AUGTYRO- repotrectinib capsule
Augtyro by
Drug Labeling and Warnings
Augtyro by is a Prescription medication manufactured, distributed, or labeled by E.R. Squibb & Sons, L.L.C.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use AUGTYRO safely and effectively. See full prescribing information for AUGTYRO.
AUGTYROTM (repotrectinib) capsules, for oral use
Initial U.S. Approval: 2023RECENT MAJOR CHANGES
INDICATIONS AND USAGE
AUGTYRO is a kinase inhibitor indicated for the treatment of
- adult patients with locally advanced or metastatic ROS1-positive non-small cell lung cancer (NSCLC). (1.1)
- adult and pediatric patients 12 years of age and older with solid tumors that:
- have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion and
- are locally advanced or metastatic or where surgical resection is likely to result in severe morbidity.
- have progressed following treatment or have no satisfactory alternative therapy.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. (1.2)
DOSAGE AND ADMINISTRATION
- Select patients for the treatment of locally advanced or metastatic NSCLC based on the presence of ROS1 rearrangement(s) in tumor specimens. (2.1)
- Select patients for treatment of locally advanced or metastatic solid tumors based on the presence of an NTRK gene fusion. (2.1)
- Recommended Dosage: 160 mg orally once daily for 14 days, then increase to 160 mg twice daily, with or without food. (2.4)
DOSAGE FORMS AND STRENGTHS
Capsules: 40 mg, 160 mg (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Central Nervous System (CNS) Effects: Can cause CNS adverse reactions including dizziness, ataxia, and cognitive impairment. Withhold and then resume at same or reduced dose upon improvement, or permanently discontinue AUGTYRO based on severity. (5.1)
- Interstitial Lung Disease (ILD)/Pneumonitis: Monitor patients for new or worsening pulmonary symptoms indicative of ILD/pneumonitis. Immediately withhold in patients with suspected ILD/pneumonitis and permanently discontinue if ILD/pneumonitis is confirmed. (5.2)
- Hepatotoxicity: Monitor liver function tests every 2 weeks during the first month of treatment, and as clinically indicated thereafter. Based on severity, withhold and then resume at same or reduced dose, or permanently discontinue. (5.3)
- Myalgia with Creatine Phosphokinase (CPK) Elevation: Monitor serum CPK levels during treatment in patients reporting unexplained muscle pain, tenderness, or weakness. Based on severity, withhold and resume at same or reduced dose upon improvement. (5.4)
- Hyperuricemia: Monitor serum uric acid levels prior to initiating and periodically during treatment. Initiate treatment with urate-lowering medications as clinically indicated. Withhold and resume at same or reduced dose, or permanently discontinue based on severity. (5.5)
- Skeletal Fractures: Promptly evaluate patients with signs or symptoms (e.g., pain, changes in mobility, deformity) of fractures. (5.6)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use an effective non-hormonal method of contraception. (5.7)
ADVERSE REACTIONS
The most common adverse reactions (≥20%) were dizziness, dysgeusia, peripheral neuropathy, constipation, dyspnea, fatigue, ataxia, cognitive impairment, muscular weakness, and nausea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Bristol-Myers Squibb at 1-800-721-5072 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Strong and Moderate CYP3A Inhibitors: Avoid concomitant use. (7.1)
- P-gp inhibitors: Avoid concomitant use. (7.1)
- Strong and Moderate CYP3A Inducers: Avoid concomitant use. (7.1)
- Certain CYP3A Substrates: Avoid concomitant use with CYP3A substrates, where minimal concentration changes can cause reduced efficacy. (7.2)
- Hormonal contraceptives: Avoid concomitant use. (7.2)
USE IN SPECIFIC POPULATIONS
- Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 6/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 ROS1-Positive Non-Small Cell Lung Cancer
1.2 NTRK Gene Fusion-Positive Solid Tumors
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
2.2 Important Information Prior to Initiating AUGTYRO
2.3 Recommended Evaluation and Testing Before Initiating AUGTYRO
2.4 Recommended Dosage
2.5 Dosage Modifications for Adverse Reactions
2.6 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Central Nervous System Adverse Reactions
5.2 Interstitial Lung Disease/Pneumonitis
5.3 Hepatotoxicity
5.4 Myalgia with Creatine Phosphokinase Elevation
5.5 Hyperuricemia
5.6 Skeletal Fractures
5.7 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on AUGTYRO
7.2 Effects of AUGTYRO on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Locally Advanced or Metastatic ROS1-Positive NSCLC
14.2 Locally Advanced or Metastatic NTRK Gene Fusion-Positive Solid Tumors
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 ROS1-Positive Non-Small Cell Lung Cancer
AUGTYRO is indicated for the treatment of adult patients with locally advanced or metastatic ROS1-positive non-small cell lung cancer (NSCLC) [see Dosage and Administration (2.1)].
1.2 NTRK Gene Fusion-Positive Solid Tumors
AUGTYRO is indicated for the treatment of adult and pediatric patients 12 years of age and older with solid tumors that:
- have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion [see Dosage and Administration (2.1)],
- are locally advanced or metastatic or where surgical resection is likely to result in severe morbidity, and
- have progressed following treatment or have no satisfactory alternative therapy.
This indication is approved under accelerated approval based on overall response rate and duration of response [seeClinical Studies (14.2)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
-
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
NSCLC
Select patients for the treatment of locally advanced or metastatic NSCLC with AUGTYRO based on the presence of ROS1 rearrangement(s) in tumor specimens [see Clinical Studies (14.1)]. An FDA-approved test to detect ROS1 rearrangements for selecting patients for treatment with AUGTYRO is not currently available.
Solid Tumors
Select patients for the treatment of solid tumors with AUGTYRO based on the presence of NTRK1/2/3 rearrangements in tumor specimens [see Clinical Studies (14.2)]. An FDA-approved test to detect NTRK1/2/3 rearrangements for selecting patients for treatment with AUGTYRO is not currently available.
- In patients with secretory breast cancer or mammary analogue secretory cancer, consider treatment without confirmation of NTRK rearrangements in tumor specimens.
2.2 Important Information Prior to Initiating AUGTYRO
Prior to initiating AUGTYRO, discontinue strong and moderate CYP3A inhibitors for 3 to 5 elimination half-lives of the CYP3A inhibitor [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
2.3 Recommended Evaluation and Testing Before Initiating AUGTYRO
Prior to initiation of AUGTYRO, evaluate:
- liver function tests including bilirubin [see Warnings and Precautions (5.3)]
- uric acid level [see Warnings and Precautions (5.5)]
2.4 Recommended Dosage
The recommended dosage of AUGTYRO for adult and pediatric patients 12 years of age and older is 160 mg taken orally once daily with or without food [see Clinical Pharmacology (12.3)] for 14 days, then increase to 160 mg twice daily and continue until disease progression or unacceptable toxicity.
2.5 Dosage Modifications for Adverse Reactions
The recommended dosage reductions of AUGTYRO for the management of adverse reactions are provided in Table 1.
Table 1: Recommended Dose Reductions for AUGTYRO Adverse Reactions Dose
Dose Reduction
First
Second
160 mg Once Daily
120 mg Once Daily
80 mg Once Daily
160 mg Twice Daily
120 mg Twice Daily
80 mg Twice Daily
Recommended dosage modifications of AUGTYRO for the management of adverse reactions are provided in Table 2.
Table 2: Recommended Dosage Modifications for AUGTYRO Adverse Reactions *Graded per Common Terminology Criteria for Adverse Events v4.03 Adverse Reaction
Severity*
Dosage Modification
Central Nervous System Effects
Intolerable
Grade 2
- Withhold AUGTYRO until ≤Grade 1 or baseline.
- Resume at same or reduced dose, as clinically appropriate.
Grade 3
- Withhold AUGTYRO until ≤Grade 1 or baseline.
- Resume at reduced dose.
Grade 4
- Permanently discontinue AUGTYRO.
Interstitial Lung Disease (ILD)/Pneumonitis
Any Grade
- Withhold AUGTYRO if ILD/pneumonitis is suspected.
- Permanently discontinue if ILD/pneumonitis is confirmed.
Hepatotoxicity
Grade 3
- Withhold AUGTYRO until ≤Grade 1 or baseline.
- Resume at same dose if resolution occurs within 4 weeks.
- Resume at a reduced dose for recurrent Grade 3 events that resolve within 4 weeks.
Grade 4
- Withhold AUGTYRO until ≤Grade 1 or baseline.
- Resume at reduced dose.
