GADOTERATE MEGLUMINE injection
Gadoterate Meglumine by
Drug Labeling and Warnings
Gadoterate Meglumine by is a Prescription medication manufactured, distributed, or labeled by Fresenius Kabi USA, LLC, Jiangsu Hengrui Pharmaceuticals Co., Ltd., Jiangsu Hengrui Pharmaceuticals Co., Ltd. (Dongjin Road Site). Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use GADOTERATE MEGLUMINE INJECTION safely and effectively. See full prescribing information for GADOTERATE MEGLUMINE INJECTION.
GADOTERATE MEGLUMINE injection for intravenous use
PHARMACY BULK PACKAGE
NOT FOR DIRECT INFUSION
Initial U.S. Approval: 2013WARNING: RISK ASSOCIATED WITH INTRATHECAL USE and NEPHROGENIC SYSTEMIC FIBROSIS
See full prescribing information for complete boxed warning
- Intrathecal administration of gadolinium-based contrast agents (GBCAs) can cause serious adverse reactions including death, coma, encephalopathy, and seizures. Gadoterate Meglumine Injection is not approved for intrathecal use. (5.1)
-
GBCAs increase the risk for nephrogenic systemic fibrosis (NSF) among patients with impaired elimination of the drugs. Avoid use of Gadoterate Meglumine Injection in these patients unless the diagnostic information is essential and not available with non-contrasted MRI or other modalities. The risk for NSF appears highest among patients with:
- o Chronic, severe kidney disease (GFR < 30 mL/min/1.73 m2), or
- o Acute kidney injury.
Screen patients for acute kidney injury and other conditions that may reduce renal function. For patients at risk for chronically reduced renal function (for example, age > 60 years, hypertension or diabetes), estimate the glomerular filtration rate (GFR) through laboratory testing (5.2).
INDICATIONS AND USAGE
Gadoterate Meglumine Injection is a gadolinium-based contrast agent indicated for intravenous use with magnetic resonance imaging (MRI) in brain (intracranial), spine and associated tissues in adult and pediatric patients (including term neonates) to detect and visualize areas with disruption of the blood brain barrier (BBB) and/or abnormal vascularity. (1)
DOSAGE AND ADMINISTRATION
Adult and pediatric patients: The recommended dose of Gadoterate Meglumine Injection is 0.2 mL/kg (0.1 mmol/kg) body weight administered as an intravenous bolus injection at a flow rate of approximately 2 mL/second for adults and 1 to 2 mL/second for pediatric patients (including term neonates). The dose is delivered by manual or power injection. (2)
DOSAGE FORMS AND STRENGTHS
Gadoterate Meglumine Injection 0.5 mmol per mL contains 376.9 mg per mL of gadoterate meglumine. Gadoterate Meglumine Injection Pharmacy Bulk Package is available in vials. (3)
CONTRAINDICATIONS
Clinically important hypersensitivity reactions to Gadoterate Meglumine Injection. (4)
WARNINGS AND PRECAUTIONS
- Hypersensitivity Reactions: Anaphylactoid/anaphylactic reactions with cardiovascular, respiratory or cutaneous manifestations, ranging from mild to severe, including death, have uncommonly occurred. Monitor patients closely for need of emergency cardiorespiratory support. (5.3)
- Gadolinium Retention: Gadolinium is retained for months or years in brain, bone, and other organs. (5.4)
ADVERSE REACTIONS
The most frequent (≥ 0.2%) adverse reactions in clinical studies were nausea, headache, injection site pain, injection site coldness, and rash. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Fresenius Kabi USA, LLC at 1-800-551-7176 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
Pregnancy: Use only if imaging is essential during pregnancy and cannot be delayed. (8.1)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 5/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: RISK ASSOCIATED WITH INTRATHECAL USE and NEPHROGENIC SYSTEMIC FIBROSIS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Guidelines
2.2 Drug Handling
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Risk Associated with Intrathecal Use
5.2 Nephrogenic Systemic Fibrosis
5.3 Hypersensitivity Reactions
5.4 Gadolinium Retention
5.5 Acute Kidney Injury
5.6 Extravasation and Injection Site Reactions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
17.1 Nephrogenic Systemic Fibrosis
17.2 Common Adverse Reactions
17.3 General Precautions
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: RISK ASSOCIATED WITH INTRATHECAL USE and NEPHROGENIC SYSTEMIC FIBROSIS
Risk Associated with Intrathecal Use
Intrathecal administration of gadolinium-based contrast agents (GBCAs) can cause serious adverse reactions including death, coma, encephalopathy, and seizures. Gadoterate Meglumine Injection is not approved for intrathecal use [see Warnings and Precautions (5.1)].
Nephrogenic Systemic Fibrosis
GBCAs increase the risk for nephrogenic systemic fibrosis (NSF) among patients with impaired elimination of the drugs. Avoid use of Gadoterate Meglumine Injection in these patients unless the diagnostic information is essential and not available with non-contrasted MRI or other modalities. NSF may result in fatal or debilitating fibrosis affecting the skin, muscle and internal organs.
-
The risk for NSF appears highest among patients with:
- - Chronic, severe kidney disease (GFR < 30 mL/min/1.73 m2), or
- - Acute kidney injury.
- Screen patients for acute kidney injury and other conditions that may reduce renal function. For patients at risk for chronically reduced renal function (e.g. age> 60 years, hypertension, diabetes), estimate the glomerular filtration rate (GFR) through laboratory testing (5.1).
- For patients at highest risk for NSF, do not exceed the recommended Gadoterate Meglumine Injection dose and allow a sufficient period of time for elimination of the drug from the body prior to any re-administration [see Warnings and Precautions (5.2)].
