RAPIVAB- peramivir solution
Rapivab by
Drug Labeling and Warnings
Rapivab by is a Prescription medication manufactured, distributed, or labeled by BioCryst Pharmaceuticals, Inc., Alcami Carolinas Corporation, Cilag Ag, Eurofins Lancaster Laboratories Inc., Galbraith Labs Inc., Jubilant HollisterStier LLC, Patheon Manufacturing Services LLC, Siegfried USA, LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use RAPIVAB safely and effectively. See full prescribing information for RAPIVAB.
RAPIVAB® (peramivir) injection, for intravenous use
Initial U.S. Approval: 2014INDICATIONS AND USAGE
RAPIVAB is an influenza virus neuraminidase inhibitor indicated for the treatment of acute uncomplicated influenza in patients 6 months and older who have been symptomatic for no more than two days. (1)
Limitations of Use:
- Efficacy based on clinical trials in which the predominant influenza virus type was influenza A; a limited number of subjects infected with influenza B virus were enrolled. (1)
- Consider available information on influenza drug susceptibility patterns and treatment effects when deciding whether to use. (1)
- Efficacy could not be established in patients with serious influenza requiring hospitalization. (1)
DOSAGE AND ADMINISTRATION
- Administer RAPIVAB as a single dose within 2 days of onset of influenza symptoms (2.1)
- Administer RAPIVAB by intravenous infusion for a minimum of 15 minutes (2.1)
Recommended Dosage Single Dose Adults and adolescents (13 years and older) 600 mg Pediatric patients (6 months to 12 years of age) 12 mg/kg
(up to 600 mg)Recommended Dosage Adjustments in Patients with Altered Creatinine Clearance Creatinine Clearance (mL/min) ≥50 30-49 10-29 a Up to maximum dose of 600 mg. Adults and adolescents
(13 years and older)600 mg 200 mg 100 mg Pediatric patientsa
(2 to 12 years of age)12 mg/kg 4 mg/kg 2 mg/kg - No recommendation for dosage adjustment can be made for pediatric patients 6 months to less than 2 years of age with creatinine clearance less than 50 mL/min (2.2)
- Hemodialysis: Administer after dialysis (2.2)
- RAPIVAB must be diluted prior to administration (2.3)
- See the Full Prescribing Information for drug compatibility information (2.4)
DOSAGE FORMS AND STRENGTHS
Injection: 200 mg in 20 mL (10 mg/mL) in a single-use vial (3)
CONTRAINDICATIONS
Patients with known serious hypersensitivity or anaphylaxis to peramivir or any component of RAPIVAB (4)
WARNINGS AND PRECAUTIONS
- Cases of anaphylaxis and serious skin/hypersensitivity reactions such as Stevens-Johnson syndrome and erythema multiforme have occurred with RAPIVAB. Discontinue RAPIVAB and initiate appropriate treatment if anaphylaxis or serious skin reaction occurs or is suspected. (5.1)
- Neuropsychiatric events: Patients with influenza may be at an increased risk of hallucinations, delirium, and abnormal behavior early in their illness. Monitor for signs of abnormal behavior. (5.2)
ADVERSE REACTIONS
Most common adverse reaction (incidence >2%) is diarrhea. (6)
To report SUSPECTED ADVERSE REACTIONS, contact BioCryst Pharmaceuticals, Inc. at 1-833-633-2279 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
DRUG INTERACTIONS
Live attenuated influenza vaccine (LAIV), intranasal: Avoid use of LAIV within 2 weeks before or 48 hours after administration of RAPIVAB, unless medically indicated (7.1)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosage in Acute Uncomplicated Influenza
2.2 Dosing in Patients with Renal Impairment
2.3 Preparation of RAPIVAB for Intravenous Infusion
2.4 Drug Compatibility
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Serious Skin/Hypersensitivity Reactions
5.2 Neuropsychiatric Events
5.3 Risk of Bacterial Infections
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Influenza Vaccines
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Patients with Impaired Renal Function
8.7 Patients with Serious Influenza Requiring Hospitalization
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Cardiac Electrophysiology
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Acute Uncomplicated Influenza in Adults
14.2 Acute Uncomplicated Influenza in Pediatric Subjects
14.3 Serious Influenza Requiring Hospitalization
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
RAPIVAB is indicated for the treatment of acute uncomplicated influenza in patients 6 months and older who have been symptomatic for no more than 2 days.
Limitations of Use:
- Efficacy of RAPIVAB is based on clinical trials of naturally occurring influenza in which the predominant influenza infections were influenza A virus; a limited number of subjects infected with influenza B virus were enrolled.
- Influenza viruses change over time. Emergence of resistance substitutions could decrease drug effectiveness. Other factors (for example, changes in viral virulence) might also diminish clinical benefit of antiviral drugs. Prescribers should consider available information on influenza drug susceptibility patterns and treatment effects when deciding whether to use RAPIVAB [see Microbiology (12.4)].
- The efficacy of RAPIVAB could not be established in patients with serious influenza requiring hospitalization [see Clinical Studies (14.3)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosage in Acute Uncomplicated Influenza
Administer RAPIVAB within 2 days of onset of symptoms of influenza.
