide, titanium dioxide. The black ink is comprised of black iron oxide, potassium hydroxide, propylene glycol, shellac.
The capsule shell for the 12 mg/25 mg strength consists of FDA/E172 yellow iron oxide, FD&C Red # 3, red iron oxide, gelatin, titanium dioxide. The black ink is comprised of black iron oxide, potassium hydroxide, propylene glycol, shellac.
The capsule shell for the 12 mg/50 mg strength consists of FDA/E172 black iron oxide, FD&C Red # 3, red iron oxide, gelatin, titanium dioxide. The black ink is comprised of black iron oxide, potassium hydroxide, propylene glycol, shellac.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Although the exact mechanism of olanzapine and fluoxetine is unknown, it has been proposed that the activation of 3 monoaminergic neural systems (serotonin, norepinephrine, and dopamine) is responsible for its enhanced antidepressant effect. In animal studies, olanzapine and fluoxetine in combination has been shown to produce synergistic increases in norepinephrine and dopamine release in the prefrontal cortex compared with either component alone, as well as increases in serotonin.
12.2 Pharmacodynamics
Olanzapine binds with high affinity to the following receptors: serotonin 5HT2A/2C, 5HT6 (Ki=4, 11, and 5 nM, respectively), dopamine D1-4 (Ki=11 to 31 nM), histamine H1 (Ki=7 nM), and adrenergic α1 receptors (Ki=19 nM). Olanzapine is an antagonist with moderate affinity binding for serotonin 5HT3 (Ki=57 nM) and muscarinic M1-5 (Ki=73, 96, 132, 32, and 48 nM, respectively). Olanzapine binds weakly to GABAA, BZD, and β-adrenergic receptors (Ki>10 μM). Fluoxetine is an inhibitor of the serotonin transporter and is a weak inhibitor of the norepinephrine and dopamine transporters.
Antagonism at receptors other than dopamine and 5HT2 may explain some of the other therapeutic and side effects of olanzapine. Olanzapine's antagonism of muscarinic M1-5 receptors may explain its anticholinergic-like effects. The antagonism of histamine H1 receptors by olanzapine may explain the somnolence observed with this drug. The antagonism of α1-adrenergic receptors by olanzapine may explain the orthostatic hypotension observed with this drug. Fluoxetine has relatively low affinity for muscarinic, α1-adrenergic, and histamine H1 receptors.
12.3 Pharmacokinetics
Olanzapine and Fluoxetine
Fluoxetine (administered as a 60 mg single dose or 60 mg daily for 8 days) caused a small increase in the mean maximum concentration of olanzapine (16%) following a 5 mg dose, an increase in the mean area under the curve (17%) and a small decrease in mean apparent clearance of olanzapine (16%). In another study, a similar decrease in apparent clearance of olanzapine of 14% was observed following olanzapine doses of 6 or 12 mg with concomitant fluoxetine doses of 25 mg or more. The decrease in clearance reflects an increase in bioavailability. The terminal half-life is not affected, and therefore the time to reach steady state should not be altered. The overall steady-state plasma concentrations of olanzapine and fluoxetine when given as the combination in the therapeutic dose ranges were comparable with those typically attained with each of the monotherapies. The small change in olanzapine clearance, observed in both studies, likely reflects the inhibition of a minor metabolic pathway for olanzapine via CYP2D6 by fluoxetine, a potent CYP2D6 inhibitor, and was not deemed clinically significant. Therefore, the pharmacokinetics of the individual components is expected to reasonably characterize the overall pharmacokinetics of the combination.
Absorption and Bioavailability
Olanzapine and Fluoxetine
Following a single oral 12 mg/50 mg dose of olanzapine and fluoxetine, peak plasma concentrations of olanzapine and fluoxetine occur at approximately 4 and 6 hours, respectively. The effect of food on the absorption and bioavailability of olanzapine and fluoxetine has not been evaluated. The bioavailability of olanzapine given as Zyprexa, and the bioavailability of fluoxetine given as Prozac were not affected by food. It is unlikely that there would be a significant food effect on the bioavailability of olanzapine and fluoxetine.
Olanzapine
Olanzapine is well absorbed and reaches peak concentration approximately 6 hours following an oral dose. Food does not affect the rate or extent of olanzapine absorption when olanzapine is given as Zyprexa. It is eliminated extensively by first pass metabolism, with approximately 40% of the dose metabolized before reaching the systemic circulation.
Fluoxetine
Following a single oral 40-mg dose, peak plasma concentrations of fluoxetine from 15 to 55 ng/mL are observed after 6 to 8 hours. Food does not appear to affect the systemic bioavailability of fluoxetine given as Prozac, although it may delay its absorption by 1 to 2 hours, which is probably not clinically significant.
Distribution
Olanzapine and Fluoxetine
The in vitro binding to human plasma proteins of olanzapine and fluoxetine in combination is similar to the binding of the individual components.
Olanzapine
Olanzapine is extensively distributed throughout the body, with a volume of distribution of approximately 1000 L. It is 93% bound to plasma proteins over the concentration range of 7 to 1100 ng/mL, binding primarily to albumin and α1-acid glycoprotein.
Fluoxetine
Over the concentration range from 200 to 1000 ng/mL, approximately 94.5% of fluoxetine is bound in vitro to human serum proteins, including albumin and α1-glycoprotein. The interaction between fluoxetine and other highly protein-bound drugs has not been fully evaluated [see DRUG INTERACTIONS (7.7)].
Metabolism and Elimination
Olanzapine and Fluoxetine
Olanzapine and fluoxetine therapy yielded steady-state concentrations of norfluoxetine similar to those seen with fluoxetine in the therapeutic dose range.
Olanzapine
Olanzapine displays linear pharmacokinetics over the clinical dosing range. Its half-life ranges from 21 to 54 hours (5th to 95th percentile; mean of 30 hr), and apparent plasma clearance ranges from 12 to 47 L/hr (5th to 95th percentile; mean of 25 L/hr). Administration of olanzapine once daily leads to steady-state concentrations in about 1 week that are approximately twice the concentrations after single doses. Plasma concentrations, half-life, and clearance of olanzapine may vary between individuals on the basis of smoking status, gender, and age [see DOSAGE AND ADMINISTRATION (2.3) and CLINICAL PHARMACOLOGY (12.4 )].
Following a single oral dose of 14C-labeled olanzapine, 7% of the dose of olanzapine was recovered in the urine as unchanged drug, indicating that olanzapine is highly metabolized. Approximately 57% and 30% of the dose was recovered in the urine and feces, respectively. In the plasma, olanzapine accounted for only 12% of the AUC for total radioactivity, indicating significant exposure to metabolites. After multiple dosing, the major circulating metabolites were the 10-N-glucuronide, present at steady state at 44% of the concentration of olanzapine, and 4′-N-desmethyl olanzapine, present at steady state at 31% of the concentration of olanzapine. Both metabolites lack pharmacological activity at the concentrations observed.
Direct glucuronidation and CYP450-mediated oxidation are the primary metabolic pathways for olanzapine. In vitro studies suggest that CYP1A2, CYP2D6, and the flavin-containing monooxygenase system are involved in olanzapine oxidation. CYP2D6-mediated oxidation appears to be a minor metabolic pathway in vivo, because the clearance of olanzapine is not reduced in subjects who are deficient in this enzyme.
Fluoxetine
Fluoxetine is a racemic mixture (50/50) of R-fluoxetine and S-fluoxetine enantiomers. In animal models, both enantiomers are specific and potent serotonin uptake inhibitors with essentially equivalent pharmacologic activity. The S-fluoxetine enantiomer is eliminated more slowly and is the predominant enantiomer present in plasma at steady state.
Fluoxetine is extensively metabolized in the liver to its only identified active metabolite, norfluoxetine, via the CYP2D6 pathway. A number of unidentified metabolites exist.
In animal models, S-norfluoxetine is a potent and selective inhibitor of serotonin uptake and has activity essentially equivalent to R- or S-fluoxetine. R-norfluoxetine is significantly less potent than the parent drug in the inhibition of serotonin uptake. The primary route of elimination appears to be hepatic metabolism to inactive metabolites excreted by the kidney.
Clinical Issues Related to Metabolism and Elimination
The complexity of the metabolism of fluoxetine has several consequences that may potentially affect the clinical use of olanzapine and fluoxetine.
