TAFINLAR- dabrafenib capsule
Tafinlar by
Drug Labeling and Warnings
Tafinlar by is a Prescription medication manufactured, distributed, or labeled by Novartis Pharmaceuticals Corporation. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TAFINLAR safely and effectively. See full prescribing information for TAFINLAR.
TAFINLAR® (dabrafenib) capsules, for oral use
Initial U.S. Approval: 2013RECENT MAJOR CHANGES
INDICATIONS AND USAGE
TAFINLAR is a kinase inhibitor indicated as a single agent for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test. (1.1, 2.1)
TAFINLAR is indicated, in combination with trametinib, for:
- the treatment of patients with unresectable or metastatic melanoma with BRAF V600E or V600K mutations as detected by an FDA-approved test. (1.2, 2.1)
- the adjuvant treatment of patients with melanoma with BRAF V600E or V600K mutations, as detected by an FDA-approved test, and involvement of lymph node(s), following complete resection. (1.3, 2.1)
- the treatment of patients with metastatic non-small cell lung cancer (NSCLC) with BRAF V600E mutation as detected by an FDA-approved test. (1.4, 2.1)
- the treatment of patients with locally advanced or metastatic anaplastic thyroid cancer (ATC) with BRAF V600E mutation and with no satisfactory locoregional treatment options. (1.5, 2.1)
Limitations of Use: TAFINLAR is not indicated for treatment of patients with wild-type BRAF melanoma, wild-type BRAF NSCLC, or wild-type BRAF ATC. (1.6, 5.2)
DOSAGE AND ADMINISTRATION
- The recommended dosage of TAFINLAR is 150 mg orally twice daily. Take TAFINLAR at least 1 hour before or at least 2 hours after a meal. (2)
DOSAGE FORMS AND STRENGTHS
Capsules: 50 mg, 75 mg (3)
CONTRAINDICATIONS
- None. (4)
WARNINGS AND PRECAUTIONS
-
New Primary Malignancies, Cutaneous, and Non-cutaneous can occur when TAFINLAR is administered as a single agent or with trametinib. Monitor patients for new malignancies prior to, or while on therapy, and following discontinuation of treatment. (5.1, 2.7)
-
Tumor Promotion in BRAF Wild-type Tumors: Increased cell proliferation can occur with BRAF inhibitors. (5.2, 2.1)
-
Hemorrhage: Major hemorrhagic events can occur in patients receiving TAFINLAR with trametinib. Monitor for signs and symptoms of bleeding. (5.3)
-
Cardiomyopathy: Assess left ventricular ejection fraction (LVEF) before treatment with TAFINLAR and trametinib, after one month of treatment, then every 2 to 3 months thereafter. (5.4, 2.7)
-
Uveitis: Perform ophthalmologic evaluation for any visual disturbances. (5.5, 2.7)
-
Serious Febrile Reactions: Incidence and severity of pyrexia are increased with TAFINLAR and trametinib. (5.6, 2.7)
-
Serious Skin Toxicities: Monitor for skin toxicities. Discontinue for intolerable Grade 2 or for Grade 3 or 4 rash not improving within 3 weeks despite interruption of TAFINLAR. Permanently discontinue for severe cutaneous adverse reactions (SCARs). (5.7, 2.7)
-
Hyperglycemia: Monitor serum glucose levels in patients with preexisting diabetes or hyperglycemia. (5.8)
-
Glucose-6-phosphate Dehydrogenase Deficiency (G6PD): Closely monitor for hemolytic anemia. (5.9)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of potential risk to a fetus and to use an effective non-hormonal method of contraception. (5.11, 8.1, 8.3)
ADVERSE REACTIONS
Most common adverse reactions (≥ 20%) for TAFINLAR as a single agent are hyperkeratosis, headache, pyrexia, arthralgia, papilloma, alopecia, and palmar-plantar erythrodysesthesia syndrome. (6.1)
Most common adverse reactions (≥ 20%) for TAFINLAR, in combination with trametinib, include:
- Unresectable or metastatic melanoma: pyrexia, rash, chills, headache, arthralgia, and cough. (6.1)
- Adjuvant treatment of melanoma: pyrexia, fatigue, nausea, headache, rash, chills, diarrhea, vomiting, arthralgia, and myalgia. (6.1)
- NSCLC: pyrexia, fatigue, nausea, vomiting, diarrhea, dry skin, decreased appetite, edema, rash, chills, hemorrhage, cough, and dyspnea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 4/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 BRAF V600E Mutation-Positive Unresectable or Metastatic Melanoma
1.2 BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma
1.3 Adjuvant Treatment of BRAF V600E or V600K Mutation-Positive Melanoma
1.4 BRAF V600E Mutation-Positive Metastatic NSCLC
1.5 BRAF V600E Mutation-Positive Locally Advanced or Metastatic Anaplastic Thyroid Cancer
1.6 Limitations of Use
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
2.2 Recommended Dosage for Unresectable or Metastatic Melanoma
2.3 Recommended Dosage for the Adjuvant Treatment of Melanoma
2.4 Recommended Dosage for NSCLC
2.5 Recommended Dosage for ATC
2.6 Administration
2.7 Dosage Modifications for Adverse Reactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 New Primary Malignancies
5.2 Tumor Promotion in BRAF Wild-Type Tumors
5.3 Hemorrhage
5.4 Cardiomyopathy
5.5 Uveitis
5.6 Serious Febrile Reactions
5.7 Serious Skin Toxicities
5.8 Hyperglycemia
5.9 Glucose-6-Phosphate Dehydrogenase Deficiency
5.10 Risks Associated With Combination Treatment
5.11 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on TAFINLAR
7.2 Effects of TAFINLAR on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 BRAF V600E Mutation-Positive Unresectable or Metastatic Melanoma – TAFINLAR As a Single Agent
14.2 BRAF V600E or V600K Unresectable or Metastatic Melanoma – TAFINLAR With Trametinib
14.3 Adjuvant Treatment of BRAF V600E or V600K Mutation-Positive Melanoma
14.4 BRAF V600E Mutation-Positive Metastatic Non-Small Cell Lung Cancer
14.5 BRAF V600E Mutation-Positive Locally Advanced or Metastatic Anaplastic Thyroid Cancer
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1
INDICATIONS AND USAGE
1.1 BRAF V600E Mutation-Positive Unresectable or Metastatic Melanoma
TAFINLAR® is indicated as a single agent for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test.
1.2 BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma
TAFINLAR is indicated, in combination with trametinib, for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E or V600K mutations, as detected by an FDA-approved test [see Dosage and Administration (2.1)].
1.3 Adjuvant Treatment of BRAF V600E or V600K Mutation-Positive Melanoma
TAFINLAR is indicated, in combination with trametinib, for the adjuvant treatment of patients with melanoma with BRAF V600E or V600K mutations, as detected by an FDA-approved test, and involvement of lymph node(s), following complete resection [see Dosage and Administration (2.1)].
1.4 BRAF V600E Mutation-Positive Metastatic NSCLC
TAFINLAR is indicated, in combination with trametinib, for the treatment of patients with metastatic non-small cell lung cancer (NSCLC) with BRAF V600E mutation as detected by an FDA-approved test [see Dosage and Administration (2.1)].
1.5 BRAF V600E Mutation-Positive Locally Advanced or Metastatic Anaplastic Thyroid Cancer
TAFINLAR is indicated, in combination with trametinib, for the treatment of patients with locally advanced or metastatic anaplastic thyroid cancer (ATC) with BRAF V600E mutation and with no satisfactory locoregional treatment options [see Dosage and Administration (2.1)].
-
2
DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Melanoma
- Confirm the presence of BRAF V600E mutation in tumor specimens prior to initiation of treatment with TAFINLAR as a single agent [see Warnings and Precautions (5.2), Clinical Studies (14.1)].
- Confirm the presence of BRAF V600E or V600K mutation in tumor specimens prior to initiation of treatment with TAFINLAR and trametinib [see Warnings and Precautions (5.2), Clinical Studies (14.2, 14.3)].
