ZAFIRLUKAST tablet, film coated
Zafirlukast by
Drug Labeling and Warnings
Zafirlukast by is a Prescription medication manufactured, distributed, or labeled by Carilion Materials Management. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
DESCRIPTION
Zafirlukast is a synthetic, selective peptide leukotriene receptor antagonist (LTRA), with the chemical name 4-(5-cyclopentyloxy-carbonylamino-1-methyl-indol-3-ylmethyl)-3-methoxy-N-o-tolylsulfonylbenzamide. The molecular weight of zafirlukast is 575.7 and the structural formula is:
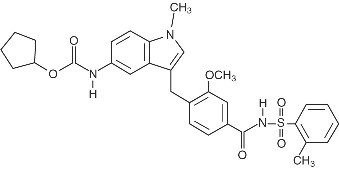
The molecular formula is: C31H33N3O6S
Zafirlukast, white to pale yellow coloured powder, freely soluble in tetrahydrofuran, and dimethylsulfoxide and practically insoluble in water.
Zafirlukast is supplied as 10 and 20 mg tablets for oral administration.
Inactive ingredients: Film coated tablets containing hydroxypropyl cellulose, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polyethylene glycol 400, sodium starch glycolate (Type-A) and titanium dioxide. .
-
CLINICAL PHARMACOLOGY
Mechanism of Action:
Zafirlukast is a selective and competitive receptor antagonist of leukotriene D4 and E4 (LTD4 and LTE4), components of slow-reacting substance of anaphylaxis (SRSA). Cysteinyl leukotriene production and receptor occupation have been correlated with the pathophysiology of asthma, including airway edema, smooth muscle constriction, and altered cellular activity associated with the inflammatory process, which contribute to the signs and symptoms of asthma. Patients with asthma were found in one study to be 25-100 times more sensitive to the bronchoconstricting activity of inhaled LTD4 than nonasthmatic subjects.
In vitro studies demonstrated that zafirlukast antagonized the contractile activity of three leukotrienes (LTC4, LTD4 and LTE4) in conducting airway smooth muscle from laboratory animals and humans. Zafirlukast prevented intradermal LTD4-induced increases in cutaneous vascular permeability and inhibited inhaled LTD4-induced influx of eosinophils into animal lungs. Inhalational challenge studies in sensitized sheep showed that zafirlukast suppressed the airway responses to antigen; this included both the early- and late-phase response and the nonspecific hyperresponsiveness.
In humans, zafirlukast inhibited bronchoconstriction caused by several kinds of inhalational challenges. Pretreatment with single oral doses of zafirlukast inhibited the bronchoconstriction caused by sulfur dioxide and cold air in patients with asthma. Pretreatment with single doses of zafirlukast attenuated the early- and late-phase reaction caused by inhalation of various antigens such as grass, cat dander, ragweed, and mixed antigens in patients with asthma. Zafirlukast also attenuated the increase in bronchial hyperresponsiveness to inhaled histamine that followed inhaled allergen challenge.
Clinical Pharmacokinetics and Bioavailability:
Absorption
Zafirlukast is rapidly absorbed following oral administration. Peak plasma concentrations are generally achieved 3 hours after oral administration. The absolute bioavailability of zafirlukast is unknown. In two separate studies, one using a high fat and the other a high protein meal, administration of zafirlukast with food reduced the mean bioavailability by approximately 40%.
Distribution
Zafirlukast is more than 99% bound to plasma proteins, predominantly albumin. The degree of binding was independent of concentration in the clinically relevant range. The apparent steady-state volume of distribution (Vss/F) is approximately 70 L, suggesting moderate distribution into tissues. Studies in rats using radiolabeled zafirlukast indicate minimal distribution across the blood-brain barrier.
Metabolism
Zafirlukast is extensively metabolized. The most common metabolic products are hydroxylated metabolites which are excreted in the feces. The metabolites of zafirlukast identified in plasma are at least 90 times less potent as LTD4 receptor antagonists than zafirlukast in a standard in vitro test of activity. In vitro studies using human liver microsomes showed that the hydroxylated metabolites of zafirlukast excreted in the feces are formed through the cytochrome P450 2C9 (CYP2C9) pathway. Additional in vitro studies utilizing human liver microsomes show that zafirlukast inhibits the cytochrome P450 CYP3A4 and CYP2C9 isoenzymes at concentrations close to the clinically achieved total plasma concentrations (see Drug Interactions).
Excretion
The apparent oral clearance (CL/f) of zafirlukast is approximately 20 L/h. Studies in the rat and dog suggest that biliary excretion is the primary route of excretion. Following oral administration of radiolabeled zafirlukast to volunteers, urinary excretion accounts for approximately 10% of the dose and the remainder is excreted in feces. Zafirlukast is not detected in urine.