-
The risk for NSF appears highest among patients with:
-
1 INDICATIONS AND USAGE
Gadoterate Meglumine Injection is a gadolinium-based contrast agent indicated for intravenous use with magnetic resonance imaging (MRI) in brain (intracranial), spine and associated tissues in adult and pediatric patients (including term neonates) to detect and visualize areas with disruption of the blood brain barrier (BBB) and/or abnormal vascularity.
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Guidelines
For adult and pediatric patients (including term neonates), the recommended dose of Gadoterate Meglumine Injection is 0.2 mL/kg (0.1 mmol/kg) body weight administered as an intravenous bolus injection, manually or by power injector, at a flow rate of approximately 2 mL/second for adults and 1 to 2 mL/second for pediatric patients. Table 1 provides weight-adjusted dose volumes.
Table 1: Volumes of Gadoterate Meglumine Injection by Body Weight Body Weight
Volume
Pounds (lb)
Kilograms (kg)
Milliliters (mL)
5.5
2.5
0.5
11
5
1
22
10
2
44
20
4
66
30
6
88
40
8
110
50
10
132
60
12
154
70
14
176
80
16
198
90
18
220
100
20
242
110
22
264
120
24
286
130
26
308
140
28
330
150
30
To ensure complete injection of Gadoterate Meglumine Injection the injection may be followed by normal saline flush. Contrast MRI can begin immediately following injection of Gadoterate Meglumine Injection.
2.2 Drug Handling
- Visually inspect Gadoterate Meglumine Injection for particulate matter prior to administration. Do not use the solution if particulate matter is present or if the container appears damaged. Gadoterate Meglumine Injection should be a clear, colorless to yellow solution.
- Do not mix with other drugs or parenteral nutrition.
- Discard any unused portions of the drug.
- When Gadoterate Meglumine Injection is to be injected using plastic disposable syringes, the contrast medium should be drawn into the syringe and used immediately.
Pharmacy Bulk Package Preparation:
- Do not use the Pharmacy Bulk Package for direct intravenous infusion.
- Perform the transfer of Gadoterate Meglumine Injection from the Pharmacy Bulk Package in an aseptic work area, such as laminar flow hood and using aseptic technique and suitable transfer device. Penetrate the closure only one time.
- Once the container closure is punctured, do not remove the Pharmacy Bulk Package from the aseptic work area.
- The Pharmacy Bulk Package is used as a multiple dose container with an appropriate transfer device for filling empty sterile syringes.
- Use each individual dose of Gadoterate Meglumine Injection promptly following withdrawal from the Pharmacy Bulk Package.
- Use the contents of the Pharmacy Bulk Package within 24 hours after initial puncture.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
History of clinically important hypersensitivity reactions to Gadoterate Meglumine Injection [see Warnings and Precautions (5.3)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Risk Associated with Intrathecal Use
Intrathecal administration of GBCAs can cause serious adverse reactions including death, coma, encephalopathy, and seizures. The safety and effectiveness of Gadoterate Meglumine Injection have not been established with intrathecal use. Gadoterate Meglumine Injection is not approved for intrathecal use [see Dosage and Administration (2.1)].
5.2 Nephrogenic Systemic Fibrosis
GBCAs increase the risk for nephrogenic systemic fibrosis (NSF) among patients with impaired elimination of the drugs. Avoid use of Gadoterate Meglumine Injection among these patients unless the diagnostic information is essential and not available with non-contrast MRI or other modalities. The GBCA-associated NSF risk appears highest for patients with chronic, severe kidney disease (GFR < 30 mL/min/1.73 m2) as well as patients with acute kidney injury. The risk appears lower for patients with chronic, moderate kidney disease (GFR 30 to 59 mL/min/1.73 m2) and little, if any, for patients with chronic, mild kidney disease (GFR 60 to 89 mL/min/1.73 m2). NSF may result in fatal or debilitating fibrosis affecting the skin, muscle and internal organs. Report any diagnosis of NSF following Gadoterate Meglumine Injection administration to Fresenius Kabi USA, LLC at 1-800-551-7176 or FDA (1-800-FDA-1088 or www.fda.gov/medwatch).
Screen patients for acute kidney injury and other conditions that may reduce renal function. Features of acute kidney injury consist of rapid (over hours to days) and usually reversible decrease in kidney function, commonly in the setting of surgery, severe infection, injury or drug-induced kidney toxicity. Serum creatinine levels and estimated GFR may not reliably assess renal function in the setting of acute kidney injury. For patients at risk for chronically reduced renal function (e.g., age > 60 years, diabetes mellitus or chronic hypertension), estimate the GFR through laboratory testing.
The factors that may increase the risk for NSF are repeated or higher than recommended doses of a GBCA and the degree of renal impairment at the time of exposure. Record the specific GBCA and the dose administered to a patient. For patients at highest risk for NSF, do not exceed the recommended Gadoterate Meglumine Injection dose and allow a sufficient period of time for elimination of the drug prior to re-administration. For patients receiving hemodialysis, physicians may consider the prompt initiation of hemodialysis following the administration of a GBCA in order to enhance the contrast agent’s elimination. The usefulness of hemodialysis in the prevention of NSF is unknown [see Dosage and Administration (2) and Clinical Pharmacology (12)].
5.3 Hypersensitivity Reactions
Anaphylactic and anaphylactoid reactions have been reported with gadoterate meglumine, involving cardiovascular, respiratory, and/or cutaneous manifestations. Some patients experienced circulatory collapse and died. In most cases, initial symptoms occurred within minutes of gadoterate meglumine administration and resolved with prompt emergency treatment [see Adverse Reactions (6)].
- Before Gadoterate Meglumine Injection administration, assess all patients for any history of a reaction to contrast media, bronchial asthma and/or allergic disorders. These patients may have an increased risk for a hypersensitivity reaction to Gadoterate Meglumine Injection.