Variability in Metabolism
A subset (about 7%) of the population has reduced activity of the drug metabolizing enzyme CYP2D6. Such individuals are referred to as “poor metabolizers” of drugs such as debrisoquin, dextromethorphan, and the tricyclic antidepressants (TCAs). In a study involving labeled and unlabeled enantiomers administered as a racemate, these individuals metabolized S-fluoxetine at a slower rate and thus achieved higher concentrations of S-fluoxetine. Consequently, concentrations of S-norfluoxetine at steady state were lower. The metabolism of R-fluoxetine in these poor metabolizers appears normal. When compared with normal metabolizers, the total sum at steady state of the plasma concentrations of the 4 enantiomers was not significantly greater among poor metabolizers. Thus, the net pharmacodynamic activities were essentially the same. Alternative nonsaturable pathways (non-CYP2D6) also contribute to the metabolism of fluoxetine. This explains how fluoxetine achieves a steady-state concentration rather than increasing without limit.
Because the metabolism of fluoxetine, like that of a number of other compounds including TCAs and other selective serotonin antidepressants, involves the CYP2D6 system, concomitant therapy with drugs also metabolized by this enzyme system (such as the TCAs) may lead to drug interactions [see DRUG INTERACTIONS (7.7 )].
Accumulation and Slow Elimination
The relatively slow elimination of fluoxetine (elimination half-life of 1 to 3 days after acute administration and 4 to 6 days after chronic administration) and its active metabolite, norfluoxetine (elimination half-life of 4 to 16 days after acute and chronic administration), leads to significant accumulation of these active species in chronic use and delayed attainment of steady state, even when a fixed dose is used. After 30 days of dosing at 40 mg/day, plasma concentrations of fluoxetine in the range of 91 to 302 ng/mL and norfluoxetine in the range of 72 to 258 ng/mL have been observed. Plasma concentrations of fluoxetine were higher than those predicted by single-dose studies, because the metabolism of fluoxetine is not proportional to dose. However, norfluoxetine appears to have linear pharmacokinetics. Its mean terminal half-life after a single dose was 8.6 days and after multiple dosing was 9.3 days. Steady-state levels after prolonged dosing are similar to levels seen at 4 to 5 weeks.
The long elimination half-lives of fluoxetine and norfluoxetine assure that, even when dosing is stopped, active drug substance will persist in the body for weeks (primarily depending on individual patient characteristics, previous dosing regimen, and length of previous therapy at discontinuation). This is of potential consequence when drug discontinuation is required or when drugs are prescribed that might interact with fluoxetine and norfluoxetine following the discontinuation of fluoxetine.
12.4 Specific Populations
Geriatric
Based on the individual pharmacokinetic profiles of olanzapine and fluoxetine, the pharmacokinetics of olanzapine and fluoxetine may be altered in geriatric patients. Caution should be used in dosing the elderly, especially if there are other factors that might additively influence drug metabolism and/or pharmacodynamic sensitivity.
In a study involving 24 healthy subjects, the mean elimination half-life of olanzapine was about 1.5 times greater in elderly subjects (≥65 years of age) than in non-elderly subjects (<65 years of age).
The disposition of single doses of fluoxetine in healthy elderly subjects (≥65 years of age) did not differ significantly from that in younger normal subjects. However, given the long half-life and nonlinear disposition of the drug, a single-dose study is not adequate to rule out the possibility of altered pharmacokinetics in the elderly, particularly if they have systemic illness or are receiving multiple drugs for concomitant diseases. The effects of age upon the metabolism of fluoxetine have been investigated in 260 elderly but otherwise healthy depressed patients (≥60 years of age) who received 20 mg fluoxetine for 6 weeks. Combined fluoxetine plus norfluoxetine plasma concentrations were 209.3 ± 85.7 ng/mL at the end of 6 weeks. No unusual age-associated pattern of adverse reactions was observed in those elderly patients.
Renal Impairment
The pharmacokinetics of olanzapine and fluoxetine has not been studied in patients with renal impairment. However, olanzapine and fluoxetine individual pharmacokinetics do not differ significantly in patients with renal impairment. Olanzapine and fluoxetine dosing adjustment based upon renal impairment is not routinely required.
Because olanzapine is highly metabolized before excretion and only 7% of the drug is excreted unchanged, renal dysfunction alone is unlikely to have a major impact on the pharmacokinetics of olanzapine. The pharmacokinetic characteristics of olanzapine were similar in patients with severe renal impairment and normal subjects, indicating that dosage adjustment based upon the degree of renal impairment is not required. In addition, olanzapine is not removed by dialysis. The effect of renal impairment on olanzapine metabolite elimination has not been studied.
In depressed patients on dialysis (N=12), fluoxetine administered as 20 mg once daily for 2 months produced steady-state fluoxetine and norfluoxetine plasma concentrations comparable with those seen in patients with normal renal function. While the possibility exists that renally excreted metabolites of fluoxetine may accumulate to higher levels in patients with severe renal dysfunction, use of a lower or less frequent dose is not routinely necessary in renally impaired patients.
Hepatic Impairment
Based on the individual pharmacokinetic profiles of olanzapine and fluoxetine, the pharmacokinetics of olanzapine and fluoxetine may be altered in patients with hepatic impairment. The lowest starting dose should be considered for patients with hepatic impairment [see DOSAGE AND ADMINISTRATION (2.3) and WARNINGS AND PRECAUTIONS (5.20)].
Although the presence of hepatic impairment may be expected to reduce the clearance of olanzapine, a study of the effect of impaired liver function in subjects (N=6) with clinically significant cirrhosis (Child-Pugh Classification A and B) revealed little effect on the pharmacokinetics of olanzapine.
As might be predicted from its primary site of metabolism, liver impairment can affect the elimination of fluoxetine. The elimination half-life of fluoxetine was prolonged in a study of cirrhotic patients, with a mean of 7.6 days compared with the range of 2 to 3 days seen in subjects without liver disease; norfluoxetine elimination was also delayed, with a mean duration of 12 days for cirrhotic patients compared with the range of 7 to 9 days in normal subjects.
Gender
Clearance of olanzapine is approximately 30% lower in women than in men. There were, however, no apparent differences between men and women in effectiveness or adverse effects. Dosage modifications based on gender should not be needed.
Smoking Status
Olanzapine clearance is about 40% higher in smokers than in nonsmokers, although dosage modifications are not routinely required.
Race
No olanzapine and fluoxetine pharmacokinetic study was conducted to investigate the effects of race. In vivo studies have shown that exposures to olanzapine are similar among Japanese, Chinese and Caucasians, especially after normalization for body weight differences. Dosage modifications for race, therefore, are not routinely required.
Combined Effects
The combined effects of age, smoking, and gender could lead to substantial pharmacokinetic differences in populations. The clearance of olanzapine in young smoking males, for example, may be 3 times higher than that in elderly nonsmoking females. Olanzapine and fluoxetine dosing modification may be necessary in patients who exhibit a combination of factors that may result in slower metabolism of the olanzapine component [see DOSAGE AND ADMINISTRATION (2.3)].
Children and Adolescents (ages 10 to 17 years)
Based on the pediatric olanzapine and fluoxetine study, steady-state olanzapine, fluoxetine, and norfluoxetine plasma concentrations were about 31%, 76%, and 38% higher, respectively, in pediatric patients with lower body weights (less than 50 kg) than in pediatric patients with high body weight (greater than or equal to 50 kg). Exposures in pediatric patients with high body weight were similar to those previously observed in adults. Dose modifications based on body weight are not required.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity, mutagenicity, or fertility studies were conducted with olanzapine and fluoxetine hydrochloride. The following data are based on findings in studies performed with the individual components, therefore all dose multiples (based on body surface area) reflect the maximum recommended human dose (MRHD) of 20 mg olanzapine, or 80 mg fluoxetine, when each drug is administered separately.
