MYQORZO- aficamten tablet, film coated
MYQORZO by
Drug Labeling and Warnings
MYQORZO by is a Prescription medication manufactured, distributed, or labeled by Cytokinetics Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use MYQORZO safely and effectively. See full prescribing information for MYQORZO.
MYQORZO™ (aficamten) tablets, for oral use
Initial U.S. Approval: 2025WARNING: RISK OF HEART FAILURE
See full prescribing information for complete boxed warning.
- MYQORZO can cause heart failure due to systolic dysfunction. (5.1)
- Echocardiogram assessments of left ventricular ejection fraction (LVEF) are required before and during MYQORZO use. (2.1)
- Initiation in patients with left ventricular ejection fraction (LVEF) <55% is not recommended. (2.1)
- Decrease dose if LVEF <50% and ≥40%. Interrupt dosing if LVEF <40% or if worsening clinical status. (2.2)
- MYQORZO is available only through a restricted program called the MYQORZO REMS Program. (5.2)
INDICATIONS AND USAGE
MYQORZO is a cardiac myosin inhibitor indicated for the treatment of adults with symptomatic obstructive hypertrophic cardiomyopathy (oHCM) to improve functional capacity and symptoms. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Film-coated tablets: 5 mg, 10 mg, 15 mg, 20 mg. (3)
CONTRAINDICATIONS
- Rifampin (4)
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
The most common adverse reaction was hypertension (8%). (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Cytokinetics at 1-833-633-2986 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 12/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: RISK OF HEART FAILURE
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Evaluation Before and During Use of MYQORZO
2.2 Recommended Dosage and Administration
2.3 Dosage Modifications for Drug Interactions
2.4 Missed Dose
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Heart Failure
5.2 MYQORZO REMS Program
5.3 Cytochrome P450 Interactions Leading to Heart Failure or Loss of Effectiveness
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1. Potential for Other Drugs to Affect Plasma Concentrations of MYQORZO
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.5 Pharmacogenomics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: RISK OF HEART FAILURE
MYQORZO reduces left ventricular ejection fraction (LVEF) and can cause heart failure due to systolic dysfunction [see Warnings and Precautions (5.1)].
Echocardiogram assessments are required prior to and during treatment with MYQORZO to monitor for systolic dysfunction. Initiation of MYQORZO in patients with LVEF <55% is not recommended. Decrease the dose of MYQORZO if LVEF is <50% and ≥40% [see Dosage and Administration (2.2) and Warnings and Precautions (5.1)]. Interrupt the dose of MYQORZO if LVEF <40% or if the patient experiences heart failure symptoms or worsening clinical status due to systolic dysfunction [see Dosage and Administration (2.2)].
Because of the risk of heart failure due to systolic dysfunction, MYQORZO is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the MYQORZO REMS Program [see Warnings and Precautions (5.2)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Evaluation Before and During Use of MYQORZO
Initiation or up-titration of MYQORZO in patients with LVEF <55% is not recommended.
Patients may develop heart failure while taking MYQORZO. Regular LVEF and Valsalva left ventricular outflow tract gradient (LVOT-G) assessment is needed for titration to achieve an appropriate target Valsalva LVOT-G, while maintaining LVEF ≥50% and avoiding heart failure symptoms.
2.2 Recommended Dosage and Administration
The recommended starting dose of MYQORZO is 5 mg orally once daily. Increase the dose every 2 to 8 weeks by 5 mg until a maintenance dose or the maximum recommended dose of 20 mg once daily is achieved. The maintenance dose of MYQORZO is individualized based on the patient's LVEF and LVOT-G. Recommendations for dosing based on LVEF and LVOT-G criteria are provided in Table 1.
Table 1: Dose Adjustment of MYQORZO LVEF Valsalva LVOT-G Dose Adjustment - * Decrease dose as follows: 20 mg to 15 mg; 15 mg to 10 mg; 10 mg to 5 mg
≥55% ≥30 mmHg Increase dose by 5 mg (up to the maximum dose of 20 mg once daily) ≥55% <30 mmHg Maintain Dose <55% and ≥50% Any Maintain Dose <50% and ≥40% Any Decrease dose by 5 mg*
If already on 5 mg, interrupt treatment for at least 7 days<40% Any Interrupt treatment for at least 7 days Perform an echocardiographic assessment 2 to 8 weeks after initiation of treatment or any dose adjustment (e.g., due to LVEF and LVOT-G criteria or drug interaction). After a treatment interruption due to low LVEF, resume treatment, no earlier than 7 days, when LVEF ≥55% and re-initiate dose titration at the starting dose of 5 mg (see Table 1).
After the maintenance dose has been established, assess LVEF and Valsalva LVOT-G every 6 months, or every 3 months in patients with LVEF <55% to ≥50%. Consider monitoring LVEF and adjust the dose per Table 1 as needed, in patients with an intercurrent illness (e.g., severe infection or COVID-19), new arrhythmia (e.g., new or uncontrolled atrial fibrillation or other uncontrolled tachyarrhythmia) or any other conditions that may impair systolic function. Do not increase the dose until the intercurrent illness or new arrhythmia has resolved or stabilized.
MYQORZO should be taken once daily with or without meals at about the same time every day. Swallow tablets whole.
2.3 Dosage Modifications for Drug Interactions
Initiate MYQORZO at the recommended starting dose of 5 mg once daily in patients who are on stable therapy with fluconazole, voriconazole, fluvoxamine, strong CYP2C9 inhibitors, or in patients discontinuing a moderate to strong CYP3A inducer.
Concomitant Administration with Fluconazole or Voriconazole
In patients who initiate fluconazole (if used for more than 3 days) or voriconazole, reduce the dose of MYQORZO to 5 mg if they are currently receiving 15 mg or 20 mg. Avoid concomitant use if patients are currently receiving MYQORZO 5 mg or 10 mg. Assess LVEF and LVOT-G 2 to 8 weeks after initiation of fluconazole or voriconazole and titrate the dose of MYQORZO according to Table 1 [see Dosage and Administration (2.2), Warnings and Precautions (5.3) and Drug Interactions (7.1)].
Concomitant Administration with Fluvoxamine or Strong CYP2C9 Inhibitors
In patients who initiate fluvoxamine or a strong CYP2C9 inhibitor, reduce the dose of MYQORZO (20 mg to 10 mg; 15 mg to 5 mg; 10 mg to 5 mg). For patients currently receiving MYQORZO 5 mg, maintain the 5 mg dose. Assess LVEF and LVOT-G 2 to 8 weeks after inhibitor initiation and titrate the dose of MYQORZO according to Table 1 [see Dosage and Administration (2.2), Warnings and Precautions (5.3) and Drug Interactions (7.1)].
Concomitant Administration with Moderate to Strong CYP3A Inducers
In patients who are on stable therapy with moderate to strong CYP3A inducers (e.g., carbamazepine), when discontinuing these medications, reduce the dose of MYQORZO (20 mg to 10 mg; 15 mg to 5 mg; 10 mg to 5 mg). For patients currently receiving MYQORZO 5 mg, maintain the 5 mg dose. Assess LVEF and LVOT-G 2 to 8 weeks after inducer discontinuation and titrate the dose of MYQORZO according to Table 1 [see Dosage and Administration (2.2), Warnings and Precautions (5.3) and Drug Interactions (7.1)].