- Information on FDA-approved tests for the detection of BRAF V600 mutations in melanoma is available at: http://www.fda.gov/CompanionDiagnostics.
NSCLC
- Confirm the presence of BRAF V600E mutation in tumor specimens prior to initiation of treatment with TAFINLAR and trametinib [see Clinical Studies (14.4)].
- Information on FDA-approved tests for the detection of BRAF V600E mutations in NSCLC is available at: http://www.fda.gov/CompanionDiagnostics.
ATC
- Confirm the presence of BRAF V600E mutation in tumor specimens prior to initiation of treatment with TAFINLAR and trametinib [see Clinical Studies (14.5)]. An FDA-approved test for the detection of BRAF V600E mutation in ATC is not currently available.
2.2 Recommended Dosage for Unresectable or Metastatic Melanoma
The recommended dosage of TAFINLAR is 150 mg orally taken twice daily, as a single agent or in combination with trametinib, until disease progression or unacceptable toxicity. Refer to the trametinib prescribing information for recommended trametinib dosing information.
2.3 Recommended Dosage for the Adjuvant Treatment of Melanoma
The recommended dosage of TAFINLAR is 150 mg orally taken twice daily in combination with trametinib until disease recurrence or unacceptable toxicity for up to 1 year. Refer to the trametinib prescribing information for recommended trametinib dosing information.
2.4 Recommended Dosage for NSCLC
The recommended dosage of TAFINLAR is 150 mg orally taken twice daily, in combination with trametinib until disease recurrence or unacceptable toxicity. Refer to the trametinib prescribing information for recommended trametinib dosing information.
2.5 Recommended Dosage for ATC
The recommended dosage of TAFINLAR is 150 mg orally taken twice daily, in combination with trametinib until disease recurrence or unacceptable toxicity. Refer to the trametinib prescribing information for recommended trametinib dosing information.
2.6 Administration
- Take TAFINLAR at doses approximately 12 hours apart.
- Take TAFINLAR at least 1 hour before or 2 hours after a meal [see Clinical Pharmacology (12.3)].
- Do not take a missed dose of TAFINLAR within 6 hours of the next dose of TAFINLAR.
- Do not open, crush, or break TAFINLAR capsules.
2.7 Dosage Modifications for Adverse Reactions
Dose reductions for adverse reactions associated with TAFINLAR are presented in Table 1.
Table 1. Recommended Dose Reductions for TAFINLAR for Adverse Reactions Action Recommended Dose First Dose Reduction 100 mg orally twice daily Second Dose Reduction 75 mg orally twice daily Third Dose Reduction 50 mg orally twice daily Subsequent Modification Permanently discontinue if unable to tolerate TAFINLAR 50 mg orally twice daily Dosage modifications for adverse reactions associated with TAFINLAR are presented in Table 2.
Table 2. Recommended Dosage Modifications for TAFINLAR for Adverse Reactions aNational Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.0.
bSee Table 1 for recommended dose reductions of TAFINLAR.
cDose modifications are not recommended for TAFINLAR when administered with trametinib for the following adverse reactions of trametinib: retinal vein occlusion (RVO), retinal pigment epithelial detachment (RPED), interstitial lung disease/pneumonitis, and uncomplicated venous thromboembolism. Dose modification of TAFINLAR is not required for new primary cutaneous malignancies.Severity of Adverse Reactiona Dosage Modification for TAFINLARb New Primary Malignancies [see Warnings and Precautions (5.1)] Non-Cutaneous RAS Mutation-positive Malignancies Permanently discontinue TAFINLAR. Cardiomyopathy [see Warnings and Precautions (5.4)] - Symptomatic congestive heart failure
- Absolute decrease in left ventricular ejection fraction (LVEF) of greater than 20% from baseline that is below lower limit of normal (LLN)
Withhold TAFINLAR until LVEF improves to at least the institutional LLN and absolute decrease to less than or equal to 10% compared to baseline, then resume at same dose. Uveitis [see Warnings and Precautions (5.5)] - Uveitis, including iritis and iridocyclitis
For mild or moderate uveitis does not respond to ocular therapy, or for severe uveitis, withhold TAFINLAR for up to 6 weeks. - If improved to Grade 0-1, then resume TAFINLAR at same or lower dose.
- If not improved, permanently discontinue TAFINLAR.
Febrile Reactions [see Warnings and Precautions (5.6)] - Fever of 101.3°F to 104°F
Withhold TAFINLAR until fever resolves, then resume at same or lower dose. - Fever higher than 104°F
- Fever complicated by rigors, hypotension, dehydration, or renal failure
- Withhold TAFINLAR until fever resolves, then resume at lower dose.
- Permanently discontinue TAFINLAR.
Skin Toxicities [see Warnings and Precautions (5.7)] - Intolerable Grade 2
- Grade 3 or 4
Withhold TAFINLAR for up to 3 weeks. - If improved, resume TAFINLAR at lower dose.
- If not improved, permanently discontinue TAFINLAR.
- Severe cutaneous adverse reactions (SCARs)
Permanently discontinue TAFINLAR. Other Adverse Reactionsc, including Hemorrhage [see Warnings and Precautions (5.3)] - Intolerable Grade 2
- Any Grade 3
Withhold TAFINLAR. - If improved to Grade 0-1, resume TAFINLAR at lower dose.
- If not improved, permanently discontinue TAFINLAR.
- First occurrence of any Grade 4
- Withhold TAFINLAR until improves to Grade 0-1, then resume at a lower dose.
- Permanently discontinue TAFINLAR.
- Recurrent Grade 4
Permanently discontinue TAFINLAR. Refer to the trametinib prescribing information for dose modifications for adverse reactions associated with trametinib.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5
WARNINGS AND PRECAUTIONS
5.1 New Primary Malignancies
Cutaneous Malignancies
Across clinical trials of TAFINLAR monotherapy, cutaneous squamous cell carcinomas (cuSCC), and keratoacanthomas occurred in 11% and 4% of patients, respectively. Basal cell carcinoma and new primary melanoma occurred in 4% and 1% of patients, respectively.
Across clinical trials of TAFINLAR administered with trametinib, the incidence of cuSCC (including keratoacanthomas) occurred in 2% of patients. Basal cell carcinoma and new primary melanoma occurred in 3% and < 1% of patients, respectively.
Perform dermatologic evaluations prior to initiation of TAFINLAR, every 2 months while on therapy, and for up to 6 months following discontinuation of TAFINLAR.
Non-cutaneous Malignancies
Based on its mechanism of action, TAFINLAR may promote the growth and development of malignancies with activation of RAS through mutation or other mechanisms [see Warnings and Precautions (5.2)]. Across clinical trials of TAFINLAR monotherapy and TAFINLAR administered with trametinib, non-cutaneous malignancies occurred in 1% of patients.
Monitor patients receiving TAFINLAR for signs or symptoms of non-cutaneous malignancies. Permanently discontinue TAFINLAR for RAS mutation-positive non-cutaneous malignancies [see Dosage and Administration (2.7)].
5.2 Tumor Promotion in BRAF Wild-Type Tumors
In vitro experiments have demonstrated paradoxical activation of MAP-kinase signaling and increased cell proliferation in BRAF wild-type cells which are exposed to BRAF inhibitors. Confirm evidence of BRAF V600E or V600K mutation status prior to initiation of TAFINLAR as a single agent or in combination with trametinib [see Indications and Usage (1.6), Dosage and Administration (2.1)].
5.3 Hemorrhage
Hemorrhage, including major hemorrhage defined as symptomatic bleeding in a critical area or organ, can occur when TAFINLAR is administered with trametinib. Fatal cases have been reported.