- Administer Gadoterate Meglumine Injection only in situations where trained personnel and therapies are promptly available for the treatment of hypersensitivity reactions, including personnel trained in resuscitation.
- During and following Gadoterate Meglumine Injection administration, observe patients for signs and symptoms of hypersensitivity reactions.
5.4 Gadolinium Retention
Gadolinium is retained for months or years in several organs. The highest concentrations (nanomoles per gram of tissue) have been identified in the bone, followed by other organs (e.g. brain, skin, kidney, liver and spleen). The duration of retention also varies by tissue and is longest in bone. Linear GBCAs cause more retention than macrocyclic GBCAs. At equivalent doses, gadolinium retention varies among the linear agents with Omniscan (gadodiamide) and Optimark (gadoversetamide) causing greater retention than other linear agents [Eovist (gadoxetate disodium), Magnevist (gadopentetate dimeglumine), MultiHance (gadobenate dimeglumine)]. Retention is lowest and similar among the macrocyclic GBCAs [Gadoterate Meglumine Injection (gadoterate meglumine), Gadavist (gadobutrol), ProHance (gadoteridol)].
Consequences of gadolinium retention in the brain have not been established. Pathologic and clinical consequences of GBCA administration and retention in skin and other organs have been established in patients with impaired renal function [see Warnings and Precautions (5.2)]. There are rare reports of pathologic skin changes in patients with normal renal function. Adverse events involving multiple organ systems have been reported in patients with normal renal function without an established causal link to gadolinium retention [see Adverse Reactions (6.2)].
While clinical consequences of gadolinium retention have not been established in patients with normal renal function, certain patients might be at higher risk. These include patients requiring multiple lifetime doses, pregnant and pediatric patients, and patients with inflammatory conditions. Consider the retention characteristics of the agent when choosing a GBCA for these patients. Minimize repetitive GBCA imaging studies, particularly closely spaced studies when possible.
5.5 Acute Kidney Injury
In patients with chronically reduced renal function, acute kidney injury requiring dialysis has occurred with the use of GBCAs. The risk of acute kidney injury may increase with increasing dose of the contrast agent; administer the lowest dose necessary for adequate imaging. Screen all patients for renal impairment by obtaining a history and/or laboratory tests. Consider follow-up renal function assessments for patients with a history of renal dysfunction.
5.6 Extravasation and Injection Site Reactions
Ensure catheter and venous patency before the injection of Gadoterate Meglumine Injection. Extravasation into tissues during Gadoterate Meglumine Injection administration may result in tissue irritation [see Nonclinical Toxicology (13.2)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
Nephrogenic systemic fibrosis [see Warnings and Precautions (5.2)].
Hypersensitivity reactions [see Warnings and Precautions (5.3)].
Gadolinium Retention [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The data described below reflect gadoterate meglumine exposure in 2,867 patients, representing 2,682 adults and 185 pediatric patients. Overall, 55% of the patients were men. In clinical trials where ethnicity was recorded, the ethnic distribution was 81% Caucasian, 11% Asian, 4% Black, and 4% others. The average age was 53 years (range from < 1 week to 97 years).
Overall, 4% of patients reported at least one adverse reaction, primarily occurring immediately or within 24 hours following gadoterate meglumine administration. Most adverse reactions were mild or moderate in severity and transient in nature.
Table 2 lists adverse reactions that occurred in ≥ 0.2% patients who received gadoterate meglumine.
Table 2: Adverse Reactions in Clinical Trials Reaction
Rate (%)
n=2,867
Nausea
0.6%
Headache
0.4%
Injection Site Pain
0.4%
Injection Site Coldness
0.2%
Rash
0.2%
Adverse reactions that occurred with a frequency < 0.2% in patients who received gadoterate meglumine include: feeling cold, feeling hot, burning sensation, somnolence, pain, dizziness, dysgeusia, blood creatinine increased, hypotension, hypertension, asthenia, fatigue, injection site reactions (inflammation, extravasation, pruritus, swelling, warmth), paresthesia, pruritus, laryngeal discomfort, pain in extremity, vomiting, anxiety and palpitations.
Adverse Reactions in Pediatric Patients
During clinical trials, 185 pediatric patients (52 aged < 24 months, 33 aged 2 to 5 years, 57 aged 6 to 11 years and 43 aged 12 to 17 years) received gadoterate meglumine. Overall, 7 pediatric patients (3.8%) reported at least one adverse reaction following gadoterate meglumine administration. The most frequently reported adverse reaction was headache (1.1%). Most adverse events were mild in intensity and transient in nature.
6.2 Postmarketing Experience
The following additional adverse reactions have been identified during postmarketing use of gadoterate meglumine or other GBCAs. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Table 3: Adverse Reactions in the Postmarketing Experience System Organ Class Adverse Reaction Cardiac Disorders
bradycardia, tachycardia, arrhythmia
Immune System Disorders
hypersensitivity /anaphylactoid reactions including cardiac arrest, respiratory arrest, cyanosis, pharyngeal edema, laryngospasm, bronchospasm, angioedema, conjunctivitis, ocular hyperemia, eyelid edema, lacrimation increased, hyperhidrosis, urticaria
Nervous System Disorders
coma, convulsion, syncope, presyncope, parosmia, tremor
Musculoskeletal and Connective Tissue Disorders
muscle contracture, muscle weakness
Gastrointestinal Disorders
diarrhea, salivary hypersecretion, acute pancreatitis with onset within 48 hours after GBCA administration
General Disorders and Administration Site Conditions
malaise, fever
Adverse reactions with variable onset and duration have been reported after GBCA administration. These include fatigue, asthenia, pain syndromes, and heterogeneous clusters of symptoms in the neurological, cutaneous, and musculoskeletal systems.
Respiratory, Thoracic and Mediastinal Disorders
Acute respiratory distress syndrome, pulmonary edema
Skin and Subcutaneous Tissue Disorders
NSF, in patients whose reports were confounded by the receipt of other GBCAs or in situations where receipt of other GBCAs could not be ruled out.