Carcinogenesis
Olanzapine
Oral carcinogenicity studies were conducted in mice and rats. Olanzapine was administered to mice in two 78-week studies at doses of 3, 10, and 30/20 mg/kg/day [equivalent to 0.8 to 5 times the maximum recommended human daily dose (MRHD) on a mg/m2 basis] and 0.25, 2, and 8 mg/kg/day (equivalent to 0.06 to 2 times the MRHD on a mg/m2 basis). Rats were dosed for 2 years at doses of 0.25, 1, 2.5, and 4 mg/kg/day (males) and 0.25, 1, 4, and 8 mg/kg/day (females) (equivalent to 0.1 to 2 and 0.1 to 4 times the MRHD on a mg/m2 basis, respectively). The incidence of liver hemangiomas and hemangiosarcomas was significantly increased in 1 mouse study in females dosed at 8 mg/kg/day (2 times the MRHD on a mg/m2 basis). These tumors were not increased in another mouse study in females dosed at 10 or 30/20 mg/kg/day (2 to 5 times the MRHD on a mg/m2 basis); in this study, there was a high incidence of early mortalities in males of the 30/20 mg/kg/day group. The incidence of mammary gland adenomas and adenocarcinomas was significantly increased in female mice dosed at ≥2 mg/kg/day and in female rats dosed at ≥4 mg/kg/day (0.5 and 2 times the MRHD on a mg/m2 basis, respectively). Antipsychotic drugs have been shown to chronically elevate prolactin levels in rodents. Serum prolactin levels were not measured during the olanzapine carcinogenicity studies; however, measurements during subchronic toxicity studies showed that olanzapine elevated serum prolactin levels up to 4-fold in rats at the same doses used in the carcinogenicity study. An increase in mammary gland neoplasms has been found in rodents after chronic administration of other antipsychotic drugs and is considered to be prolactin-mediated. The relevance for human risk of the finding of prolactin-mediated endocrine tumors in rodents is unknown [see WARNINGS AND PRECAUTIONS (5.22)].
Fluoxetine
The dietary administration of fluoxetine to rats and mice for 2 years at doses of up to 10 and 12 mg/kg/day, respectively (approximately 1.2 and 0.7 times, respectively, the MRHD on a mg/m2 basis), produced no evidence of carcinogenicity.
Mutagenesis
Olanzapine
No evidence of genotoxic potential for olanzapine was found in the Ames reverse mutation test, in vivo micronucleus test in mice, the chromosomal aberration test in Chinese hamster ovary cells, unscheduled DNA synthesis test in rat hepatocytes, induction of forward mutation test in mouse lymphoma cells, or in vivo sister chromatid exchange test in bone marrow of Chinese hamsters.
Fluoxetine
Fluoxetine and norfluoxetine have been shown to have no genotoxic effects based on the following assays: bacterial mutation assay, DNA repair assay in cultured rat hepatocytes, mouse lymphoma assay, and in vivo sister chromatid exchange assay in Chinese hamster bone marrow cells.
Impairment of Fertility
Olanzapine and Fluoxetine Hydrochloride
Fertility studies were not conducted with olanzapine and fluoxetine hydrochloride. However, in a repeat-dose rat toxicology study of 3 months duration, ovary weight was decreased in females treated with the low-dose [2 and 4 mg/kg/day (1 and 0.5 times the MRHD on a mg/m2 basis), respectively] and high-dose [4 and 8 mg/kg/day (2 and 1 times the MRHD on a mg/m2 basis), respectively] combinations of olanzapine and fluoxetine. Decreased ovary weight, and corpora luteal depletion and uterine atrophy were observed to a greater extent in the females receiving the high-dose combination than in females receiving either olanzapine or fluoxetine alone. In a 3-month repeat-dose dog toxicology study, reduced epididymal sperm and reduced testicular and prostate weights were observed with the high-dose combination of olanzapine and fluoxetine [5 and 5 mg/kg/day (9 and 2 times the MRHD on a mg/m2 basis), respectively] and with olanzapine alone (5 mg/kg/day or 9 times the MRHD on a mg/m2 basis).
Olanzapine
In an oral fertility and reproductive performance study in rats, male mating performance, but not fertility, was impaired at a dose of 22.4 mg/kg/day and female fertility was decreased at a dose of 3 mg/kg/day (11 and 1.5 times the MRHD on a mg/m2 basis, respectively). Discontinuance of olanzapine treatment reversed the effects on male-mating performance. In female rats, the precoital period was increased and the mating index reduced at 5 mg/kg/day (2.5 times the MRHD on a mg/m2 basis). Diestrous was prolonged and estrous was delayed at 1.1 mg/kg/day (0.6 times the MRHD on a mg/m2 basis); therefore, olanzapine may produce a delay in ovulation.
Fluoxetine
Two fertility studies conducted in adult rats at doses of up to 7.5 and 12.5 mg/kg/day (approximately 0.9 and 1.5 times the MRHD on a mg/m2 basis) indicated that fluoxetine had no adverse effects on fertility. However, adverse effects on fertility were seen when juvenile rats were treated with fluoxetine at a high dose (30 mg/kg) associated with significant toxicity [see USE IN SPECIFIC POPULATIONS (8.4)].
-
14 CLINICAL STUDIES
Efficacy for olanzapine and fluoxetine was established for the:
- Acute treatment of depressive episodes in Bipolar I Disorder in adults, and children and adolescents (10 to 17 years) in 3 short-term, placebo-controlled trials (Studies 1, 2, 3) [see CLINICAL STUDIES (14.1)]
14.1 Depressive Episodes Associated with Bipolar I Disorder
Adults
The efficacy of olanzapine and fluoxetine for the acute treatment of depressive episodes associated with Bipolar I Disorder was established in 2 identically designed, 8-week, randomized, double-blind, controlled studies of patients who met Diagnostic and Statistical Manual 4th edition (DSM-IV) criteria for Bipolar I Disorder, Depressed utilizing flexible dosing of olanzapine and fluoxetine (6/25, 6/50, or 12/50 mg/day), olanzapine (5 to 20 mg/day), and placebo. These studies included patients (≥18 years of age [n=788]) with or without psychotic symptoms and with or without a rapid cycling course.
The primary rating instrument used to assess depressive symptoms in these studies was the Montgomery-Asberg Depression Rating Scale (MADRS), a 10-item clinician-rated scale with total scores ranging from 0 to 60. The primary outcome measure of these studies was the change from baseline to endpoint in the MADRS total score. In both studies, olanzapine and fluoxetine was statistically significantly superior to both olanzapine monotherapy and placebo in reduction of the MADRS total score. Refer to Table 18 (Studies 1 and 2).
Children and Adolescents
The efficacy of olanzapine and fluoxetine for the acute treatment of depressive episodes associated with Bipolar I Disorder was established in a single 8-week, randomized, double-blind, placebo-controlled study of patients, 10 to 17 years of age [N=255], who met Diagnostic and Statistical Manual 4th edition-Text Revision (DSM-IV-TR) criteria for Bipolar I Disorder, Depressed. Patients were initiated at a dose of 3/25 mg/day and force-titrated to the maximum dose of 12/50 mg/day over two weeks. After Week 2, there was flexible dosing of olanzapine and fluoxetine in the range of 6/25, 6/50, 12/25, or 12/50 mg/day. The average daily dose was olanzapine 7.7 mg and fluoxetine 37.6 mg. The recommended starting dose for children and adolescents is 3/25 mg per day. Flexible dosing is recommended, rather than the forced titration used in the study [see DOSAGE AND ADMINISTRATION (2.1)]. This study included patients with or without psychotic symptoms.
The primary rating instrument used to assess depressive symptoms in these studies was the Children's Depressive Rating Scale-Revised (CDRS-R), a 17-item clinician-rated scale with total scores ranging from 17 to 113. The primary outcome measure of this study was the change from baseline to Week 8 in the CDRS-R total score. In this study, olanzapine and fluoxetine was statistically significantly superior to placebo in reduction of the CDRS-R total score. Refer to Table 18 (Study 3).
Table 18: Summary of the Primary Efficacy Result for Studies in Bipolar Depression*
Study Number
(Primary Efficacy Measure)Treatment group
Mean baseline score (SD)
LS mean change from baseline (SE)
Difference† from olanzapine and fluoxetine (95% CI)
Study 1
(MADRS)Olanzapine and fluoxetine hydrochloride
29.9 (5)
-18.7 (1.8)
Olanzapine
32.4 (6.3)
-14.4 (1)
-4.4 (NA)
Placebo
31.2 (5.7)
-13.3 (1)
-5.5 (NA)
Study 2
(MADRS)Olanzapine and fluoxetine hydrochloride
31.7 (6.8)
-18.44 (1.7)
Olanzapine
32.8 (6.1)
-15.81 (1)
-2.6 (NA)
Placebo
31.4 (6.6)
-10.68 (1)
-7.8 (NA)
Study 3
(CDRS R)
Olanzapine and fluoxetine hydrochloride
54.6 (10)
-28.43 (1.1)
_
Placebo
53.7 (8.2)
-23.40 (1.5)
-5 (-8.3, -1.8)
*SD – standard deviation; SE – standard error; LS mean – least-squares mean estimate; CI – unadjusted confidence interval; NA – not available.