Across clinical trials of TAFINLAR administered with trametinib, hemorrhagic events occurred in 17% of patients. Gastrointestinal hemorrhage occurred in 3% of patients who received TAFINLAR administered with trametinib. Intracranial hemorrhage occurred in 0.6% of patients who received TAFINLAR administered with trametinib. Fatal hemorrhage occurred in 0.5% of patients who received TAFINLAR administered with trametinib. The fatal events were cerebral hemorrhage and brainstem hemorrhage. Permanently discontinue TAFINLAR for all Grade 4 hemorrhagic events and for any Grade 3 hemorrhagic events that do not improve. Withhold TAFINLAR for Grade 3 hemorrhagic events; if improved, resume at the next lower dose level.
5.4 Cardiomyopathy
Across clinical trials of TAFINLAR administered with trametinib, cardiomyopathy, defined as a decrease in left ventricular ejection fraction (LVEF) ≥ 10% from baseline and below the institutional lower limit of normal (LLN), occurred in 6% of patients. Development of cardiomyopathy resulted in dose interruption or discontinuation of TAFINLAR in 3% and < 1% of patients, respectively. Cardiomyopathy resolved in 45 of 50 patients who received TAFINLAR administered with trametinib.
Assess LVEF by echocardiogram or multigated acquisition (MUGA) scan before initiation of TAFINLAR with trametinib, one month after initiation of TAFINLAR, and then at 2- to 3-month intervals while on treatment. Withhold TAFINLAR for symptomatic cardiomyopathy or asymptomatic LV dysfunction of > 20% from baseline that is below institutional LLN. Resume TAFINLAR at the same dose level upon recovery of cardiac function to at least the institutional LLN for LVEF and absolute decrease ≤ 10% compared to baseline [see Dosage and Administration (2.7)].
5.5 Uveitis
Across clinical trials, uveitis occurred in 1% of patients who received TAFINLAR monotherapy and in 2% of patients who received TAFINLAR administered with trametinib. Treatment employed in clinical trials included steroid and mydriatic ophthalmic drops.
Monitor patients for visual signs and symptoms of uveitis (e.g., change in vision, photophobia, eye pain). If iritis is diagnosed, administer ocular therapy and continue TAFINLAR without dose modification. If severe uveitis (i.e., iridocyclitis) or if mild or moderate uveitis does not respond to ocular therapy, withhold TAFINLAR and treat as clinically indicated. Resume TAFINLAR at the same or lower dose if improves to Grade 0 or 1. Permanently discontinue TAFINLAR for persistent Grade 2 or greater uveitis of > 6 weeks [see Dosage and Administration (2.7)].
5.6 Serious Febrile Reactions
Serious febrile reactions and fever of any severity complicated by hypotension, rigors or chills, dehydration, or renal failure, can occur with TAFINLAR.
The incidence and severity of pyrexia are increased when TAFINLAR is administered with trametinib compared with TAFINLAR as a single agent [see Adverse Reactions (6.1)].
Across clinical trials of TAFINLAR monotherapy, fever (serious and non-serious) occurred in 30% of patients. Approximately 13% of these patients experienced 3 or more discrete episodes. Serious febrile reactions and fever of any severity complicated by hypotension, rigors or chills occurred in 6% of patients.
Across clinical trials of TAFINLAR administered with trametinib, fever occurred in 58% of patients. Serious febrile reactions and fever of any severity complicated by hypotension, rigors or chills, dehydration or renal failure occurred in 5% of patients. Fever was complicated by hypotension in 4%, dehydration in 3%, syncope in 2%, renal failure in 1%, and severe chills/rigors in < 1% of patients. Withhold TAFINLAR for fever of greater than or equal to 101.3ºF, or fever complicated by hypotension, rigors or chills, dehydration, or renal failure, and evaluate for signs and symptoms of infection. Monitor serum creatinine and other evidence of renal function during and following severe pyrexia. Refer to Table 2 for recommended dose modifications for adverse reactions [see Dosage and Administration (2.7)]. Administer antipyretics as secondary prophylaxis when resuming TAFINLAR if patient had a prior episode of severe febrile reaction or fever associated with complications. Administer corticosteroids (e.g., prednisone 10 mg daily) for at least 5 days for second or subsequent pyrexia if temperature does not return to baseline within 3 days of onset of pyrexia, or for pyrexia associated with complications, such as dehydration, hypotension, renal failure or severe chills/rigors, and there is no evidence of active infection.
5.7 Serious Skin Toxicities
Severe cutaneous adverse reactions (SCARs), including Stevens-Johnson syndrome (SJS) and drug reaction with eosinophilia and systemic symptoms (DRESS), which can be life-threatening or fatal, have been reported during treatment with TAFINLAR administered with trametinib [see Adverse Reactions (6.2)].
Across clinical trials of TAFINLAR administered with trametinib, other serious skin toxicity occurred in < 1% of patients.
Monitor for new or worsening serious skin reactions. Permanently discontinue TAFINLAR for SCARs. For other skin toxicities, withhold TAFINLAR for intolerable or severe skin toxicity. Resume TAFINLAR at a lower dose in patients with improvement or recovery from skin toxicity within 3 weeks. Permanently discontinue TAFINLAR if skin toxicity has not improved within 3 weeks [see Dosage and Administration (2.7)].
5.8 Hyperglycemia
Across clinical trials of TAFINLAR monotherapy, 14% of patients with a history of diabetes that received TAFINLAR required more intensive hypoglycemic therapy. Grade 3 and Grade 4 hyperglycemia occurred in 3% of patients.
Across clinical trials of TAFINLAR administered with trametinib, 15% of patients with a history of diabetes who had received TAFINLAR with trametinib required more intensive hypoglycemic therapy. Grade 3 and Grade 4 hyperglycemia occurred in 2% of patients. Monitor serum glucose levels upon initiation and as clinically appropriate when TAFINLAR is administered in patients with preexisting diabetes or hyperglycemia. Initiate or optimize anti-hyperglycemic medications as clinically indicated.
5.9 Glucose-6-Phosphate Dehydrogenase Deficiency
TAFINLAR, which contains a sulfonamide moiety, confers a potential risk of hemolytic anemia in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency. Monitor patients with G6PD deficiency for signs of hemolytic anemia while taking TAFINLAR.
5.10 Risks Associated With Combination Treatment
TAFINLAR is indicated for use in combination with trametinib. Review the Prescribing Information for trametinib for information on the serious risks of trametinib prior to initiation of TAFINLAR with trametinib.
5.11 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, TAFINLAR can cause fetal harm when administered to a pregnant woman. Dabrafenib was teratogenic and embryotoxic in rats at doses three times greater than the human exposure at the recommended clinical dose. Advise pregnant women of the potential risk to a fetus. Advise female patients of reproductive potential to use effective non-hormonal contraception, since TAFINLAR can render hormonal contraceptives ineffective, during treatment with TAFINLAR and for 2 weeks after the last dose [see Drug Interactions (7.2), Use in Specific Populations (8.1, 8.3)].
-
6
ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- New Primary Malignancies [see Warnings and Precautions (5.1)]
- Tumor Promotion in BRAF Wild-Type Melanoma [see Warnings and Precautions (5.2)]
- Hemorrhage [see Warnings and Precautions (5.3)]
- Cardiomyopathy [see Warnings and Precautions (5.4)]
- Uveitis [see Warnings and Precautions (5.5)]
- Serious Febrile Reactions [see Warnings and Precautions (5.6)]
- Serious Skin Toxicities [see Warnings and Precautions (5.7)]
- Hyperglycemia [see Warnings and Precautions (5.8)]
- Glucose-6-Phosphate Dehydrogenase Deficiency [see Warnings and Precautions (5.9)]
There are additional adverse reactions associated with trametinib. Refer to the trametinib prescribing information for additional information.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety populations described in the WARNINGS AND PRECAUTIONS reflect exposure to TAFINLAR as a single agent in 586 patients with various solid tumors, including BRAF V600 mutation-positive unresectable or metastatic melanoma, enrolled in BREAK-2, BREAK-3, BREAK-MB, BRF113220, and BRF112680 and that to TAFINLAR administered with trametinib in 1087 patients enrolled in METRIC, MEK113583, MEK111504, COMBI-d, COMBI-v, COMBI-AD, and BRF113928 with unresectable or metastatic melanoma, adjuvant melanoma or NSCLC. Among these 586 patients who received TAFINLAR as a single agent, 46% were exposed for 6 months or longer and 15% were exposed for greater than one year. Among the 1087 patients who received TAFINLAR administered with trametinib, 70% were exposed for 6 months or longer and 21% were exposed for greater than one year.