- Permanently discontinue if adverse reaction does not resolve within 4 weeks.
- Permanently discontinue for recurrent Grade 4 events.
ALT or AST greater than 3 times ULN with concurrent total bilirubin greater than 1.5 times ULN (in the absence of cholestasis or hemolysis)
- Permanently discontinue AUGTYRO.
Creatine Phosphokinase (CPK) Elevation [see Warnings and Precautions (5.4)]
CPK elevation greater than 5 times ULN
- Withhold until recovery to baseline or to less than or equal to 2.5 times ULN, then resume at same dose.
CPK elevation greater than 10 times ULN or second occurrence of CPK elevation of greater than 5 times ULN
- Withhold until recovery to baseline or to less than or equal to 2.5 times ULN, then resume at reduced dose.
Hyperuricemia [see Warnings and Precautions (5.5)]
Grade 3 or Grade 4
- Withhold AUGTYRO until improvement of signs or symptoms.
- Resume AUGTYRO at same or reduced dose.
Other Clinically Relevant Adverse Reactions [see Adverse Reactions (6.1)]
Intolerable
Grade 2 or Grade 3 or Grade 4
- Withhold AUGTYRO until ≤Grade 1 or baseline.
- Resume at the same or reduced dose if resolution occurs within 4 weeks.
- Permanently discontinue if adverse reaction does not resolve within 4 weeks.
- Permanently discontinue for recurrent Grade 4 events.
2.6 Administration
Take AUGTYRO at approximately the same time each day with or without food [see Pharmacokinetics (12.3)].
Swallow AUGTYRO capsules whole. Do not open, chew, crush, or dissolve the capsule prior to swallowing. Do not take any AUGTYRO capsules that are broken, cracked, or damaged.
If a dose of AUGTYRO is missed or if vomiting occurs at any time after taking a dose, skip the dose and resume AUGTYRO at its regularly scheduled time.
-
3 DOSAGE FORMS AND STRENGTHS
Capsules: 40 mg, white, opaque, immediate release, Size 0, hard shell capsule, filled with white to off-white powder which may appear as a plug, imprinted with “REP 40” in blue text on the cap.
Capsules: 160 mg, blue, opaque, immediate release, Size 0, hard shell capsule, filled with white to off-white powder which may appear as a plug, imprinted with “REP 160” in white text on the cap.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Central Nervous System Adverse Reactions
AUGTYRO can cause central nervous system adverse reactions.
Among the 426 patients who received AUGTYRO in Study TRIDENT-1, a broad spectrum of central nervous system (CNS) adverse reactions including dizziness, ataxia, and cognitive disorders occurred in 77% of patients, with Grade 3 or 4 events occurring in 4.5% of patients.
Dizziness, including vertigo, occurred in 65% of patients; Grade 3 dizziness occurred in 2.8% of patients. The median time to onset was 7 days (1 day to 1.4 years). Dose interruption was required in 9% of patients, and 11% required dose reduction of AUGTYRO due to dizziness.
Ataxia, including gait disturbance and balance disorder, occurred in 28% of patients; Grade 3 ataxia occurred in 0.5% of patients. The median time to onset was 15 days (1 day to 1.4 years). Dose interruption was required in 5% of patients, 8% required dose reduction, and one patient (0.2%) permanently discontinued AUGTYRO due to ataxia.
Cognitive impairment, including memory impairment and disturbance in attention, occurred in 25% of patients. Cognitive impairment included memory impairment (15%), disturbance in attention (12%), and confusional state (2%); Grade 3 cognitive impairment occurred in 0.9% of patients. The median time to onset of cognitive disorders was 37 days (1 day to 1.4 years). Dose interruption was required in 2% of patients, 2.1% required dose reduction, and 0.5% patients permanently discontinued AUGTYRO due to cognitive adverse reactions.
Mood disorders occurred in 6% of patients. Mood disorders occurring in > 1% of patients included anxiety (2.6%); Grade 4 mood disorders (mania) occurred in 0.2% of patients. Dose interruption was required in 0.2% of patients and 0.2% of patients required a dose reduction due to mood disorders.
Sleep disorders including insomnia and hypersomnia occurred in 18% of patients. Sleep disorders observed in > 1% of patients were somnolence (9%), insomnia (6%) and hypersomnia (1.6%). Dose interruption was required in 0.7% of patients, and 0.2% of patients required a dose reduction due to sleep disorders.
The incidences of CNS adverse reactions observed were similar in patients with and without CNS metastases.
Advise patients and caregivers of the risk of CNS adverse reactions with AUGTYRO. Advise patients not to drive or use machines if they are experiencing CNS adverse reactions. Withhold and then resume at same or reduced dose upon improvement, or permanently discontinue AUGTYRO based on severity [see Dosage and Administration (2.5)].
5.2 Interstitial Lung Disease/Pneumonitis
AUGTYRO can cause interstitial lung disease (ILD)/pneumonitis.
Among the 426 patients treated with AUGTYRO, ILD/pneumonitis (pneumonitis [2.8%] and ILD [0.2%]) occurred in 3.1% of patients; Grade 3 ILD/pneumonitis occurred in 1.2% of patients. The median time to onset was 45 days (19 days to 0.9 years). Dose interruption was required in 1.4% of patients, 0.5% of patients required dose reduction, and 1.1% of patients permanently discontinued AUGTYRO due to ILD/pneumonitis.
Monitor patients for new or worsening pulmonary symptoms indicative of ILD/pneumonitis. Immediately withhold AUGTYRO in patients with suspected ILD/pneumonitis and permanently discontinue AUGTYRO if ILD/pneumonitis is confirmed [see Dosage and Administration (2.5)].
5.3 Hepatotoxicity
AUGTYRO can cause hepatotoxicity.
Among the 426 patients treated with AUGTYRO, increased alanine transaminase (ALT) occurred in 38%, increased aspartate aminotransferase (AST) occurred in 41%, including Grade 3 or 4 increased ALT in 3.3% and increased AST in 2.9%. The median time to onset of increased ALT or AST was 15 days (range: 1 day to 1.9 years). Increased ALT or AST leading to dose interruptions or reductions occurred in 2.8% and 1.2% of patients, respectively. Hyperbilirubinemia leading to dose interruptions occurred in 0.5%.
Monitor liver function tests, including ALT, AST and bilirubin, every 2 weeks during the first month of treatment, then monthly thereafter and as clinically indicated. Withhold and then resume at the same or reduced dose upon improvement, or permanently discontinue AUGTYRO based on the severity [see Dosage and Administration (2.5)].
5.4 Myalgia with Creatine Phosphokinase Elevation
AUGTYRO can cause myalgia with or without creatine phosphokinase (CPK) elevation.
Among the 426 patients treated with AUGTYRO, myalgia occurred in 13% of patients, with Grade 3 in 0.7%. Median time to onset of myalgia was 19 days (range: 1 day to 2 years). Concurrent increased CPK within a 7-day window was observed in 3.7% of patients. AUGTYRO was interrupted in one patient with myalgia and concurrent CPK elevation.
Advise patients to report any unexplained muscle pain, tenderness, or weakness.
Monitor serum CPK levels during AUGTYRO treatment and monitor CPK levels every 2 weeks during the first month of treatment and as needed in patients reporting unexplained muscle pain, tenderness, or weakness. Initiate supportive care as clinically indicated. Based on severity, withhold and then resume AUGTYRO at the same or reduced dose upon improvement [see Dosage and Administration (2.5)].
5.5 Hyperuricemia
AUGTYRO can cause hyperuricemia.
Among the 426 patients treated with AUGTYRO, 21 patients (5%) experienced hyperuricemia reported as an adverse reaction and 0.7% of patients experienced Grade 3 or 4 hyperuricemia. One patient without pre-existing gout required urate-lowering medication.
Monitor serum uric acid levels prior to initiating AUGTYRO and periodically during treatment. Initiate treatment with urate-lowering medications as clinically indicated. Withhold and then resume at the same or reduced dose upon improvement, or permanently discontinue AUGTYRO based on severity [see Dosage and Administration (2.5)].
5.6 Skeletal Fractures
AUGTYRO can cause skeletal fractures.
Among 426 adult patients who received AUGTYRO, fractures occurred in 2.3%. Fractures involved the ribs (0.5%), feet (0.5%), spine (0.2%), acetabulum (0.2%), sternum (0.2%), and ankles (0.2%). Some fractures occurred at sites of disease and prior radiation therapy. The median time to fracture was 71 days (range: 31 days to 1.4 years). AUGTYRO was interrupted in 0.3% of patients.