No unconfounded cases of NSF have been reported with gadoterate meglumine.
Gadolinium-associated plaques
Vascular Disorders
superficial phlebitis
- 7 DRUG INTERACTIONS
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
GBCAs cross the human placenta and result in fetal exposure and gadolinium retention. The human data on the association between GBCAs and adverse fetal outcomes are limited and inconclusive (see Data). In animal reproduction studies, there were no adverse developmental effects observed in rats or rabbits with intravenous administration of gadoterate meglumine during organogenesis at doses up to 16 and 10 times, respectively, the recommended human dose (see Data). Because of the potential risks of gadolinium to the fetus, use Gadoterate Meglumine Injection only if imaging is essential during pregnancy and cannot be delayed.
The estimated background risk of major birth defects and miscarriage for the indicated population(s) are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20% respectively.
Data
Risk Summary
GBCAs cross the human placenta and result in fetal exposure and gadolinium retention. The human data on the association between GBCAs and adverse fetal outcomes are limited and inconclusive (see Data). In animal reproduction studies, there were no adverse developmental effects observed in rats or rabbits with intravenous administration of gadoterate meglumine during organogenesis at doses up to 16 and 10 times, respectively, the recommended human dose (see Data). Because of the potential risks of gadolinium to the fetus, use Gadoterate Meglumine Injection only if imaging is essential during pregnancy and cannot be delayed.
The estimated background risk of major birth defects and miscarriage for the indicated population(s) are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20% respectively.
Human Data
Contrast enhancement is visualized in the placenta and fetal tissues after maternal GBCA administration.
Cohort studies and case reports on exposure to GBCAs during pregnancy have not reported a clear association between GBCAs and adverse effects in the exposed neonates. However, a retrospective cohort study, comparing pregnant women who had a GBCA MRI to pregnant women who did not have an MRI, reported a higher occurrence of stillbirths and neonatal deaths in the group receiving GBCA MRI. Limitations of this study include a lack of comparison with non-contrast MRI and lack of information about the material indication for MRI. Overall, these data preclude a reliable evaluation of the potential risk of adverse fetal outcomes with the use of GBCAs in pregnancy.
Animal Data
Gadolinium Retention
GBCAs administered to pregnant non-human primates (0.1 mmol/kg on gestational days 85 and 135) result in measurable gadolinium concentration in the offspring in bone, brain, skin, liver, kidney, and spleen for at least 7 months. GBCAs administered to pregnant mice (2 mmol/kg daily on gestational days 16 through 19) result in measurable gadolinium concentrations in the pups in bone, brain, kidney, liver, blood, muscle, and spleen at one month postnatal age.
Reproductive Toxicology
Gadoterate meglumine was administered in intravenous doses of 0, 2, 4 and 10 mmol/kg/day [3, 7 and 16 times the recommended human dose (RHD) based on body surface area (BSA)] to female rats for 14 days before mating, throughout the mating period and until gestation day (GD) 17. Pregnant rabbits were administered gadoterate meglumine in intravenous doses of 0, 1, 3 and 7 mmol/kg/day ( 3, 10 and 23 times the RHD based on BSA) from GD6 to GD19. No effects on embryo-fetal development were observed at doses up to 10 mmol/kg/day in rats and 3 mmol/kg/day in rabbits. Maternal toxicity was observed in rats at 10 mmol/kg/day and in rabbits at 7 mmol/kg/day. This maternal toxicity was characterized in rats by a slightly lower litter size and gravid uterus weight compared to the control group, and in rabbits by a reduction in body weight and food consumption.
8.2 Lactation
Risk Summary
There are no data on the presence of gadoterate in human milk, the effects on the breastfed infant, or the effects on milk production. However, published lactation data on other GBCAs indicate that 0.01 to 0.04% of the maternal gadolinium dose is present in breast milk. Additionally, there is limited GBCA gastrointestinal absorption in the breast-fed infant. Gadoterate is present in goat milk (see Data). The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Gadoterate Meglumine Injection and any potential adverse effects on the breastfed infant from Gadoterate Meglumine Injection or from the underlying maternal condition.8.4 Pediatric Use
The safety and efficacy of gadoterate meglumine at a single dose of 0.1 mmol/kg have been established in pediatric patients from birth (term neonates ≥ 37 weeks gestational age) to 17 years of age based on clinical data in 133 pediatric patients 2 years of age and older, and clinical data in 52 pediatric patients birth to less than 2 years of age that supported extrapolation from adult data [see Clinical Studies (14)]. Adverse reactions in pediatric patients were similar to those reported in adults [see Adverse Reactions (6.1)]. No dosage adjustment according to age is necessary in pediatric patients [see Dosage and Administration (2.1), Pharmacokinetics (12.3)]. The safety of gadoterate meglumine has not been established in preterm neonates.
No cases of NSF associated with gadoterate meglumine or any other GBCA have been identified in pediatric patients age 6 years and younger [see Warnings and Precautions (5.2)]. Normal estimated GFR (eGFR) is approximately 30 mL/minute/1.73m2 at birth and increases to adult values by 2 years of age.
Juvenile Animal Data
Single and repeat-dose toxicity studies in neonatal and juvenile rats did not reveal findings suggestive of a specific risk for use in pediatric patients including term neonates and infants.
8.5 Geriatric Use
In clinical studies of gadoterate meglumine, 900 patients were 65 years of age and over, and 304 patients were 75 years of age and over. No overall differences in safety or efficacy were observed between these subjects and younger subjects. In general, use of Gadoterate Meglumine Injection in elderly patients should be cautious, reflecting the greater frequency of impaired renal function and concomitant disease or other drug therapy. No age-related dosage adjustment is necessary.