In the pivotal bioequivalence study, the mean terminal half-life of zafirlukast is approximately 10 hours in both normal adult subjects and patients with asthma. In other studies, the mean plasma half-life of zafirlukast ranged from approximately 8 to 16 hours in both normal subjects and patients with asthma. The pharmacokinetics of zafirlukast are approximately linear over the range from 5 mg to 80 mg. Steady-state plasma concentrations of zafirlukast are proportional to the dose and predictable from single-dose pharmacokinetic data. Accumulation of zafirlukast in the plasma following twice-daily dosing is approximately 45%.
The pharmacokinetic parameters of zafirlukast 20 mg administered as a single dose to 36 male volunteers are shown with the table below.
Special Populations
Gender: The pharmacokinetics of zafirlukast are similar in males and females. Weight-adjusted apparent oral clearance does not differ due to gender.
Race: No differences in the pharmacokinetics of zafirlukast due to race have been observed.
Elderly: The apparent oral clearance of zafirlukast decreases with age. In patients above 65 years of age, there is an approximately 2-3 fold greater Cmax and AUC compared to young adult patients.
Children: Following administration of a single 20 mg dose of zafirlukast to 20 boys and girls between 7 and 11 years of age, and in a second study, to 29 boys and girls between 5 and 6 years of age, the following pharmacokinetic parameters were obtained:
Parameter
Children age 5-6 years Mean (%Coefficient of Variation)
Children age 7-11 years Mean
(% Coefficient of Variation)
Cmax (ng/mL)
756 (39%)
601 (45%)
AUC (ngh/mL)
2458 (34%)
2027 (38%)
tmax (h)
2.1 (61%)
2.5 (55%)
CL/f (L/h)
9.2 (37%)
11.4 (42%)
Weight unadjusted apparent clearance was 11.4 L/h (42%) in the 7-11 year old children and 9.2 L/h (37%) in the 5-6 year old children, which resulted in greater systemic drug exposures than that obtained in adults for an identical dose. To maintain similar exposure levels in children compared to adults, a dose of 10 mg twice daily is recommended in children 5-11 years of age (see DOSAGE AND ADMINISTRATION).
Zafirlukast disposition was unchanged after multiple dosing (20 mg twice daily) in children and the degree of accumulation in plasma was similar to that observed in adults.
Hepatic Insufficiency: In a study of patients with hepatic impairment (biopsy-proven cirrhosis), there was a reduced clearance of zafirlukast resulting in a 50-60% greater Cmax and AUC compared to normal subjects.
Renal Insufficiency: Based on a cross-study comparison, there are no apparent differences in the pharmacokinetics of zafirlukast between renally-impaired patients and normal subjects.
Drug-Drug Interactions: The following drug interaction studies have been conducted with zafirlukast (see PRECAUTIONS, Drug Interactions).
●Coadministration of multiple doses of zafirlukast (160 mg/day) to steady-state with a single 25 mg dose of warfarin (a substrate of CYP2C9) resulted in a significant increase in the mean AUC (+63%) and half-life (+36%) of S-warfarin. The mean prothrombin time increased by approximately 35%. The pharmacokinetics of zafirlukast were unaffected by coadministration with warfarin.
●Coadministration of zafirlukast (80 mg/day) at steady-state with a single dose of a liquid theophylline preparation (6 mg/kg) in 13 asthmatic patients, 18 to 44 years of age, resulted in decreased mean plasma concentrations of zafirlukast by approximately 30%, but no effect on plasma theophylline concentrations was observed.
●Coadministration of zafirlukast (20 mg/day) or placebo at steady-state with a single dose of sustained release theophylline preparation (16 mg/kg) in 16 healthy boys and girls (6 through 11 years of age) resulted in no significant differences in the pharmacokinetic parameters of theophylline.
●Coadministration of zafirlukast dosed at 40 mg twice daily in a single-blind, parallel-group, 3-week study in 39 healthy female subjects taking oral contraceptives, resulted in no significant effect on ethinyl estradiol plasma concentrations or contraceptive efficacy.
●Coadministration of zafirlukast (40 mg/day) with aspirin (650 mg four times daily) resulted in mean increased plasma concentrations of zafirlukast by approximately 45%.
● Coadministration of a single dose of zafirlukast (40 mg) with erythromycin (500 mg three times daily for 5 days) to steady-state in 11 asthmatic patients resulted in decreased mean plasma concentrations of zafirlukast by approximately 40% due to a decrease in zafirlukast bioavailability.
Clinical Studies:
Three U.S. double-blind, randomized, placebo-controlled, 13-week clinical trials in 1380 adults and children 12 years of age and older with mild-to-moderate asthma demonstrated that zafirlukast improved daytime asthma symptoms, nighttime awakenings, mornings with asthma symptoms, rescue beta2-agonist use, FEV1, and morning peak expiratory flow rate. In these studies, the patients had a mean baseline FEV1 of approximately 75% of predicted normal and a mean baseline beta2-agonist requirement of approximately 4-5 puffs of albuterol per day. The results of the largest of the trials are shown in the table below.