Metastatic or Unresectable BRAF V600E or V600K Mutation-Positive Melanoma
TAFINLAR as a Single Agent
The safety of TAFINLAR was evaluated in BREAK-3, a multicenter, international, open-label, randomized (3:1), controlled trial allocated 250 patients with unresectable or metastatic BRAF V600E mutation-positive melanoma to receive TAFINLAR 150 mg orally twice daily (n = 187) or dacarbazine 1000 mg/m2 intravenously every 3 weeks (n = 63) [see Clinical Studies (14.1)]. The trial excluded patients with abnormal left ventricular ejection fraction (LVEF) or cardiac valve morphology (≥ Grade 2), corrected QT interval ≥ 480 milliseconds on electrocardiogram, or a known history of glucose-6-phosphate dehydrogenase deficiency. The median duration on treatment was 4.9 months for patients treated with TAFINLAR and 2.8 months for dacarbazine-treated patients. The population exposed to TAFINLAR was 60% male, 99% white, and had a median age of 53 years.
The most common adverse reactions (≥ 20%) in patients treated with TAFINLAR were, in order of decreasing frequency: hyperkeratosis, headache, pyrexia, arthralgia, papilloma, alopecia, and palmar-plantar erythrodysesthesia syndrome (PPES).
The incidence of adverse events resulting in permanent discontinuation of study medication in the BREAK-3 study was 3% for patients treated with TAFINLAR and 3% for patients treated with dacarbazine. The most frequent (≥ 2%) adverse reactions leading to dose reduction of TAFINLAR were pyrexia (9%), PPES (3%), chills (3%), fatigue (2%), and headache (2%). Table 3 and Table 4 present adverse reactions and laboratory abnormalities, respectively, of TAFINLAR as a single agent in the BREAK-3 study.
Table 3. Select Adverse Reactions Occurring in ≥ 10% (All Grades) or ≥ 2% (Grades 3 or 4) of Patients Treated With TAFINLAR in the BREAK-3 Studya Abbreviations: cuSCC, cutaneous squamous cell carcinoma, includes squamous cell carcinoma of the skin and keratoacanthoma; NA, not applicable.
aAdverse reactions, reported using MedDRA and graded using NCI CTCAE version 4.0 for assessment of toxicity.
bGrade 4 adverse reactions limited to hyperkeratosis (n = 1) and constipation (n = 1).
cIncludes skin papilloma and papilloma.
dCases of cuSCC were required to be reported as Grade 3 per protocol.TAFINLAR
N = 187Dacarbazine
N = 59Adverse Reactions All
Grades
(%)Grades
3 and 4b
(%)All
Grades
(%)Grades
3 and 4
(%)Skin and subcutaneous tissue Hyperkeratosis 37 1 0 0 Alopecia 22 NA 2 NA Palmar-plantar erythrodysesthesia syndrome 20 2 2 0 Rash 17 0 0 0 Nervous system Headache 32 0 8 0 General Pyrexia 28 3 10 0 Musculoskeletal Arthralgia 27 1 2 0 Back pain 12 3 7 0 Myalgia 11 0 0 0 Neoplasms Papillomac 27 0 2 0 cuSCCd 7 4 0 0 Respiratory Cough 12 0 5 0 Gastrointestinal Constipation 11 2 14 0 Infections Nasopharyngitis 10 0 3 0 Table 4. Laboratory Abnormalities Worsening from Baseline Occurring at a Higher Incidence in Patients Treated With TAFINLAR in the BREAK-3 Study [Between-Arm Difference of ≥ 5% (All Grades) or ≥ 2% (Grades 3 or 4)]a aAdverse reactions, reported using MedDRA and graded using NCI CTCAE version 4.0 for assessment of toxicity.
bGrade 4 laboratory abnormality limited to hypophosphatemia (n = 1).TAFINLAR
N = 187Dacarbazine N = 59 Laboratory Abnormality All
Grades
(%)Grades
3 and 4
(%)All
Grades
(%)Grades
3 and 4
(%)Hyperglycemia 50 6 43 0 Hypophosphatemia 37 6b 14 2 Increased alkaline phosphatase 19 0 14 2 Hyponatremia 8 2 3 0 Other clinically important adverse reactions observed in less than 10% of patients (N = 586) treated with TAFINLAR were:
Gastrointestinal: Pancreatitis
Immune System: Hypersensitivity manifesting as bullous rash
Renal and Urinary: Interstitial nephritis
TAFINLAR with Trametinib
The safety of TAFINLAR when administered with trametinib was evaluated in 559 patients with previously untreated, unresectable or metastatic, BRAF V600E or V600K mutation-positive melanoma who received TAFINLAR in two trials, the COMBI-d study (n = 209) a multicenter, double-blind, randomized (1:1), active controlled trial and the COMBI-v study (n = 350) a multicenter, open-label, randomized (1:1), active controlled trial. In the COMBI-d and COMBI-v studies, patients received TAFINLAR 150 mg orally twice daily and trametinib 2 mg orally once daily until disease progression or unacceptable toxicity. Both trials excluded patients with abnormal LVEF, history of acute coronary syndrome within 6 months, history of Class II or greater congestive heart failure (New York Heart Association), history of retinal vein occlusion (RVO) or retinal pigment epithelial detachment (RPED), QTcB interval ≥ 480 msec, treatment refractory hypertension, uncontrolled arrhythmias, active brain metastases, or a known history of G6PD deficiency [see Clinical Studies (14.2)].
Among these 559 patients, 199 (36%) were exposed to TAFINLAR for > 6 months to 12 months while 185 (33%) were exposed to TAFINLAR for ≥ 1 year. The median age was 55 years (range: 18 to 91), 57% were male, 98% were white, 72% had baseline ECOG performance status 0 and 28% had ECOG performance status 1, 64% had M1c stage disease, 35% had elevated lactate dehydrogenase (LDH) at baseline and 0.5% had a history of brain metastases.
The most common adverse reactions (≥ 20%) for TAFINLAR in patients who received TAFINLAR plus trametinib in the COMBI-d and COMBI-v studies were: pyrexia, rash, chills, headache, arthralgia, and cough. The demographics and baseline tumor characteristics of patients enrolled in the COMBI-d study are summarized in Clinical Studies [see Clinical Studies (14.2)]. Patients who received TAFINLAR plus trametinib had a median duration of exposure of 11 months (range: 3 days to 30 months) to TAFINLAR. Among the 209 patients who received TAFINLAR plus trametinib, 26% were exposed to TAFINLAR for > 6 months to 12 months while 46% were exposed to TAFINLAR for > 1 year.
In the COMBI-d study, adverse reactions resulting in discontinuation of TAFINLAR occurred in 11% of patients who received TAFINLAR plus trametinib; the most frequent was pyrexia (1.9%). Adverse reactions leading to dose reductions of TAFINLAR occurred in 26% of patients who received TAFINLAR plus trametinib; the most frequent were pyrexia (14%), neutropenia (1.9%), rash (1.9%), and chills (1.9%). Adverse reactions leading to dose interruptions of TAFINLAR occurred in 56% of patients who received TAFINLAR plus trametinib; the most frequent were pyrexia (35%), chills (11%), vomiting (7%), nausea (5%), and decreased ejection fraction (5%).
Table 5 and Table 6 present adverse reactions and laboratory abnormalities, respectively, observed in the COMBI-d study.