†Difference (olanzapine and fluoxetine minus active comparator or placebo) in least squares estimates.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Olanzapine and fluoxetine capsules, USP are available as follows:
3 mg/25 mg, is yellow ivory opaque body and peach opaque cap hard gelatin capsule imprinted with ‘
 SANDOZ’ ‘651’ in black ink on both cap and body filled with yellow colored powder.
SANDOZ’ ‘651’ in black ink on both cap and body filled with yellow colored powder.NDC: 0781-2195-31, bottles of 30 capsules
6 mg/25 mg, is yellow ivory opaque body and orange opaque cap hard gelatin capsule imprinted with ‘
 SANDOZ’ ‘664’ in black ink on both cap and body filled with yellow colored powder.
SANDOZ’ ‘664’ in black ink on both cap and body filled with yellow colored powder.NDC: 0781-2191-31, bottle of 30 capsules
6 mg/50 mg, is light gray opaque body and orange opaque cap hard gelatin capsule imprinted with ‘
 SANDOZ’ ‘666’ in black ink on both cap and body filled with yellow colored powder.
SANDOZ’ ‘666’ in black ink on both cap and body filled with yellow colored powder.NDC: 0781-2193-31, bottle of 30 capsules
12 mg/25 mg, is yellow ivory opaque body and red opaque cap hard gelatin capsule imprinted with ‘
 SANDOZ’ ‘665’ in black ink on both cap and body filled with yellow colored powder.
SANDOZ’ ‘665’ in black ink on both cap and body filled with yellow colored powder.NDC: 0781-2192-31, bottle of 30 capsules
12 mg/50 mg, is light grey opaque body and red opaque cap hard gelatin capsule imprinted with ‘
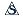 SANDOZ’ ‘667’ in black ink on both cap and body filled with yellow colored powder.
SANDOZ’ ‘667’ in black ink on both cap and body filled with yellow colored powder.NDC: 0781-2194-31, bottle of 30 capsules
-
17 PATIENT COUNSELING INFORMATION
See FDA-approved Medication Guide.
Patients should be advised of the following issues and asked to alert their prescriber if these occur while taking olanzapine and fluoxetine capsules.
17.1 Information on Medication Guide
Prescribers or other health professionals should inform patients, their families, and their caregivers about the potential benefits and potential risks associated with treatment with olanzapine and fluoxetine capsules and should counsel them in its appropriate use. A patient Medication Guide is available for olanzapine and fluoxetine capsules. The prescribers or other health professionals should instruct patients, their families, and their caregivers to read the Medication Guide and should assist them in understanding its contents. Patients should be given the opportunity to discuss the contents of the Medication Guide and to obtain answers to any questions they may have.
17.2 Suicidal Thoughts and Behaviors in Children, Adolescents, and Young Adults
Patients, their families, and their caregivers should be encouraged to be alert to the emergence of anxiety, agitation, panic attacks, insomnia, irritability, hostility, aggressiveness, impulsivity, akathisia (psychomotor restlessness), hypomania, mania, other unusual changes in behavior, worsening of depression, and suicidal ideation, especially early during antidepressant treatment and when the dose is adjusted up or down. Families and caregivers of patients should be advised to look for the emergence of such symptoms on a day-to-day basis, since changes may be abrupt. Such symptoms should be reported to the patient's prescriber or health professional, especially if they are severe, abrupt in onset, or were not part of the patient's presenting symptoms. Symptoms such as these may be associated with an increased risk for suicidal thinking and behavior and indicate a need for very close monitoring and possibly changes in the medication [see BOXED WARNING and WARNINGS AND PRECAUTIONS (5.1)].
17.3 Elderly Patients with Dementia-Related Psychosis: Increased Mortality and Cerebrovascular Adverse Events (CVAE), Including Stroke
Patients and caregivers should be advised that elderly patients with dementia-related psychosis treated with antipsychotic drugs are at increased risk of death. Patients and caregivers should be advised that elderly patients with dementia-related psychosis treated with olanzapine had a significantly higher incidence of cerebrovascular adverse events (e.g., stroke, transient ischemic attack) compared with placebo. Olanzapine and fluoxetine capsules are not approved for elderly patients with dementia-related psychosis [see BOXED WARNING and WARNINGS AND PRECAUTIONS (5.2)].
17.4 Neuroleptic Malignant Syndrome (NMS)
Patients and caregivers should be counseled that a potentially fatal symptom complex sometimes referred to as NMS has been reported in association with administration of antipsychotic drugs, including olanzapine, a component of olanzapine and fluoxetine capsules. Signs and symptoms of NMS include hyperpyrexia, muscle rigidity, altered mental status, and evidence of autonomic instability (irregular pulse or blood pressure, tachycardia, diaphoresis, and cardiac dysrhythmia) [see WARNINGS AND PRECAUTIONS (5.3)].
17.5 Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)
Patients should be advised to report to their health care provider at the earliest onset of any signs and symptoms that may be associated with Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) [see WARNINGS AND PRECAUTIONS (5.4)]
17.6 Hyperglycemia and Diabetes Mellitus
Patients should be advised of the potential risk of hyperglycemia-related adverse reactions. Patients should be monitored regularly for worsening of glucose control. Patients and caregivers should be counseled that metabolic changes have occurred during treatment with olanzapine and fluoxetine hydrochloride capsules. Patients who have diabetes should follow their doctor's instructions about how often to check their blood sugar while taking olanzapine and fluoxetine hydrochloride capsules [see WARNINGS AND PRECAUTIONS (5.5)].
17.7 Dyslipidemia
Patients should be counseled that dyslipidemia has occurred during treatment with olanzapine and fluoxetine capsules. Patients should have their lipid profile monitored regularly [see WARNINGS AND PRECAUTIONS (5.5)].
17.8 Weight Gain
Patients should be counseled that weight gain has occurred during treatment with olanzapine and fluoxetine capsules. Patients should have their weight monitored regularly [see WARNINGS AND PRECAUTIONS (5.5)].
17.9 Serotonin Syndrome
Patients should be cautioned about the risk of serotonin syndrome with the concomitant use of olanzapine and fluoxetine capsules and other serotonergic agents including triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, tryptophan, buspirone, , amphetamines, and St. John’s Wort [see CONTRAINDICATIONS (4.1) and WARNINGS AND PRECAUTIONS (5.6), and DRUG INTERACTIONS (7.3)]. Patients should be advised of the signs and symptoms associated with serotonin syndrome that may include mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular changes (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). Patients should be cautioned to seek medical care immediately if they experience these symptoms.
17.10 Angle-Closure Glaucoma
Patients should be advised that taking olanzapine and fluoxetine capsules can cause mild pupillary dilation, which in susceptible individuals, can lead to an episode of angle-closure glaucoma. Pre-existing glaucoma is almost always open-angle glaucoma because angle-closure glaucoma, when diagnosed, can be treated definitively with iridectomy. Open-angle glaucoma is not a risk factor for angle-closure glaucoma. Patients may wish to be examined to determine whether they are susceptible to angle-closure, and have a prophylactic procedure (e.g., iridectomy), if they are susceptible [see WARNINGS AND PRECAUTIONS (5.7)].
17.11 Allergic Reactions and Rash
Patients should be advised to notify their physician if they develop a rash or hives [see WARNINGS AND PRECAUTIONS (5.8)]. Patients should also be advised of the signs and symptoms associated with a severe allergic reaction, including swelling of the face, eyes, or mouth, or have trouble breathing. Patients should be cautioned to seek medical care immediately if they experience these symptoms.
17.12 Orthostatic Hypotension
Patients should be advised of the risk of orthostatic hypotension, especially during the period of initial dose titration and in association with the use of concomitant drugs that may potentiate the orthostatic effect of olanzapine, e.g., diazepam or alcohol [see WARNINGS AND PRECAUTIONS (5.11) and DRUG INTERACTIONS (7.6, 7.7 )]. Patients should be advised to change positions carefully to help prevent orthostatic hypotension, and to lie down if they feel dizzy or faint, until they feel better. Patients should be advised to call their doctor if they experience any of the following signs and symptoms associated with orthostatic hypotension: dizziness, fast or slow heart beat, or fainting.