2.2 Dosing in Patients with Renal Impairment
Significantly increased drug exposures were observed when RAPIVAB was administered to adult subjects with renal dysfunction [see Clinical Pharmacology (12.3)]. Therefore, the RAPIVAB dosage should be reduced for patients with baseline creatinine clearance below 50 mL/min using the recommendations in Table 1 and Table 2. No dosage adjustment is required for single administration of RAPIVAB in patients with creatinine clearance of 50 mL/min or higher [see Clinical Pharmacology (12.3)].
In patients with chronic renal impairment maintained on hemodialysis, RAPIVAB should be administered after dialysis at a dose adjusted based on renal function (Table 1 and Table 2) [see Clinical Pharmacology (12.3)].
Table 1. Dosage Adjustment for Adults and Adolescents (13 Years and Older) with Altered Creatinine Clearance Creatinine Clearancea (mL/min) ≥50 30 to 49 10 to 29 a Calculated using the Cockcroft and Gault equation. Recommended Dose (mg) 600 mg 200 mg 100 mg Table 2. Dosage Adjustment for Pediatric Patients (2 to 12 Years of Age) with Altered Creatinine Clearance Creatinine Clearancea (mL/min) ≥50 30 to 49 10 to 29 a Calculated using the Cockcroft and Gault equation. b Up to maximum dose of 600 mg. Recommended Dose (mg/kg)b 12 mg/kg 4 mg/kg 2 mg/kg No data are available to inform a recommendation for dosage adjustment with RAPIVAB in pediatric patients 6 months to less than 2 years of age with creatinine clearance less than 50 mL/min [see Use in Specific Populations (8.4, 8.6), Clinical Pharmacology (12.3)].
2.3 Preparation of RAPIVAB for Intravenous Infusion
Use aseptic technique during the preparation of RAPIVAB to prevent inadvertent microbial contamination. There is no preservative or bacteriostatic agent present in the solution.
Follow the steps below to prepare a diluted solution of RAPIVAB:
- (a) Do not use if seal over bottle opening is broken or missing.
- (b) Visually inspect RAPIVAB for particulate matter and discoloration prior to administration.
- (c)
Dilute an appropriate dose of RAPIVAB 10 mg/mL solution [see Dosage and Administration (2.1, 2.2)] in 0.9% or 0.45% sodium chloride, 5% dextrose, or lactated Ringer's. The maximum infusion volume is provided in Table 3. The final concentration of diluted RAPIVAB for administration should be between 1 mg/mL and 6 mg/mL.
Table 3. Maximum Infusion Volume by Age and Weight Age Weight (kg) Maximum Infusion Volumea (mL) aInfusion volume is the total volume of RAPIVAB 10 mg/mL solution and diluent. The final concentration of diluted RAPIVAB for administration should be between 1 mg/mL and 6 mg/mL. Infants 6 months to 1 year of age Any 25 mL Adults and pediatric patients 1 year and older 5 kg to less than 10 kg 25 mL 10 kg to less than 15 kg 50 mL 15 kg to less than 20 kg 75 mL At least 20 kg 100 mL - (d) Administer the diluted solution via intravenous infusion for 15 to 30 minutes.
- (e) Discard any unused diluted solution of RAPIVAB after 24 hours.
Once a diluted solution of RAPIVAB has been prepared, administer immediately or store under refrigerated conditions (2° to 8°C or 36° to 46°F) for up to 24 hours. If refrigerated, allow the diluted solution of RAPIVAB to reach room temperature then administer immediately.
2.4 Drug Compatibility
RAPIVAB injection is compatible with 0.9% or 0.45% sodium chloride, 5% dextrose, or lactated Ringer's. Do not mix or co-infuse RAPIVAB with other intravenous medications.
RAPIVAB injection is compatible with materials commonly used for administration such as polyvinylchloride (PVC) bags and PVC-free bags, polypropylene syringes, and polyethylene tubing.
-
3 DOSAGE FORMS AND STRENGTHS
Each vial of RAPIVAB injection contains 200 mg per 20 mL (10 mg per mL) as a clear, colorless solution [see How Supplied/Storage and Handling (16)].
-
4 CONTRAINDICATIONS
RAPIVAB is contraindicated in patients with known serious hypersensitivity or anaphylaxis to peramivir or any component of the product. Severe allergic reactions have included anaphylaxis, erythema multiforme, and Stevens-Johnson syndrome [see Warnings and Precautions (5.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Serious Skin/Hypersensitivity Reactions
Rare cases of serious skin reactions, including erythema multiforme, have been reported with RAPIVAB in clinical studies and in postmarketing experience. Cases of anaphylaxis and Stevens-Johnson syndrome have been reported in postmarketing experience with RAPIVAB. Discontinue RAPIVAB and institute appropriate treatment if anaphylaxis or a serious skin reaction occurs or is suspected. The use of RAPIVAB is contraindicated in patients with known serious hypersensitivity or anaphylaxis to RAPIVAB [see Contraindications (4), Adverse Reactions (6.2)].
5.2 Neuropsychiatric Events
Influenza can be associated with a variety of neurologic and behavioral symptoms that can include events such as hallucinations, delirium, and abnormal behavior, in some cases resulting in fatal outcomes. These events may occur in the setting of encephalitis or encephalopathy but can occur in uncomplicated influenza as well.
There have been postmarketing reports of delirium and abnormal behavior leading to injury in patients with influenza who were receiving neuraminidase inhibitors, including RAPIVAB. Because these events were reported voluntarily during clinical practice, estimates of frequency cannot be made, but they appear to be uncommon. These events were reported primarily among pediatric patients and often had an abrupt onset and rapid resolution. The contribution of RAPIVAB to these events has not been established. Patients with influenza should be closely monitored for signs of abnormal behavior.