Mean Change from Baseline at Study End Point - * p<0.05, compared to placebo
Zafirlukast 20 mg twice daily
N=514
Placebo
N=248
Daytime Asthma symptom score
(0-3 scale)
-0.44*
-0.25
Nightime Awakenings
(number per week)
-1.27*
-0.43
Mornings with Asthma Symptoms
(days per week)
-1.32*
-0.75
Rescue β2-agonist use
(puffs per day)
-1.15*
-0.24
FEV1 (L)
+0.15*
+0.05
Morning PEFR (L/min)
+22.06*
+7.63
Evening PEFR (L/min)
+13.12
+10.14
In a second and smaller study, the effect of zafirlukast on most efficacy parameters was comparable to the active control (inhaled cromolyn sodium 1600 mcg four times per day) and superior to placebo at end point for decreasing rescue beta2-agonist use (figure below).
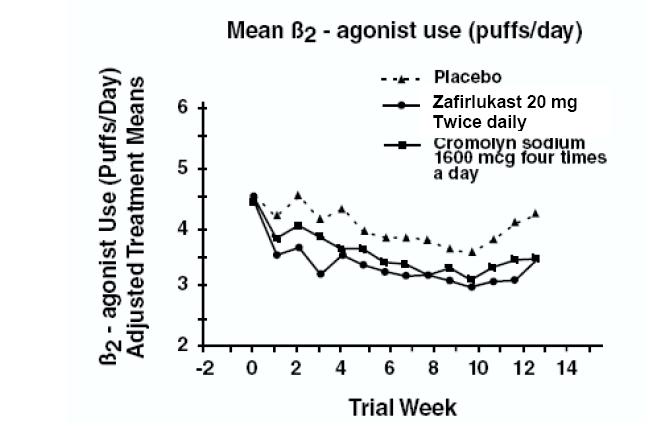
In these trials, improvement in asthma symptoms occurred within one week of initiating treatment with zafirlukast. The role of zafirlukast in the management of patients with more severe asthma, patients receiving antiasthma therapy other than as-needed, inhaled beta2-agonists, or as an oral or inhaled corticosteroid-sparing agent remains to be fully characterized.
- INDICATIONS AND USAGE
- CONTRAINDICATIONS
-
WARNINGS
Hepatotoxicity:
Cases of life-threatening hepatic failure have been reported in patients treated with zafirlukast. Cases of liver injury without other attributable cause have been reported from post-marketing adverse event surveillance of patients who have received the recommended dose of zafirlukast (40 mg/day). In most, but not all post-marketing reports, the patient’s symptoms abated and the liver enzymes returned to normal or near normal after stopping zafirlukast. In rare cases, patients have either presented with fulminant hepatitis or progressed to hepatic failure, liver transplantation and death. In extremely rare post-marketing cases, no clinical symptoms or signs suggestive of liver dysfunction were reported to precede the latter observations.
Physicians may consider the value of liver function testing. Periodic serum transaminase testing has not proven to prevent serious injury but it is generally believed that early detection of drug-induced hepatic injury along with immediate withdrawal of the suspect drug enhances the likelihood for recovery.
Patients should be advised to be alert for signs and symptoms of liver dysfunction (eg, right upper quadrant abdominal pain, nausea, fatigue, lethargy, pruritus, jaundice, flu-like symptoms, and anorexia) and to contact their physician immediately if they occur. Ongoing clinical assessment of patients should govern physician interventions, including diagnostic evaluations and treatment.
If liver dysfunction is suspected based upon clinical signs or symptoms (eg, right upper quadrant abdominal pain, nausea, fatigue, lethargy, pruritus, jaundice, flu-like symptoms, anorexia, and enlarged liver), zafirlukast should be discontinued. Liver function tests, in particular serum ALT, should be measured immediately and the patient managed accordingly. If liver function tests are consistent with hepatic dysfunction, zafirlukast therapy should not be resumed. Patients in whom zafirlukast was withdrawn because of hepatic dysfunction where no other attributable cause is identified should not be re-exposed to zafirlukast (see PRECAUTIONS, Information for Patients and ADVERSE REACTIONS).
Bronchospasm:
Zafirlukast tablets are not indicated for use in the reversal of bronchospasm in acute asthma attacks, including status asthmaticus. Therapy with zafirlukast can be continued during acute exacerbations of asthma.
Concomitant Warfarin Administration:
Coadministration of zafirlukast with warfarin results in a clinically significant increase in prothrombin time (PT). Patients on oral warfarin anticoagulant therapy and zafirlukast should have their prothrombin times monitored closely and anticoagulant dose adjusted accordingly (see PRECAUTIONS, Drug Interactions).
-
PRECAUTIONS
Information for Patients:
Patients should be told that a rare side effect of zafirlukast is hepatic dysfunction, and to contact their physician immediately if they experience symptoms of hepatic dysfunction (eg. right upper quadrant abdominal pain, nausea, fatigue, lethargy, pruritus, jaundice, flu-like symptoms, and anorexia). Liver failure resulting in liver transplantation and death has occurred in patients taking zafirlukast (see WARNINGS, Hepatotoxicity and ADVERSE REACTIONS).