Of 26 evaluable patients in an ongoing open-label study in pediatric patients, fractures occurred in one 12-year-old patient (ankle/foot) and one 10-year-old patient (stress fracture). AUGTYRO was interrupted in both patients. AUGTYRO is not approved for use in pediatric patients less than 12 years of age [see Pediatric Use (8.4)].
Promptly evaluate patients with signs or symptoms (e.g., pain, changes in mobility, deformity) of fractures. There are no data on the effects of AUGTYRO on healing of known fractures and risk of future fractures.
5.7 Embryo-Fetal Toxicity
Based on literature reports in humans with congenital mutations leading to changes in tropomyosin receptor tyrosine kinase (TRK) signaling, findings from animal studies, and its mechanism of action, AUGTYRO can cause fetal harm when administered to a pregnant woman.
Oral administration of repotrectinib to pregnant rats during the period of organogenesis resulted in fetal malformations at doses approximately 0.3 times the recommended 160 mg twice daily dose based on body surface area (BSA).
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective non-hormonal contraception during treatment with AUGTYRO and for 2 months following the last dose, since AUGTYRO can render some hormonal contraceptives ineffective [see Drug Interactions (7.2)].Advise male patients with female partners of reproductive potential to use effective contraception during treatment with AUGTYRO and for 4 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Central Nervous System Adverse Reactions [see Warnings and Precautions (5.1)]
- Interstitial Lung Disease (ILD)/Pneumonitis [see Warnings and Precautions (5.2)]
- Hepatotoxicity [see Warnings and Precautions (5.3)]
- Myalgia with Creatine Phosphokinase Elevation [see Warnings and Precautions (5.4)]
- Hyperuricemia [see Warnings and Precautions (5.5)]
- Skeletal Fractures [see Warnings and Precautions (5.6)]
- Embryo-Fetal Toxicity [see Warnings and Precautions (5.7)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates reported in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in WARNINGS AND PRECAUTIONS and below reflects exposure to AUGTYRO in 426 patients with ROS1-positive NSCLC (n=320), NTRK1/2/3-positive solid tumors (n=104), or other solid tumors (n=2) in TRIDENT-1. Patients received AUGTYRO at a dose of 160 mg orally once daily for the first 14 days, then increased to 160 mg orally twice daily until disease progression or unacceptable toxicity [see Clinical Studies (14.1), (14.2)]. Eligible patients had an ECOG status of ≤1. Patients with a history of ILD, drug-related pneumonitis, significant, uncontrolled, active cardiovascular disease, or prolonged QTc interval were excluded from enrollment in this trial. Forty-eight percent of patients were exposed to AUGTYRO for at least 6 months, and 28% were exposed for greater than 1 year.
The median age of patients who received AUGTYRO was 57 years (range: 18 to 93); 59% female; 43% White, 47% Asian, 2.8% Black, 0.5% Native Hawaiian or Other Pacific Islander, 0.5% American Indian or Alaska Native, 6.1% race not reported or other, and 0.7% unknown.
Serious adverse reactions occurred in 35% of patients who received AUGTYRO. Serious adverse reactions in ≥2% of patients included pneumonia (6.3%), dyspnea (3.1%), pleural effusion (2.8%), and hypoxia (2.6%). Fatal adverse reactions occurred in 3.5% of patients who received AUGTYRO, including pneumonia, pneumonia aspiration, cardiac arrest, sudden cardiac death, cardiac failure, hypoxia, dyspnea, respiratory failure, tremor, and disseminated intravascular coagulation.
Permanent discontinuation of AUGTYRO due to an adverse reaction occurred in 7% of patients. There were no specific adverse reactions that accounted for ≥1% of permanent discontinuations.
Dosage interruptions of AUGTYRO due to an adverse reaction occurred in 50% of patients. Adverse reactions that required dosage interruption in ≥2% of patients were dizziness, dyspnea, muscular weakness, ataxia, pneumonia, peripheral neuropathy, anemia, and vomiting.
Dose reductions of AUGTYRO due to an adverse reaction occurred in 38% of patients. Adverse reactions that required dosage reductions in ≥2% of patients included dizziness, ataxia, muscular weakness, peripheral neuropathy, and cognitive impairment.
The most common (≥20%) adverse reactions that occurred in patients receiving AUGTYRO were dizziness, dysgeusia, peripheral neuropathy, constipation, dyspnea, fatigue, ataxia, cognitive impairment, muscular weakness and nausea.
Table 3 summarizes the adverse reactions that occurred in TRIDENT-1.
Table 3: Adverse Reactions (≥10%) in Patients with ROS1-Positive NSCLC or NTRK-Positive Solid Tumors Who Received AUGTYRO in TRIDENT-1 1 Based on NCI CTCAE v4.03 a Includes terms dizziness, vertigo, dizziness postural, dizziness exertional, vertigo positional b Includes terms dysgeusia, ageusia, anosmia, hypogeusia c Includes terms neuralgia, neuropathy peripheral, peripheral sensory neuropathy, dysesthesia, peripheral motor neuropathy, polyneuropathy, paresthesia, hypoesthesia, hyperesthesia d Includes terms ataxia, gait disturbance, balance disorder, cerebellar ataxia and coordination abnormal e Includes terms memory impairment, disturbance in attention, cognitive disorder, confusional state, amnesia, attention deficit hyperactivity disorder, delirium, altered state of consciousness, aphasia, delusion, depressed level of consciousness, hallucination, mental status changes, neurological decompensation f Includes terms headache, migraine, tension headache g Includes terms dyspnea and dyspnea exertional h Includes terms productive cough, cough, and upper-airway cough syndrome i Includes terms pneumonia, pneumonia aspiration, lower respiratory tract infection, pneumonia viral, pneumonia bacterial, lower respiratory tract infection bacterial, pneumonia klebsiella j Includes terms fatigue and asthenia k Includes terms generalized edema, periorbital edema, localized edema, face edema, edema peripheral, edema, eye edema, scrotal edema l Includes terms myalgia, myositis, musculoskeletal discomfort, musculoskeletal pain m Includes terms vision blurred, dry eye, visual impairment, visual field defect, cataract, conjunctivitis, eye pain, photophobia, photosensitivity reaction, visual acuity reduced, vitreous floaters, blepharospasm, color blindness, diplopia, eye hematoma, eye swelling, eyelid disorder, eyelid injury, eyelids pruritus, glaucoma, night blindness, ophthalmic herpes zoster Adverse Reaction1
AUGTYRO
N=426
All Grades
(%)Grade 3 or 4
(%)Nervous System Disorders
Dizzinessa
65
2.8
Dysgeusiab
54
0
Peripheral neuropathyc
49
1.4
Ataxiad
28
0.5
Cognitive impairmente
25
0.9
Headachef
19
0
Gastrointestinal Disorders
Constipation
38
0.2
Nausea
20
0.7
Diarrhea
14
0.7
Vomiting
12
1.2
Respiratory, Thoracic, and Mediastinal Disorders
Dyspneag
30
6
Coughh
18
0.2
Pneumoniai
11
6
General Disorders
Fatiguej
30
1.2
Edemak
15
0.5
Decreased appetite
11
0.2
Musculoskeletal and Connective Tissue Disorders
Muscular weakness
20
2
Myalgial
13
0.7
Metabolism and Nutritional
Increased weight
16
3
Eye Disorders
Vision disordersm
12
0.5
Clinically relevant adverse reactions occurring in <10% of patients receiving AUGTYRO were pyrexia (9.2%) and fall (3.8%).
Table 4 summarizes the laboratory abnormalities in TRIDENT-1.
Table 4: Laboratory Abnormalities (≥20%) that Worsened from Baseline in Patients with ROS1-Positive NSCLC or NTRK-Positive Solid Tumors Who Received AUGTYRO in TRIDENT-1 Laboratory Abnormality1 AUGTYRO2
N=426All Grades
(%)Grade 3 or 4
(%)1 Based on NCI CTCAE v4.03 2 The denominator used to calculate the rate varied from 233 to 423 based on the number of patients with a baseline value and at least one post-treatment value. Hematology
Decreased hemoglobin
79
8.4
Decreased lymphocytes
43
10
Decreased neutrophils
34
9
Increased activated partial thromboplastin time
26
0.3
Increased INR
24
0
Chemistry
Increased creatine phosphokinase
61
7
Increased gamma glutamyl transferase
50
13
Increased aspartate aminotransferase
41
2.9
Increased alanine aminotransferase
38
3.3
Increased sodium
33
0.2
Increased alkaline phosphatase
29
2.1
Increased glucose
26
2.4
Increased urate
23
12
Decreased phosphate
22
6
Increased potassium
22
0.7
Decreased glucose
20
0.2
-
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on AUGTYRO
Strong and Moderate CYP3A Inhibitors
Avoid concomitant use with strong or moderate CYP3A inhibitors. Concomitant use of AUGTYRO with a strong or a moderate CYP3A inhibitor may increase repotrectinib exposure, which may increase the incidence and severity of adverse reactions of AUGTYRO. Discontinue CYP3A inhibitors for 3 to 5 elimination half-lives of the CYP3A inhibitor prior to initiating AUGTYRO [see Clinical Pharmacology (12.3)].