-
3 DOSAGE FORMS AND STRENGTHS
MYQORZO is available as tablets in the following strengths:
Tablets: 5 mg - purple, round tablet debossed with "5" on one side and "CK" on the other side.
Tablets: 10 mg - purple, triangular tablet debossed with "10" on one side and "CK" on the other side.
Tablets: 15 mg - purple, pentagonal tablet debossed with "15" on one side and "CK" on the other side.
Tablets: 20 mg - purple, oval tablet debossed with "20" on one side and "CK" on the other side.
-
4 CONTRAINDICATIONS
MYQORZO is contraindicated with concomitant use of rifampin [see Warnings and Precautions (5.3) and Drug Interactions (7.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Heart Failure
MYQORZO reduces cardiac contractility, which can reduce LVEF and cause heart failure.
Patients who experience a serious intercurrent illness (e.g., serious infection) or arrhythmia (e.g., new or uncontrolled atrial fibrillation) may be at greater risk of developing systolic dysfunction and heart failure [see Clinical Trial Experience (6.1)]. Asymptomatic LVEF reduction, intercurrent illnesses, and arrhythmias require additional monitoring considerations [see Dosage and Administration (2.2)].
Assess the patient's clinical status and LVEF prior to and regularly during treatment and adjust the MYQORZO dose accordingly [see Dosage and Administration (2.3)]. New or worsening arrhythmia, dyspnea, chest pain, fatigue, leg edema, or elevations in N-terminal pro-B-type natriuretic peptide (NT-proBNP) may be signs and symptoms of heart failure and should prompt an evaluation of cardiac function.
Initiation of MYQORZO in patients with LVEF <55% is not recommended.
5.2 MYQORZO REMS Program
MYQORZO is available only through a restricted program called the MYQORZO REMS Program, because of the risk of heart failure due to systolic dysfunction [see Warnings and Precautions (5.1)].
Notable requirements of the MYQORZO REMS Program include the following:
- Prescribers must be certified by enrolling in the MYQORZO REMS Program.
- Patients must enroll in the MYQORZO REMS Program and comply with ongoing monitoring requirements [see Dosage and Administration (2.1)].
- Pharmacies must be certified by enrolling in the MYQORZO REMS Program and must only dispense to patients who are authorized to receive MYQORZO.
- Wholesalers and distributors must only distribute to certified pharmacies.
Further information is available at www.MYQORZOREMS.com or by telephone at 1-844-285-7367.
5.3 Cytochrome P450 Interactions Leading to Heart Failure or Loss of Effectiveness
MYQORZO is metabolized primarily by CYP2C9, and to a lesser extent by CYP3A, CYP2D6, and CYP2C19 enzymes. Initiation of medications that inhibit multiple P450 pathways of MYQORZO elimination (e.g., fluconazole, voriconazole, fluvoxamine) or strong CYP2C9 inhibitors, and discontinuation of moderate-to-strong CYP3A inducers may lead to increased blood concentrations of aficamten and increase the risk of heart failure due to systolic dysfunction [see Contraindications (4), Warnings and Precautions (5.1), and Drug Interactions (7.1)]. Conversely, initiation of medications that induce P450 pathways of MYQORZO (e.g., rifampin, moderate-to-strong CYP3A inducers) may lead to decreased blood concentrations of aficamten and potential loss of effectiveness [see Contraindications (4) and Drug Interactions (7.1)]. Assess LVEF 2 to 8 weeks after initiation of such inhibitors or after discontinuation of such inducers and adjust the dose of MYQORZO accordingly [see Dosage and Administration (2.3)].
Advise patients of the potential for drug interactions. Advise patients to inform their healthcare provider of all concomitant medications prior to and during MYQORZO treatment [see Drug Interactions (7.1) and Patient Counseling Information (17)].
-
6 ADVERSE REACTIONS
The following adverse reaction is discussed in other sections of labeling:
- Heart Failure [see Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of MYQORZO was evaluated in SEQUOIA-HCM, a phase 3, randomized, double-blind, placebo-controlled study [see Clinical Studies (14)]. Of the 282 adults with oHCM, 142 patients received daily doses of MYQORZO (initiated at 5 mg and titrated up to a maximum dose of 20 mg) and 140 patients received placebo. The median treatment duration for patients receiving MYQORZO was ~24 weeks (range 4 to 29 weeks).
Hypertension (8% vs. 2%) was the only adverse reaction occurring in >5% of patients and more commonly on MYQORZO than on placebo.
Eligible oHCM patients were able to participate in an ongoing, open-label, single-arm, long-term safety study (FOREST-HCM). Based on available data, the safety profile of MYQORZO in FOREST-HCM was similar to that observed in SEQUOIA-HCM.
Effects on Systolic Function
In SEQUOIA-HCM, the mean (SD) resting LVEF at baseline was 75% (6) in both treatment groups. Consistent with the mechanism of action of MYQORZO, LS mean (SE) change from baseline in LVEF was -7% (0.6) in the MYQORZO group and -2% (0.6) in the placebo group at the end of the 24-week treatment period. Four weeks after the end of treatment, mean LVEF was similar between the MYQORZO and placebo groups. During the 24-week treatment period, 5 (4%) patients in the MYQORZO group and 1 (1%) patient in the placebo group experienced a reversible reduction in LVEF to <50% (median LVEF: 47%; range 34% - 49% for these 5 patients in the MYQORZO group). Reductions in LVEF to <50% did not require treatment interruption and were not associated with clinical heart failure [see Warnings and Precautions (5.1)].
Effects on Blood Pressure
In SEQUOIA-HCM, the mean (SD) systolic/diastolic blood pressure (SBP/DBP) at baseline was 125(16)/75(11) mmHg for patients in the MYQORZO group and 126(16)/74(11) mmHg in the placebo group. There was a greater mean change from baseline in SBP/DBP (SD) in the MYQORZO group compared to the placebo group at the end of the 24-week treatment period [2(13)/3(8) mmHg and -3(14)/-1(9) mmHg, respectively]. Four weeks after the end of treatment, the mean SBP/DBP was similar between the MYQORZO and placebo groups. In SEQUOIA-HCM, SBP ≥160 mmHg was observed in approximately 16% of patients in the MYQORZO group and 8% of patients in the placebo group. MYQORZO-associated increases in blood pressure are consistent with relief of LVOT obstruction and improved cardiac output.
-
7 DRUG INTERACTIONS
7.1. Potential for Other Drugs to Affect Plasma Concentrations of MYQORZO
Aficamten is primarily metabolized by CYP2C9 and, to a lesser extent by CYP3A, CYP2D6, and CYP2C19. Concomitant administration of drugs that inhibit multiple P450 pathways of aficamten elimination, strong inhibitors of CYP2C9, and moderate-to-strong inducers of CYP3A, may affect the exposure of aficamten [see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)] (see Table 2).
Table 2: Established and Potentially Significant Pharmacokinetic Drug-Drug Interactions with MYQORZO Inhibitors of Multiple CYPs Fluvoxamine Clinical Impact Fluvoxamine is a strong CYP2C19 inhibitor, weak to moderate CYP3A inhibitor, weak CYP2C9 inhibitor, and weak CYP2D6 inhibitor.
Coadministration of fluvoxamine is predicted to increase aficamten exposure, which may increase the risk of developing heart failure due to systolic dysfunction.Prevention or Management Initiate MYQORZO at 5 mg in patients on stable therapy with fluvoxamine.