Zafirlukast tablets are indicated for the chronic treatment of asthma and should be taken regularly as prescribed, even during symptom-free periods. Zafirlukast is not a bronchodilator and should not be used to treat acute episodes of asthma. Patients receiving zafirlukast should be instructed not to decrease the dose or stop taking any other antiasthma medications unless instructed by a physician. Patients should be instructed to notify their physician if neuropsychiatric events occur while using zafirlukast (see PRECAUTIONS, Neuropsychiatric Events).Women who are breast-feeding should be instructed not to take zafirlukast (see PRECAUTIONS, Nursing Mothers). Alternative antiasthma medication should be considered in such patients.
The bioavailability of zafirlukast may be decreased when taken with food. Patients should be instructed to take zafirlukast at least 1 hour before or 2 hours after meals.
Eosinophilic Conditions: In rare cases, patients with asthma on zafirlukast may present with systemic eosinophilia, eosinophilic pneumonia, or clinical features of vasculitis consistent with Churg-Strauss syndrome, a condition which is often treated with systemic steroid therapy. Physicians should be alert to eosinophilia, vasculitic rash, worsening pulmonary symptoms, cardiac complications, and/or neuropathy presenting in their patients. These events have usually, but not always, been associated with the reduction and/or withdrawal of steroid therapy. The possibility that zafirlukast may be associated with emergence of Churg-Strauss syndrome can neither be excluded nor established (see ADVERSE REACTIONS).
Neuropsychiatric Events: Neuropsychiatric events have been reported in adult, adolescent and pediatric patients taking zafirlukast. Post-marketing reports with zafirlukast include insomnia and depression. The clinical details of some post-marketing reports involving zafirlukast appear consistent with a drug-induced effect. Patients and prescribers should be alert for neuropsychiatric events. Patients should be instructed to notify their prescriber if these changes occur. Prescribers should carefully evaluate the risks and benefits of continuing treatment with zafirlukast if such events occur (see ADVERSE REACTIONS).
Drug Interactions:
In a drug interaction study in 16 healthy male volunteers, coadministration of multiple doses of zafirlukast (160 mg/day) to steady-state with a single 25 mg dose of warfarin resulted in a significant increase in the mean AUC (+ 63%) and half-life (+36%) of S-warfarin. The mean prothrombin time (PT) increased by approximately 35%. This interaction is probably due to an inhibition by zafirlukast of the cytochrome P450 2C9 isoenzyme system. Patients on oral warfarin anticoagulant therapy and zafirlukast should have their prothrombin times monitored closely and anticoagulant dose adjusted accordingly (see WARNINGS, Concomitant Warfarin Administration). No formal drug-drug interaction studies with zafirlukast and other drugs known to be metabolized by the cytochrome P450 2C9 isoenzyme (eg, tolbutamide, phenytoin, carbamazepine) have been conducted; however, care should be exercised when zafirlukast is coadministered with these drugs.
In a drug interaction study in 11 asthmatic patients, coadministration of a single dose of zafirlukast (40 mg) with erythromycin (500 mg three times daily for 5 days) to steady-state resulted in decreased mean plasma levels of zafirlukast by approximately 40% due to a decrease in zafirlukast bioavailability.
Coadministration of zafirlukast (20 mg/day) or placebo at steady-state with a single dose of sustained release theophylline preparation (16 mg/kg) in 16 healthy boys and girls (6 through 11 years of age) resulted in no significant differences in the pharmacokinetic parameters of theophylline.
Coadministration of zafirlukast (80 mg/day) at steady-state with a single dose of a liquid theophylline preparation (6 mg/kg) in 13 asthmatic patients, 18 to 44 years of age, resulted in decreased mean plasma levels of zafirlukast by approximately 30%, but no effect on plasma theophylline levels was observed.
Rare cases of patients experiencing increased theophylline levels with or without clinical signs or symptoms of theophylline toxicity after the addition of zafirlukast to an existing theophylline regimen have been reported. The mechanism of the interaction between zafirlukast and theophylline in these patients is unknown (see ADVERSE REACTIONS).
Coadministration of zafirlukast (40 mg/day) with aspirin (650 mg four times daily) resulted in mean increased plasma levels of zafirlukast by approximately 45%.
In a single-blind, parallel-group, 3-week study in 39 healthy female subjects taking oral contraceptives, 40 mg twice daily of zafirlukast had no significant effect on ethinyl estradiol plasma concentrations or contraceptive efficacy.
No formal drug-drug interaction studies between zafirlukast and marketed drugs known to be metabolized by the P450 3A4 (CYP3A4) isoenzyme (eg, dihydropyridine calcium-channel blockers, cyclosporin, cisapride) have been conducted. As zafirlukast is known to be an inhibitor of CYP3A4 in vitro, it is reasonable to employ appropriate clinical monitoring when these drugs are coadministered with zafirlukast.