P-gp Inhibitors
Avoid concomitant use with P-gp inhibitors. Concomitant use of AUGTYRO with a P-gp inhibitor may increase repotrectinib exposure, which may increase the incidence and severity of adverse reactions of AUGTYRO [see Clinical Pharmacology (12.3)].
Strong and Moderate CYP3A Inducers
Avoid concomitant use with strong or moderate CYP3A inducers. Concomitant use of AUGTYRO with a strong or moderate CYP3A inducer may decrease repotrectinib plasma concentrations, which may decrease efficacy of AUGTYRO [see Clinical Pharmacology (12.3)].
7.2 Effects of AUGTYRO on Other Drugs
Certain CYP3A4 Substrates
Avoid concomitant use unless otherwise recommended in the Prescribing Information for CYP3A substrates, where minimal concentration changes can cause reduced efficacy. If concomitant use is unavoidable, increase the CYP3A4 substrate dosage in accordance with approved product labeling.
Repotrectinib is a CYP3A4 inducer. Concomitant use of repotrectinib decreases the concentration of CYP3A4 substrates [see Clinical Pharmacology (12.3)], which can reduce the efficacy of these substrates.
Contraceptives
Repotrectinib is a CYP3A4 inducer, which can decrease progestin or estrogen exposure to an extent that could reduce the effectiveness of hormonal contraceptives.
Avoid concomitant use of AUGTYRO with hormonal contraceptives [see Use in Specific Populations (8.1, 8.3)]. Advise females of reproductive potential to use an effective nonhormonal contraceptive [see Use in Specific Populations (8.1, 8.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on literature reports in humans with congenital mutations leading to changes in TRK signaling, findings from animal studies, and its mechanism of action [see Clinical Pharmacology (12.1)], AUGTYRO can cause fetal harm when administered to a pregnant woman. There are no available data on AUGTYRO use in pregnant women. Oral administration of repotrectinib to pregnant rats during the period of organogenesis resulted in fetal malformations at doses approximately 0.3 times the recommended dose of 160 mg twice daily based on BSA (see Data). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Human Data
Published reports of individuals with congenital mutations in TRK pathway proteins suggest that decreases in TRK-mediated signaling are correlated with obesity, developmental delays, cognitive impairment, insensitivity to pain, and anhidrosis.
Animal Data
In an embryo-fetal development study, once daily oral administration of repotrectinib to pregnant rats during the period of organogenesis from gestation day 6 to 17 resulted in maternal effects of increased body weight and skin abrasions/ulcerations at doses ≥6 mg/kg, fetal malformations of malrotated hindlimbs and lower fetal body weights at doses ≥12 mg/kg [approximately 0.3 times the recommended dose of 160 mg twice daily based on BSA]. No embryolethality was observed.
8.2 Lactation
Risk Summary
There are no data on the presence of AUGTYRO in human milk or its effects on either the breastfed child or on milk production. Because of the potential for serious adverse reactions in breastfed children from AUGTYRO, advise a lactating woman to discontinue breastfeeding during treatment with AUGTYRO and for 10 days after the last dose.
8.3 Females and Males of Reproductive Potential
AUGTYRO can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status of females of childbearing potential prior to initiating AUGTYRO [see Use in Specific Populations (8.1)].
Contraception
AUGTYRO can cause embryo-fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Females
Advise females of childbearing potential to use effective non-hormonal contraception during treatment with AUGTYRO and for 2 months following the last dose. AUGTYRO can render some hormonal contraceptives ineffective [see Drug Interactions (7.2)].
Males
Based on genotoxicity findings, advise male patients with female partners of childbearing potential to use effective contraception during treatment with AUGTYRO and for 4 months following the last dose [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of AUGTYRO in pediatric patients with ROS1-positive NSCLC have not been established.
The safety and effectiveness of AUGTYRO have not been established in pediatric patients younger than 12 years of age with solid tumors who have an NTRK gene fusion.
The safety and effectiveness of AUGTYRO for the treatment of locally advanced or metastatic NTRK-positive solid tumors have been established in pediatric patients 12 years of age or older. Use of AUGTYRO in this age group is supported by evidence from an adequate and well-controlled study in adult patients with additional pharmacokinetic and safety data in pediatric patients aged 12 years and older. This includes data demonstrating that the exposure of repotrectinib in pediatric patients 12 years of age and older is expected to result in similar safety and efficacy to that of adults, and that the course of locally advanced or metastatic NTRK-positive solid tumors is sufficiently similar in adults and pediatric patients 12 years of age or older to allow extrapolation of data in adult to pediatric patients 12 years of age or older [see Dosage and Administration (2.4), Warnings and Precautions (5.7), Adverse Reactions (6.1) and Clinical Pharmacology (12.3)].
Juvenile Animal Data
Daily oral administration of repotrectinib to juvenile rats for 8 weeks starting on postnatal day 12 (approximately equal to a human pediatric age of a newborn) resulted in toxicities similar to those observed in adult rats, though juvenile animals showed decreased body weight gain at doses ≥1 mg/kg (approximately ≥0.04 times the human exposure based on AUC at the recommended clinical dose of 160 mg BID) and decreased femur lengths at 3 mg/kg (approximately 0.1 times the human exposure based on AUC at the recommended clinical dose of 160 mg BID). Decreased body weight gain and decreased femur lengths persisted following 4 weeks of recovery.
8.5 Geriatric Use
Of the 426 patients who received AUGTYRO, in the TRIDENT-1 study for ROS1-positive non-small cell lung cancer or NTRK gene fusion-positive solid tumors, 19% were 65 to 75 years old, and 6% were 75 years of age or older. There were no clinically meaningful differences in safety and efficacy between patients younger than 65 years of age and patients 65 years of age or older.
8.6 Renal Impairment
The recommended dosage of AUGTYRO has not been established in patients with severe renal impairment or kidney failure (eGFR <30 mL/min) and patients on dialysis [see Clinical Pharmacology (12.3)].
No dosage modification is recommended for patients with mild or moderate renal impairment (eGFR 30 to 90 mL/min).
8.7 Hepatic Impairment
The recommended dosage of AUGTYRO has not been established in patients with moderate (total bilirubin >1.5 to 3 times upper limit of normal [ULN] with any AST) or severe (total bilirubin >3 times ULN with any AST) hepatic impairment [see Clinical Pharmacology (12.3)].
No dosage modification is recommended for patients with mild (total bilirubin >1 to 1.5 times ULN or AST > ULN) hepatic impairment.
-
11 DESCRIPTION
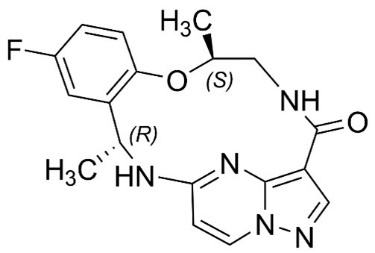
Repotrectinib is a kinase inhibitor. The molecular formula for repotrectinib is C18H18FN5O2 and the molecular weight is 355.37 Daltons. The chemical name is (3R,11S)-6-Fluoro-3,11-dimethyl-10-oxa-2,13,17,18,21-pentaazatetracyclo[13.5.2.04,9.018,22]docosa-1(21),4,6,8,15(22),16,19-heptaen-14-one. The chemical structure of repotrectinib is as follows:
Repotrectinib is a white to off-white powder.
AUGTYRO (repotrectinib) capsules for oral use are supplied as printed hard shell capsules containing 40 mg of repotrectinib. Inactive ingredients are microcrystalline cellulose, sodium lauryl sulfate, croscarmellose sodium, and colloidal silicon dioxide.
The white opaque capsule shell contains gelatin and titanium dioxide. The printing ink contains shellac and FD & C blue #2 aluminum lake.