-
10 OVERDOSAGE
Gadoterate Meglumine Injection administered to healthy volunteers and to adult patients at cumulative doses up to 0.3 mmol/kg was tolerated in a manner similar to lower doses. Adverse reactions to overdosage with gadoterate meglumine have not been reported. Gadoterate meglumine can be removed from the body by hemodialysis [See Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
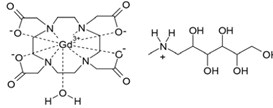
Gadoterate Meglumine Injection, USP is a paramagnetic macrocyclic ionic contrast agent administered for magnetic resonance imaging. The chemical name for gadoterate meglumine is D-glucitol, 1-deoxy-1-(methylamino)-,[1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraaceto(4-)-.kappa.N1, .kappa.N4, .kappa.N7, .kappa.N10, .kappa.O1, .kappa.O4, .kappa.O7, .kappa.O10]gadolinate(1-)(1:1); it has a formula weight of 753.9 g/mol and empirical formula of C23H42O13N5Gd (anhydrous basis).
The structural formula of gadoterate meglumine in solution is as follows:
CAS Registry No. 92943-93-6
Gadoterate Meglumine Injection, USP is a sterile, nonpyrogenic, clear, colorless to yellow, aqueous solution of 0.5 mmol per mL of gadoterate meglumine. No preservative is added. Each mL of Gadoterate Meglumine Injection, USP contains 376.9 mg of gadoterate meglumine, 0.25 mg of DOTA and water for injection. Gadoterate Meglumine Injection, USP has a pH of 6.5 to 8.0.
The main physiochemical properties of Gadoterate Meglumine Injection, USP are provided below:
Table 4: Physicochemical Properties Parameter
Value
Density @ 20°C
1.1753 g/cm3
Viscosity @ 20°C
3.4 mPa·s
Viscosity @ 37°C
2.4 mPa·s
Osmolality
1,350 mOsm/kg water
The thermodynamic stability constants for gadoterate (log Ktherm and log Kcond at pH 7.4) are 25.6 and 19.3, respectively.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Gadoterate is a paramagnetic molecule that develops a magnetic moment when placed in a magnetic field. The magnetic moment enhances the relaxation rates of water protons in its vicinity, leading to an increase in signal intensity (brightness) of tissues.
In magnetic resonance imaging (MRI), visualization of normal and pathological tissue depends in part on variations in the radiofrequency signal intensity that occurs with:
- 1. differences in proton density
- 2. differences of the spin-lattice or longitudinal relaxation times (T1)
- 3. differences in the spin-spin or transverse relaxation time (T2)
When placed in a magnetic field, gadoterate shortens the T1 and T2 relaxation times in target tissues. At recommended doses, the effect is observed with greatest sensitivity in the T1-weighted sequences.
12.2 Pharmacodynamics
Gadoterate affects proton relaxation times and consequently the MR signal, and the contrast obtained is characterized by the relaxivity of the gadoterate molecule. The relaxivity values for gadoterate are similar across the spectrum of magnetic field strengths used in clinical MRI (0.2 to 1.5 T).
Disruption of the blood-brain barrier or abnormal vascularity allows distribution of gadoterate in lesions such as neoplasms, abscesses, and infarcts.
12.3 Pharmacokinetics
The pharmacokinetics of total gadolinium assessed up to 48 hours following an intravenously administered 0.1 mmol/kg dose of gadoterate meglumine in healthy adult subjects demonstrated a mean elimination half-life (reported as mean ± SD) of about 1.4 ± 0.2 hr and 2.0 ± 0.7 hr in female and male subjects, respectively. Similar pharmacokinetic profile and elimination half-life values were observed after intravenous injection of 0.1 mmol/kg of gadoterate meglumine followed 20 minutes later by a second injection of 0.2 mmol/kg (1.7 ± 0.3 hr and 1.9 ± 0.2 hr in female and male subjects, respectively).
Distribution
The volume of distribution at steady state of total gadolinium in healthy subjects is 179 ± 26 and 211 ± 35 mL/kg in female and male subjects respectively, roughly equivalent to that of extracellular water. Gadoterate does not undergo protein binding in vitro. The extent of blood cell partitioning of gadoterate is not known.
Following GBCA administration, gadolinium is present for months or years in brain, bone, skin and other organs [see Warnings and Precautions (5.4)].
Metabolism
Gadoterate is not known to be metabolized.
Elimination
Following a 0.1 mmol/kg dose of gadoterate meglumine, total gadolinium is excreted primarily in the urine with 72.9 ± 17.0 % and 85.4 ± 9.7 % (mean ± SD) eliminated within 48 hours, in female and male subjects, respectively. Similar values were achieved after a cumulative dose of 0.3 mmol/kg (0.1 + 0.2 mmol/kg, 20 minutes later), with 85.5 ± 13.2 % and 92.0 ± 12.0 % recovered in urine within 48 hrs in female and male subjects respectively.
In healthy subjects, the renal and total clearance rates of total gadolinium are comparable (1.27 ± 0.32 and 1.74 ± 0.12 mL/min/kg in females; and 1.40 ± 0.31 and 1.64 ± 0.35 mL/min/kg in males, respectively) indicating that the drug is primarily cleared through the kidneys. Within the studied dose range (0.1 to 0.3 mmol/kg), the kinetics of total gadolinium appear to be linear.
Specific Populations
Renal Impairment
A single intravenous dose of 0.1 mmol/kg of gadoterate meglumine was administered to 8 patients (5 men and 3 women) with impaired renal function (mean serum creatinine of 498 ± 98 µmol/L in the 10 to 30 mL/min creatinine clearance group and 192 ± 62 µmol/L in the 30 to 60 mL/min creatinine clearance group). Renal impairment delayed the elimination of total gadolinium. Total clearance decreased as a function of the degree of renal impairment. The distribution volume was unaffected by the severity of renal impairment (Table 5). No changes in renal function test parameters were observed after gadoterate meglumine injection. The mean cumulative urinary excretion of total gadolinium was approximately 76.9 ± 4.5% in 48 hrs in patients with moderate renal impairment, 68.4 ± 3.5% in 72 hrs in patients with severe renal impairment and 93.3 ± 4.7% in 24 hrs for subjects with normal renal function.