Table 5. Select Adverse Reactions Occurring in ≥ 10% (All Grades) of Patients Treated With TAFINLAR Administered With Trametinib in the COMBI-d Studya aNCI CTCAE version 4.0.
bGrade 4 adverse reactions limited to headache (n = 1).
cIncludes rash generalized, rash pruritic, rash erythematous, rash papular, rash vesicular, rash macular, rash maculo-papular, and rash folliculitis.Pooled TAFINLAR plus
Trametinib
N = 559COMBI-d Study TAFINLAR plus Trametinib N = 209 TAFINLAR
N = 211Adverse Reactions All
Grades
(%)Grades
3 and 4b
(%)All
Grades
(%)Grades
3 and 4
(%)All
Grades
(%)Grades
3 and 4
(%)General Pyrexia 54 5 57 7 33 1.9 Chills 31 0.5 31 0 17 0.5 Skin Rashc 32 1.1 42 0 27 1.4 Dry skin 10 0 12 0 16 0 Nervous system Headache 30 0.9 33 0.5 30 1.4 Dizziness 11 0.2 14 0 7 0 Musculoskeletal Arthralgia 25 0.9 26 0.9 31 0 Myalgia 15 0.2 13 0.5 13 0 Respiratory Cough 20 0 21 0 21 0 Gastrointestinal Constipation 13 0.2 13 0.5 10 0 Infections Nasopharyngitis 12 0 12 0 10 0 Other clinically important adverse reactions for TAFINLAR across the COMBI-d and COMBI-v studies (N = 559) observed in less than 10% of patients who received TAFINLAR administered with trametinib were:
Gastrointestinal: Colitis, Gastrointestinal perforation, Pancreatitis
Subcutaneous Tissue: Panniculitis
Table 6. Select Laboratory Abnormalities Worsening from Baseline Occurring at ≥ 10% (All Grades) of Patients Who Received TAFINLAR With Trametinib in the COMBI-d Study aFor these laboratory tests the denominator is 556.
bFor these laboratory tests the denominator is 208 for the combination arm, 208-209 for the TAFINLAR arm.
cGrade 4 adverse reactions limited to hyperglycemia (n = 4), hyponatremia and hypophosphatemia (each n = 1), in the pooled combination arm; hyperglycemia (n = 1) in the COMBI-d study combination arm; hypophosphatemia (n = 1) in the TAFINLAR arm.Laboratory Abnormality Pooled TAFINLAR
plus Trametinib
N = 559aCOMBI-d Study TAFINLAR plus Trametinib
N = 209bTAFINLAR
N = 211bAll
Grades
(%)Grades
3 and 4c
(%)All
Grades
(%)Grades
3 and 4c
(%)All
Grades
(%)Grades
3 and 4c
(%)Chemistry Hyperglycemia 60 4.7 65 6 57 4.3 Hypophosphatemia 38 6 38 3.8 35 7 Hyponatremia 25 8 24 6 14 2.9 Hepatic Increased blood alkaline
phosphatase49 2.7 50 1.0 25 0.5 Adjuvant Treatment of BRAF V600E or V600K Mutation-Positive Melanoma
The safety of TAFINLAR when administered with trametinib was evaluated in 435 patients with Stage III melanoma with BRAF V600E or V600K mutations following complete resection who received at least one dose of study therapy in the COMBI-AD study [see Clinical Studies (14.3)]. Patients received TAFINLAR 150 mg orally twice daily and trametinib 2 mg orally once daily for 12 months. The trial excluded patients with abnormal left ventricular ejection fraction; history of acute coronary syndromes, coronary angioplasty, or stenting within 6 months; Class II or greater congestive heart failure (New York Heart Association); QTc interval ≥ 480 msec; treatment-refractory hypertension; uncontrolled arrhythmias; or history of retinal vein occlusion. The median age of patients who received TAFINLAR administered with trametinib was 50 years (range: 18 to 89), 56% were male, 99% were white, 92% had baseline ECOG performance status 0, and 8% had baseline ECOG performance status of 1. Patients who received TAFINLAR in combination with trametinib had a median duration of exposure of 11 months (range: 0 to 12) to TAFINLAR. Among the 435 patients receiving TAFINLAR in combination with trametinib, 71% were exposed to TAFINLAR for > 6 months.
The most common adverse reactions (≥ 20%) in patients who received TAFINLAR administered with trametinib were: pyrexia, fatigue, nausea, headache, rash, chills, diarrhea, vomiting, arthralgia, and myalgia.
Adverse reactions resulting in discontinuation, dose reduction, or dose interruption of TAFINLAR occurred in 25%, 35%, and 66% of patients, respectively; the most frequent for each were pyrexia and chills.
Table 7 summarizes adverse reactions that occurred in at least 20% of patients who received TAFINLAR administered with trametinib.
Table 7. Adverse Reactions Occurring in ≥ 20% of Patients in the COMBI-AD Studya aNCI CTCAE version 4.0.
bIncludes pyrexia and hyperpyrexia.
cIncludes fatigue, asthenia, and malaise.
dIncludes headache and tension headache.
eIncludes rash, rash maculo-papular, rash macular, rash generalized, rash erythematous, rash papular, rash pruritic, nodular rash, rash vesicular, and rash pustular.
fIncludes myalgia, musculoskeletal pain, and musculoskeletal chest pain.Adverse Reactions TAFINLAR plus Trametinib
N = 435Placebo
N = 432All
Grades
(%)Grades
3 and 4
(%)All
Grades
(%)Grades
3 and 4
(%)General Pyrexiab 63 5 11 < 1 Fatiguec 59 5 37 < 1 Chills 37 1 4 0 Gastrointestinal Nausea 40 < 1 20 0 Diarrhea 33 < 1 15 < 1 Vomiting 28 < 1 10 0 Nervous system Headached 39 1 24 0 Skin Rashe 37 < 1 16 < 1 Musculoskeletal Arthralgia 28 < 1 14 0 Myalgiaf 20 < 1 14 0 Other clinically important adverse reactions observed in less than 20% of patients in the COMBI-AD study who received TAFINLAR administered with trametinib were blurred vision (6%), ejection fraction decreased (5%), and rhabdomyolysis (< 1%).
The laboratory abnormalities are summarized in Table 8.
Table 8. Laboratory Abnormalities Worsening from Baseline Occurring in ≥ 20% of Patients in the COMBI-AD Study Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase.
aThe incidence is based on the number of patients who had both a baseline and at least one on-study laboratory measurement:
TAFINLAR plus Trametinib (range: 429 to 431) and placebo arm (range: 426 to 428).Laboratory Abnormality TAFINLAR plus Trametiniba
N = 435Placeboa
N = 432All
Grades
(%)Grades
3 and 4
(%)All
Grades
(%)Grades
3 and 4
(%)Chemistry Hyperglycemia 63 3 47 2 Hypophosphatemia 42 7 10 < 1 Hypoalbuminemia 25 < 1 < 1 0 Hepatic Increased AST 57 6 11 < 1 Increased ALT 48 5 18 < 1 Increased blood alkaline phosphatase 38 1 6 < 1 Hematology Neutropenia 47 6 12 < 1 Lymphopenia 26 5 6 < 1 Anemia 25 < 1 6 < 1 Metastatic, BRAF V600E-Mutation Positive Non-Small Cell Lung Cancer
The safety of TAFINLAR when administered with trametinib was evaluated in 93 patients with previously untreated (n = 36) and previously treated (n = 57) metastatic BRAF V600E mutation-positive NSCLC in a multicenter, multi-cohort, non-randomized, open-label trial (Study BRF113928). Patients received TAFINLAR 150 mg orally twice daily and trametinib 2 mg orally once daily until disease progression or unacceptable toxicity. The trial excluded patients with abnormal left ventricular ejection fraction, history of acute coronary syndrome within 6 months, history of Class II or greater congestive heart failure (New York Heart Association), QTc interval ≥ 480 msec, treatment refractory hypertension, uncontrolled arrhythmias, active brain metastases, history of interstitial lung disease or pneumonitis, or history or current retinal vein occlusion [see Clinical Studies (14.4)].
Among these 93 patients, 53 (57%) were exposed to TAFINLAR and trametinib for > 6 months and 27 (29%) were exposed to TAFINLAR and trametinib for ≥ 1 year. The median age was 65 years (range: 41 to 91); 46% were male; 85% were white; 32% had baseline ECOG performance status 0 and 61% had ECOG performance status 1; 98% had non-squamous histology; and 12% were current smokers, 60% were former smokers, and 28% had never smoked.