In patients who are on MYQORZO treatment and intend to initiate fluvoxamine: Reduce dose of MYQORZO (i.e., 20 mg to 10 mg, 15 mg to 5 mg, or 10 mg to 5 mg) or continue 5 mg [see Dosage and Administration (2.3)].Fluconazole or Voriconazole Clinical Impact Fluconazole is a moderate CYP2C9 inhibitor, moderate CYP3A inhibitor, and strong CYP2C19 inhibitor.
Voriconazole is a strong CYP3A inhibitor, moderate CYP2C19 inhibitor, and weak CYP2C9 inhibitor.
Coadministration with fluconazole is observed, and voriconazole is predicted, to increase aficamten exposure, which may increase the risk of developing heart failure due to systolic dysfunction [see Clinical Pharmacology (12.3)].Prevention or Management Initiate MYQORZO at 5 mg in patients on stable therapy with fluconazole or voriconazole.
In patients currently receiving MYQORZO who intend to initiate fluconazole (if used more than 3 days) or voriconazole: Reduce dose of MYQORZO to 5 mg if they are currently receiving MYQORZO 15 mg or 20 mg. Avoid concomitant use if currently receiving MYQORZO 5 mg or 10 mg [see Dosage and Administration (2.3)] .Strong CYP2C9 inhibitors Clinical Impact Coadministration with a strong CYP2C9 inhibitor is predicted to increase the exposure of aficamten, which may increase the risk of developing heart failure due to systolic dysfunction [see Clinical Pharmacology (12.3)]. Prevention or Management Initiate MYQORZO at 5 mg in patients who are on stable therapy with a strong CYP2C9 inhibitor.
In patients who are on MYQORZO treatment and intend to initiate a strong CYP2C9 inhibitor: Reduce dose of MYQORZO (i.e., 20 mg to 10 mg, 15 mg to 5 mg, or 10 mg to 5 mg) or continue 5 mg [see Dosage and administration (2.3)].CYP Inducers Rifampin Clinical Impact Rifampin is a strong CYP3A inducer, strong CYP2C19 inducer, and moderate CYP2C9 inducer. Coadministration of rifampin decreases aficamten exposure, which may reduce the effectiveness of MYQORZO [see Contraindications (4) and Clinical Pharmacology (12.3)]. Prevention of Management Concomitant use with rifampin is contraindicated. Other moderate-to-strong CYP3A inducers Clinical Impact Concomitant use with a moderate to strong CYP3A inducer decreases aficamten exposure, which may reduce MYQORZO effectiveness [see Clinical Pharmacology (12.3)].
The risk of heart failure due to systolic dysfunction may increase with discontinuation of these inducers.Prevention or Management In patients who intend to discontinue a moderate to strong CYP3A Inducer: Reduce dose of MYQORZO (20 mg to 10 mg; 15 mg to 5 mg; 10 mg to 5 mg) or continue 5 mg [see Dosage and Administration (2.3)]. -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on the use of MYQORZO during pregnancy to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. The underlying maternal condition during pregnancy poses a risk to the mother and fetus (see Clinical Considerations). In embryo-fetal and pre- and postnatal development studies, when pregnant rats were administered aficamten during the period of organogenesis, aficamten increased the incidence of structural malformations at exposures ≥4-times the maximum recommended human dose (MRHD) of 20 mg based on free area under the concentration curve (AUC). No adverse effects on development were seen at 3-times the MRHD exposure (see Data).
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defects, loss, and other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
There is a pregnancy safety study for MYQORZO. If MYQORZO is administered during pregnancy, or if a patient becomes pregnant while receiving MYQORZO or within 3 weeks after the last dose of MYQORZO, healthcare providers should report MYQORZO exposure by contacting Cytokinetics, Inc. at 1-833-633-2986.
Disease-Associated Maternal and Embryo-Fetal Risk
Obstructive HCM in pregnancy has been associated with increased risk for preterm birth.
Animal Data
Aficamten given to pregnant rats (2, 6 and 9 mg/kg/day) during the period of organogenesis was associated with increased external fetal malformations (kinked tail) at aficamten exposures 5-times the clinical exposures at the MRHD based on free AUC, with uncertain human relevance. Increased post-implantation loss (early and late resorptions) and decreased mean fetal body weight occurred in the presence of significant maternal toxicity at exposures 5-times the clinical exposures at the MRHD. No adverse effects on embryofetal development were observed at 6 mg/kg/day, representing 3-times the clinical exposure at the MRHD.
Pregnant rabbits given aficamten (5, 10 and 20/15 mg/kg/day) during the period of embryofetal organogenesis showed a numerical increase in post-implantation loss (late resorptions) and fetuses with cardiac malformations at 10 mg/kg/day. However, no increase in malformations was observed at the high dose of 20/15 mg/kg/day, a dose that caused significant maternal toxicity (leading to dose reduction from 20 to 15 mg/kg/day during the study) at clinically relevant exposures.
Pregnant rats given aficamten (0.5, 1.5, 6 mg/kg/day) during embryofetal organogenesis through offspring weaning in a pre- and postnatal development study had reduced offspring viability at the 6 mg/kg/day in the presence of maternal toxicity, at 4-times the clinical exposure at the MRHD based on free AUC. An increased number of offspring with bent tails was also observed at the 6 mg/kg/day. No adverse maternal toxicity or developmental effects occurred at 1.5 mg/kg/day, which corresponds to clinical exposure at the MRHD. Aficamten did not affect neurobehavioral parameters, sexual maturation, or reproductive function of the offspring at any dose.
8.2 Lactation
Risk Summary
There are no data on the presence of aficamten in human milk, the effects on the breastfed infant, or the effects on milk production. Aficamten was detected in the plasma of rat pups when dams were dosed with the drug orally during the lactation period. When a drug is present in animal milk, it is likely that the drug will be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for MYQORZO and any potential adverse effects on the breastfed infant from MYQORZO or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of MYQORZO in pediatric patients have not been established.
8.5 Geriatric Use
There were 57 (40%) MYQORZO-treated patients, aged 65 years and older, in SEQUOIA-HCM [see Clinical Studies (14)]. Of the total number of MYQORZO-treated patients in SEQUOIA-HCM, 45 (32%) patients were 65 to 74 years of age, and 12 (8%) patients were 75 years of age and older.
No overall differences in safety or effectiveness of MYQORZO have been observed between patients 65 years of age and older and younger adult patients.
-
10 OVERDOSAGE
Clinical Experience and Effects
There have been no reports of overdose with MYQORZO. Cardiovascular effects of overdose may include reduced LVEF, heart failure, and hypotension. Signs and symptoms of heart failure such as dyspnea, fatigue, leg edema, or elevations in NT-proBNP should prompt an evaluation of cardiac function [see Warnings and Precautions (5.1)].
Management
Discontinue MYQORZO treatment. Provide medically supportive measures to maintain hemodynamic stability and monitor left ventricular function.
Consider contacting the Poison Help line (1-800-222-1222) or a medical toxicologist for additional overdose management recommendations.
-
11 DESCRIPTION
MYQORZO tablets for oral use contain aficamten, a cardiac myosin inhibitor.
The chemical name of aficamten is (R)-N-(5-(5-ethyl-1,2,4-oxadiazol-3-yl)-2,3-dihydro-1H-inden-1-yl)-1-methyl-1H-pyrazole-4-carboxamide. The molecular formula is C18H19N5O2 and the molecular weight is 337.38 g/mol.