17.13 Abnormal Bleeding
Patients should be cautioned about the concomitant use of olanzapine and fluoxetine capsules and NSAIDs, aspirin, warfarin, or other drugs that affect coagulation since the combined use of psychotropic drugs that interfere with serotonin reuptake and these agents have been associated with an increased risk of bleeding [see WARNINGS AND PRECAUTIONS (5.16)]. Patients should be advised to call their doctor if they experience any increased or unusual bruising or bleeding while taking olanzapine and fluoxetine capsules.
17.14 Hyponatremia
Patients should be advised that hyponatremia has been reported during treatment with SNRIs and SSRIs, including olanzapine and fluoxetine capsules. Signs and symptoms of hyponatremia include headache, difficulty concentrating, memory impairment, confusion, weakness, and unsteadiness, which may lead to falls. More severe and/or acute cases have been associated with hallucination, syncope, seizure, coma, respiratory arrest, and death [see WARNINGS AND PRECAUTIONS (5.17)].
17.15 Potential for Cognitive and Motor Impairment
Olanzapine and fluoxetine hydrochloride capsules have the potential to impair judgment, thinking, or motor skills. Patients should be cautioned about operating hazardous machinery, including automobiles, until they are reasonably certain that olanzapine and fluoxetine capsules therapy does not affect them adversely [see WARNINGS AND PRECAUTIONS (5.18)].
17.16 Body Temperature Dysregulation
Patients should be advised regarding appropriate care in avoiding overheating and dehydration. Patients should be advised to call their doctor right away if they become severely ill and have some or all of these symptoms of dehydration: sweating too much or not at all, dry mouth, feeling very hot, feeling thirsty, not able to produce urine [see WARNINGS AND PRECAUTIONS (5.19)].
17.17 Concomitant Medication
Patients should be advised to inform their physician if they are taking Prozac, Prozac Weekly, Sarafem, fluoxetine, Zyprexa, Zyprexa Zydis or Zyprexa Relprevv. Patients should be advised to inform their physicians if they are taking, plan to take, or have stopped taking any prescription or over-the-counter drugs, including herbal supplements, since there is a potential for interactions. Patients should also be advised to inform their physicians if they plan to discontinue any medications they are taking while taking olanzapine and fluoxetine capsules, as stopping a medication may also impact the overall blood level of olanzapine and fluoxetine [see WARNINGS AND PRECAUTIONS (5.23)].
17.18 Discontinuation of Treatment with Olanzapine and Fluoxetine Capsules
Patients should be advised to take olanzapine and fluoxetine capsules exactly as prescribed, and to continue taking olanzapine and fluoxetine capsules as prescribed even after their mood symptoms improve. Patients should be advised that they should not alter their dosing regimen, or stop taking olanzapine and fluoxetine capsules, without consulting their physician [see WARNINGS AND PRECAUTIONS (5.25)].
17.19 Alcohol
Patients should be advised to avoid alcohol while taking olanzapine and fluoxetine capsules [see DRUG INTERACTIONS (7.6, 7.7)].
17.20 Use in Specific Populations
Pregnancy
Patients should be advised to notify their physician if they become pregnant or intend to become pregnant during olanzapine and fluoxetine capsules therapy. Olanzapine and fluoxetine capsules should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus [see USE IN SPECIFIC POPULATIONS (8.1)].
Nursing Mothers
Patients, if taking olanzapine and fluoxetine capsules, should be advised not to breast-feed [see USE IN SPECIFIC POPULATIONS (8.3)].
Pediatric Use
Safety and efficacy of olanzapine and fluoxetine capsules in patients 10 to 17 years of age have been established for the acute treatment of Depressive Episodes Associated with Bipolar I Disorder.
The types of adverse reactions observed with olanzapine and fluoxetine hydrochloride capsules in children and adolescents were generally similar to those observed in adults. However, the magnitude and frequency of some changes were greater in children and adolescents than adults. These included increases in lipids, hepatic enzymes, and prolactin, as well as increases in the QT interval. Educate patients, families, and caregivers about these risks [see WARNINGS AND PRECAUTIONS (5.5, 5.18, 5.20), ADVERSE REACTIONS (6.2), and USE IN SPECIFIC POPULATIONS (8.4)].
The frequency of weight gain ≥7%, and the magnitude and frequency of increases in lipids, hepatic analytes, and prolactin in children and adolescents treated with olanzapine and fluoxetine hydrochloride capsules were similar to those observed in adolescents treated with olanzapine monotherapy [see WARNINGS AND PRECAUTIONS (5.5, 5.20), ADVERSE REACTIONS (6.2), and USE IN SPECIFIC POPULATIONS (8.4)].
The safety and effectiveness of olanzapine and fluoxetine hydrochloride capsules for the treatment of bipolar I depression in patients under 10 years of age have not been established.
17.21 QT Prolongation
Patients should be advised that QT interval prolongation and ventricular arrhythmia including Torsade de Pointes have been reported in patients treated with fluoxetine. Signs and symptoms of ventricular arrhythmia include fast, slow, or irregular heart rate, dyspnea, syncope, or dizziness, which may indicate serious cardiac arrhythmia [see WARNINGS AND PRECAUTIONS (5.20)].
The brands listed are trademarks of their respective owners and are not trademarks of Sandoz Inc.
Manufactured in India by Sandoz Private Ltd.
for Sandoz Inc., Princeton, NJ 08540
Rev. April 2017
-
MEDICATION GUIDE
Olanzapine and Fluoxetine Capsules, USP
(oh-LAN-zah-peen and floo-OX-eh-teen)
Read the Medication Guide that comes with olanzapine and fluoxetine capsules before you start taking it and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking to your doctor about your medical condition or treatment. Talk with your doctor or pharmacist if there is something you do not understand or you want to learn more about olanzapine and fluoxetine capsules.
What is the most important information I should know about olanzapine and fluoxetine capsules?
Olanzapine and fluoxetine capsules may cause serious side effects, including:
- 1. Suicidal thoughts or actions.
- 2. Increased risk of death in elderly people who are confused, have memory loss and have lost touch with reality (dementia-related psychosis).
- 3. High blood sugar (hyperglycemia).
- 4. High fat levels in your blood (increased cholesterol and triglycerides), especially in children and adolescents age 10 to 17.
- 5. Weight gain, especially in children and adolescents age 10 to 17.
These serious side effects are described below.
- 1. Suicidal thoughts or actions.
Antidepressant medicines, depression and other serious mental illnesses, and suicidal thoughts or actions:
Talk to your, or your family member's, healthcare provider about:
-
- ∘ all risks and benefits of treatment with antidepressant medicines.
- ∘ all treatment choices for depression or other serious mental illness.
- Antidepressant medicines may increase suicidal thoughts or actions in some children, teenagers, and young adults within the first few months of treatment.
- Depression and other serious mental illnesses are the most important causes of suicidal thoughts and actions. Some people may have a particularly high risk of having suicidal thoughts or actions. These include people who have (or have a family history of) bipolar illness (also called manic-depressive illness) or suicidal thoughts or actions.
- How can I watch for and try to prevent suicidal thoughts and actions in myself or a family member?
- Pay close attention to any changes, especially sudden changes, in mood, behaviors, thoughts, or feelings. This is very important when an antidepressant medicine is started or when the dose is changed.
- Call the healthcare provider right away to report new or sudden changes in mood, behavior, thoughts, or feelings.
- Keep all follow-up visits with the healthcare provider as scheduled. Call the healthcare provider between visits as needed, especially if you have concerns about symptoms.
Call a healthcare provider right away if you or your family member has any of the following symptoms, especially if they are new, worse, or worry you:
- thoughts about suicide or dying
- attempts to commit suicide
- new or worse depression
- new or worse anxiety
- feeling very agitated or restless
- panic attacks
- trouble sleeping (insomnia)
- new or worse irritability
- acting aggressive, being angry, or violent
- acting on dangerous impulses
- an extreme increase in activity and talking (mania)
- or other unusual changes in behavior or mood.
What else do I need to know about antidepressant medicines?