Carcinogenesis, Mutagenesis, Impairment of Fertility:
In two-year carcinogenicity studies, zafirlukast was administered at dietary doses of 10, 100, and 300 mg/kg to mice and 40, 400, and 2000 mg/kg to rats. Male mice at an oral dose of 300 mg/kg/day (approximately 30 times the maximum recommended daily oral dose in adults and in children on a mg/m2 basis) showed an increased incidence of hepatocellular adenomas; female mice at this dose showed a greater incidence of whole body histocytic sarcomas. Male and female rats at an oral dose of 2000 mg/kg/day (resulting in approximately 160 times the exposure to drug plus metabolites from the maximum recommended daily oral dose in adults and in children based on a comparison of the plasma area-under the curve [AUC] values) of zafirlukast showed an increased incidence of urinary bladder transitional cell papillomas. Zafirlukast was not tumorigenic at oral doses up to 100 mg/kg (approximately 10 times the maximum recommended daily oral dose in adults and in children on a mg/m2 basis) in mice and at oral doses up to 400 mg/kg (resulting in approximately 140 times the exposure to drug plus metabolites from the maximum recommended daily oral dose in adults and in children based on a comparison of the plasma AUC values) in rats. The clinical significance of these findings for the long-term use of zafirlukast is unknown.
Zafirlukast showed no evidence of mutagenic potential in the reverse microbial assay, in 2 forward point mutation (CHO-HGPRT and mouse lymphoma) assays or in two assays for chromosomal aberrations (the in vitro human peripheral blood lymphocyte clastogenic assay and the in vivo rat bone marrow micronucleus assay).
No evidence of impairment of fertility and reproduction was seen in male and female rats treated with zafirlukast at oral doses up to 2000 mg/kg (approximately 410 times the maximum recommended daily oral dose in adults on a mg/m2 basis).
Pregnancy Category B:
No teratogenicity was observed at oral doses up to 1600 mg/kg/day in mice (approximately 160 times the maximum recommended daily oral dose in adults on a mg/m2 basis), up to 2000 mg/kg/day in rats (approximately 410 times the maximum recommended daily oral dose in adults on a mg/m2 basis) and up to 2000 mg/kg/day in cynomolgus monkeys (which resulted in approximately 20 times the exposure to drug plus metabolites compared to that from the maximum recommended daily oral dose in adults based on comparison of the AUC values). At an oral dose of 2000 mg/kg/day in rats, maternal toxicity and deaths were seen with increased incidence of early fetal resorption. Spontaneous abortions occurred in cynomolgus monkeys at the maternally toxic oral dose of 2000 mg/kg/day. There are no adequate and well-controlled trials in pregnant women. Because animal reproductive studies are not always predictive of human response, zafirlukast should be used during pregnancy only if clearly needed.
Nursing Mothers:
Zafirlukast is excreted in breast milk. Following repeated 40 mg twice-a-day dosing in healthy women, average steady-state concentrations of zafirlukast in breast milk were 50 ng/mL compared to 255 ng/mL in plasma. Because of the potential for tumorigenicity shown for zafirlukast in mouse and rat studies and the enhanced sensitivity of neonatal rats and dogs to the adverse effects of zafirlukast, zafirlukast should not be administered to mothers who are breast-feeding.
Pediatric Use:
The safety of zafirlukast at doses of 10 mg twice daily has been demonstrated in 205 pediatric patients 5 through 11 years of age in placebo-controlled trials lasting up to six weeks and with 179 patients in this age range participating in 52 weeks of treatment in an open label extension.
The effectiveness of zafirlukast for the prophylaxis and chronic treatment of asthma in pediatric patients 5 through 11 years of age is based on an extrapolation of the demonstrated efficacy of zafirlukast in adults with asthma and the likelihood that the disease course, and pathophysiology and the drug’s effect are substantially similar between the two populations. The recommended dose for the patients 5 through 11 years of age is based upon a cross-study comparison of the pharmacokinetics of zafirlukast in adults and pediatric subjects, and on the safety profile of zafirlukast in both adult and pediatric patients at doses equal to or higher than the recommended dose.
The safety and effectiveness of zafirlukast for pediatric patients less than 5 years of age has not been established. The effect of zafirlukast on growth in children has not been determined.
Geriatric Use:
Based on cross-study comparison, the clearance of zafirlukast is reduced in patients 65 years of age and older such that Cmax and AUC are approximately 2- to 3-fold greater than those of younger patients (see DOSAGE AND ADMINISTRATION and CLINICAL PHARMACOLOGY).
A total of 8094 patients were exposed to zafirlukast in North American and European short-term placebo-controlled clinical trials. Of these, 243 patients were elderly (age 65 years and older). No overall difference in adverse events was seen in the elderly patients, except for an increase in the frequency of infections among zafirlukast-treated elderly patients compared to placebo-treated elderly patients (7.0% vs. 2.9%). The infections were not severe, occurred mostly in the lower respiratory tract, and did not necessitate withdrawal of therapy.