5.3 Risk of Bacterial Infections
There is no evidence for efficacy of RAPIVAB in any illness caused by agents other than influenza viruses. Serious bacterial infections may begin with influenza-like symptoms or may coexist with or occur as complications during the course of influenza. RAPIVAB has not been shown to prevent such complications.
Prescribers should be alert to the potential for secondary bacterial infections and treat with antibiotics as appropriate.
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of the labeling:
- Serious skin and hypersensitivity reactions [see Warnings and Precautions (5.1)]
- Neuropsychiatric events [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions in Adults (18 years of age and older)
In 5 randomized, double-blind, controlled trials, 1,399 subjects with acute uncomplicated influenza received a single dose of RAPIVAB, administered intravenously or intramuscularly, at doses up to 600 mg. Among the 664 subjects receiving RAPIVAB 600 mg (intravenous or intramuscular), the most commonly observed adverse reaction was diarrhea, occurring at a rate of 8% versus 7% in subjects receiving placebo. No subject receiving RAPIVAB 600 mg experienced a serious adverse event and <1% discontinued study because of an adverse reaction.
Clinically significant laboratory abnormalities (DAIDS Grades 2 to 4) listed in Table 4 occurred more frequently in subjects treated with RAPIVAB 600 mg (intravenous or intramuscular) than placebo. Only events occurring at ≥2% are included.
Table 4: Laboratory Abnormalities Occurring in ≥2% of Subjects Treated with RAPIVAB 600 mg Laboratory Parameter Abnormalitya RAPIVAB 600 mg Placebo a Frequencies based on treatment-emergent laboratory abnormalities. Alanine Aminotransferase (>2.5 × ULN) (n = 654)
3%(n = 430)
2%Serum Glucose (>160 mg/dL) (n = 660)
5%(n = 433)
3%Creatine Phosphokinase (≥6.0 × ULN) (n = 654)
4%(n = 431)
2%Neutrophils (<1.000 ×109/L) (n = 654)
8%(n = 430)
6%In a subset of subjects with serious influenza requiring hospitalization treated with RAPIVAB 600 mg as monotherapy (n = 101), the following adverse reactions were also reported more frequently with RAPIVAB as compared to placebo: constipation (4% versus 2%), insomnia (3% versus 0%), AST increased (3% versus 2%), and hypertension (2% versus 0%).
Adverse Reactions in Adolescent and Pediatric Subjects (6 months to 17 years of age)
Assessment of adverse reactions is based on a randomized, active-controlled study in which 130 adolescent and pediatric subjects ages 6 months to 17 years of age with acute uncomplicated influenza received open-label treatment with a single dose of RAPIVAB (n = 107), or 5 days of treatment with oseltamivir (n = 23) [see Use in Specific Populations (8.4), Clinical Studies (14.2)].
The safety profile of RAPIVAB in subjects 6 months to 17 years of age was generally similar to that observed in adults. The only adverse reaction reported in pediatric subjects treated with RAPIVAB (occurring in ≥2% of subjects) and not reported in adults was vomiting (3% versus 9% for oseltamivir). The only clinically significant laboratory abnormality (DAIDS Grade 2) occurring in ≥2% of pediatric subjects treated with RAPIVAB (and not previously reported in adults) was proteinuria by dipstick analysis (3% versus 0% for oseltamivir).
6.2 Postmarketing Experience
The following additional adverse reactions have been identified during postapproval use of RAPIVAB. Because postmarketing reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Dermatologic: Stevens-Johnson syndrome, exfoliative dermatitis, rash [see Warnings and Precautions (5.1)].
General Disorders and Administration Site Conditions: Anaphylactic/anaphylactoid reactions [see Warnings and Precautions (5.1)].
Psychiatric: Abnormal behavior, hallucination [see Warnings and Precautions (5.2)].
-
7 DRUG INTERACTIONS
This section describes clinically relevant drug interactions with RAPIVAB. Drug-drug interaction studies are described elsewhere in the labeling [see Clinical Pharmacology (12.3)].
7.1 Influenza Vaccines
Inactivated influenza vaccine can be administered at any time relative to use of RAPIVAB. For live attenuated influenza vaccine (LAIV), antiviral drugs may inhibit viral replication and thus may reduce vaccine efficacy. The concurrent use of RAPIVAB with LAIV intranasal has not been evaluated. Because of the potential for interference between these two products, avoid use of LAIV within 2 weeks before or 48 hours after administration of RAPIVAB unless medically indicated.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Limited available data with RAPIVAB use in pregnant women are insufficient to determine a drug-associated risk of adverse developmental outcomes. There are risks to the mother and fetus associated with influenza in pregnancy [see Clinical Considerations]. In animal reproduction studies, no adverse developmental effects were observed in rats when peramivir was administered by intravenous bolus injection during organogenesis at the maximum feasible dose, resulting in systemic drug exposures (AUC) approximately 8 times those in humans at the recommended dose. However, when peramivir was administered to rats by continuous intravenous infusion during the same gestation period, fetal abnormalities of reduced renal papilla and dilated ureters were observed. In rabbits, administration of peramivir during organogenesis at exposures 8 times those in humans at the recommended dose resulted in developmental toxicity (abortion or premature delivery) at a maternally toxic dose [see Data].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Pregnant women are at higher risk of severe complications from influenza, which may lead to adverse pregnancy and/or fetal outcomes including maternal death, stillbirths, birth defects, preterm delivery, low birthweight, and small for gestational age.