- Never stop an antidepressant medicine without first talking to a healthcare provider. Stopping an antidepressant medicine suddenly can cause other symptoms.
- Antidepressants are medicines used to treat depression and other illnesses. It is important to discuss all the risks of treating depression and also the risks of not treating it. Patients and their families or other caregivers should discuss all treatment choices with the healthcare provider, not just the use of antidepressants.
- Antidepressant medicines have other side effects. Talk to the healthcare provider about the side effects of the medicine prescribed for you or your family member.
- Antidepressant medicines can interact with other medicines. Know all of the medicines that you or your family member takes. Keep a list of all medicines to show the healthcare provider. Do not start new medicines without first checking with your healthcare provider.
- Not all antidepressant medicines prescribed for children are FDA approved for use in children. Talk to your child's healthcare provider for more information.
- 2. Increased risk of death in elderly people who are confused, have memory loss and have lost touch with reality (dementia-related psychosis). Olanzapine and fluoxetine capsules are not approved for treating psychosis in elderly people with dementia.
- 3. High blood sugar (hyperglycemia): High blood sugar can happen if you have diabetes already or if you have never had diabetes. High blood sugar could lead to:
- build up of acid in your blood due to ketones (ketoacidosis)
- coma
- death
Your doctor should do tests to check your blood sugar before you start taking olanzapine and fluoxetine capsules and during treatment. In people who do not have diabetes, sometimes high blood sugar goes away when olanzapine and fluoxetine capsules are stopped. People with diabetes and some people who did not have diabetes before taking olanzapine and fluoxetine capsules need to take medicine for high blood sugar even after they stop taking olanzapine and fluoxetine capsules.
If you have diabetes, follow your doctor’s instructions about how often to check your blood sugar while taking olanzapine and fluoxetine capsules.
Call your doctor if you have any of these symptoms of high blood sugar (hyperglycemia) while taking olanzapine and fluoxetine capsules:
- feel very thirsty
- need to urinate more than usual
- feel very hungry
- feel weak or tired
- feel sick to your stomach
- feel confused, or your breath smells fruity.
4. High fat levels in your blood (increased cholesterol and triglycerides). High fat levels may happen in people treated with olanzapine and fluoxetine capsules, especially in children and adolescents (10 to 17 years old). You may not have any symptoms, so your doctor should do blood tests to check your cholesterol and triglyceride levels before you start taking olanzapine and fluoxetine capsules and during treatment.
5. Increase in weight (weight gain): Weight gain is common in people who take olanzapine and fluoxetine capsules. Children and adolescents (10 to 17 years old) who received olanzapine and fluoxetine capsules, were more likely to gain weight and to gain more weight than adults. Some people may gain a lot of weight while taking olanzapine and fluoxetine capsules, so you and your doctor should check your weight regularly. Talk to your doctor about ways to control weight gain, such as eating a healthy, balanced diet, and exercising
What are olanzapine and fluoxetine capsules?
Olanzapine and fluoxetine capsules are a prescription medicine used for:
- short-term treatment of episodes of depression that happen with Bipolar I Disorder.
Olanzapine and fluoxetine capsules contain two medicines, olanzapine and fluoxetine hydrochloride.
It is not known if olanzapine and fluoxetine capsules are safe and effective in children under the age of 10.
The symptoms of Bipolar I Disorder include alternating periods of depression and high or irritable mood, increased activity and restlessness, racing thoughts, talking fast, impulsive behavior, and a decreased need for sleep. With treatment, some of your symptoms of Bipolar I Disorder may improve.
If you do not think you are getting better, call your doctor.
Who should not take olanzapine and fluoxetine capsules?
- Do not take olanzapine and fluoxetine capsules if you take a Monoamine Oxidase Inhibitor (MAOI). Ask your healthcare provider or pharmacist if you are not sure if you take an MAOI, including the antibiotic linezolid.
- Do not take an MAOI within 5 weeks of stopping olanzapine and fluoxetine capsules. unless directed to do so by your physician.
- Do not start olanzapine and fluoxetine capsules. if you stopped taking an MAOI in the last 2 weeks unless directed to do so by your physician.
People who take olanzapine and fluoxetine capsules close in time to an MAOI can have serious and life-threatening side effects, with symptoms including:
- high fever
- continued muscle spasms that you cannot control
- rigid muscles
- changes in heart rate and blood pressure that happen fast
- confusion
- unconsciousness.
- Do not take olanzapine and fluoxetine capsules if you take Mellaril® (thioridazine). Do not take Mellaril®within 5 weeks of stopping olanzapine and fluoxetine capsules. Mellaril can cause serious heart rhythm problems and you could die suddenly.
- Do not take olanzapine and fluoxetine capsules if you take the antipsychotic medicine pimozide (Orap®). Do not take pimozide (Orap®) within 5 weeks of stopping olanzapine and fluoxetine capsules.
What should I tell my doctor before taking olanzapine and fluoxetine capsules?
Olanzapine and fluoxetine capsules may not be right for you. Before starting olanzapine and fluoxetine capsules, tell your doctor about all your medical conditions, including if you have or had any of the following:
- heart problems
- seizures (convulsions)
- diabetes or high blood sugar levels (hyperglycemia)
- high cholesterol or triglyceride levels in your blood
- liver problems
- low or high blood pressure
- strokes or “mini-strokes” also called transient ischemic attacks (TIAs)
- bleeding problems
- Alzheimer's disease
- angle-closure glaucoma
- enlarged prostate in men
- bowel obstruction
- breast cancer
- are pregnant or plan to become pregnant. It is not known if olanzapine and fluoxetine capsules will harm your unborn baby.
- are breast-feeding or plan to breast-feed. Olanzapine and fluoxetine can pass into your breast milk and may harm your baby. You should not breast-feed while taking olanzapine and fluoxetine capsules. Talk to your doctor about the best way to feed your baby if you take olanzapine and fluoxetine capsules.
Before starting olanzapine and fluoxetine capsules, tell your doctor about all the medicines that you take, including:
- Prescription and non-prescription medicines
- Vitamins, and herbal supplements.
- Triptans used to treat migraine headache
- Medicines used to treat mood, anxiety, psychotic or thought disorders, including tricyclics, lithium, buspirone, SSRIs, SNRIs, MAOIs, or antipsychotics
- Tramadol and fentanyl
- Amphetamines
- Over-the-counter supplements such as tryptophan or St. John’s Wort
- Electroconvulsive therapy (ECT)
Olanzapine and fluoxetine capsules and some medicines may interact with each other and may not work as well, or cause possible serious side effects. Your doctor can tell you if it is safe to take olanzapine and fluoxetine capsules with your other medicines. Do not start or stop any medicine while taking olanzapine and fluoxetine capsules without talking to your doctor first.
If you take olanzapine and fluoxetine capsules, you should not take any other medicines that contain:
- olanzapine (the active ingredient in Zyprexa® and Zyprexa® Zydis®) or
- fluoxetine hydrochloride (the active ingredient in Prozac®, Prozac® Weekly™, and Sarafem®).
You could take too much medicine (overdose).
How should I take olanzapine and fluoxetine capsules?
- Take olanzapine and fluoxetine capsules exactly as prescribed. Your doctor may need to change (adjust) the dose of olanzapine and fluoxetine capsules until it is right for you.
- If you miss a dose of olanzapine and fluoxetine capsules, take the missed dose as soon as you remember. If it is almost time for the next dose, skip the missed dose and take your next dose at the regular time. Do not take two doses of olanzapine and fluoxetine capsules at the same time.
- To prevent serious side effects, do not stop taking olanzapine and fluoxetine capsules suddenly. If you need to stop taking olanzapine and fluoxetine capsules, your doctor can tell you how to safely stop taking it.
- If you take too many olanzapine and fluoxetine capsules, call your doctor or poison control center right away, or get emergency treatment.
- Olanzapine and fluoxetine capsules can be taken with or without food.
- Olanzapine and fluoxetine capsules are usually taken one time each day, in the evening.
- If you do not think you are getting better or have any concerns about your condition while taking olanzapine and fluoxetine capsules, call your doctor.
What should I avoid while taking olanzapine and fluoxetine capsules?
- Olanzapine and fluoxetine capsules can cause sleepiness and may affect your ability to make decisions, think clearly, or react quickly. You should not drive, operate heavy machinery, or do other dangerous activities until you know how olanzapine and fluoxetine capsules affects you.