The structural formula of aficamten is:
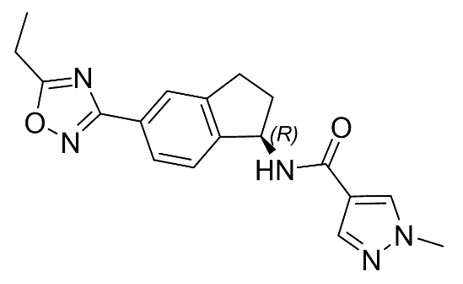
Aficamten is a white to off-white to yellow to grey solid that is practically insoluble in water and aqueous buffers (pH 2 – 9), sparingly soluble in acetone and ethanol, and soluble in N-methylpyrrolidone (NMP).
MYQORZO is supplied as tablets containing 5 mg, 10 mg, 15 mg, or 20 mg of aficamten per tablet as the active ingredient and the following inactive ingredients: croscarmellose sodium, hydroxypropyl cellulose, magnesium stearate (non-bovine), mannitol, microcrystalline cellulose, and sodium lauryl sulfate. The tablet coating contains carmine, FD&C Blue No. 2, glyceryl mono and dicaprylate, polyvinyl alcohol, polyvinyl alcohol graft polyethylene copolymer, talc and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Aficamten is an allosteric and reversible inhibitor of cardiac myosin motor activity. Aficamten reduces the force generated by myosin at the cardiac sarcomere, which contributes to the pathophysiology of HCM. In patients with HCM, myosin inhibition with aficamten reduces cardiac contractility and left ventricular outflow tract (LVOT) obstruction.
12.2 Pharmacodynamics
Left Ventricular Ejection Fraction and Left Ventricular Outflow Tract Obstruction
In SEQUOIA-HCM, reductions in LVOT-G were observed at 2 weeks after initiating treatment, and LVOT-G continued to decrease through Week 8. The reduction in LVOT-G was sustained through the remainder of the 24-week trial. At Week 24, the mean (SD) change from baseline in resting and Valsalva LVOT-G was -35 (29) mmHg and -48 (37) mmHg, respectively, for the MYQORZO group and 4 (35) mmHg and 2 (35) mmHg, respectively, for the placebo group. The reduction in LVOT-G was accompanied by a greater decrease in LVEF in the MYQORZO group compared to the placebo group with mean (SD) change from baseline in LVEF of -7 (7)% and -2 (6)%, respectively. Four weeks after discontinuation of treatment, LVEF and Valsalva LVOT-G returned to baseline.
Cardiac Structure
In SEQUOIA-HCM, maximal left ventricular wall thickness, left ventricular mass index (LVMI) and left atrial volume index (LAVI) were measured at baseline and Week 24. At Week 24, the mean (SD) maximal left ventricular wall thickness decreased in the MYQORZO group by -0.2 (0.3) cm and in the placebo group by -0.1 (0.3) cm. LVMI decreased in the MYQORZO group by -5.3 (24) g/m2 and increased in the placebo group by 5.8 (25) g/m2. At Week 24, LAVI decreased in the MYQORZO group by -2.6 (8) mL/m2 and increased in the placebo group by 1.4 (8) mL/m2. The clinical significance of these findings is unknown.
Cardiac Biomarkers
In SEQUOIA-HCM, reductions in NT-proBNP were observed in the MYQORZO group starting at Week 2 and were sustained through Week 24. At Week 24, NT-proBNP was 80% lower with MYQORZO compared to placebo (proportion of geometric mean ratio between MYQORZO and placebo, 0.20 [95% CI: 0.17, 0.23]). At both Week 12 and 24, troponin I was 40% lower with MYQORZO compared to placebo (proportion of geometric mean ratio between the two groups, 0.57 [95% CI: 0.51, 0.64]).
The clinical significance of the NT-proBNP and troponin findings is unknown.
Cardiac Electrophysiology
The results of a thorough QT study demonstrated a lack of QTc prolongation across the therapeutic concentration range of MYQORZO. At a single dose of 50 mg (similar exposure to 20 mg daily dosing to steady-state), the upper limit of the predicted placebo-corrected change from baseline in QT interval corrected by Fridericia (ΔΔQTcF) 90% confidence interval for MYQORZO was <10 msec.
12.3 Pharmacokinetics
Aficamten exposure increases dose proportionally following single doses of 1 mg to 75 mg and multiple once daily doses of 5 mg to 20 mg. Aficamten pharmacokinetics was comparable between healthy subjects and patients with oHCM. Geometric mean accumulation ratios for aficamten were similar across dose levels and ranged from 4.6 to 4.8. Steady state is predicted to be reached following approximately 17 days of once daily dosing of MYQORZO in patients with oHCM.
Absorption
Aficamten is rapidly absorbed with a median time to maximum concentration (Tmax) of 1.5 to 2.0 hours. The bioavailability of aficamten after oral administration is unknown.
Effect of Food
No clinically significant differences in aficamten AUC and Cmax were observed following its administration with a high-fat, high-calorie meal.
Distribution
Aficamten is approximately 90% bound to plasma proteins and demonstrates a volume of distribution of 313 L. The blood to plasma ratio of aficamten was 0.94.
Elimination
Aficamten has a median terminal half-life (t½) of approximately 80 hours in patients with oHCM. At steady-state, the peak-to-trough plasma concentration ratio with once daily dosing is approximately 1.2. The total clearance is 2.6 L/hr and the renal clearance is <0.1% of total clearance.
Metabolism
Aficamten is extensively metabolized in humans, primarily through CYP2C9 with contributions by CYP3A, CYP2D6, and CYP2C19. Aficamten is primarily metabolized to two pharmacologically inactive metabolites, CK-3834282 and CK-3834283, that circulate at approximately 56% and 103% of parent in plasma, respectively.
Excretion
Following a single 20 mg radiolabeled dose of aficamten, 32% (0.06% unchanged aficamten) was excreted in urine and 58% (5.1% unchanged aficamten) was excreted in feces.
Specific Populations
No clinically meaningful differences in the pharmacokinetics of aficamten were observed based on age (18 – 83 years), race, mild to moderate hepatic impairment (Child-Pugh Classes A and B), or mild to moderate renal impairment (estimated glomerular filtration rate (eGFR) ≥30 mL/min normalized by body surface area and based on the modification of diet in renal disease (MDRD) equation). The effects of severe renal (eGFR <30 mL/min based on MDRD equation) or hepatic (Child-Pugh Class C) impairment are unknown.
Small differences in aficamten exposure with sex and body weight were observed. Female patients demonstrated a 31% higher mean exposure (AUC) compared to male patients. The highest body weight quartile of patients (median: 105 kg) demonstrated a 34% lower aficamten mean exposure (AUC) than the lowest body weight quartile of patients (median: 64 kg). These differences are not clinically significant.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Inhibitors and Inducers of CYP enzymes: The predicted and observed effect of coadministration of select CYP inhibitors and inducers on the plasma exposure of aficamten is provided in Table 3.
Table 3: In Vivo Observed and Predicted Effects of CYP Inhibitors and Inducers on Aficamten Plasma Exposure Concomitant Medication Dosage Effect on Aficamten Mean AUC - * Predicted via physiologically-based pharmacokinetic modeling.
- † Observed.