Data
Animal Data
Reproductive toxicity studies have been performed in rats and rabbits. In rats, peramivir was administered once daily by intravenous bolus injection at doses of 200, 400, and 600 mg/kg/day on Gestational Days 6 to 17. No treatment-related fetal toxicities were observed when peramivir was administered by intravenous bolus injection at the maximum feasible dose of 600 mg/kg, resulting in exposures approximately 8 times those in humans at the recommended dose.
Peramivir was also administered by continuous intravenous infusion to rats at daily doses of 50, 400, and 1000 mg/kg/day on Gestational Days 6 to 17. Dose related increases in the incidence of fetal abnormalities of reduced renal papilla and dilated ureters were observed at 400 and 1000 mg/kg/day. The systemic drug exposure in rats at a dose without fetal effects was less than the exposures in humans at the recommended dose.
In rabbits, peramivir was administered once daily by intravenous bolus injection at doses of 25, 50, 100, and 200 mg/kg/day on Gestational Days 7 to 19. Developmental toxicity (abortion or premature delivery) was observed at maternally toxic dose levels (100 and 200 mg/kg/day) resulting in exposures approximately 8 times those in humans at the recommended dose. The exposure in rabbits at doses without developmental toxicity was less than the exposure in humans at the recommended dose.
A pre/post-natal developmental toxicity study was performed in pregnant rats administered peramivir once daily by intravenous infusion at doses of 50, 200, 400, and 600 mg/kg/day on Gestational Day 6 through Lactation Day 20. No significant effects of peramivir on developmental outcomes were observed in nursing pups at up to the highest dose tested.
8.2 Lactation
Risk Summary
There are no data on the presence of RAPIVAB in human milk, the effects on the breastfed infant, or the effects on milk production. Peramivir is present in rat milk [see Data]. Limited clinical data during lactation preclude a clear determination of the risk of RAPIVAB to an infant during lactation; therefore, the developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for RAPIVAB and any potential adverse effects on the breastfed child from the drug or from the underlying maternal condition.
Data
A pharmacokinetic study was performed in lactating rats administered a single intravenous dose of peramivir (10 mg/kg) on Lactation/Postpartum Days 11 to 13. The maximum concentration of peramivir in milk was reached at 0.75 hours post-dose. The milk to plasma AUC ratio of peramivir was approximately 0.5.
8.4 Pediatric Use
The safety and effectiveness of RAPIVAB for the treatment of influenza has been established in pediatric patients 6 months to 17 years of age. Use of RAPIVAB for this indication is supported by evidence from adequate and well-controlled trials of RAPIVAB in adults with additional data from Study 305, a randomized, active-controlled trial of 130 adolescent and pediatric subjects with acute uncomplicated influenza who received open-label treatment with a single dose of RAPIVAB or 5 days of treatment with oseltamivir administered within 48 hours of onset of symptoms of influenza [see Dosage and Administration (2.1, 2.2, 2.3), Adverse Reactions (6.1), Clinical Pharmacology (12.3), Clinical Studies (14.2)]. Study 305 included:
- 13 to 17 years of age: 21 subjects treated with RAPIVAB 600 mg
- 6 months to 12 years of age: 86 subjects treated with RAPIVAB 12 mg/kg (up to a maximum dose of 600 mg)
Safety and effectiveness of RAPIVAB in pediatric patients less than 6 months of age have not been established. No data are available for RAPIVAB use in pediatric patients 6 months to less than 2 years with creatinine clearance <50 mL/min to inform a recommendation for dosage adjustment [see Dosage and Administration (2.2), Clinical Pharmacology (12.3)].
8.5 Geriatric Use
Clinical trials of RAPIVAB did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in exposures between the elderly and younger subjects [see Clinical Pharmacology (12.3)].
8.6 Patients with Impaired Renal Function
A reduced dose of RAPIVAB is recommended for patients 2 years and older with creatinine clearance <50 mL/min [see Dosage and Administration (2.2), Clinical Pharmacology (12.3)]. Dose adjustment is not required for a single administration of RAPIVAB for patients with creatinine clearance ≥50 mL/min [see Dosage and Administration (2.2), Clinical Pharmacology (12.3)].
In patients with chronic renal impairment maintained on hemodialysis, RAPIVAB should be administered after dialysis at a dose adjusted based on renal function [see Dosage and Administration (2.2), Clinical Pharmacology (12.3)].
No data are available for RAPIVAB use in pediatric patients 6 months to less than 2 years with creatinine clearance <50 mL/min to inform a recommendation for dosage adjustment [see Dosage and Administration (2.2), Clinical Pharmacology (12.3)].
8.7 Patients with Serious Influenza Requiring Hospitalization
The use of RAPIVAB was not shown to provide benefit in patients with serious influenza requiring hospitalization [see Indications and Usage (1), Clinical Studies (14.2)].
-
10 OVERDOSAGE
There is no human experience of acute overdosage with RAPIVAB. Treatment of overdosage with RAPIVAB should consist of general supportive measures including monitoring of vital signs and observation of the clinical status of the patient. There is no specific antidote for overdose with RAPIVAB.
RAPIVAB is cleared by renal excretion and can be cleared by hemodialysis.