- Avoid drinking alcohol while taking olanzapine and fluoxetine capsules. Drinking alcohol while you take olanzapine and fluoxetine capsules may make you sleepier than if you take olanzapine and fluoxetine capsules alone.
What are the possible side effects of olanzapine and fluoxetine capsules?
Other possible serious risks:
- Increased risk of death and increased incidence of stroke or “mini-strokes” called transient ischemic attacks (TIAs) in elderly people with psychosis related to dementia (a brain disorder that lessens the ability to remember, think, and reason). Olanzapine and fluoxetine capsules are not approved for these patients.
-
Severe allergic reactions: Tell your doctor right away if you get red itchy welts (hives) or, a rash alone or with fever and joint pain, while taking olanzapine and fluoxetine capsules. Call your doctor right away if you become severely ill and have some or all of these symptoms:
- ∘ swelling of your face, eyes, or mouth
- ∘ trouble breathing
-
Neuroleptic malignant syndrome (NMS): NMS is a rare but very serious condition that can happen in people who take antipsychotic medicines, including olanzapine and fluoxetine capsules. NMS can cause death and must be treated in a hospital. Call your doctor right away if you become severely ill and have some or all of these symptoms:
- ∘ high fever
- ∘ excessive sweating
- ∘ rigid muscles
- ∘ confusion
- ∘ changes in your breathing, heartbeat, and blood pressure
- Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS): DRESS can occur. Features of DRESS may include rash, fever, swollen glands and other internal organ involvement such as liver, kidney, lung and heart. DRESS is sometimes fatal; therefore, tell your doctor immediately if you experience any of these signs.
- Tardive Dyskinesia: This condition causes body movements that keep happening and that you cannot control. These movements usually affect the face and tongue. Tardive dyskinesia may not go away, even if you stop taking olanzapine and fluoxetine capsules. It may also start after you stop taking olanzapine and fluoxetine capsules. Tell your doctor if you get any body movements that you cannot control.
-
Serotonin Syndrome: This is a condition that can be life threatening. Call your doctor right away if you become severely ill and have some or all of these symptoms:
- ∘ agitation, hallucinations, coma or other changes in mental status
- ∘ coordination problems or muscle twitching (overactive reflexes)
- ∘ racing heartbeat, high or low blood pressure
- ∘ sweating or fever
- ∘ nausea, vomiting, and diarrhea
- ∘ muscle rigidity
- ∘ dizziness
- ∘ flushing
- ∘ tremor
- ∘ seizures
-
Visual problems:
- ∘ eye pain
- ∘ changes in vision
- ∘ swelling or redness in or around the eye
Only some people are at risk for these problems. You may want to undergo an eye examination to see if you are at risk and receive preventative treatment if you are.
-
Abnormal bleeding: Tell your doctor if you notice any increased or unusual bruising or bleeding while taking olanzapine and fluoxetine capsules, especially if you take one of these medicines:
- ∘ the blood thinner warfarin (Coumadin, Jantoven)
- ∘ a non-steroidal anti-inflammatory drug (NSAID)
- ∘ aspirin
-
Low salt (sodium) levels in the blood (hyponatremia): Call your doctor right away if you become severely ill and have some or all of these symptoms:
- ∘ headache
- ∘ feel weak
- ∘ confusion
- ∘ problems concentrating
- ∘ memory problems
- ∘ feel unsteady
-
Changes in the electrical activity of your heart (QT prolongation and ventricular arrhythmia including Torsade de Pointes). This condition can be life threatening. The symptoms may include:
- ∘ fast, slow, or irregular heartbeat
- ∘ shortness of breath
- ∘ dizziness or fainting
- Decreased blood pressure when you change positions, with symptoms of dizziness, fast or slow heart beat, or fainting
- Difficulty swallowing
- Seizures
-
Problems with control of body temperature: You could become very hot, for instance when you exercise a lot or stay in an area that is very hot. It is important for you to drink water to avoid dehydration. Call your doctor right away if you become severely ill and have some or all of these symptoms of dehydration:
- ∘ sweating too much or not at all
- ∘ dry mouth
- ∘ feeling very hot
- ∘ feeling thirsty
- ∘ not able to produce urine
Common possible side effects of olanzapine and fluoxetine capsules include: dry mouth, tiredness, sleeping for long period of time, increased appetite, swelling of your hands and feet, drowsiness, tremors (shakes), or blurred vision.
Tell your doctor about any side effect that bothers you or that does not go away.
These are not all the possible side effects with olanzapine and fluoxetine capsules. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to Sandoz Inc. at 1-800-525-8747 or FDA at 1-800-FDA-1088.
How should I store olanzapine and fluoxetine capsules?
- Store olanzapine and fluoxetine capsules at 68° to 77°F (20° to 25°C).
- Keep olanzapine and fluoxetine capsules away from light.
- Keep olanzapine and fluoxetine capsules dry and away from moisture. Keep the bottle closed tightly.
Keep olanzapine and fluoxetine capsules and all medicines out of the reach of children.
General information about olanzapine and fluoxetine capsules
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use olanzapine and fluoxetine capsules for a condition for which it was not prescribed. Do not give olanzapine and fluoxetine capsules to other people, even if they have the same condition. It may harm them.
This Medication Guide summarizes the most important information about olanzapine and fluoxetine capsules. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about olanzapine and fluoxetine capsules that was written for healthcare professionals. For more information about olanzapine and fluoxetine capsules, call Sandoz Inc. at 1-800-525-8747.
What are the ingredients in olanzapine and fluoxetine capsules?
Active ingredients: Olanzapine and fluoxetine hydrochloride
Inactive ingredients: Each capsule contains pregelatinized starch.
The capsule shell for the 3 mg/25 mg strength consists of gelatin, FDA/E172 yellow iron oxide, FDA/E172 red iron oxide, titanium dioxide. The black ink is comprised of black iron oxide, potassium hydroxide, propylene glycol, shellac.
The capsule shell for the 6 mg/25 mg strength consists of D&C red # 28, D&C yellow # 10, FDA/E172 yellow iron oxide, gelatin, titanium dioxide. The black ink is comprised of black iron oxide, potassium hydroxide, propylene glycol, shellac.
The capsule shell for the 6 mg/50 mg strength consists of D&C red # 28, D&C yellow # 10, gelatin, FDA/E172 black iron oxide, titanium dioxide. The black ink is comprised of black iron oxide, potassium hydroxide, propylene glycol, shellac.
The capsule shell for the 12 mg/25 mg strength consists of FDA/E172 yellow iron oxide, FD&C Red # 3, red iron oxide, gelatin, titanium dioxide. The black ink is comprised of black iron oxide, potassium hydroxide, propylene glycol, shellac.
The capsule shell for the 12 mg/50 mg strength consists of FDA/E172 black iron oxide, FD&C Red # 3, red iron oxide, gelatin, titanium dioxide. The black ink is comprised of black iron oxide, potassium hydroxide, propylene glycol, shellac.
The brands listed are trademarks of their respective owners and are not trademarks of Sandoz Inc.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Manufactured in India by Sandoz Private Ltd.