An open-label, uncontrolled, 4-week trial of 3759 asthma patients compared the safety and efficacy of zafirlukast 20 mg given twice daily in three patient age groups, adolescents (12-17 years), adults (18-65 years), and elderly (greater than 65 years). A higher percentage of elderly patients (n=384) reported adverse events when compared to adults and adolescents. These elderly patients showed less improvement in efficacy measures. In the elderly patients, adverse events occurring in greater than 1% of the population included headache (4.7%), diarrhea and nausea (1.8%), and pharyngitis (1.3%). The elderly reported the lowest percentage of infections of all three age groups in this study.
-
ADVERSE REACTIONS
Adults and Children 12 years of age and older
The safety database for zafirlukast consists of more than 4000 healthy volunteers and patients who received zafirlukast, of which 1723 were asthmatics enrolled in trials of 13 weeks duration or longer. A total of 671 patients received zafirlukast for 1 year or longer. The majority of the patients were 18 years of age or older; however, 222 patients between the age of 12 and 18 years received zafirlukast.
A comparison of adverse events reported by ≥1% of zafirlukast-treated patients, and at rates numerically greater than in placebo-treated patients, is shown for all trials in the table below.
Zafirlukast PLACEBO
Adverse Event
N=4058
N=2032
Headache
12.9%
11.7%
Infection
3.5%
3.4%
Nausea
3.1%
2.0%
Diarrhea
2.8%
2.1%
Pain (generalized)
1.9%
1.7%
Asthenia
1.8%
1.6%
Abdominal Pain
1.8%
1.1%
Accidental Injury
1.6%
1.5%
Dizziness
1.6%
1.5%
Myalgia
1.6%
1.5%
Fever
1.6%
1.1%
Back Pain
1.5%
1.2%
Vomiting
1.5%
1.1%
SGPT Elevation
1.5%
1.1%
Dyspepsia
1.3%
1.2%
The frequency of less common adverse events was comparable between zafirlukast and placebo.
Rarely, elevations of one or more liver enzymes have occurred in patients receiving zafirlukast in controlled clinical trials. In clinical trials, most of these have been observed at doses four times higher than the recommended dose. The following hepatic events (which have occurred predominantly in females) have been reported from postmarketing adverse event surveillance of patients who have received the recommended dose of zafirlukast (40 mg/day): cases of symptomatic hepatitis (with or without hyperbilirubinemia) without other attributable cause; and rarely, hyperbilirubinemia without other elevated liver function tests. In most, but not all postmarketing reports, the patient’s symptoms abated and the liver enzymes returned to normal or near normal after stopping zafirlukast. In rare cases, patients have presented with fulminant hepatitis or progressed to hepatic failure, liver transplantation and death (see WARNINGS, Hepatotoxicity and PRECAUTIONS, Information for Patients).
In clinical trials, an increased proportion of zafirlukast patients over the age of 55 years reported infections as compared to placebo-treated patients. A similar finding was not observed in other age groups studied. These infections were mostly mild or moderate in intensity and predominantly affected the respiratory tract. Infections occurred equally in both sexes, were dose-proportional to total milligrams of zafirlukast exposure, and were associated with coadministration of inhaled corticosteroids. The clinical significance of this finding is unknown.
In rare cases, patients with asthma on zafirlukast may present with systemic eosinophilia, eosinophilic pneumonia, or clinical features of vasculitis consistent with Churg-Strauss syndrome, a condition which is often treated with systemic steroid therapy. Physicians should be alert to eosinophilia, vasculitic rash, worsening pulmonary symptoms, cardiac complications, and/or neuropathy presenting in their patients. These events have usually, but not always, been associated with the reduction and/or withdrawal of steroid therapy. The possibility that zafirlukast may be associated with emergence of Churg-Strauss syndrome can neither be excluded nor established (see PRECAUTIONS, Eosinophilic Conditions).
Neuropsychiatric adverse events, including insomnia and depression, have been reported in association with zafirlukast therapy, (see PRECAUTIONS, Neuropsychiatric Events), Hypersensitivity reactions, including urticaria, angioedema and rashes, with or without blistering, have been reported in association with zafirlukast therapy. Additionally, there have been reports of patients experiencing agranulocytosis, bleeding, bruising, or edema, arthralgia, myalgia, malaise, and pruritus in association with zafirlukast therapy.
Rare cases of patients experiencing increased theophylline levels with or without clinical signs or symptoms of theophylline toxicity after the addition of zafirlukast to an existing theophylline regimen have been reported. The mechanism of the interaction between zafirlukast and theophylline in these patients is unknown and not predicted by available in vitro metabolism data and the results of two clinical drug interaction studies (see CLINICAL PHARMACOLOGY and PRECAUTIONS, Drug Interactions).