-
11 DESCRIPTION
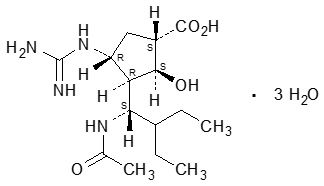
RAPIVAB (peramivir) is an inhibitor of influenza virus neuraminidase. The chemical name is (1S,2S,3R,4R)-3-[(1S)-1-(acetylamino)-2-ethylbutyl]-4-(carbamimidoylamino)-2-hydroxycyclopentanecarboxylic acid, trihydrate. The chemical formula is C15H28N4O4 ∙ 3H2O, representing a molecular weight of 382.45. The molecular structure is as follows:
RAPIVAB injection is a clear, colorless, sterile, isotonic solution (200 mg per 20 mL) in glass vials fitted with rubber stoppers and royal blue flip-off seals. Each mL contains 10 mg peramivir (on an anhydrous basis) in 0.9% sodium chloride solution. The pH may have been adjusted with sodium hydroxide, USP and/or hydrochloric acid, USP. The pH is 5.5 to 8.5.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Peramivir is an antiviral drug with activity against influenza virus [see Microbiology (12.4)].
12.2 Cardiac Electrophysiology
At twice the maximum recommended dose, RAPIVAB did not prolong the QTc interval to any clinically relevant extent.
12.3 Pharmacokinetics
The pharmacokinetics of RAPIVAB was evaluated in Phase 1 trials in adults. The pharmacokinetic parameters following intravenous administration of RAPIVAB (0.17 to 2 times the recommended dose) showed a linear relationship between dose and exposure parameters (Cmax and AUC).
Following intravenous administration of a single dose of RAPIVAB 600 mg over 30 minutes, a maximum plasma concentration (Cmax) of 46,800 ng/mL (46.8 µg/mL) was reached at the end of infusion. AUC0-∞ values were 102,700 ng∙hr/mL.
Distribution
In vitro binding of peramivir to human plasma proteins is <30%.
Based on a population pharmacokinetic analysis, the central volume of distribution was 12.56 L.
Metabolism and Elimination
Peramivir is not a substrate for cytochrome P450 (CYP) enzymes, does not affect glucuronidation, and is not a substrate or inhibitor of P-glycoprotein mediated transport.
Peramivir is not significantly metabolized in humans.
The elimination half-life of RAPIVAB following intravenous administration to healthy subjects of 600 mg as a single dose is approximately 20 hours. The major route of elimination of RAPIVAB is via the kidney. Renal clearance of unchanged peramivir accounts for approximately 90% of total clearance. Negligible accumulation was observed following multiple doses, either once or twice daily, for up to 10 days.
Specific Populations
Race: Pharmacokinetics of peramivir was evaluated primarily in Caucasians and Asians. Based on a population pharmacokinetic analysis including race as a covariate, volume of distribution was dependent on weight and Asian race. No dose adjustment is required based on weight or Asian race.
Pediatric Patients: The pharmacokinetics of peramivir has been evaluated in a study in pediatric subjects 6 months to 17 years of age with acute uncomplicated influenza. Pharmacokinetic sampling in this study was limited to approximately 3 hours after administration of peramivir. Geometric mean (GM) PK parameters are provided in Table 5.
Table 5. Geometric Mean (%CV) Cmax and AUC0-3 by Age Group in Comparison to Adults Age Group GM Cmax (ng/mL) (%CV) GM AUC0-3 (ng.h/mL) (%CV) 6 months to <2 years 38,000 (73.7) 46,200 (35.8) 2 to <7 years 47,400 (48.4) 62,700 (39.7) 7 to <13 years 61,200 (53) 76,300 (43.1) 13 to <18 years 51,500 (33) 65,500 (28.1) Healthy Adults (Study 113) 45,700 (21.5) 68,500 (19.1) Peramivir pharmacokinetics in subjects 2 to 17 years of age was similar to adults. In pediatric patients 6 months to less than 2 years of age, the GM AUC0-3 and Cmax were lower than that of healthy adult subjects, with GM ratios (90% CI) of 0.68 (0.52 to 0.88) and 0.83 (0.59 to 1.18), respectively. The difference in exposure is not considered to be clinically significant.
Geriatric Patients: Peramivir pharmacokinetics in elderly subjects was similar to non-elderly subjects. Peak concentrations of peramivir after a single 4 mg/kg intravenous dose were approximately 10% higher in elderly subjects when compared to young adults (22,647 vs 20,490 ng/mL, respectively). Exposure (AUC0-12) to peramivir at steady state was roughly 34% higher in elderly subjects compared to young adults (61,572 vs 46,000 ng∙hr/mL, respectively). Dose adjustment is not required for elderly patients.
Patients with Impaired Renal Function: A trial was conducted in adult subjects with various degrees of renal impairment. When compared to a concurrent cohort with normal renal function, no change in mean Cmax was observed (6 subjects per cohort). However, mean AUC0-∞ after a single 2 mg/kg intravenous dose was increased by 28%, by 302%, and by 412% in subjects with creatinine clearance 50 to 79, 30 to 49, and 10 to 29 mL/min, respectively.
Hemodialysis was effective in reducing systemic exposure of peramivir by 73% to 81%.
A reduced dose of RAPIVAB is recommended for adult and adolescent patients 13 years and older with creatinine clearance <50 mL/min [see Dosage and Administration (2.2)].