The most common adverse reactions (≥ 20%) in these 93 patients were: pyrexia, fatigue, nausea, vomiting, diarrhea, dry skin, decreased appetite, edema, rash, chills, hemorrhage, cough, and dyspnea.
Adverse reactions resulting in discontinuation of TAFINLAR occurred in 18% of patients; the most frequent were pyrexia (2.2%), decreased ejection fraction (2.2%), and respiratory distress (2.2%). Adverse reactions leading to dose reductions of TAFINLAR occurred in 35% of patients; the most frequent were pyrexia (10%), diarrhea (4.3%), nausea (4.3%), vomiting (4.3%), and neutropenia (3.2%). Adverse reactions leading to dose interruptions of TAFINLAR occurred in 62% of patients; the most frequent were pyrexia (27%), vomiting (11%), neutropenia (8%), and chills (6%).
Table 9 and Table 10 present adverse reactions and laboratory abnormalities, respectively, of TAFINLAR administered with trametinib in Study BRF113928.
Table 9. Adverse Reactions Occurring in ≥ 20% (All Grades) of Patients Treated With TAFINLAR Administered with Trametinib in Study BRF113928a aNCI CTCAE version 4.0.
bIncludes fatigue, malaise, and asthenia.
cIncludes peripheral edema, edema, and generalized edema.
dIncludes rash, rash generalized, rash papular, rash macular, rash maculo-papular, and rash pustular.
eIncludes hemoptysis, hematoma, epistaxis, purpura, hematuria, subarachnoid hemorrhage, gastric hemorrhage, urinary bladder hemorrhage, contusion, hematochezia, injection site hemorrhage, pulmonary hemorrhage, and retroperitoneal hemorrhage.Adverse Reactions TAFINLAR plus Trametinib
N = 93All
Grades
(%)Grades
3 and 4b
(%)General Pyrexia 55 5 Fatigueb 51 5 Edemac 28 0 Chills 23 1.1 Gastrointestinal Nausea 45 0 Vomiting 33 3.2 Diarrhea 32 2.2 Decreased appetite 29 0 Skin Dry skin 31 1.1 Rashd 28 3.2 Vascular Hemorrhagee 23 3.2 Respiratory system Cough 22 0 Dyspnea 20 5 Other clinically important adverse reactions for TAFINLAR observed in less than 10% of patients with NSCLC receiving TAFINLAR administered with trametinib were:
Gastrointestinal: Pancreatitis
Renal and Urinary: Tubulointerstitial nephritis
Table 10. Treatment-Emergent Laboratory Abnormalities Occurring in ≥ 20% (All Grades) of Patients Who Received TAFINLAR With Trametinib in Study BRF113928 Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase.
aFor these laboratory tests the denominator is 90.
bFor these laboratory tests the denominator is 91.Laboratory Abnormality TAFINLAR plus Trametinib
N = 93All
Grades
(%)Grades
3 and 4
(%)Chemistrya Hyperglycemia 71 9 Hyponatremia 57 17 Hypophosphatemia 36 7 Increased creatinine 21 1.1 Hepatica Increased blood alkaline phosphatase 64 0 Increased AST 61 4.4 Increased ALT 32 6 Hematologyb Leukopenia 48 8 Anemia 46 10 Neutropenia 44 8 Lymphopenia 42 14 Locally Advanced or Metastatic, BRAF V600E-Mutation Positive Anaplastic Thyroid Cancer
The safety of TAFINLAR when administered with trametinib was evaluated in a nine-cohort, multicenter, non-randomized, open-label study in patients with rare cancers with the BRAF V600E mutation, including locally advanced or metastatic ATC (Study BRF117019). At the time of the safety analysis, a total of 100 patients were enrolled in the trial, 16 of whom were enrolled in the ATC cohort. The primary safety population included all patients who received at least one dose of TAFINLAR or trametinib. Patients received TAFINLAR 150 mg orally twice daily and trametinib 2 mg orally once daily until disease progression or unacceptable toxicity.
Among these 100 patients, 46 (46%) were exposed to TAFINLAR and trametinib for > 6 months and 23 (23%) were exposed to TAFINLAR and trametinib for ≥ 1 year. The median age was 59.5 years (range: 18 to 85); 62% were male; 85% were white; and 31% had baseline ECOG performance status 0 and 59% had ECOG performance status 1.
The adverse reaction profile among all patients and among patients in the ATC cohort was similar to that observed in other approved indications.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of TAFINLAR in combination with trametinib. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Dermatologic: SCAR (including DRESS and SJS)
- New Primary Malignancies [see Warnings and Precautions (5.1)]
-
7
DRUG INTERACTIONS
7.1 Effects of Other Drugs on TAFINLAR
Strong inhibitors of CYP3A4 or CYP2C8 may increase the concentration of dabrafenib [see Clinical Pharmacology (12.3)]. Substitution of strong inhibitors of CYP3A4 or CYP2C8 is recommended during treatment with TAFINLAR. If concomitant use of strong inhibitors of CYP3A4 or CYP2C8 is unavoidable, monitor patients closely for adverse reactions when taking strong inhibitors.
7.2 Effects of TAFINLAR on Other Drugs
Dabrafenib decreased the systemic exposures of midazolam (a CYP3A4 substrate), S-warfarin (a CYP2C9 substrate), and R-warfarin (a CYP3A4/CYP1A2 substrate) [see Clinical Pharmacology (12.3)]. Monitor international normalized ratio (INR) levels more frequently in patients receiving warfarin during initiation or discontinuation of dabrafenib. Coadministration of TAFINLAR with other substrates of these enzymes, including dexamethasone or hormonal contraceptives, can result in decreased concentrations and loss of efficacy [see Use in Specific Populations (8.1, 8.3)]. Substitute for these medications or monitor patients for loss of efficacy if use of these medications is unavoidable.
-
8
USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal reproduction studies and its mechanism of action [see Clinical Pharmacology (12.1)], TAFINLAR can cause fetal harm when administered to a pregnant woman. There is insufficient data in pregnant women exposed to TAFINLAR to assess the risks. Dabrafenib was teratogenic and embryotoxic in rats at doses three times greater than the human exposure at the recommended clinical dose of 150 mg twice daily (see Data). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%-4% and 15%-20%, respectively.
Data
Animal Data
In a combined female fertility and embryo-fetal development study in rats conducted during the period of organogenesis, developmental toxicity consisted of embryo-lethality, ventricular septal defects, and variation in thymic shape at a dabrafenib dose of 300 mg/kg/day [approximately three times the human exposure at the recommended dose based on area under the curve (AUC)]. At doses of 20 mg/kg/day or greater (equivalent to the human exposure at the recommended dose based on AUC), rats demonstrated delays in skeletal development and reduced fetal body weight.
8.2 Lactation
Risk Summary
There are no data on the presence of dabrafenib in human milk, or the effects of dabrafenib on the breastfed infant, or on milk production. Because of the potential for serious adverse reactions in breastfed infants, advise women not to breastfeed during treatment with TAFINLAR and for 2 weeks following the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating TAFINLAR.
Contraception
Based on data from animal studies and its mechanism of action, TAFINLAR can cause fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
Females
Advise female patients of reproductive potential to use effective contraception during treatment with TAFINLAR and for 2 weeks after the last dose. Counsel patients to use a non-hormonal method of contraception since TAFINLAR can render hormonal contraceptives ineffective [see Drug Interactions (7.2)].
Males
To avoid potential drug exposure to pregnant partners and female partners of reproductive potential, advise male patients (including those who have had vasectomies) with female partners of reproductive potential to use condoms during treatment with TAFINLAR and for at least 2 weeks after the last dose.