AUGTYRO (repotrectinib) capsules for oral use are supplied as printed hard shell capsules containing 160 mg of repotrectinib. Inactive ingredients are microcrystalline cellulose, sodium lauryl sulfate, croscarmellose sodium, magnesium stearate, and colloidal silicon dioxide.
The blue opaque capsule shell contains gelatin, titanium dioxide and FD & C blue #1. The printing ink contains shellac and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Repotrectinib is an inhibitor of proto-oncogene tyrosine-protein kinase ROS1 (ROS1) and of the tropomyosin receptor tyrosine kinases (TRKs) TRKA, TRKB, and TRKC.
Fusion proteins that include ROS1 or TRK domains can drive tumorigenic potential through hyperactivation of downstream signaling pathways leading to unconstrained cell proliferation. Repotrectinib exhibited anti-tumor activity in cultured cells expressing ROS1 fusions and mutations including SDC4-ROS1, SDC4-ROS1G2032R, CD74-ROS1, CD74-ROS1G2032R, CD74-ROS1D2033N, and CD74-ROS1L2026M. Repotrectinib also inhibited cell proliferation in cultured cells expressing NTRK fusions and mutations including LMNA-TRKA, LMNA-TRKAG595R, EVT6-TRKBG639R, and ETV6-TRKCG623R.
12.2 Pharmacodynamics
Repotrectinib exposure-response relationships and the time course of pharmacodynamic responses are not fully characterized.
Cardiac Electrophysiology
AUGTYRO does not cause a mean increase in the QTc interval > 20 milliseconds (ms) at 160 mg QD followed by 160 mg BID, the approved recommended dosage.
12.3 Pharmacokinetics
The geometric mean (CV%) of repotrectinib steady state peak concentration (Cmax,ss) is 713 (32.5%) ng/mL and the area under the time concentration curve (AUC0-24h,ss) is 7210 (40.1%) ngh/mL following the approved recommended twice daily dosage in patients with cancer. Repotrectinib Cmax and AUC0-inf increases approximately dose proportional (but less than linear with estimated slopes of 0.78 and 0.70, respectively) over the single dose range of 40 mg to 240 mg (0.25 to 1.5 times the approved recommended dosage). Steady state PK was time-dependent with an autoinduction of CYP3A4. Steady state is achieved within 14 days of daily administration of 160 mg.
Absorption
The geometric mean (CV%) absolute bioavailability of repotrectinib is 45.7% (19.6%). Peak repotrectinib concentration occurred at approximately 2 to 3 hours post a single oral dose of 40 mg to 240 mg (0.25 to 1.5 times the approved recommended dosage) under fasted conditions.
Effect of Food
No clinically significant differences in repotrectinib pharmacokinetics were observed in patients with cancer following administration of a high-fat meal (approximately 800-1000 calories, 50% fat).
Distribution
The geometric mean (CV%) apparent volume of distribution (Vz/F) was 432 L (55.9 %) in patients with cancer following a single 160 mg oral dose of AUGTYRO.
AUGTYRO binding to plasma protein was 95.4% invitro. The blood-to-plasma ratio was 0.56 in vitro.
Elimination
Repotrectinib elimination is time-dependent due to autoinduction of CYP3A4.
The repotrectinib mean terminal half-life is approximately 60.7 h for patients with cancer following a single dose. The steady state repotrectinib terminal half-life is approximately 40.3 h for patients with cancer.
The geometric mean (CV%) apparent oral clearance (CL/F) was 15.9 L/h (45.5%) in patients with cancer following a single 160 mg oral dose of AUGTYRO.
Metabolism
Repotrectinib is primarily metabolized by CYP3A4 followed by secondary glucuronidation.
Excretion
Following a single oral 160 mg dose of radiolabeled repotrectinib, 4.84% (0.56% as unchanged) was recovered in urine and 88.8% (50.6% unchanged) in feces.
Specific Populations
No clinically significant differences in the pharmacokinetics of repotrectinib were observed based on age (12 to 93 years), sex, race (White 50%, Asian 38%, Black 7%), mild to moderate renal impairment (eGFR 30 to < 90 mL/min), or mild hepatic impairment (total bilirubin >1 to 1.5 times ULN or AST > ULN). The effect of moderate (total bilirubin >1.5 to 3 times ULN with any AST) or severe (total bilirubin >3 x ULN with any AST) hepatic impairment, severe renal impairment, kidney failure (eGFR < 30 mL/min), or dialysis on repotrectinib pharmacokinetics is unknown or not fully characterized.
Drug Interaction Studies
Clinical Studies
Strong CYP3A and P-gp inhibitors: Repotrectinib AUC0-inf increased by 5.9-fold and Cmax by 1.7-fold following concomitant use with itraconazole (strong CYP3A and P-gp inhibitor).
Strong CYP3A and P-gp inducers: Repotrectinib AUC0-inf decreased by 92% and Cmax by 79% following concomitant use with rifampin (strong CYP3A and P-gp inducer).
CYP3A substrates: Midazolam (CYP3A substrate AUC0-inf decreased by 69% and Cmax by 48% following concomitant use in subjects who were previously administered 160 mg repotrectinib once daily for 14 days followed by 160 mg twice daily for 7 days.
In vitro Studies
CYP Enzymes: Repotrectinib induces CYP3A4, CYP2B6, CYP2C8, CYP2C19, CYP2C9 and inhibits CYP3A4/5 (GI tract). Repotrectinib does not induce CYP1A2.
Other Metabolic Pathways: Repotrectinib inhibits UGT1A1.
Transporter Systems: Repotrectinib inhibits P-gp, BCRP, OATP1B1, and MATE2-K. Repotrectinib is a substrate for P-gp.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with repotrectinib were not conducted.
Repotrectinib was genotoxic in an in vitro assay in human lymphoblastoid TK6 cells and in an in vivo rat bone marrow micronucleus assay via an aneugenic mechanism of action. Repotrectinib was not mutagenic in vitro in the bacterial reverse mutation (Ames) assay.
Dedicated fertility studies were not conducted with repotrectinib. There were no effects on male and female reproductive organs observed in general repeat-dose toxicology studies of up to 3 months in duration in rats and monkeys at any dose level tested, which equated to exposures of up to approximately 3 times the human exposure at the 160 mg twice daily dose based on AUC.
-
14 CLINICAL STUDIES
14.1 Locally Advanced or Metastatic ROS1-Positive NSCLC
The efficacy of AUGTYRO was evaluated in TRIDENT-1, a multicenter, single-arm, open-label, multi-cohort clinical trial (NCT03093116). Patients were required to have ROS1-positive locally advanced or metastatic NSCLC, ECOG performance status ≤1, measurable disease per RECIST v 1.1, and ≥8 months from first dose. All patients were assessed for CNS lesions at baseline, and patients with symptomatic brain metastases were excluded from the trial. Patients received AUGTYRO 160 mg orally once daily for 14 days, then increased to 160 mg twice daily until disease progression or unacceptable toxicity. Tumor assessments were performed at least every 8 weeks. Identification of ROS1 gene fusions in tumor specimens was prospectively determined in local laboratories using next-generation sequencing (NGS), polymerase chain reaction (PCR) or fluorescence in situ hybridization (FISH) tests. All ROS1-positive patients by local FISH testing required central laboratory confirmation of ROS1 fusion using an analytically validated NGS test. ROS1 fusions were identified by NGS in 51%, FISH in 26%, and PCR in 23%. The major efficacy outcome measures were overall response rate (ORR) and duration of response (DOR) according to RECIST v1.1 as assessed by blinded independent central review (BICR). Intracranial response according to modified RECIST v1.1 was assessed by BICR. Tumor assessments with imaging were performed every 8 weeks. The efficacy populations included 71 ROS1 TKI-naïve patients who received up to 1 prior line of platinum-based chemotherapy and/or immunotherapy and 56 patients who received 1 prior ROS1 TKI with no prior platinum-based chemotherapy or immunotherapy.
Among the 71 ROS1 TKI-naïve patients, the median age was 57 years (range: 28 to 80); female (60.6%); Asian (67.6%), White (25.4%), Hispanic or Latino (4.2%), Black or African American (1.4%); never smoked (63.4%); and ECOG performance status of 1 at baseline (66.2%). At baseline, 94.4% of patients had metastatic disease, 25.4% of patients had CNS metastases by BICR; 97.2% had adenocarcinoma; and 28.2% patients had prior chemotherapy consisting of platinum-based chemotherapy and/or immunotherapy for locally advanced or metastatic disease.