The pharmacokinetics of peramivir has not been studied in pediatric subjects with renal impairment. Given that the pharmacokinetics in pediatric subjects with normal renal function is comparable to that observed in adults, the same proportional dose reduction is recommended in pediatric patients with renal impairment >2 years of age [see Dosage and Administration (2.2)].
In pediatric patients with renal impairment less than 2 years of age, given the developmental immaturity of renal function in this age group, a recommendation for dose reduction cannot be made [see Dosage and Administration (2.2)].
Assessment of Drug Interactions
The potential for CYP-mediated interactions involving RAPIVAB with other drugs is low, based on the known elimination pathway of RAPIVAB, and data from in vitro studies indicating RAPIVAB does not induce or inhibit CYP P450.
There was no evidence of drug-drug interactions when RAPIVAB was administered with oral rimantadine, oseltamivir, or oral contraceptives containing ethinyl estradiol and levonorgestrel; or when peramivir IM was administered with oral probenecid.
RAPIVAB is primarily cleared in the urine by glomerular filtration.
12.4 Microbiology
Mechanism of Action
Peramivir is an inhibitor of influenza virus neuraminidase, an enzyme that releases viral particles from the plasma membrane of infected cells. The median neuraminidase inhibitory activities (IC50 values) of peramivir in biochemical assays against influenza A/H1N1 virus, influenza A/H3N2 virus, and influenza B virus clinical isolates were 0.16 nM (n = 44; range: 0.01 to 1.77 nM), 0.13 nM (n = 32; range: 0.05 to 11 nM), and 0.99 nM (n = 39; range: 0.04 to 54.2 nM), respectively, in a neuraminidase assay with a fluorescently labeled MUNANA substrate.
Antiviral Activity
The antiviral activity of peramivir against laboratory strains and clinical isolates of influenza virus was determined in cell culture. The concentrations of peramivir required for inhibition of influenza virus in cell culture varied depending on the assay method used and the virus tested. The median 50% effective concentrations (EC50 values) of peramivir in cell culture assays were 2.6 nM (n = 13; range: 0.09 to 21 nM), 0.08 nM (n = 17; range: 0.01 to 1.9 nM) and 4.8 nM (n = 11; range: 0.06 to 120 nM) for influenza A/H1N1 virus, A/H3N2 virus, and B virus strains, respectively.
The relationship between the antiviral activity in cell culture, inhibitory activity in the neuraminidase assay, and the inhibition of influenza virus replication in humans has not been established.
Resistance
Cell culture: Influenza A and B virus isolates with reduced susceptibility to peramivir were recovered by serial passage of virus in cell culture in the presence of increasing concentrations of peramivir. Reduced susceptibility of influenza virus to inhibition by peramivir may be conferred by amino acid substitutions in the viral neuraminidase or hemagglutinin proteins (Table 6).
Table 6: Amino Acid Substitutions Selected by Peramivir in Cell Culture Studies Influenza Virus Type/Subtype Protein A/H1N1a A/H3N2b Bc a Numbering based on A/California/04/2009. b Numbering based on A/Texas/50/2012. c Numbering based on B/Massachusetts/02/2012. d Numbering begins after the predicted signal peptide. HAd D125S, R208K N63K, G78D, N145D, K189E T139N, G141E, R162M, D195N, T198N, Y319H NA N58D, I211T, H275Y - H273Y In vivo: Influenza A and B virus isolates with amino acid substitutions associated with reduced susceptibility to peramivir were observed in clinical isolates collected during clinical trials with peramivir (Table 7). Amino acid substitutions have also been observed in viral isolates sampled during community surveillance studies which may be associated with reduced susceptibility to peramivir (Table 7). The clinical impact of this reduced susceptibility is unknown and may be strain dependent.
Table 7: Neuraminidase and Hemagglutinin Amino Acid Substitutions Associated with Reduced Susceptibility to Peramivir in Clinical Virus Isolates Influenza Virus Type / Subtype Protein A/H1N1a A/H3N2b Bc a Numbering based on A/California/04/2009. b Numbering based on A/Texas/50/2012. c Numbering based on B/Massachusetts/02/2012. d Substitutions at this site may be potential cell culture artifacts. NA Clinical Trial R152K, H275Y R292K, N294S - Community Surveillance Studies G147R, I223R/V, S247N/R, H275Y, N295S, I427T E119V, Q136K, V143M+S315R, D151A/E/G/N/Vd, V313A, Q391K G104E, E105K, I115T, H134N/Y, P139S, G145E/R, D197E/N/Y, A200T, I221T/V, G243S, A245T, G247D+I361V, H273Y, N294S, K360E, R374K, A395E, D432G/N HA Clinical Trial V479F - - Circulating seasonal influenza strains expressing neuraminidase resistance-associated substitutions have been observed in individuals who have not received RAPIVAB. Prescribers should consider available information from the CDC on influenza virus drug susceptibility patterns and treatment effects when deciding whether to use RAPIVAB.
Zoonotic Viruses: Amino acid substitutions have been observed in H5N1 and H7N9 clinical viral isolates that conferred reduced susceptibility to peramivir in neuraminidase biochemical assays (Table 8). The clinical impact of reduced susceptibility in these viruses is unknown, and the effects of specific substitutions on virus susceptibility to peramivir may be strain dependent.