Table 5: Pharmacokinetic Profile of Total Gadolinium in Normal and Renally Impaired Patients Population
Elimination Half-life
(hr)
Plasma Clearance
(L/h/kg)
Distribution Volume
(L/kg)
Healthy volunteers
1.6 ± 0.2
0.10 ± 0.01
0.246 ± 0.03
Patients with moderate renal impairment
5.1 ± 1.0
0.036 ± 0.007
0.236 ± 0.01
Patients with severe renal impairment
13.9 ± 1.2
0.012 ± 0.001
0.234 ± 0.01
Gadoterate was shown to be dialyzable after an IV injection of gadoterate meglumine in 10 patients with end-stage renal failure who required hemodialysis treatment. Gd serum concentration decreased over time by 88%, 93% and 97% at 0.5 hr, 1.5 hr, and 4 hrs after start of dialysis, respectively. A second and third hemodialysis session further removed Gd. After the third dialysis, Gd serum concentration decreased by 99.7%.
Pediatric population
The pharmacokinetics of gadoterate in pediatric patients receiving gadoterate meglumine aged birth (term neonates) to 23 months, was investigated in an open label, multicenter study, using a population pharmacokinetics approach. A total of 45 subjects (22 males, 23 females) received a single intravenous dose of gadoterate meglumine 0.1 mmol/kg (0.2 mL/kg). The age ranged from less than one week to 23.8 months (mean 9.9 months) and body weight ranged from 3 to 15 kg (mean 8.1 kg). Individual level of renal maturity in the study population, as expressed by eGFR ranged between 52 and 281 mL/min/1.73 m2 and 11 patients had an eGFR below 100 mL/min/1.73 m2 (range 52 to 95 mL/min/1.73 m2).Gadoterate concentrations obtained up to 8 hours after gadoterate meglumine administration were best fitted using a biphasic model with linear elimination from the intravascular space. The mean clearance adjusted to body weight was estimated at 0.16 ± 0.07 L/h/kg and increased with eGFR. The estimated mean elimination half-life was 1.47 ± 0.45 hr.
The body weight adjusted clearance of gadoterate after single intravenous injection of 0.1 mmol/kg of gadoterate meglumine in pediatric subjects aged less than 2 years was similar to that observed in healthy adults.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal studies have not been performed to evaluate the carcinogenic potential of gadoterate meglumine.
Gadoterate meglumine did not demonstrate mutagenic potential in in vitro bacterial reverse mutation assays (Ames test) using Salmonella typhimurium, in an in vitro chromosome aberration assay in Chinese hamster ovary cells, in an in vitro gene mutation assay in Chinese hamster lung cells, nor in an in vivo mouse micronucleus assay.
No impairment of male or female fertility and reproductive performance was observed in rats after intravenous administration of gadoterate meglumine at the maximum tested dose of 10 mmol/kg/day (16 times the maximum human dose based on surface area), given during more than 9 weeks in males and more than 4 weeks in females. Sperm counts and sperm motility were not adversely affected by treatment with the drug.
13.2 Animal Toxicology and/or Pharmacology
Local intolerance reactions, including moderate irritation associated with infiltration of inflammatory cells were observed after perivenous injection in rabbits suggesting the possibility of local irritation if the contrast medium leaks around the veins in a clinical setting [see Warnings and Precautions (5.5)].
Toxicity of gadoterate meglumine was evaluated in neonatal and juvenile (pre- and post-weaning) rats following a single or repeated intravenous administration at doses 1, 2, and 4 times the MHD based on BSA. Gadoterate meglumine was well tolerated at all dose levels tested and had no effect on growth, pre-weaning development, behavior and sexual maturation.
-
14 CLINICAL STUDIES
CNS Imaging
Efficacy and safety of gadoterate meglumine were evaluated in a multi-center clinical trial (Study A) that enrolled 364 adult and 38 pediatric patients (aged ≥ 2 years) with known or suspected CNS lesions. Adults were randomized 2 to 1 to receive either gadoterate meglumine or gadopentetate dimeglumine, each administered at a dose of 0.1 mmol/kg. All pediatric patients received gadoterate meglumine, also at a dose of 0.1 mmol/kg. In the trial, patients first underwent a baseline (pre-contrast) MRI examination followed by the assigned GBCA administration and a post-contrast MR examination. The images (pre-contrast, post-contrast and “paired pre- and post-contrast”) were interpreted by three independent off-site readers blinded to clinical information. The primary efficacy analysis compared three patient-level visualization scores (paired images) to baseline MRI (pre-contrast images) for adults who received gadoterate meglumine. The three primary visualization components were: contrast enhancement, border delineation and internal morphology. For each of these components there was a pre-defined scoring scale. Lesion counting (up to five per patient) was also reflected within each component’s patient-level visualization score.
Among the adult patients, 245 received gadoterate meglumine and their data comprised the primary efficacy population. There were 114 (47%) men and 131 (53%) women with a mean age of 53 years (range 18 to 85 years), the racial and ethnic representations were 84% Caucasian, 11% Asian, 4% Black, and 1% other.
Table 6 displays a comparison of paired images (pre-and post-contrast) to pre-contrast images with respect to the proportion of patients who had paired image scores that were greater “better”, or same/worse “not better” than the pre-contrast scores and with respect to the difference in the mean patient level visualization score. Across the three readers 56% to 94% of patients had improved lesion visualization for paired images compared to pre-contrast images. Gadoterate meglumine provided a statistically significant improvement for all three primary visualization components. More lesions were seen on the paired images than the pre-contrast images.