Infertility
Females
Advise female patients of reproductive potential that TAFINLAR may impair fertility. A reduction in fertility was observed in female rats at dose exposures equivalent to the human exposure at the recommended dose. A reduction in the number of corpora lutea was noted in pregnant rats at dose exposures approximately three times the human exposure at the recommended dose [see Nonclinical Toxicology (13.1)].
Males
Advise male patients of the potential risk for impaired spermatogenesis which may be irreversible. Effects on spermatogenesis have been observed in animals treated with dabrafenib at dose exposures up to three times the human exposure at the recommended dose [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of TAFINLAR as a single agent or with trametinib have not been established in pediatric patients.
Juvenile Animal Toxicity Data
In a repeat-dose toxicity study in juvenile rats, an increased incidence of kidney cysts and tubular deposits were noted at doses as low as 0.2 times the human exposure at the recommended adult dose based on AUC. Additionally, forestomach hyperplasia, decreased bone length, and early vaginal opening were noted at doses as low as 0.8 times the human exposure at the recommended adult dose based on AUC.
8.5 Geriatric Use
Of the 586 patients with various solid tumors who received single agent TAFINLAR, 22% were aged 65 years and older. Of the 187 patients with melanoma who received single-agent TAFINLAR in the BREAK-3 study, 21% were aged 65 years or older [see Clinical Studies (14.1)]. No overall differences in the effectiveness or safety of TAFINLAR were observed between geriatric patients as compared to younger adults in the BREAK-3 study.
Of the 994 patients with melanoma who received TAFINLAR plus trametinib in the COMBI-d, COMBI-v, and COMBI-AD studies [see Clinical Studies (14.2, 14.3)], 21% were aged 65 years and older and 5% were aged 75 years and older. No overall differences in the effectiveness of TAFINLAR plus trametinib were observed between geriatric patients as compared to younger adults across these melanoma studies. The incidences of peripheral edema (26% vs. 12%) and anorexia (21% vs. 9%) were increased in geriatric patients as compared to younger adults in these studies.
Of the 171 patients with NSCLC who received TAFINLAR in Study BRF113928, there were insufficient numbers of geriatric patients to determine whether they respond differently from younger adults [see Clinical Studies (14.4)].
Of the 26 patients with ATC who received TAFINLAR in Study BRF117019, 77% were aged 65 years and older, and 31% were aged 75 years and older [see Clinical Studies (14.5)]. This study in ATC did not include sufficient numbers of younger adults to determine whether they respond differently compared to geriatric patients.
8.6 Renal Impairment
Dose adjustment is not recommended for patients with mild (GFR 60 to 89 mL/min/1.73 m2) or moderate (GFR 30 to 59 mL/min/1.73 m2) renal impairment. An appropriate dose has not been established for patients with severe (GFR ≤ 30 mL/min/1.73 m2) renal impairment [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Dose adjustment is not recommended for patients with mild (bilirubin ≤ upper limit of normal (ULN) and alanine aminotransferase (AST) > ULN or bilirubin > 1x to 1.5x ULN and any AST) hepatic impairment. As hepatic metabolism and biliary secretion are the primary routes of elimination of dabrafenib and its metabolites, patients with moderate (bilirubin > 1.5x to 3x ULN and any AST) to severe (bilirubin > 3x to 10x ULN and any AST) hepatic impairment may have increased exposure. An appropriate dose has not been established for patients with moderate to severe hepatic impairment [see Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11
DESCRIPTION
Dabrafenib mesylate is a kinase inhibitor. The chemical name for dabrafenib mesylate is N-{3-[5-(2-amino-4-pyrimidinyl)-2-(1,1-dimethylethyl)-1,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzene sulfonamide, methanesulfonate salt. It has the molecular formula C23H20F3N5O2S2CH4O3S and a molecular weight of 615.68 g/mol. Dabrafenib mesylate has the following chemical structure:
![The following chemical structure for Dabrafenib mesylate is a kinase inhibitor. The chemical name for dabrafenib mesylate is N-{3-[5-(2-amino-4-pyrimidinyl)-2-(1,1-dimethylethyl)-1,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzene sulfonamide, methanesulfonate salt. It has the molecular formula C23H20F3N5O2S2CH4O3S and a molecular weight of 615.68.](https://fda.report/DailyMed/fee1e6b1-e1a5-4254-9f2e-a70e0f8dbdea/tafinlar-01.jpg)
Dabrafenib mesylate is a white to slightly colored solid with three pKas: 6.6, 2.2, and -1.5. It is very slightly soluble at pH 1 and practically insoluble above pH 4 in aqueous media.
TAFINLAR (dabrafenib) capsules for oral use are supplied as 50 mg and 75 mg capsules for oral administration. Each 50 mg capsule contains 59.25 mg dabrafenib mesylate equivalent to 50 mg of dabrafenib free base. Each 75 mg capsule contains 88.88 mg dabrafenib mesylate equivalent to 75 mg of dabrafenib free base. The inactive ingredients of TAFINLAR are colloidal silicon dioxide, magnesium stearate, and microcrystalline cellulose. Capsule shells contain hypromellose, red iron oxide (E172), and titanium dioxide (E171).
-
12
CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Dabrafenib is an inhibitor of some mutated forms of BRAF kinases with in vitro IC50 values of 0.65, 0.5, and 1.84 nM for BRAF V600E, BRAF V600K, and BRAF V600D enzymes, respectively. Dabrafenib also inhibits wild-type BRAF and CRAF kinases with IC50 values of 3.2 and 5.0 nM, respectively, and other kinases, such as SIK1, NEK11, and LIMK1 at higher concentrations. Some mutations in the BRAF gene, including those that result in BRAF V600E, can result in constitutively activated BRAF kinases that may stimulate tumor cell growth [see Indications and Usage (1)]. Dabrafenib inhibits cell growth of various BRAF V600 mutation-positive tumors in vitro and in vivo.
Dabrafenib and trametinib target two different kinases in the RAS/RAF/MEK/ERK pathway. Use of dabrafenib and trametinib in combination resulted in greater growth inhibition of BRAF V600 mutation-positive tumor cell lines in vitro and prolonged inhibition of tumor growth in BRAF V600 mutation positive tumor xenografts compared with either drug alone.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The potential effect of TAFINLAR on QT interval was assessed in a dedicated multiple-dose study in 32 patients with BRAF V600 mutation-positive tumors. No large changes in the mean QT interval (i.e., > 20 ms) were detected with dabrafenib 300 mg administered twice daily (two times the recommended dosage).
In clinical trials, QTc (heart rate-corrected QT) prolongation to ≥ 500 ms occurred in 0.8% (2/264) of patients who received TAFINLAR plus trametinib and in 1.5% (4/264) of patients who received TAFINLAR as a single agent. The QTc was increased > 60 ms from baseline in 3.8% (10/264) of patients who received TAFINLAR plus trametinib and 3% (8/264) of patients treated with TAFINLAR as a single agent.
12.3 Pharmacokinetics
Absorption
After oral administration, median time to achieve peak plasma concentration (Tmax) is 2 hours. Mean absolute bioavailability of oral dabrafenib is 95%. Following a single dose, dabrafenib exposure (Cmax and AUC) increased in a dose-proportional manner across the dose range of 12 mg to 300 mg, but the increase was less than dose-proportional after repeat twice-daily dosing. After repeat twice-daily dosing of 150 mg, the mean accumulation ratio was 0.73, and the inter-subject variability (CV%) of AUC at steady-state was 38%.
Effect of Food
Administration of dabrafenib with a high-fat meal (approximately 1000 calories, 58-75 grams fat, 58 grams carbohydrates, and 33 grams protein) decreased Cmax by 51%, decreased AUC by 31%, and delayed median Tmax by 3.6 hours as compared with the fasted state.
Distribution
Dabrafenib is 99.7% bound to human plasma proteins. The apparent volume of distribution (Vc/F) is 70.3 L.
Elimination
The mean terminal half-life of dabrafenib is 8 hours after oral administration. Hydroxy-dabrafenib terminal half-life (10 hours) parallels that of dabrafenib while the carboxy- and desmethyl-dabrafenib metabolites exhibit longer half-lives (21 to 22 hours). The apparent clearance of dabrafenib is 17.0 L/h after single dosing and 34.4 L/h after 2 weeks of twice-daily dosing.