Inhibitors of Individual CYP Enzymes Sulfaphenazole* - Strong CYP2C9 Inhibitor 2000 mg once daily AUC: ↑ 113% Itraconazole† - Strong CYP3A Inhibitor 200 mg once daily AUC: ↑ 26% Paroxetine† – Strong CYP2D6 Inhibitor 40 mg once daily AUC: ↑ 27% Inhibitors of Multiple CYP Enzymes Fluconazole† - Strong CYP2C19, Moderate CYP3A, and Moderate CYP2C9 Inhibitor 400 mg once daily AUC: ↑ 278% 150 mg × 2 doses; separated by 48 hours AUC: No change Fluvoxamine* - Strong CYP2C19, Weak-to-Moderate CYP3A, Weak CYP2C9, and Weak CYP2D6 Inhibitor 150 mg twice daily AUC: ↑ 116% Voriconazole* - Strong CYP3A, Moderate CYP2C19, and Weak CYP2C9 inhibitor 200 mg twice daily AUC: ↑ 94% Fluoxetine† - Strong CYP2D6 and Strong CYP2C19 Inhibitor 40 mg once daily AUC: ↑ 32% CYP Enzyme Inducers Rifampin* - Strong CYP3A, Strong CYP2C19, and Moderate CYP2C9 Inducer 600 mg once daily AUC: ↓ 79% Carbamazepine† – Moderate-to-strong CYP3A and Weak CYP2C9 Inducer 300 mg twice daily AUC: ↓ 51% P-gp Transporter Substrates: Concomitant administration of a single 20 mg dose of aficamten increased dabigatran (administered as dabigatran etexilate) mean AUCinf by 26%.
In Vitro Drug Interaction Studies
CYP enzymes: Aficamten is not expected to inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C19, CYP2D6, or CYP3A4 at clinically relevant concentrations. Aficamten is not expected to induce CYP1A2, CYP2B6 or CYP3A4 at clinically relevant concentrations.
Transporter systems: Aficamten is not a sensitive substrate of drug transporters. Aficamten is not expected to inhibit BCRP, OCT2, OCT3, OAT1, OAT3, OATP1B1, OATP1B3, MATE1 or MATE2-K drug transporters at clinically relevant concentrations.
12.5 Pharmacogenomics
Aficamten is primarily metabolized by the polymorphic enzyme, CYP2C9, with lesser contributions by other polymorphic enzymes (e.g., CYP2D6 and CYP2C19) [see Clinical Pharmacology (12.3)]. The impact of CYP2C9 genetic variants on the pharmacokinetics of aficamten has not been directly evaluated, though, a similar effect on exposure is expected as seen with strong CYP2C9 inhibitors [see Drug Interactions (7.1)].
No additional dosage modifications are required for patients who are CYP2C9 poor metabolizers, as MYQORZO dosage titration and modifications account for CYP2C9 metabolizer status [see Dosage and Administration (2.2) and (2.3)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
The carcinogenic potential of aficamten was assessed in a 26-week study in transgenic rasH2 mice and a 2-year study in Sprague Dawley rats. There was no evidence of carcinogenicity with oral administration of aficamten at dose levels up to 2.0 mg/kg/day in the mouse or 3 mg/kg/day in the rat (corresponding to 4-fold the MRHD based on free AUC).
Mutagenesis
Aficamten was not genotoxic in the in vitro bacterial (Ames) mutagenicity assay or the combined in vivo micronucleus assay and comet assay in rat.
Impairment of Fertility
In a fertility and early embryonic development toxicology study in the rat, aficamten was administered at doses up to 6 mg/kg/day in females and 3 mg/kg/day in males, with no drug-related effects on mating, fertility, estrous cycling, male reproductive assessments, or early embryonic survival.
-
14 CLINICAL STUDIES
The efficacy of MYQORZO was evaluated in SEQUOIA-HCM (NCT05186818), a phase 3, multicenter, randomized, double-blind, placebo-controlled study in 282 adults (142 aficamten, 140 placebo) with symptomatic New York Heart Association (NYHA) class II and III oHCM, LVEF ≥60%, and resting and post-Valsalva peak LVOT-G ≥30 and ≥50 mmHg at screening, respectively. Patients who completed SEQUOIA-HCM were eligible to participate in an ongoing, open-label, single-arm extension study (FOREST-HCM). Patients with a known infiltrative or storage disorder causing cardiac hypertrophy such as Noonan syndrome, Fabry disease or amyloidosis were excluded.
Patients were randomized in a 1:1 ratio to receive either MYQORZO or placebo once daily for 24 weeks. Randomization was stratified by use of beta-blockers (yes or no) and cardiopulmonary exercise testing (CPET) exercise modality (treadmill or bicycle).
At baseline, 76% of the randomized patients were NYHA class II and 24% were NYHA class III. The mean peak oxygen uptake (pVO2) by CPET at baseline was 18.5 mL/kg/min with 55% using treadmill and 45% using bicycle, respectively. At baseline, the median LVEF was 76%, the mean resting LVOT-G was 55 mmHg, the mean Valsalva LVOTG was 83 mmHg, and mean Kansas City Cardiomyopathy Questionnaire – Clinical Summary Score (KCCQ-CSS) was 74. At baseline, 61% of patients were on beta-blockers, 29% were on non-dihydropyridine calcium channel blockers, 13% were on disopyramide, and 15% were not taking any background medication for oHCM.
Groups were balanced with respect to age (mean 59 years; range 18 to 84 years), sex (59% male), race (79% White, 19% Asian and 1% Black or African American), body mass index (mean 28 kg/m2), heart rate (mean 66 bpm) and blood pressure (mean 124/74 mmHg).
Patients were initiated on MYQORZO at a dose of 5 mg once daily. Doses were individually titrated (or sham-titrated if on placebo) at Week 2, 4 and 6 if Valsalva LVOT-G was ≥30 mmHg and LVEF was ≥55% in 5 mg dose increments up to a maximum dose of 20 mg once daily. At Week 24, in the MYQORZO group, 46% of patients were on the 20 mg dose, 35% were on the 15 mg dose, 15% were on the 10 mg dose and 4% were on the 5 mg dose.
Primary Endpoint - Peak Oxygen Uptake (pVO2)
In SEQUOIA-HCM, the primary endpoint of change from baseline in pVO2 to Week 24 was greater with MYQORZO compared to placebo, as shown in Table 4.
Table 4: Change from Baseline in pVO2 by CPET at Week 24 in SEQUOIA-HCM pVO2 MYQORZO
(N = 142)Placebo
(N = 140)- * LS mean (SE), and LS mean difference (95% CI) were estimated from an ANCOVA model. A placebo-based multiple imputation approach was used to impute the missing data.
Baseline (mL/min/kg), mean (SD) 18.4 (4.4) 18.6 (4.5) Change from baseline to Week 24 LS mean (SE)* 1.7 (0.3) 0.0 (0.3) LS mean difference vs. placebo (95% CI)* 1.7 (1.0, 2.4) p-value <0.0001 A range of demographic characteristics, baseline disease characteristics, and baseline concomitant medications (e.g., use of beta-blockers) were examined for their influence on outcomes. Results of the primary endpoint analysis consistently favored MYQORZO across all subgroups analyzed (Figure 1).