Among the 56 patients who had received 1 prior ROS1 TKI (including crizotinib [82%] and entrectinib [16%]) with no prior platinum-based chemotherapy or immunotherapy, the median age was 57 years (range: 33 – 78); female (67.9%); Asian (48.2%), White (44.6%), Black or African American and Hispanic or Latino (1.8% each); never smoked (64.3%); and ECOG performance status of 1 at baseline (67.9%). At baseline, 98.2% patients had metastatic disease, 42.9% with CNS metastases by BICR, and 94.6% had adenocarcinoma.
Efficacy results are summarized in Table 5.
Table 5: Efficacy Results for Patients with ROS1-Positive NSCLC in TRIDENT-1 Abbreviations: CI = confidence interval; NE = not evaluable; “+” indicates ongoing response a DOR results are based on the updated data as of 19 December 2022. b Median DOR (95% CI) are based on Kaplan-Meier estimates. c DOR landmark analysis is based on the observed DOR. Efficacy Parameters
ROS1 Inhibitor
Naïve Patients(N=71)
ROS1 Inhibitor Pretreated Patients
(N=56)
Confirmed Overall Response Rate, % (95% CI)
79% (68, 88)
38% (25, 52)
Complete Response
6%
5%
Partial Response
73%
32%
Duration of Response (DOR)a
Median in Months (95% CI)b
34.1 (25.6, NE)
14.8 (7.6, NE)
Range (months)
1.4+, 42.4+
3.6, 22.9+
% DOR ≥12 monthsc
70
48
Among TKI-naïve patients, 8 had measurable CNS metastases at baseline as assessed by BICR; responses in intracranial lesions were observed in 7 of these 8 patients. Among the TKI pretreated patients with no prior platinum-based chemotherapy, 12 had measurable CNS metastases at baseline as assessed by BICR; responses in intracranial lesions were observed in 5 of these 12 patients.
Among the 56 ROS1 inhibitor-pretreated patients, 8 had resistance mutations following TKI therapy. Responses were observed in 6 of these 8 patients; responders included patients with solvent front (ROS1G2032R), gatekeeper (ROS1L2026M), and other mutations (ROS1S1986F/Y).
14.2 Locally Advanced or Metastatic NTRK Gene Fusion-Positive Solid Tumors
The efficacy of AUGTYRO was evaluated in TRIDENT-1 (NCT03093116), a multi-center, single-arm, open-label, multi-cohort clinical trial in 88 adult patients with locally advanced or metastatic NTRK gene fusion-positive (NTRK1/2/3) solid tumors who had either received a prior TKI treatment or were TKI-naïve. All patients were assessed for CNS lesions at baseline, and patients with symptomatic brain metastases were excluded from the trial. Patients received AUGTYRO 160 mg orally once daily for 14 days, then increased to 160 mg twice daily until disease progression or unacceptable toxicity. Tumor assessments were performed every 8 weeks. NTRK gene fusions were identified prospectively using NGS in 94%, FISH in 5%, and PCR in 1%. NTRK gene fusion-positive tumors identified by local FISH testing required central laboratory confirmation using an analytically validated NGS test. The major efficacy outcome measures were ORR and DOR according to RECIST v1.1 as assessed by BICR. Intracranial response according to modified RECIST v1.1 was assessed by BICR.
Among the 40 TRK TKI-naïve patients, the median age was 61 years (range: 25 to 84); 60% were female patients; race was Asian 53%, White 25%, Black or African American 5%, and other or not reported 18%; ethnicity was Hispanic or Latino 5%, not Hispanic or Latino 87%, and not reported 8%; and ECOG performance status of 1 at baseline was 55%. At baseline, 98% of patients had metastatic disease and 23% of patients had CNS metastases by BICR. Seventy percent (n=28) of patients received prior systemic therapy with a median of one prior systemic regimen, and 7.5% (n=3) received three or more prior systemic regimens.
Among the 48 TRK TKI-pretreated patients, the median age was 58 years (range: 20 to 81); 48% were female patients; race was White 65%, Asian 25%, Black or African American 2%, and not reported 8%; ethnicity was not Hispanic or Latino 92%, and missing 8%; and ECOG performance status of 1 at baseline was 60%. At baseline, 96% of patients had metastatic disease and 25% of patients had CNS metastases by BICR. Seventy-seven percent (n=37) of patients received 2 or more prior systemic regimens, and 46% (n=22) received three or more prior systemic regimens, and 7 patients (15%) received 2 prior TKI therapies.
Efficacy results are summarized in Table 6.
Table 6: Efficacy Results for Patients with NTRK Gene Fusion-Positive Tumors in TRIDENT-1 NE = not evaluable; “+” indicates ongoing response a Median DOR (95% CI) are based on Kaplan-Meier estimates. b DOR landmark analysis is based on the observed DOR. Efficacy Parameters
TKI-Naïve Patients
(n=40)
TKI-Pretreated Patients
(n=48)
Confirmed Overall Response Rate, % (95% CI)
58
(41, 73)
50
(35, 65)
Complete Response, %
15
0
Partial Response, %
43
50
Median Duration of Response (mDOR)a,
in Months (95% CI)
NE
(NE, NE)
9.9
(7.4, 13.0)
Range (months)
3.7+, 43.9+
1.8, 26.5+
% with DOR ≥ 6 monthsb
87
71
% with DOR ≥ 9 monthsb
83
63
% with DOR ≥ 12 monthsb
83
42
Among the 88 patients, 5 had measurable CNS metastases at baseline as assessed by BICR. Responses were seen in 2 (100%) TKI-naïve patients and 3 (100%) TKI-pretreated patients. One out of 2 TKI-naïve and 2 out of 3 TKI-pretreated patients received prior radiotherapy to the brain, all more than 2 months prior to study entry.
Twenty-six of the TRK TKI-pretreated patients had a resistance mutation at baseline, including 24 with solvent front mutations (NTRK1G595R and NTRK3G623L/R/E/V mutations), one with both a solvent front mutation and a gatekeeper mutation (NTRK1F589L), and one with another mutation (NTRK1G667C). In the 25 TKI-pretreated patients with solvent front mutations at baseline, ORR was 60% (95% CI: 39, 79).
ORR and DOR by tumor type in adult patients with NTRK gene fusion-positive solid tumors are presented in Tables 7 and 8 below.
Table 7: Efficacy Results by Tumor Type in TKI-naïve NTRK Gene Fusion Patients * Includes esophageal cancer and head and neck cancer PD: progressive disease; PR: partial response; SD: stable disease; NA: not applicable “+” indicates ongoing response Tumor type
Patients (n=40)
ORR
DOR
n (%)
95% CI
Range (months)
NSCLC
21
13 (62)
38, 82
3.7+, 31.3+
Thyroid Cancer
5
5 (100)
48, 100
4.7, 43.9+
Salivary Gland Cancer
3
3 (100.0)
29, 100
17.7+, 31.4+
Secretory carcinoma
1
PR
NA
23.0+
Sarcoma, Soft tissue
3
1 (33)
0.8, 91
14.7+
Breast Cancer (adenocarcinoma)
2
PD, PD
NA
NA
Other*
2
SD, SD
NA
NA
Glioblastoma
1
SD
NA
NA
Cholangiocarcinoma
1
PD
NA
NA
Colorectal cancer
1
SD
NA
NA
Peripheral Nerve Sheath Tumor
1
PR
NA
23.0+
Table 8: Efficacy Results by Tumor Type in TKI-pretreated NTRK Gene Fusion-Positive Patients * Includes gallbladder cancer and unknown primary cancer PD: progressive disease; PR: partial response; SD: stable disease; NA: not applicable “+” indicates ongoing response Tumor type
Patients (n=48)
ORR
DOR
n (%)
95% CI
Range (months)
NSCLC
14
6 (43)
18, 71
1.9, 23.0+
Salivary Gland Cancer
8
7 (88)
47, 100
3.7, 26.5+
Secretory carcinoma
3
3 (100)
29, 100
7.9, 26.5+
Sarcoma, Soft tissue
6
1 (17)
0.4, 64
5.6
Thyroid Cancer
4
2 (50)
7, 93
2.0, 9.6
Glioblastoma
3
1 (33.3)
0.8, 91
23.5
Cholangiocarcinoma
2
PR, PD
NA
1.8
Colorectal cancer
2
PR, SD
NA
17.5
Peripheral Nerve Sheath Tumor
2
PR, PR
NA
5.5, 11.1
Neuroendocrine Tumor
2
PR, PR
NA
5.5, 9.1
Pancreatic Cancer
2
PD, PD
NA
NA
Other*
2
SD, PD
NA
NA
Breast Cancer (adenocarcinoma)
1
PR
NA
15.6+
ORR and DOR in adult patients are presented by NTRK gene fusion partner Tables 9 and 10 below.