for Sandoz Inc., Princeton, NJ 08540
Rev. April 2017
-
Package/Label Display Panel
NDC: 0781-2195-31
Olanzapine and Fluoxetine Capsules, USP
3 mg/25 mg
PHARMACIST: Dispense the Medication Guide provided separately to each patient
30 capsules
-
Package/Label Display Panel
NDC: 0781-2194-31
Olanzapine and Fluoxetine Capsules, USP
12 mg/50 mg
PHARMACIST: Dispense the Medication Guide provided separately to each patient
30 capsules
-
Package/Label Display Panel
NDC: 0781-2191-31
Olanzapine and Fluoxetine Capsules, USP
6 mg/25 mg
PHARMACIST: Dispense the Medication Guide provided separately to each patient
30 capsules
-
Package/Label Display Panel
NDC: 0781-2193-31
Olanzapine and Fluoxetine Capsules, USP
6 mg/50 mg
PHARMACIST: Dispense the Medication Guide provided separately to each patient
30 capsules
-
Package/Label Display Panel
NDC: 0781-2192-31
Olanzapine and Fluoxetine Capsules, USP
12 mg/25 mg
PHARMACIST: Dispense the Medication Guide provided separately to each patient
30 capsules
-
INGREDIENTS AND APPEARANCE
OLANZAPINE AND FLUOXETINE
olanzapine and fluoxetine capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0781-2195 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OLANZAPINE (UNII: N7U69T4SZR) (OLANZAPINE - UNII:N7U69T4SZR) OLANZAPINE 3 mg FLUOXETINE HYDROCHLORIDE (UNII: I9W7N6B1KJ) (FLUOXETINE - UNII:01K63SUP8D) FLUOXETINE 25 mg Inactive Ingredients Ingredient Name Strength STARCH, CORN (UNII: O8232NY3SJ) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SHELLAC (UNII: 46N107B71O) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) FERROSOFERRIC OXIDE (UNII: XM0M87F357) POTASSIUM HYDROXIDE (UNII: WZH3C48M4T) Product Characteristics Color YELLOW (ivory) , ORANGE (peach) Score no score Shape CAPSULE Size 16mm Flavor Imprint Code Sandoz;651 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0781-2195-31 30 in 1 BOTTLE; Type 0: Not a Combination Product 11/30/2012 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA078901 11/30/2012 OLANZAPINE AND FLUOXETINE
olanzapine and fluoxetine capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0781-2191 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OLANZAPINE (UNII: N7U69T4SZR) (OLANZAPINE - UNII:N7U69T4SZR) OLANZAPINE 6 mg FLUOXETINE HYDROCHLORIDE (UNII: I9W7N6B1KJ) (FLUOXETINE - UNII:01K63SUP8D) FLUOXETINE 25 mg Inactive Ingredients Ingredient Name Strength STARCH, CORN (UNII: O8232NY3SJ) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERROSOFERRIC OXIDE (UNII: XM0M87F357) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SHELLAC (UNII: 46N107B71O) D&C RED NO. 28 (UNII: 767IP0Y5NH) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) POTASSIUM HYDROXIDE (UNII: WZH3C48M4T) Product Characteristics Color YELLOW (ivory) , ORANGE Score no score Shape CAPSULE Size 16mm Flavor Imprint Code Sandoz;664 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0781-2191-31 30 in 1 BOTTLE; Type 0: Not a Combination Product 11/30/2012 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA078901 11/30/2012 OLANZAPINE AND FLUOXETINE
olanzapine and fluoxetine capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0781-2193 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OLANZAPINE (UNII: N7U69T4SZR) (OLANZAPINE - UNII:N7U69T4SZR) OLANZAPINE 6 mg FLUOXETINE HYDROCHLORIDE (UNII: I9W7N6B1KJ) (FLUOXETINE - UNII:01K63SUP8D) FLUOXETINE 50 mg Inactive Ingredients Ingredient Name Strength STARCH, CORN (UNII: O8232NY3SJ) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERROSOFERRIC OXIDE (UNII: XM0M87F357) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SHELLAC (UNII: 46N107B71O) D&C RED NO. 28 (UNII: 767IP0Y5NH) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) POTASSIUM HYDROXIDE (UNII: WZH3C48M4T) Product Characteristics Color GRAY (light) , ORANGE Score no score Shape CAPSULE Size 18mm Flavor Imprint Code Sandoz;666 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0781-2193-31 30 in 1 BOTTLE; Type 0: Not a Combination Product 11/30/2012 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA078901 11/30/2012 OLANZAPINE AND FLUOXETINE
olanzapine and fluoxetine capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0781-2192 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OLANZAPINE (UNII: N7U69T4SZR) (OLANZAPINE - UNII:N7U69T4SZR) OLANZAPINE 12 mg FLUOXETINE HYDROCHLORIDE (UNII: I9W7N6B1KJ) (FLUOXETINE - UNII:01K63SUP8D) FLUOXETINE 25 mg Inactive Ingredients Ingredient Name Strength STARCH, CORN (UNII: O8232NY3SJ) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERROSOFERRIC OXIDE (UNII: XM0M87F357) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SHELLAC (UNII: 46N107B71O) FD&C RED NO. 3 (UNII: PN2ZH5LOQY) FERRIC OXIDE RED (UNII: 1K09F3G675) POTASSIUM HYDROXIDE (UNII: WZH3C48M4T) Product Characteristics Color YELLOW (ivory) , RED Score no score Shape CAPSULE Size 16mm Flavor Imprint Code Sandoz;665 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0781-2192-31 30 in 1 BOTTLE; Type 0: Not a Combination Product 11/30/2012 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA078901 11/30/2012 OLANZAPINE AND FLUOXETINE
olanzapine and fluoxetine capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0781-2194 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OLANZAPINE (UNII: N7U69T4SZR) (OLANZAPINE - UNII:N7U69T4SZR) OLANZAPINE 12 mg FLUOXETINE HYDROCHLORIDE (UNII: I9W7N6B1KJ) (FLUOXETINE - UNII:01K63SUP8D) FLUOXETINE 50 mg Inactive Ingredients Ingredient Name Strength STARCH, CORN (UNII: O8232NY3SJ) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERROSOFERRIC OXIDE (UNII: XM0M87F357) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SHELLAC (UNII: 46N107B71O) FD&C RED NO. 3 (UNII: PN2ZH5LOQY) FERRIC OXIDE RED (UNII: 1K09F3G675) POTASSIUM HYDROXIDE (UNII: WZH3C48M4T) Product Characteristics Color GRAY (light) , RED Score no score Shape CAPSULE Size 18mm Flavor Imprint Code Sandoz;667 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0781-2194-31 30 in 1 BOTTLE; Type 0: Not a Combination Product 11/30/2012 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA078901 11/30/2012 Labeler - Sandoz Inc (005387188)
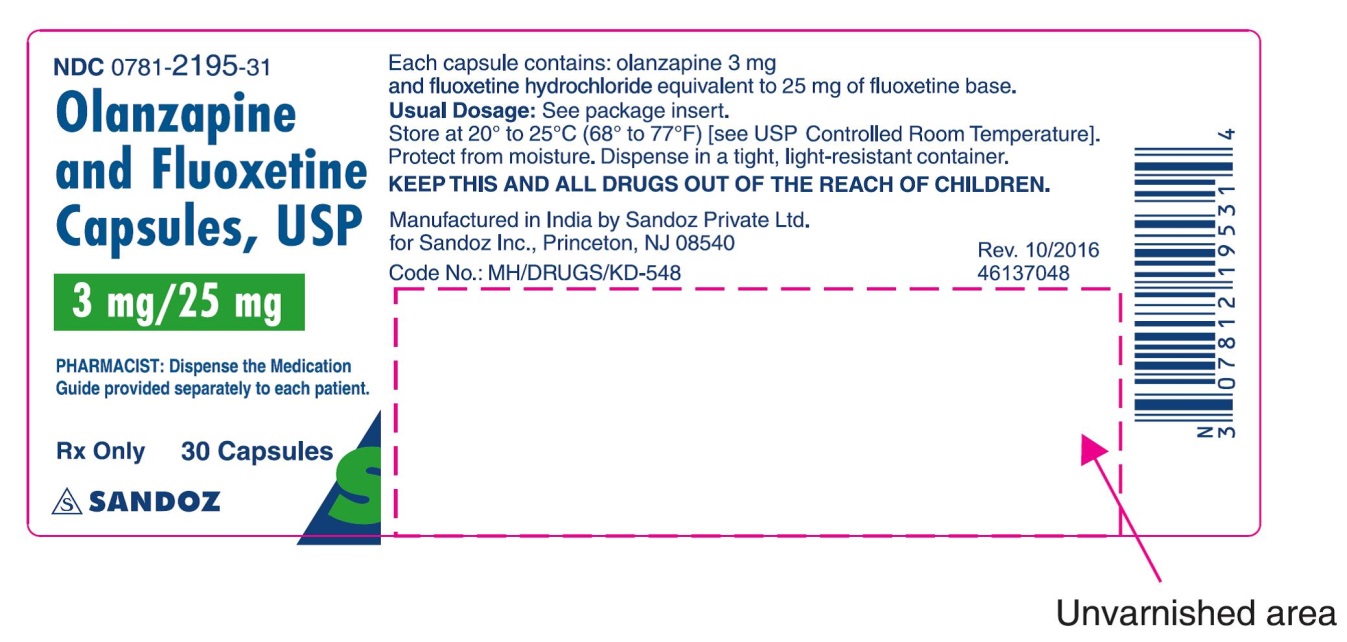




© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.