Figure 1: Forest Plot for Change in Baseline pVO2 by CPET at Week 24 by Subgroups in SEQUOIA-HCM
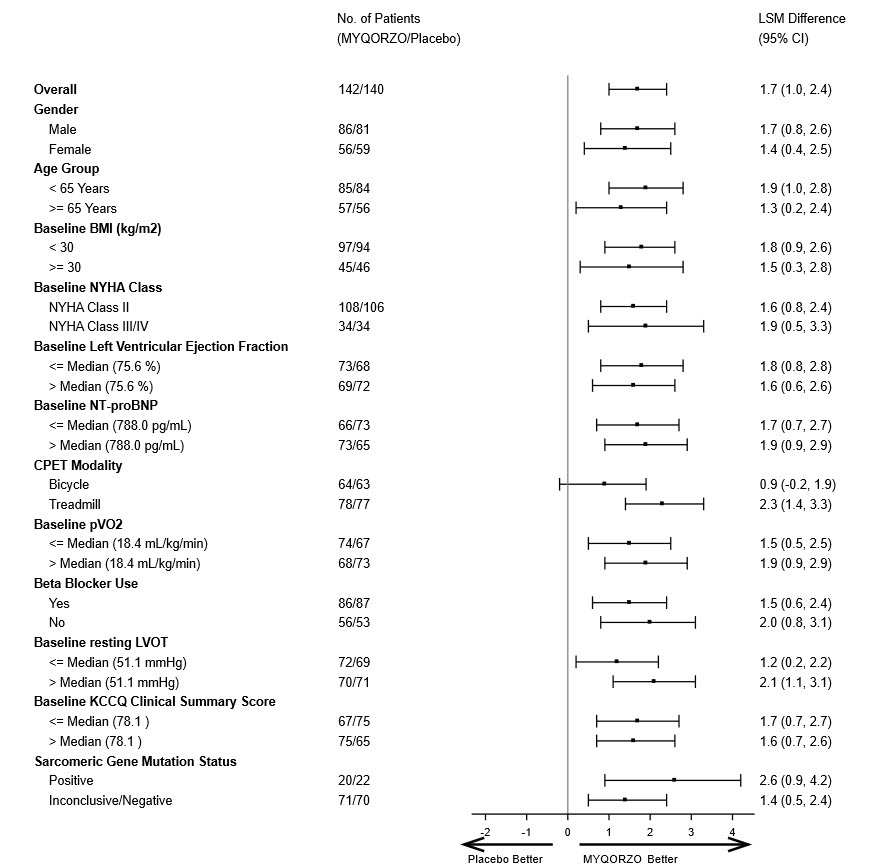
BMI: Body Mass Index; NYHA: New York Heart Association; NT-proBNP: N-terminal pro-B-type natriuretic peptic; CPET: Cardiac Pulmonary Exercise Test; pVO2: Peak Oxygen Uptake; LVOT: left ventricular outflow tract; KCCQ CSS: Kansas City Cardiomyopathy Questionnaire – Clinical Summary Score.
Note: The figure above presents effects in various subgroups, all of which are baseline characteristics. The 95% confidence limits that are shown do not take into account the number of comparisons made and may not reflect the effect of a particular factor after adjustment for all other factors. Apparent homogeneity or heterogeneity among groups should not be over-interpreted.
Secondary Endpoints
The treatment effects of MYQORZO on health status, functional capacity, and LVOT obstruction were assessed by change in the KCCQ-CSS, proportion of patients with ≥1 class improvement in NYHA functional class, change from baseline in Valsalva LVOT-G, proportion of patients with Valsalva LVOT-G ≤30 mmHg, duration of eligibility for septal reduction therapy (SRT), and change in total workload during CPET. At Week 24, patients receiving MYQORZO had greater improvement compared to the placebo group across all secondary points (Table 5, Figure 2 and Figure 3).
Table 5. Results of Secondary Efficacy Endpoints in SEQUOIA-HCM Endpoints MYQORZO
N=142Placebo
N=140LS Mean Difference
(95% CI)p-Value CPET: Cardiac Pulmonary Exercise Test; KCCQ CSS: Kansas City Cardiomyopathy Questionnaire – Clinical Summary Score; NYHA: New York Heart Association; LVOT-G: left ventricular outflow tract gradient; SRT: septal reduction therapy. - * LS means (SE) and LS mean difference (95% CI) was estimated from an ANCOVA model (mixed model with repeated measures was used for longitudinal data) for continuous endpoints. Placebo-based multiple imputation approach was used to impute the missing data.
- † The KCCQ-CSS score ranges from 0 to 100, with higher scores indicating better health status.
- ‡ The number (percentage) of responders and common Odds Ratio (OR) obtained using Cochran-Mantel-Haenszel method (95% CI) are presented for binary endpoints.
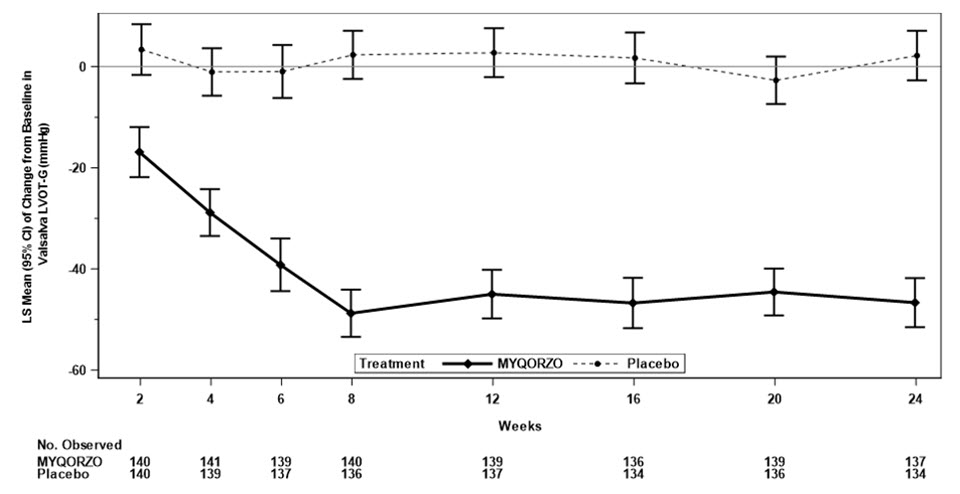
Mean Change from Baseline in KCCQ-CSS*,† Week 12 11.1 (0.9) 4.0 (0.9) 7.0 (4.6, 9.5) <0.0001 Week 24 11.6 (1.0) 4.3 (1.0) 7.3 (4.6, 10.1) <0.0001 Mean Change from Baseline in Valsalva LVOT-G (mmHg)* Week 12 -45.0 (2.4) 2.7 (2.4) -47.7
(-54.5, -41.0)<0.0001 Week 24 -46.7 (2.5) 2.2 (2.5) -48.8
(-55.7, -42.0)<0.0001 Mean Duration of SRT Eligibility* Days spent SRT-eligible during 24 Weeks of Treatment 35 (8) 113 (8) −78
(−100, −56)<0.0001 Mean Change from Baseline in Total Workload (Watts) during CPET* Week 24 13.1 (2.1) 0.9 (2.2) 12.2 (6.3, 18.1) <0.0001 Endpoints MYQORZO
N=142Placebo
N=140Odds Ratio
(95% CI)p-Value Proportion of Patients with ≥1 NYHA Class Improvement‡ Week 12 69 (48.6) 25 (17.9) OR: 4.6
(2.6, 8.4)<0.0001 Week 24 83 (58.5) 34 (24.3) OR: 4.4
(2.6, 7.6)<0.0001 Proportion of Patients with Valsalva LVOT-G <30 (mmHg)‡ Week 12 73 (51.4) 8 (5.7) OR: 16.9
(7.7, 37.2)<0.0001 Week 24 70 (49.3) 5 (3.6) OR: 22.9
(8.8, 59.6)<0.0001 Figure 2: Change from Baseline in KCCQ-CSS through Week 24
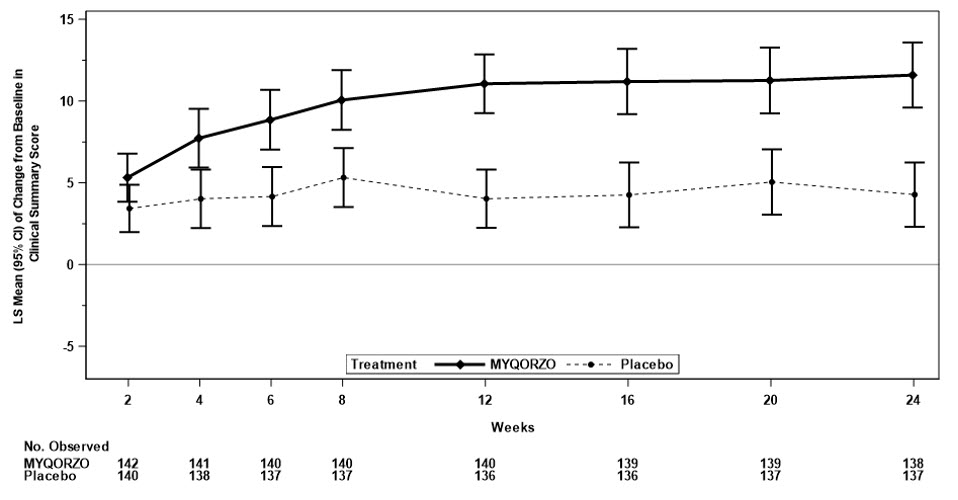
Figure 3: Change from Baseline in Valsalva LVOT-G through Week 24
-
16 HOW SUPPLIED/STORAGE AND HANDLING
MYQORZO is supplied as purple, film-coated tablets containing 5 mg, 10 mg, 15 mg, or 20 mg aficamten. The tablets are debossed on one side with "CK" and the other side with "5", "10", "15" and "20" for the 5, 10, 15, and 20 mg strength, respectively.
Tablets are supplied in bottles with child-resistant closure as follows:
5 mg round tablets Bottles of 30 tablets NDC: 82112-105-30 10 mg triangular tablets Bottles of 30 tablets NDC: 82112-110-30 15 mg pentagonal tablets Bottles of 30 tablets NDC: 82112-115-30 20 mg oval tablets Bottles of 30 tablets NDC: 82112-120-30 -
17 PATIENT COUNSELING INFORMATION
Advise the patient and/or caregiver to read the FDA-approved patient labeling (Medication Guide).
Heart Failure
Inform patients that cardiac function monitoring using echocardiography must be performed before and while on treatment to monitor for signs and symptoms of heart failure [see Dosage and Administration (2.1), Warnings and Precautions (5.1)]. Advise patients to report any signs or symptoms of heart failure immediately to their healthcare provider.