Table 9: Efficacy Results by NTRK Gene Fusion Partner in TRK TKI-Naïve Patients PD: progressive disease; PR: partial response; SD: stable disease; NA: not applicable “+” indicates ongoing response NTRK Partner
Subjects
(n=40)ORR
DOR
n (%)
95% CI
Range (Months)
ETV6-NTRK3
12
9 (75)
(43, 95)
4.7, 31.4+
TPM3-NTRK1
7
5 (71)
(29, 96)
3.8, 23.1+
EML4-NTRK3
2
Missing, PR
NA
14.8+
IRF2BP2-NTRK1
2
PR, PR
NA
3.7+, 20.3+
PEAR1-NTRK1
2
Missing, PD
NA
NA
Unknown
2
PD, SD
NA
NA
ATP2B2-IT2-NTRK1
1
SD
NA
NA
GOLGB1-NTRK1
1
SD
NA
NA
IL1RL2-NTRK2
1
SD
NA
NA
LRPPRC-NTRK3
1
SD
NA
NA
LRRC71-NTRK1
1
Missing
NA
NA
Multiple
1
PR
NA
28.6+
RBPMS-NTRK3
1
PR
NA
34.3+
SLC28A3-NTRK2
1
PD
NA
NA
SQSTM1-NTRK1
1
PR
NA
15.7+
STRN3-NTRK1
1
PR
NA
23.9+
TMED3-NTRK3
1
PD
NA
NA
TPR-NTRK1
1
PR
NA
43.9+
TRIM33-NTRK1
1
CR
NA
17.8+
Table 10: Efficacy Results by NTRK Gene Fusion Partner in TRK TKI-Pretreated Subjects PR = partial response; PD = progressive disease; SD = stable disease; NA = not applicable; NE = not evaluable “+” indicates ongoing response NTRK Partner
Subjects
(n=48)ORR
DOR
n (%)
95% CI
Range (Months)
ETV6-NTRK3
24
16 (67)
(45, 84)
1.8, 26.5+
EML4-NTRK3
5
4 (80)
(28, 99)
1.9, 12.9
LMNA-NTRK1
4
1 (25)
(0.6, 81)
5.6
TPM3-NTRK1
3
0 (0)
(0, 71)
NA
ATP1B1-NTRK1
1
PD
NA
NA
BCR-NTRK2
1
SD
NA
NA
ETV6-NTRK2
1
NE
NA
NA
GP2-NTRK1
1
PD
NA
NA
IRF2BP2-NTRK1
1
Missing
NA
NA
KANK2-NTRK2
1
PR
NA
23.5
Multiple
1
PD
NA
NA
PRDX1-NTRK1
1
Missing
NA
NA
RBPMS-NTRK3
1
PD
NA
NA
SEL1L-NTRK1
1
PD
NA
NA
SQSTM1-NTRK3
1
PR
NA
5.5
STRN3-NTRK3
1
PR
NA
11.1
-
16 HOW SUPPLIED/STORAGE AND HANDLING
AUGTYRO (repotrectinib) 40 mg, Size 0, white opaque cap, white opaque body, hard shell capsules, filled with a white to off-white powder which may appear as a plug, imprinted with “REP 40” in blue text on the cap are supplied as follows:
- Bottles of 60 capsules (NDC: 0003-4040-60)
- Bottles of 120 capsules (NDC: 0003-4040-12)
AUGTYRO (repotrectinib) 160 mg, Size 0, blue opaque cap, blue opaque body, hard shell capsules, filled with a white to off-white powder which may appear as a plug, imprinted with “REP 160” in white text on the cap are supplied as follows:
- Bottles of 14 capsules (NDC: 0003-4160-14)
- Bottles of 60 capsules (NDC: 0003-4160-60)
Store at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C to 30°C (59°F to 86°F) [See USP Controlled Room Temperature].
-
17 PATIENT COUNSELING INFORMATION
Advise patients to read the FDA-approved patient labeling (Patient Information).
Central Nervous System (CNS) Effects
- Advise patients to inform their healthcare provider if they experience new or worsening CNS symptoms. Instruct patients not to drive or operate hazardous machinery if they are experiencing CNS adverse reactions [see Warnings and Precautions (5.1)].
Interstitial Lung Disease (ILD)/Pneumonitis
- Advise patients to inform their healthcare provider if they experience new or worsening pulmonary symptoms indicative of ILD/pneumonitis [see Warnings and Precautions (5.2)].
Hepatotoxicity
- Advise patients of the need for laboratory tests to monitor liver function and to immediately report symptoms of hepatotoxicity [see Warnings and Precautions (5.3)].
Myalgia with Creatine Phosphokinase Elevation
- Advise patients to inform their healthcare provider if they experience muscle pain [see Warnings and Precautions (5.4)].
Hyperuricemia
- Advise patients to inform their healthcare provider if they experience signs or symptoms associated with hyperuricemia [see Warnings and Precautions (5.5)].
Skeletal Fractures
- Inform patients that bone fractures have occurred in patients taking AUGTYRO and advise patients to report symptoms to their healthcare provider [see Warnings and Precautions (5.6)].
Embryo-Fetal Toxicity
- Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.7), Use in Specific Populations (8.1, 8.3)].
- Advise females of reproductive potential to use effective non-hormonal contraception during treatment with AUGTYRO and for 2 months after the last dose, since AUGTYRO can render some hormonal contraceptives ineffective [see Drug Interactions (7.2)].
- Advise male patients with female partners of reproductive potential to use effective contraception during treatment with AUGTYRO and for 4 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
Lactation
- Advise females not to breastfeed during treatment with AUGTYRO and for 10 days after the last dose [see Use in Specific Populations (8.2)].
Drug Interactions
- Advise patients to inform their healthcare providers of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products [see Drug Interactions (7)].
- Advise patients to avoid grapefruit or grapefruit juice while taking AUGTYRO [see Drug Interactions (7)].
- Advise patients that hormonal contraceptives can be ineffective while taking AUGTYRO [see Drug Interactions (7)].
Administration
- Advise patients to swallow AUGTYRO capsules whole with or without food [see Dosage and Administration (2.6), Pharmacokinetics (12.3)].
- Instruct patients if they miss a dose, or vomit at any time after taking a dose of AUGTYRO, not to “make it up,” but take the next dose of AUGTYRO at the next scheduled time [see Dosage and Administration (2.6)].
For more information, go to www.AUGTYRO.com or call 1-877-284-8976.
Distributed by:
Bristol-Myers Squibb Company
Princeton, NJ 08543 USA
U.S. License No. 1713AUGTYROTM is a trademark of Turning Point Therapeutics, Inc., a Bristol Myers Squibb company.
-
Patient Package Insert
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 06/2024 PATIENT INFORMATION
AUGTYRO™ [Aug-TYE-ro](repotrectinib)
capsules
What is the most important information I should know about AUGTYRO?
AUGTYRO may cause serious side effects, including:
- Central nervous system (CNS) effects. Tell your healthcare provider right away if you experience any new or worsening symptoms of CNS effects during treatment with AUGTYRO, including:
-
- o dizziness
- o vertigo
- o changes in mood, such as anxiety, irritability, and depression
- o balance or coordination problems
- o problems with thinking, such as forgetfulness or confusion
- o seeing or hearing things that are not real (hallucinations)
- o problems with concentration, attention, memory, and sleep
- Lung problems (pneumonitis). Tell your healthcare provider if you have any new or worsening symptoms of lung problems, including a dry cough (without mucus), productive cough (with mucus), wheezing, or trouble breathing.
- Liver problems. Your healthcare provider will do blood tests to check your liver function before starting treatment with AUGTYRO, every 2 weeks for the first month and as needed during treatment. Tell your healthcare provider right away if you develop symptoms of liver problems including:
-
- o your skin or the white part of your eyes turns yellow
- o dark or “tea-colored” urine
- o light-colored stools (bowel movements)
- o loss of appetite
- o nausea or vomiting
- o pain on the upper right side of your stomach area
- Muscle problems. Your healthcare provider will do blood tests before starting treatment with AUGTYRO, every 2 weeks for the first month and as needed during treatment. Tell your healthcare provider right away if you get new or worsening signs and symptoms of muscle problems, including unexplained muscle pain or muscle pain that does not go away, tenderness, or weakness.