Table 6: Study A. Improvement in Patient-level Lesion Visualization Scores, Paired versus Pre-contrast Images(a) (a) Better: number of patients with paired (pre-and post-contrast) score greater than the pre-contrast score
Not better: number of patients with paired score same as or worse than the pre-contrast score
Missing: number of patients with missing score
(b) Difference = paired mean score minus pre-contrast mean score
*Statistically significant improvement by paired t-testLesion Scores
Reader 1
Reader 2
Reader 3
n = 231
n = 232
n = 237
Border Delineation
Better
195 (84%)
215 (93%)
132 (56%)
Not Better
28 (12%)
7 (3%)
88 (37%)
Missing
8 (4%)
10 (4%)
17 (7%)
Difference in Mean Score (b)
2.26*
2.89*
1.17*
Internal Morphology
Better
218 (94%)
214 (93%)
187 (79%)
Not Better
5 (2%)
8 (3%)
33 (14%)
Missing
8 (4%)
10 (4%)
17 (7%)
Difference in Mean Score (b)
2.74*
2.75*
1.54*
Contrast Enhancement
Better
208 (90%)
216 (93%)
208 (88%)
Not Better
15 (6%)
6 (3%)
12 (5%)
Missing
8 (4%)
10 (4%)
17 (7%)
Difference in Mean Score (b)
3.09*
3.69*
2.92*
In secondary analyses, post-contrast images were improved in comparison to pre-contrast images. Gadoterate meglumine lesion visualization scores were similar to those for gadopentetate dimeglumine. Gadoterate meglumine imaging results in the pediatric patients were also similar to those seen in adults.
In a second clinical trial (Study B), MR images were reread from 150 adult patients with known CNS lesions who had participated in previously conducted clinical trial. Gadoterate meglumine administration and image interpretation was performed in the same manner as in Study A. Similar to Study A, this trial also demonstrated improved lesion visualization with gadoterate meglumine.
CNS Imaging in the Sub-population of Pediatric Patients < 2 years old
A non-randomized study (Study C) with 28 pediatric patients under 2 years of age who were referred for contrast MRI of the CNS supported extrapolation of CNS efficacy findings from adults and older children. CNS lesions were identified in 16 of these 28 patients on paired pre- and post-contrast images compared to 15 patients on pre-contrast images alone. In the 16 patients who had identifiable lesions, the scores for the co-endpoints of lesion visualization were improved for at least one lesion on paired pre- and post-contrast images compared to pre-contrast images in 8 out of 16 (50%) patients for lesion border delineation, 8 out of 16 (50%) patients for lesion internal morphology, and 14 out of 16 (88%) patients for lesion contrast enhancement.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Gadoterate Meglumine Injection, USP is a clear, colorless to yellow solution containing 0.5 mmol per mL of gadoterate meglumine. Gadoterate Meglumine Injection, USP Pharmacy Bulk Package is available in vials.
- Gadoterate Meglumine Injection, USP Pharmacy Bulk Package is supplied in 100 mL vials containing 100 mL of solution.
Each Pharmacy Bulk Package vial is closed with a rubber stopper and sealed with an aluminum cap and the contents are sterile.
Gadoterate Meglumine Injection, USP Pharmacy Bulk Package vials are individually packaged in a box of 6, in the following configurations:
Product Code
Unit of Sale
Each
800150
NDC: 65219-088-50
Unit of 6NDC: 65219-088-06
100 mL in glass vialStorage
Store at 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].
Should solidification occur in the vial because of exposure to the cold, bring Gadoterate Meglumine Injection, USP to room temperature before use. If allowed to stand at room temperature for a minimum of 90 minutes, Gadoterate Meglumine Injection, USP should return to a clear, colorless to yellow solution. Before use, examine the product to assure that all solids are dissolved and that the container and closure have not been damaged. Discard the vial if solids persist.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide)
17.1 Nephrogenic Systemic Fibrosis
Instruct patients to inform their healthcare provider if they:
- 1. have a history of kidney disease, or
- 2. have recently received a GBCA.
GBCAs increase the risk for NSF among patients with impaired elimination of the drugs. To counsel patients at risk for NSF:
- Describe the clinical manifestations of NSF.
- Describe procedures to screen for the detection of renal impairment.
Instruct the patients to contact their physician if they develop signs or symptoms of NSF following Gadoterate Meglumine Injection administration, such as burning, itching, swelling, scaling, hardening and tightening of the skin; red or dark patches on the skin; stiffness in joints with trouble moving, bending or straightening the arms, hands, legs or feet; pain in the hip bones or ribs; or muscle weakness.
17.2 Common Adverse Reactions
Inform patients that they may experience:
- Reactions along the venous injection site, such as mild and transient burning or pain or feeling of warmth or coldness at the injection site.
- Side effects of headache, nausea, abnormal taste and feeling hot.
17.3 General Precautions
- Pregnancy: Advise pregnant women of the potential risk of fetal exposure to gadoterate [see Use in Specific Populations (8.1)]
- Gadolinium Retention: Advise patients that gadolinium is retained for months or years in brain, bone, skin, and other organs in patients with normal renal function. The clinical consequences of retention are unknown. Retention depends on multiple factors and is greater following administration of linear GBCAs than following administration of macrocyclic GBCAs [see Warnings and Precautions (5.4)].
-
MEDICATION GUIDE
MEDICATION GUIDE
Gadoterate Meglumine (GAD oh TER ate MEG loo meen) Injection
(gadoterate meglumine)
Injection for intravenous use
What is the most important information I should know about Gadoterate Meglumine Injection?