MYQORZO REMS Program
MYQORZO is available only through a restricted program called the MYQORZO REMS Program [see Warnings and Precautions (5.2)]. Inform the patient of the following notable requirements:
Patients must enroll in the program and comply with ongoing monitoring requirements [see Warnings and Precautions (5.1) and (5.2)]
MYQORZO is only prescribed by certified healthcare providers and only dispensed from certified pharmacies participating in the program. Provide patients with the telephone number and website for information on how to obtain the product [see Warnings and Precautions (5.2)].
Pregnancy
Advise patients that there is a pregnancy safety study for MYQORZO. Advise patients to report pregnancy to their prescriber if they become pregnant while receiving MYQORZO or within 3 weeks after their last dose of MYQORZO [see Use in Specific Populations (8.1)].
Drug Interactions
Advise patients to inform their healthcare provider of all concomitant medications they take, prior to and during MYQORZO treatment.
Instructions for Taking MYQORZO
MYQORZO tablets should be swallowed whole. Advise patients that if they miss a dose of MYQORZO, it should be taken as soon as possible on the same day. The next scheduled dose should be taken at the usual time the following day. The patient should not take two doses on the same day.
-
SPL UNCLASSIFIED SECTION
Distributed By:
Cytokinetics Incorporated
South San Francisco, CA 94080MYQORZO and the MYQORZO logo are trademarks of Cytokinetics in the U.S.
Cytokinetics® and the C-shaped logo are trademarks of Cytokinetics in the U.S.
Patent Information: https://cytokinetics.com/patents
©2025 Cytokinetics, Incorporated, All Rights Reserved. -
MEDICATION GUIDE
MEDICATION GUIDE
MYQORZO (my-kor-zo)
(aficamten)
tablets, for oral useThis Medication Guide has been approved by the U.S. Food and Drug Administration. Issued: 12/2025 What is the most important information I should know about MYQORZO?
MYQORZO can cause serious side effects, including:-
Heart failure, a condition where the heart cannot pump with enough force. Heart failure is a serious condition that can lead to death. You must have echocardiograms before and during treatment with MYQORZO and monitor for signs and symptoms of heart failure. People who develop a serious illness such as a serious infection or who develop a new or worsening irregular heartbeat have a greater risk of heart failure during treatment with MYQORZO.
Tell your healthcare provider or get medical help right away if you develop new or worsening:
- shortness of breath
- swelling in your legs
- a racing sensation in your heart (palpitations)
- fatigue
- chest pain
- rapid weight gain
- The risk of heart failure is also increased when MYQORZO is taken with certain other medicines. Tell your healthcare provider about the medicines you take, both prescribed and obtained over-the-counter, before and during your treatment with MYQORZO.
-
Because of the risk of heart failure, MYQORZO is only available through a restricted distribution program called the MYQORZO Risk Evaluation and Mitigation Strategy (REMS) Program.
- Your healthcare provider must be enrolled in the MYQORZO REMS Program for you to be prescribed MYQORZO.
- Before you start treatment with MYQORZO, you must enroll in the MYQORZO REMS Program. Talk to your healthcare provider about how to enroll in the program. You will be given information about the program when you enroll.
- Before you take MYQORZO, your healthcare provider and pharmacist will make sure you understand how to take MYQORZO safely, which will include returning for echocardiograms when advised by your healthcare provider.
MYQORZO can only be dispensed by a certified pharmacy that participates in the MYQORZO REMS Program.
Your healthcare provider can give you information on how to find a certified pharmacy. - If you have any questions about the MYQORZO REMS Program, ask your healthcare provider, go to www.MYQORZOREMS.com, or call 1-844-285-7367.
- See "What are the possible side effects of MYQORZO?" for information about side effects.
What is MYQORZO?
MYQORZO is a prescription medicine used to treat adults with symptomatic obstructive cardiomyopathy (oHCM) to improve functional capacity and symptoms.
It is not known if MYQORZO is safe and effective in children.Who should not take MYQORZO?