Table 8. Amino Acid Substitutions Observed in Avian Influenza Viruses with Zoonotic Potential and Associated with Reduced Susceptibility to Peramivir Influenza Virus Type/Subtype Protein A/H5N1a A/H7N9b a Numbering based on A/California/04/2009. b Numbering based on A/Texas/50/2012. NA H275Y R292K Cross Resistance
Cross-resistance between peramivir, oseltamivir, and zanamivir was observed in neuraminidase biochemical assays and cell culture assays. The amino acid substitutions that resulted in reduced susceptibility to peramivir and either oseltamivir or zanamivir are summarized in Table 9. The clinical impact of this reduced susceptibility is unknown and may be strain dependent.
Table 9: Summary of Amino Acid Substitutions with Cross-Resistance between Peramivir and Oseltamivir or Zanamivir in Susceptibility Assays Influenza Virus Type/Subtype Protein A/H1N1a A/H3N2b Bc a Numbering based on A/California/04/2009. b Numbering based on A/Texas/50/2012. c Numbering based on B/Massachusetts/02/2012. d Numbering begins after the predicted signal peptide. e Substitutions at this site may be potential cell culture artifacts. Oseltamivir HAd - N63K, N145D - NA E119D/V, D151G/N, R152K, Y155H, D199G, I223R/T/V, S247N, G249R+I267V, H275Y, N295S, Q313R, R368K, I427T E119I/V, I222V, S247P, R292K, N294S, V313A G104E, E105K, G108E, P139S, G140R, G145R, D197E/N/Y, A200T, I221T/V/L, G243S, A245T, H273Y, N294S, R374K, A395E, G407S Zanamivir HAd - N63K, N145D - NA E119D/G, Q136Ke, R152K, Y155H, D199G, I223T, I223R+H275Y, S247N, G249R+I267V, N295S, Q313R, R368K, I427T E119G/V, T148I, D151A/G/N/V, I222V, S247P, R292K, N294S G104E, E105K, G108E, E117A/D/G, H134N, P139S, G145R, R150K, D197E/N/Y, A200T, I221T/L, G243S, A245T, G247D+I361V, R292K, R374K, G407S Cross-resistance between neuraminidase (NA) inhibitors and M2 ion channel inhibitors (adamantanes), or between NA inhibitors and polymerase acidic (PA) endonuclease inhibitors (baloxavir), is not expected because these classes of drugs target different viral proteins. However, a virus may carry multiple substitutions that each confer resistance to a different inhibitor class and together can confer multi-class resistance. The clinical relevance of phenotypic cross-resistance evaluations has not been established and may be strain dependent.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity studies by intravenous injection of peramivir were not performed. However, in an oral carcinogenicity study in Sprague-Dawley rats no drug-related neoplasms were observed at drug exposures 0.2- to 0.5-fold that of humans at the clinically recommended dose of 600 mg/day.
13.2 Animal Toxicology and/or Pharmacology
Peramivir caused renal tubular necrosis and abnormal renal function in rabbits. Toxicities included tubular dilatation and necrosis with protein casts in cortical areas, dilated tubules with mineralization in corticomedullary junction areas, and multifocal tubular regeneration. The rabbit appeared to be the sensitive species for peramivir renal toxicity, which was noted at exposures approximately 2- to 4-fold those in humans at the clinically recommended dose.
-
14 CLINICAL STUDIES
14.1 Acute Uncomplicated Influenza in Adults
Study 621 was a randomized, multicenter, blinded study conducted in Japan that evaluated a single intravenous administration of RAPIVAB 300 mg, RAPIVAB 600 mg, or placebo administered over 30 minutes in subjects 20 to 65 years of age with acute uncomplicated influenza. Subjects were eligible if they had fever ≥38°C (axillary) and a positive rapid antigen test for influenza virus, accompanied by at least 2 symptoms (cough, nasal symptoms, sore throat, myalgia, chills/sweats, malaise, fatigue, or headache). In addition, all subjects enrolled were allowed to take fever-reducing medications.
Study treatment was started within 48 hours of onset of symptoms. Subjects participating in the study were required to self-assess their influenza symptoms as "none", "mild", "moderate", or "severe" twice daily. The primary endpoint, time to alleviation of symptoms, was defined as the number of hours from initiation of study drug until the start of the 24-hour period in which all 7 symptoms of influenza (cough, sore throat, nasal congestion, headache, feverishness, myalgia, and fatigue) were either absent or present at a level no greater than mild for at least 21.5 hours.
The overall efficacy population, consisting of subjects with confirmed influenza and administered study drug, totaled 297 subjects. Among the 98 subjects enrolled in the RAPIVAB 600 mg dose group, the mean age was 34 years; 55% were male; 34% were smokers; 99% were infected with influenza A virus and 1% were infected with influenza B virus. The majority of subjects (53%) had influenza illness lasting <24 hours at the time of presentation.
Overall, subjects receiving RAPIVAB 600 mg experienced alleviation of their combined influenza symptoms a median of 21 hours sooner than those receiving placebo. The median time to recovery to normal temperature (<37°C) in the 600 mg group was approximately 12 hours sooner compared to placebo.
Insufficient numbers of subjects infected with influenza B virus were enrolled to determine efficacy of RAPIVAB in this influenza type.