- GBCAs like Gadoterate Meglumine Injection may cause serious side effects including death, coma, encephalopathy, and seizures when it is given intrathecally (injection given into the spinal canal). It is not known if Gadoterate Meglumine Injection is safe and effective with intrathecal use. Gadoterate Meglumine Injection is not approved for this use.
- Gadoterate Meglumine Injection contains a metal called gadolinium. Small amounts of gadolinium can stay in your body including the brain, bones, skin and other parts of your body for a long time (several months to years).
- It is not known how gadolinium may affect you, but so far, studies have not found harmful effects in patients with normal kidneys.
- Rarely patients have reported pains, tiredness, and skin, muscle or bone ailments for a long time, but these symptoms have not been directly linked to gadolinium.
- There are different GBCAs that can be used for your MRI exam. The amount of gadolinium that stays in the body is different for different gadolinium medicines. Gadolinium stays in the body more after Omniscan or Optimark than after Eovist, Magnevist or MultiHance. Gadolinium stays in the body the least after Gadoterate Meglumine Injection, Gadavist or ProHance.
- People who get many doses of gadolinium medicines, women who are pregnant and young children may be at increased risk from gadolinium staying in the body.
- Some people with kidney problems who get gadolinium medicines can develop a condition with severe thickening of the skin, muscles and other organs in the body (nephrogenic systemic fibrosis). Your healthcare provider should screen you to see how well your kidneys are working before you receive Gadoterate Meglumine Injection.
What is Gadoterate Meglumine Injection?
- Gadoterate Meglumine Injection is a prescription medicine called a gadolinium-based contrast agent (GBCA). Gadoterate Meglumine Injection, like other GBCAs, is injected into your vein and used with a magnetic resonance imaging (MRI) scanner.
- An MRI exam with a GBCA, including Gadoterate Meglumine Injection, helps your doctor to see problems better than an MRI exam without a GBCA.
- Your doctor has reviewed your medical records and has determined that you would benefit from using a GBCA with your MRI exam.
Do not receive Gadoterate Meglumine Injection if you have had a severe allergic reaction to Gadoterate Meglumine Injection.
Before receiving Gadoterate Meglumine Injection, tell your healthcare provider about all your medical conditions, including if you:
- have had any MRI procedures in the past where you received a GBCA. Your healthcare provider may ask you for more information including the dates of these MRI procedures.
- are pregnant or plan to become pregnant. It is not known if Gadoterate Meglumine Injection can harm your unborn baby. Talk to your healthcare provider about the possible risks to an unborn baby if a GBCA such as Gadoterate Meglumine Injection is received during pregnancy.
- have kidney problems, diabetes, or high blood pressure.
- have had an allergic reaction to dyes (contrast agents) including GBCAs.
What are possible side effects of Gadoterate Meglumine Injection?
- See "What is the most important information I should know about Gadoterate Meglumine Injection?"
- Allergic reactions. Gadoterate Meglumine Injection can cause allergic reactions that can sometimes be serious. Your healthcare provider will monitor you closely for symptoms of an allergic reaction.
The most common side effects of Gadoterate Meglumine Injection include: nausea, headache, pain, or cold feeling at the injection site, and rash.
These are not all the possible side effects of Gadoterate Meglumine Injection.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about the safe and effective uses of Gadoterate Meglumine Injection.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your healthcare provider for information about Gadoterate Meglumine Injection that is written for health professionals.What are the ingredients in Gadoterate Meglumine Injection?
Active ingredient: gadoterate meglumine
Inactive ingredients: DOTA, water for injection
Manufactured for: Fresenius Kabi USA, LLC
For more information, call 1-800-551-7176.This Medication Guide has been approved by the U.S. Food and Drug Administration.
451748C 24EUG04 Rev. 05/2025 - PRINCIPAL DISPLAY PANEL – 50 mmol per 100 mL Tray Label
-
PRINCIPAL DISPLAY PANEL – 50 mmol per 100 mL Carton
NDC 65219-088-06
Gadoterate Meglumine
Injection, USP50 mmol per 100 mL
(0.5 mmol per mL)For Intravenous Administration
Dispense the Medication Guide provided
separately to each patient.Sterile Solution
Withdraw Contents Within 24 HoursPharmacy Bulk Package
Not for Direct InfusionOne 100 mL Multiple Dose Vial
Rx only
-
PRINCIPAL DISPLAY PANEL – 50 mmol per 100 mL Vial Label
NDC 65219-088-06
Gadoterate Meglumine
Injection, USP50 mmol per 100 mL
(0.5 mmol per mL)For Intravenous Administration
Dispense the Medication Guide
provided separately to each patient.Sterile Solution
Withdraw Contents Within 24 HoursPharmacy Bulk Package
Not for Direct Infusion100 mL Multiple Dose Vial
Rx only
-
INGREDIENTS AND APPEARANCE
GADOTERATE MEGLUMINE
gadoterate meglumine injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 65219-088 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength gadoterate meglumine (UNII: L0ND3981AG) (GADOLINIUM CATION (3+) - UNII:AZV954TZ9N) GADOLINIUM CATION (3+) 376.9 mg in 1 mL Inactive Ingredients Ingredient Name Strength WATER (UNII: 059QF0KO0R) 1 mL in 1 mL TETRAXETAN (UNII: 1HTE449DGZ) 0.25 mg in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 65219-088-50 6 in 1 BOX 06/20/2022 1 NDC: 65219-088-06 1 in 1 CARTON 1 100 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA215982 06/20/2022 Labeler - Fresenius Kabi USA, LLC (013547657) Registrant - Jiangsu Hengrui Pharmaceuticals Co., Ltd. (654147255) Establishment Name Address ID/FEI Business Operations Jiangsu Hengrui Pharmaceuticals Co., Ltd. (Dongjin Road Site) 421324417 MANUFACTURE(65219-088)




© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.