Pediatric Patients 5 through 11 years of age
Zafirlukast has been evaluated for safety in 788 pediatric patients 5 through 11 years of age. Cumulatively, 313 pediatric patients were treated with zafirlukast 10 mg twice daily or higher for at least 6 months, and 113 of them were treated for one year or longer in clinical trials. The safety profile of zafirlukast 10 mg twice daily-versus placebo in the 4- and 6-week double-blind trials was generally similar to that observed in the adult clinical trials with zafirlukast 20 mg twice daily.
In pediatric patients receiving zafirlukast in multi-dose clinical trials, the following events occurred with a frequency of ≥ 2% and more frequently than in pediatric patients who received placebo, regardless of causality assessment: headache (4.5 vs. 4.2%) and abdominal pain (2.8 vs. 2.3%).
The post-marketing experience in this age group is similar to that seen in adults, including hepatic dysfunction, which may lead to liver failure.
-
OVERDOSAGE
No deaths occurred at oral zafirlukast doses of 2000 mg/kg in mice (approximately 210 times the maximum recommended daily oral dose in adults and children on a mg/m2 basis), 2000 mg/kg in rats (approximately 420 times the maximum recommended daily oral dose in adults and children on a mg/m2 basis), and 500 mg/kg in dogs (approximately 350 times the maximum recommended daily oral dose in adults and children on a mg/m2 basis).
Overdosage with zafirlukast has been reported in four patients surviving reported doses as high as 200 mg. The predominant symptoms reported following zafirlukast overdose were rash and upset stomach. There were no acute toxic effects in humans that could be consistently ascribed to the administration of zafirlukast. It is reasonable to employ the usual supportive measures in the event of an overdose; eg, remove unabsorbed material from the gastrointestinal tract, employ clinical monitoring, and institute supportive therapy, if required.
-
DOSAGE AND ADMINISTRATION
Because food can reduce the bioavailability of zafirlukast, zafirlukast tablets should be taken at least 1 hour before or 2 hours after meals.
Adults and Children 12 years of age and older
The recommended dose of zafirlukast tablets in adults and children 12 years and older is 20 mg twice daily.
Pediatric Patients 5 through 11 years of age
The recommended dose of zafirlukast tablets in children 5 through 11 years of age is 10 mg twice daily.
Elderly Patients: Based on cross-study comparisons, the clearance of zafirlukast is reduced in elderly patients (65 years of age and older), such that Cmax and AUC are approximately twice those of younger adults. In clinical trials, a dose of 20 mg twice daily was not associated with an increase in the overall incidence of adverse events or withdrawals because of adverse events in elderly patients.
Patients with Hepatic Impairment: The clearance of zafirlukast is reduced in patients with stable alcoholic cirrhosis such that the Cmax and AUC are approximately 50 - 60% greater than those of normal adults. Zafirlukast has not been evaluated in patients with hepatitis or in long-term studies of patients with cirrhosis.
Patients with Renal Impairment: Dosage adjustment is not required for patients with renal impairment.
- HOW SUPPLIED
-
PATIENT INFORMATION
ZAFIRLUKAST TABLETS
Read the Patient Information leaflet before you start taking zafirlukast tablets and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
What are zafirlukast tablets?
Zafirlukast tablets are a prescription medicine used to help prevent asthma attacks and for the long-term treatment of asthma symptoms in adults and children 5 years and older. It is not known if zafirlukast tablets are safe and effective when used in children under 5 years old.
The effect of zafirlukast tablets on growth in children has not been determined.
Do not take zafirlukast tablets if you need relief right away for a sudden asthma attack. If you get an asthma attack, you should follow the instructions your healthcare provider gave you for treating asthma attacks.
Who should not take zafirlukast tablets?
Do not take zafirlukast tablets if you are allergic to zafirlukast or any of the ingredients in zafirlukast tablets. See the end of this leaflet for a complete list of ingredients in zafirlukast tablets.
What should I tell my healthcare provider before taking zafirlukast tablets?
Before you take zafirlukast tablets, tell your healthcare provider if you:
- have liver problems
- have any other medical conditions
- are pregnant or plan to become pregnant. It is not known if zafirlukast tablets will harm your unborn baby. Talk to your healthcare provider if you are pregnant or plan to become pregnant.
- are breastfeeding or plan to breastfeed. Zafirlukast tablets can pass into your milk; it is not known whether zafirlukast tablets may harm your baby. Women who are breastfeeding should not take zafirlukast tablets.
Tell your healthcare provider about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements.
Zafirlukast tablets may affect the way other medicines work, and other medicines may affect how zafirlukast tablets works.
Especially tell your healthcare provider if you take:
- warfarin sodium (Coumadin, Jantoven)
- erythromycin (ERYC, ERY-TAB, PCE)
- theophylline (Elixophyllin, Theo-24, Theochron, Theolair, Uniphyl)
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.
How should I take zafirlukast tablets?
- Take zafirlukast tablets exactly as your healthcare provider tells you to take it.