Do not take MYQORZO if you take a medicine called rifampin.Before taking MYQORZO, tell your healthcare provider about all of your medical conditions, including if you: - are pregnant or plan to become pregnant. It is not known if MYQORZO can cause harm to your unborn baby. Tell your healthcare provider if you become pregnant during treatment or within 3 weeks after the last dose of MYQORZO. There is a pregnancy safety study for MYQORZO. Your healthcare provider should report your pregnancy exposure to Cytokinetics, Inc.
- are breastfeeding or plan to breastfeed. It is not known if MYQORZO passes into your breast milk. Talk to your healthcare provider about the best way to feed your baby during treatment with MYQORZO.
Especially tell your healthcare provider if you take the medicines:- fluconazole (if used for more than 3 days)
- voriconazole
- fluvoxamine
How should I take MYQORZO? - Take MYQORZO exactly as your healthcare provider tells you to take it.
- Do not change your dose of MYQORZO without talking to your healthcare provider first.
- Take MYQORZO 1 time a day, at about the same time each day,
- Take MYQORZO with or without meals.
- Swallow the MYQORZO tablet whole.
- If you miss a dose of MYQORZO, take it as soon as possible and take your next dose at your regularly scheduled time the next day. Do not take 2 doses on the same day to make up for a missed dose.
- Your healthcare provider may change your dose or temporarily stop your treatment with MYQORZO if you have certain side effects.
- If you take too much MYQORZO, call your healthcare provider or Poison Control Help Line at 1-800-222-1222 or go to the nearest hospital emergency room right away.
What are the possible side effects of MYQORZO?
MYQORZO can cause serious side effects including:- Heart failure. See "What is the most important information I should know about MYQORZO?
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
You may also report side effects to Cytokinetics at 1-833-633-2986.How should I store MYQORZO? - Store MYQORZO at room temperature between 68°F to 77°F (20°C to 25°C).
- MYQORZO comes in a bottle with a child-resistant closure.
General information about the safe and effective use of MYQORZO.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use MYQORZO for a condition for which it was not prescribed. Do not give MYQORZO to other people, even if they have the same symptoms that you have; it may harm them. You can ask your pharmacist or healthcare provider for information about MYQORZO that is written for health professionals.What are the ingredients in MYQORZO?
Active ingredients: aficamten
Inactive ingredients: croscarmellose sodium, hydroxypropyl cellulose, magnesium stearate (non-bovine), mannitol, microcrystalline cellulose, and sodium lauryl sulfate.
The tablet coating contains: carmine, FD&C Blue No. 2, glyceryl mono and dicaprylate, polyvinyl alcohol, polyvinyl alcohol graft polyethylene copolymer, talc and titanium dioxide.
Distributed by: Cytokinetics, Incorporated, South San Francisco, CA 94080
Patent information: https://cytokinetics.com/patents
MYQORZO and the MYQORZO logo are trademarks of Cytokinetics in the U.S. CYTOKINETICS® and the C-shaped logo are trademarks of Cytokinetics in the U.S.
© 2025 Cytokinetics, Incorporated, All Rights Reserved. -
Heart failure, a condition where the heart cannot pump with enough force. Heart failure is a serious condition that can lead to death. You must have echocardiograms before and during treatment with MYQORZO and monitor for signs and symptoms of heart failure. People who develop a serious illness such as a serious infection or who develop a new or worsening irregular heartbeat have a greater risk of heart failure during treatment with MYQORZO.
-
PRINCIPAL DISPLAY PANEL - 5 mg Tablet Bottle Label
NDC: 82112-105-30
MYQORZO™
(aficamten) tablets5 mg
Dispense with attached
Medication GuideRx Only
30 tablets

-
PRINCIPAL DISPLAY PANEL - 10 mg Tablet Bottle Label
NDC: 82112-110-30
MYQORZO™
(aficamten) tablets10 mg
Dispense with attached
Medication GuideRx Only
30 tablets

-
PRINCIPAL DISPLAY PANEL - 15 mg Tablet Bottle Label
NDC: 82112-115-30
MYQORZO™
(aficamten) tablets15 mg
Dispense with attached
Medication GuideRx Only
30 tablets

-
PRINCIPAL DISPLAY PANEL - 20 mg Tablet Bottle Label
NDC: 82112-120-30
MYQORZO™
(aficamten) tablets20 mg
Dispense with attached
Medication GuideRx Only
30 tablets

-
INGREDIENTS AND APPEARANCE
MYQORZO
aficamten tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 82112-105 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength AFICAMTEN (UNII: B1I77MH6K1) (AFICAMTEN - UNII:B1I77MH6K1) AFICAMTEN 5 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) MANNITOL (UNII: 3OWL53L36A) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) SODIUM LAURYL SULFATE (UNII: 368GB5141J) MAGNESIUM SULFATE, UNSPECIFIED FORM (UNII: DE08037SAB) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) GLYCERYL MONOCAPRYLOCAPRATE (UNII: G7515SW10N) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) Product Characteristics Color PURPLE Score no score Shape ROUND Size 5mm Flavor Imprint Code 5;CK Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 82112-105-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 01/12/2026 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA219083 01/12/2026 MYQORZO
aficamten tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 82112-110 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength AFICAMTEN (UNII: B1I77MH6K1) (AFICAMTEN - UNII:B1I77MH6K1) AFICAMTEN 10 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) MANNITOL (UNII: 3OWL53L36A) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) SODIUM LAURYL SULFATE (UNII: 368GB5141J) MAGNESIUM SULFATE, UNSPECIFIED FORM (UNII: DE08037SAB) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) GLYCERYL MONOCAPRYLOCAPRATE (UNII: G7515SW10N) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) Product Characteristics Color PURPLE Score no score Shape TRIANGLE Size 7mm Flavor Imprint Code 10;CK Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 82112-110-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 01/12/2026 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA219083 01/12/2026 MYQORZO
aficamten tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 82112-115 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength AFICAMTEN (UNII: B1I77MH6K1) (AFICAMTEN - UNII:B1I77MH6K1) AFICAMTEN 15 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) MANNITOL (UNII: 3OWL53L36A) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) SODIUM LAURYL SULFATE (UNII: 368GB5141J) MAGNESIUM SULFATE, UNSPECIFIED FORM (UNII: DE08037SAB) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) GLYCERYL MONOCAPRYLOCAPRATE (UNII: G7515SW10N) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) Product Characteristics Color PURPLE Score no score Shape PENTAGON (5 sided) Size 7mm Flavor Imprint Code 15;CK Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 82112-115-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 01/12/2026 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA219083 01/12/2026 MYQORZO
aficamten tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 82112-120 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength AFICAMTEN (UNII: B1I77MH6K1) (AFICAMTEN - UNII:B1I77MH6K1) AFICAMTEN 20 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) MANNITOL (UNII: 3OWL53L36A) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) SODIUM LAURYL SULFATE (UNII: 368GB5141J) MAGNESIUM SULFATE, UNSPECIFIED FORM (UNII: DE08037SAB) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) GLYCERYL MONOCAPRYLOCAPRATE (UNII: G7515SW10N) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) Product Characteristics Color PURPLE Score no score Shape OVAL Size 10mm Flavor Imprint Code 20;CK Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 82112-120-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 01/12/2026 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA219083 01/12/2026 Labeler - Cytokinetics Inc. (022799402)
Trademark Results [MYQORZO]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 MYQORZO 97619234 not registered Live/Pending |
Cytokinetics, Incorporated 2022-10-04 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.