- Increased uric acid level in your blood (hyperuricemia). AUGTYRO may cause an excess of uric acid in your blood. Your healthcare provider will do tests before and during your treatment with AUGTYRO to check the uric acid level in your blood. Your healthcare provider may prescribe medicines if you have high blood uric acid levels. Tell your healthcare provider if you experience symptoms of increased uric acid including:
-
- o red, hot, tender, or swollen joints, especially in your big toe
- o pain in your stomach-area or sides
- o decrease in your amount of urine or no urine at all
- o nausea or vomiting
- o pink or brown urine or blood in your urine
- Bone fractures. AUGTYRO may increase your risk for bone fractures. Bone fractures may happen with or without a fall or other injury. Tell your healthcare provider right way if you have pain, changes in movement, or bone abnormalities.
See “What are the possible side effects of AUGTYRO?” for more information about side effects.
What is AUGTYRO?
AUGTYRO is a prescription medicine used to treat:
- adults with non-small cell lung cancer (NSCLC) that has spread within your chest or to other parts of the body and is caused by an abnormal ROS1 gene.
- adults and children 12 years of age and older with solid tumors (cancer) that:
- are caused by certain abnormal NTRK genes, and
- have spread to other parts of the body, or if surgery to remove your cancer is likely to cause severe complications, and
- have grown or spread after other treatment or there is no satisfactory alternative treatment option.
It is not known if AUGTYRO is safe and effective in children with ROS1-positive NSCLC or in children younger than 12 years of age with NTRK-positive solid tumors.
Before taking AUGTYRO, tell your healthcare provider about all your medical conditions, including if you:
- have nervous system (neurological) problems.
- have lung or breathing problems other than lung cancer.
- have liver or kidney problems.
- are pregnant or plan to become pregnant. AUGTYRO can harm your unborn baby. Tell your healthcare provider right away if you become pregnant or think you may be pregnant during treatment with AUGTYRO.
Females who are able to become pregnant:
- o Your healthcare provider should do a pregnancy test before you start treatment with AUGTYRO.
- o You should use effective non-hormonal birth control (contraception) during treatment and for 2 months after the last dose of AUGTYRO.
- o Birth control methods that contain hormones (such as birth control pills, injections, or transdermal system patches) may not work as well during treatment with AUGTYRO.
- o Talk to your healthcare provider about birth control methods that may be right for you.
Males with female partners who are able to become pregnant:
- o You should use effective birth control during treatment with AUGTYRO and for 4 months after the last dose.
- are breastfeeding or plan to breastfeed. It is not known if AUGTYRO passes into your breast milk. Do not breastfeed during treatment and for 10 days after the last dose of AUGTYRO. Talk to your healthcare provider about the best way to feed your baby during this time.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, or herbal supplements.
Taking AUGTYRO with certain other medicines may affect the amount of AUGTYRO or other medicines in your blood and may cause side effects or affect the way that AUGTYRO or other medicines work. Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.
How should I take AUGTYRO?
- Take AUGTYRO exactly as your healthcare provider tells you to take it. Do not change your dose or stop taking AUGTYRO unless your healthcare provider tells you to.
- Your healthcare provider may change your dose, temporarily stop, or permanently stop treatment with AUGTYRO if you develop side effects.
- Take AUGTYRO at about the same time each day with or without food.
- Swallow AUGTYRO capsules whole with water. Do not open, crush, chew or dissolve the capsule. Do not take a capsule if it is broken, cracked, or damaged.
- If you miss a dose, or vomit at any time after taking a dose of AUGTYRO, do not take an extra dose. Just skip the dose and take your next dose at the regularly scheduled time. Do not take 2 doses at the same time to make up a missed or vomited dose.
What should I avoid while taking AUGTYRO?
- You should not drink grapefruit juice or eat grapefruit during your treatment with AUGTYRO. It may increase the amount of AUGTYRO in your blood to a harmful level.
- Do not drive or operate machinery until you know how AUGTYRO affects you. If you experience dizziness, blurred vision, memory loss, changes in mental status, confusion, hallucinations or have trouble with balance or coordination or problems with concentration and attention, do not drive or operate machinery until your symptoms have resolved.
What are the possible side effects of AUGTYRO?
AUGTYRO may cause serious side effects, including:
- See “What is the most important information I should know about AUGTYRO?”
The most common side effects of AUGTYRO include:
- dizziness
- change in sense of taste
- feeling of numbness or tingling in your arms or legs
- constipation
- shortness of breath
- tiredness
- trouble with balance, coordination, and walking
- problems with thinking, such as forgetfulness or confusion, memory problems and hallucinations
- muscle weakness
- nausea
These are not all of the possible side effects of AUGTYRO.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store AUGTYRO?
- Store AUGTYRO at room temperature between 68°F to 77°F (20°C to 25°C).
Keep AUGTYRO and all medicines out of the reach of children.
General information about the safe and effective use of AUGTYRO.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use AUGTYRO for a condition for which it was not prescribed. Do not give AUGTYRO to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about AUGTYRO that is written for health professionals.
What are the ingredients of AUGTYRO?
Active ingredient: repotrectinib
Inactive ingredients: microcrystalline cellulose, sodium lauryl sulfate, croscarmellose sodium, and colloidal silicon dioxide. Capsule shell contains gelatin and titanium dioxide. Printing ink contains shellac.
- White opaque capsules, printing ink contains in addition FD & C blue #2 aluminum lake.
- Blue opaque capsules contain in addition magnesium stearate and FD & C blue #1. Printing ink contains in addition titanium dioxide.
Distributed by: Bristol-Myers Squibb Company, Princeton, NJ 08543 USA; U.S. License No. 1713
AUGTYROTM is a trademark of Turning Point Therapeutics, Inc., a Bristol Myers Squibb company.
For more information, go to www.AUGTYRO.com or call 1-877-284-8976.
-
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
NDC: 0003-4160-14
14 CapsulesAUGTYROTM
(repotrectinib)
capsules
160mg
Swallow capsules whole
Rx only
Bristol Myers SquibbNDC: 0003-4160-14
14 CapsulesAUGTYROTM
(repotrectinib)
capsules
160mg
Swallow capsules whole
Rx only
Bristol Myers SquibbNDC: 0003-4040-60
60 CapsulesAUGTYROTM
(repotrectinib)
capsules
40mg
Swallow capsules whole
Rx only
Bristol Myers SquibbNDC: 0003-4040-60
60 CapsulesAUGTYROTM
(repotrectinib)
capsules
40mg
Swallow capsules whole
Rx only
Bristol Myers SquibbNDC: 0003-4040-12
120 CapsulesAUGTYROTM
(repotrectinib)
capsules
40mg
Swallow capsules whole
Rx only
Bristol Myers Squibb -
INGREDIENTS AND APPEARANCE
AUGTYRO
repotrectinib capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0003-4040 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength REPOTRECTINIB (UNII: 08O3FQ4UNP) (REPOTRECTINIB - UNII:08O3FQ4UNP) REPOTRECTINIB 40 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SODIUM LAURYL SULFATE (UNII: 368GB5141J) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) Product Characteristics Color WHITE (OPAQUE) Score no score Shape CAPSULE Size 22mm Flavor Imprint Code REP;40 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0003-4040-60 60 in 1 BOTTLE; Type 0: Not a Combination Product 12/05/2023 2 NDC: 0003-4040-12 120 in 1 BOTTLE; Type 0: Not a Combination Product 12/05/2023 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA218213 12/05/2023 AUGTYRO
repotrectinib capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0003-4160 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength REPOTRECTINIB (UNII: 08O3FQ4UNP) (REPOTRECTINIB - UNII:08O3FQ4UNP) REPOTRECTINIB 160 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SODIUM LAURYL SULFATE (UNII: 368GB5141J) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) Product Characteristics Color BLUE (OPAQUE) Score no score Shape CAPSULE Size 22mm Flavor Imprint Code REP;160 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0003-4160-14 14 in 1 BOTTLE; Type 0: Not a Combination Product 11/01/2024 2 NDC: 0003-4160-60 60 in 1 BOTTLE; Type 0: Not a Combination Product 11/01/2024 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA218213 11/01/2024 Labeler - E.R. Squibb & Sons, L.L.C. (011550092)




Trademark Results [Augtyro]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 AUGTYRO 98620301 not registered Live/Pending |
Turning Point Therapeutics, Inc. 2024-06-26 |
 AUGTYRO 97272829 not registered Live/Pending |
Turning Point Therapeutics, Inc. 2022-02-17 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.