- Take zafirlukast tablets regularly, even if you do not have asthma symptoms. Do not change your dose or stop taking zafirlukast tablets without talking to your healthcare provider.
- Do not stop taking or change the dose of your other asthma medicines unless your healthcare provider tells you to.
- Take your prescribed dose of zafirlukast tablets by mouth at least 1 hour before or 2 hours after meals.
- Zafirlukast tablets does not treat the symptoms of a sudden asthma attack. Always have a short-acting beta2-agonist medicine (rescue inhaler) with you to treat sudden symptoms. If you do not have a rescue inhaler medicine, talk to your healthcare provider to have one prescribed for you.
- If you take too much zafirlukast tablets, call your healthcare provider or go to the nearest hospital emergency room right away.
What are the possible side effects of zafirlukast tablets?
Zafirlukast tablets may cause serious side effects, including:
- Severe liver problems. In some cases, these liver problems can lead to liver failure, the need for a liver transplant or death. Tell your healthcare provider right away if you have:
- pain or tenderness in the right upper side of your stomach area (abdomen)
- nausea
- tiredness
- itchiness
- yellowing of your skin or the whites of your eyes
- flu-like symptoms
- loss of appetite
- dark (tea colored) urine
- Inflammation of your blood vessels. Rarely, this can happen in people with asthma who take zafirlukast tablets. This usually, but not always, happens in people who also take a steroid medicine by mouth that is being stopped or the dose is being lowered. Tell your healthcare provider right away if you have:
- a feeling of pins and needles or numbness of your arms or legs
- flu like symptoms
- rash
- pain and swelling of your sinuses
- Changes in behaviour or mood. Tell your healthcare provider if you have changes in your behaviour, problems sleeping or feel very sad.
- Hypersensitivity reactions. Tell your healthcare provider if you have severe itching, breathing problems, skin rash, skin blisters, or skin redness, or swelling.
The most common side effects of zafirlukast tablets in people 12 years and older include:
- headache
- infection
- nausea
- diarrhea
- pain (generalized)
The most common side effects of zafirlukast tablets in children 5 to 11 years include:
- headache
- stomach pain
Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all of the possible side effects of zafirlukast tablets. For more information, ask your healthcare provider or pharmacist. Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store zafirlukast tablets?
- Store zafirlukast tablets at 68°F to 77°F (20°C -25°C ).
- Keep zafirlukast tablets dry.
- Keep zafirlukast tablets in a tight closed container and keep zafirlukast tablets out of the light.
- Keep zafirlukast tablets and all medicines out of the reach of children.
General information about the safe and effective use of zafirlukast tablets.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use zafirlukast tablets for a condition for which it was not prescribed. Do not give zafirlukast tablets to other people, even if they have the same symptoms that you have. It may harm them.
This Patient Information leaflet summarizes the most important information about zafirlukast tablets. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about zafirlukast tablets that is written for healthcare professionals. For more information, call 1-888-375-3784.
What are the ingredients in zafirlukast tablets?
Active ingredient: zafirlukast
Inactive ingredients: hydroxypropyl cellulose, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polyethylene glycol 400, sodium starch glycolate (Type-A) and titanium dioxide.
What do zafirlukast tablets look like?
- the 10 mg tablet is white to light pink, round, film coated, debossed with ‘R’ on one side and ‘625’ on other side.
- the 20 mg tablet is white to light pink, round film coated, debossed with ‘R’ on one side and ‘626’ on other side.
Rx Only
Manufactured by:Dr. Reddy’s Laboratories Limited
Bachepalli - 502 325 INDIA
Issued: 1010
- SPL UNCLASSIFIED SECTION
- SPL UNCLASSIFIED SECTION
- 20 mg Labels
- SPL UNCLASSIFIED SECTION
- SPL UNCLASSIFIED SECTION
- Zafirlukast 20 mg tabs
-
INGREDIENTS AND APPEARANCE
ZAFIRLUKAST
zafirlukast tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68151-1977(NDC:55111-626) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ZAFIRLUKAST (UNII: XZ629S5L50) (ZAFIRLUKAST - UNII:XZ629S5L50) ZAFIRLUKAST 20 mg Inactive Ingredients Ingredient Name Strength HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) HYPROMELLOSE 2910 (5 MPA.S) (UNII: R75537T0T4) lactose monohydrate (UNII: EWQ57Q8I5X) magnesium stearate (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) polyethylene glycol 400 (UNII: B697894SGQ) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) titanium dioxide (UNII: 15FIX9V2JP) Product Characteristics Color WHITE Score no score Shape ROUND Size 8mm Flavor Imprint Code R;626 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68151-1977-6 1 in 1 PACKAGE; Type 0: Not a Combination Product 11/18/2010 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090372 11/18/2010 Labeler - Carilion Materials Management (079239644) Establishment Name Address ID/FEI Business Operations Carilion Materials Management 079239644 REPACK(68151-1977)




© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.