14.2 Acute Uncomplicated Influenza in Pediatric Subjects
Study 305 was a randomized, multicenter, open-label, active-controlled trial to evaluate the safety, pharmacokinetics, and efficacy of a single intravenous dose of RAPIVAB administered for a minimum of 15 minutes in subjects 6 months to 17 years of age with acute uncomplicated influenza who had fever ≥37.8°C (oral) with at least one respiratory symptom (cough or rhinitis) or a positive influenza rapid antigen test. Study treatment was started within 48 hours of onset of symptoms. Subjects were randomized to receive RAPIVAB 600 mg (13 to 17 years of age), RAPIVAB 12 mg/kg up to a maximum dose of 600 mg (6 months to 12 years of age), or oral oseltamivir taken twice daily for 5 days. In addition, all enrolled subjects were allowed to take fever-reducing medications.
The overall efficacy population, consisting of subjects with confirmed influenza who were administered study drug, totaled 97 subjects. Among the 81 subjects treated with RAPIVAB, the median age was 7.5 years; 52% were male; 60% were infected with influenza A virus, 33% were infected with influenza B virus, and 6% were co-infected with influenza A and B viruses.
The primary endpoint was the safety of peramivir compared to oseltamivir as measured by adverse events, laboratory analysis, vital signs, and physical exams. Secondary endpoints included efficacy outcomes such as time to resolution of influenza symptoms and time to resolution of fever; however, the study was not powered to detect statistically significant differences in these secondary endpoints. Subjects receiving RAPIVAB experienced a median time to alleviation of their combined influenza symptoms of 79 hours (interquartile range: 31 to 126 hours) compared to 100 hours (interquartile range: 57 to 145 hours) in subjects receiving oseltamivir. The median time to recovery to normal temperature (<37°C) was 40 hours (interquartile range: 21 to 68 hours) and 35 hours (interquartile range: 16 to 42 hours) in subjects receiving RAPIVAB and oseltamivir, respectively [see Use in Specific Populations (8.4)].
14.3 Serious Influenza Requiring Hospitalization
The efficacy of RAPIVAB could not be established in patients with serious influenza requiring hospitalization [see Indications and Usage (1)].
A randomized, double-blind, multicenter, placebo-controlled trial (Study 301) was conducted in 398 subjects with serious influenza requiring hospitalization. Subjects were randomized to receive RAPIVAB 600 mg daily for 5 days plus standard of care versus standard of care plus placebo within 72 hours of start of symptoms. The primary endpoint was time to clinical resolution defined as the time in hours from initiation of study treatment until resolution of at least 4 of 5 signs (temperature, oxygen saturation, respiration rate, heart rate, or systolic blood pressure), maintained for at least 24 hours. RAPIVAB plus standard of care did not improve median time to clinical resolution compared with standard of care alone.
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Advise patients of the following:
- There is a risk of severe allergic reactions (including anaphylaxis) or serious skin reactions with RAPIVAB use. Advise patients to seek immediate medical attention if an allergic-like reaction occurs or is suspected [see Warnings and Precautions (5.1)].
- There is a risk of neuropsychiatric events in patients with influenza. Patients should contact their physician if they experience signs of abnormal behavior after receiving RAPIVAB [see Warnings and Precautions (5.2)].
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL - 200 mg/20 mL Vial Carton
Rx Only
NDC: 72769-181-03
Rapivab®
peramivir injection200 mg/20 mL per vial
(10 mg/mL)For Intravenous
Infusion Only
Dilute Before UseCarton contains 3 vials

-
INGREDIENTS AND APPEARANCE
RAPIVAB
peramivir solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72769-181 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Peramivir (UNII: QW7Y7ZR15U) (Peramivir Anhydrous - UNII:9ZS94HQO3B) Peramivir Anhydrous 600 mg in 60 mL Inactive Ingredients Ingredient Name Strength Sodium Chloride (UNII: 451W47IQ8X) Hydrochloric Acid (UNII: QTT17582CB) Sodium Hydroxide (UNII: 55X04QC32I) Water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72769-181-03 3 in 1 CARTON 12/20/2014 1 NDC: 72769-181-01 20 mL in 1 VIAL, GLASS; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA206426 12/20/2014 Labeler - BioCryst Pharmaceuticals, Inc. (618194609) Establishment Name Address ID/FEI Business Operations Alcami Carolinas Corporation 117877975 ANALYSIS(72769-181) Establishment Name Address ID/FEI Business Operations Cilag Ag 483237103 API MANUFACTURE(72769-181) , ANALYSIS(72769-181) Establishment Name Address ID/FEI Business Operations Eurofins Lancaster Laboratories Inc. 069777290 ANALYSIS(72769-181) Establishment Name Address ID/FEI Business Operations Galbraith Laboratories Inc. 042456145 ANALYSIS(72769-181) Establishment Name Address ID/FEI Business Operations Jubilant HollisterStier LLC 069263643 MANUFACTURE(72769-181) , PACK(72769-181) , LABEL(72769-181) , ANALYSIS(72769-181) Establishment Name Address ID/FEI Business Operations Patheon Manufacturing Services LLC 079415560 MANUFACTURE(72769-181) , PACK(72769-181) , LABEL(72769-181) , ANALYSIS(72769-181) Establishment Name Address ID/FEI Business Operations Siegfried USA, LLC 001213784 API MANUFACTURE(72769-181) , ANALYSIS(72769-181)
Trademark Results [Rapivab]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 RAPIVAB 86141883 4778059 Live/Registered |
SEQIRUS UK LIMITED 2013-12-12 |
 RAPIVAB 85105828 not registered Dead/Abandoned |
BioCryst Pharmaceuticals, Inc. 2010-08-12 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.