Metabolism
The metabolism of dabrafenib is primarily mediated by CYP2C8 and CYP3A4 to form hydroxy-dabrafenib. Hydroxy-dabrafenib is further oxidized via CYP3A4 to form carboxy-dabrafenib and subsequently excreted in bile and urine. Carboxy-dabrafenib is decarboxylated to form desmethyl-dabrafenib; desmethyl-dabrafenib may be reabsorbed from the gut. Desmethyl-dabrafenib is further metabolized by CYP3A4 to oxidative metabolites. Mean metabolite-to-parent AUC ratios following repeat-dose administration are 0.9, 11, and 0.7 for hydroxy-, carboxy-, and desmethyl-dabrafenib, respectively. Based on systemic exposure, relative potency, and pharmacokinetic properties, both hydroxy- and desmethyl-dabrafenib are likely to contribute to the clinical activity of dabrafenib.
Excretion
Fecal excretion is the major route of elimination accounting for 71% of radioactive dose while urinary excretion accounted for 23% of total radioactivity as metabolites only.
Specific Populations
Age, Body Weight, and Sex
Age has no effect on dabrafenib pharmacokinetics. Pharmacokinetic differences based on sex and on weight are not clinically relevant.
Patients with Renal Impairment
The pharmacokinetics of dabrafenib were evaluated using a population analysis in 233 patients with mild renal impairment (GFR 60 to 89 mL/min/1.73 m2) and 30 patients with moderate renal impairment (GFR 30 to 59 mL/min/1.73 m2) enrolled in clinical trials. Mild or moderate renal impairment has no effect on systemic exposure to dabrafenib and its metabolites. No data are available in patients with severe renal impairment.
Patients with Hepatic Impairment
The pharmacokinetics of dabrafenib was evaluated using a population analysis in 65 patients with mild (bilirubin ≤ ULN and AST > ULN or bilirubin > 1x to 1.5x ULN and any AST) hepatic impairment enrolled in clinical trials. Mild hepatic impairment has no effect on systemic exposure to dabrafenib and its metabolites. No data are available in patients with moderate (bilirubin > 1.5x to 3x ULN and any AST) to severe (bilirubin > 3x to 10x ULN and any AST) hepatic impairment.
Drug Interaction Studies
Effect of Trametinib on Dabrafenib: Coadministration of trametinib 2 mg daily with dabrafenib 150 mg twice daily resulted in a 23% increase in AUC of dabrafenib, a 33% increase in AUC of desmethyl-dabrafenib, and no change in AUC of hydroxy-dabrafenib as compared with administration of dabrafenib.
Effect of Strong Inhibitors of CYP3A4 or CYP2C8 on Dabrafenib: Coadministration of dabrafenib 75 mg twice daily and ketoconazole 400 mg once daily (a strong CYP3A4 inhibitor) for 4 days increased dabrafenib AUC by 71%, hydroxy-dabrafenib AUC by 82%, and desmethyl-dabrafenib AUC by 68%. Coadministration of dabrafenib 75 mg twice daily and gemfibrozil 600 mg twice daily (a strong CYP2C8 inhibitor) for 4 days increased dabrafenib AUC by 47%, with no change in the AUC of dabrafenib metabolites.
Effect of Strong Inducers of CYP3A4 or Moderate Inducers CYP2C8 on Dabrafenib: Coadministration of dabrafenib 150 mg twice daily and rifampin 600 mg once daily (a strong CYP3A4 and moderate CYP2C8 inducer) for 10 days decreased dabrafenib AUC by 34%, had no effect on hydroxy-dabrafenib AUC, and decreased desmethyl-dabrafenib AUC by 30%.
Effect of Acid Reducing Agents on Dabrafenib: Coadministration of dabrafenib 150 mg twice daily and rabeprazole 40 mg once daily for 4 days resulted in a 3% increase in AUC of dabrafenib, a 15% decrease in AUC of desmethyl-dabrafenib, and a 5% increase in AUC of hydroxy-dabrafenib as compared to administration of dabrafenib alone. The changes in exposure of dabrafenib and its metabolites were not clinically relevant.
Effect of Dabrafenib on CYP Substrates: In vitro data demonstrate that dabrafenib is an inducer of CYP3A4 and CYP2B6 via activation of the pregnane X receptor (PXR) and constitutive androstane receptor (CAR) nuclear receptors. Dabrafenib may also induce CYP2C enzymes via the same mechanism. Coadministration of TAFINLAR 150 mg twice daily for 15 days and a single dose of midazolam 3 mg (a CYP3A4 substrate) decreased midazolam AUC by 65%. Coadministration of dabrafenib 150 mg twice daily for 15 days and a single dose of warfarin 15 mg decreased the AUC of S-warfarin (a CYP2C9 substrate) by 37% and the AUC of R-warfarin (CYP3A4/CYP1A2 substrate) by 33%.
Effect of Transporters on Dabrafenib: Dabrafenib and its metabolites, hydroxyl-dabrafenib and desmethyl-dabrafenib, are substrates of human P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), but are not substrates of organic cation transporter (OCT1) or organic anion transporting polypeptide (OATP1A2, OATP1B1, OATP1B3, OATP2B1) in vitro.
Effect of Dabrafenib on Transporters: Dabrafenib and its metabolites, hydroxy-dabrafenib, carboxy-dabrafenib, and desmethyl-dabrafenib, are inhibitors of OATP1B1, OATP1B3, and organic anion transporter (OAT1 and OAT3) in vitro. Dabrafenib and desmethyl-dabrafenib are inhibitors of OCT2 and BCRP in vitro. Coadministration of TAFINLAR 150 mg twice daily with a single dose of rosuvastatin (a sensitive OATP1B1 and OATP1B3 substrate) increased rosuvastatin Cmax by 2.6-fold, but did not change its AUC.
-
13
NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with dabrafenib have not been conducted. TAFINLAR increased the risk of cuSCCs in patients in clinical trials.
Dabrafenib was not mutagenic in vitro in the bacterial reverse mutation assay (Ames test) or the mouse lymphoma assay, and was not clastogenic in an in vivo rat bone marrow micronucleus test.
In a combined female fertility and embryo-fetal development study in rats, a reduction in fertility was noted at doses greater than or equal to 20 mg/kg/day (equivalent to the human exposure at the recommended dose based on AUC). A reduction in the number of ovarian corpora lutea was noted in pregnant females at 300 mg/kg/day (which is approximately three times the human exposure at the recommended dose based on AUC).
Male fertility studies with dabrafenib have not been conducted; however, in repeat-dose studies, testicular degeneration/depletion was seen in rats and dogs at doses equivalent to and three times the human exposure at the recommended dose based on AUC, respectively.
13.2 Animal Toxicology and/or Pharmacology
Adverse cardiovascular effects were noted in dogs at dabrafenib doses of 50 mg/kg/day (approximately five times the human exposure at the recommended dose based on AUC) or greater, when administered for up to 4 weeks. Adverse effects consisted of coronary arterial degeneration/necrosis and hemorrhage, as well as cardiac atrioventricular valve hypertrophy/hemorrhage.
-
14
CLINICAL STUDIES
14.1 BRAF V600E Mutation-Positive Unresectable or Metastatic Melanoma – TAFINLAR As a Single Agent
BREAK-3 Study
The safety and efficacy of TAFINLAR as a single agent were evaluated in an international, multicenter, randomized (3:1), open-label, active-controlled trial (the BREAK-3 study; NCT01227889) conducted in 250 patients with previously untreated BRAF V600E mutation-positive, unresectable or metastatic melanoma. Patients with any prior use of BRAF inhibitors or MEK inhibitors were excluded. Patients were randomized to receive TAFINLAR 150 mg orally twice daily (n = 187) or dacarbazine 1000 mg/m2 intravenously every 3 weeks (n = 63). Randomization was stratified by disease stage at baseline [unresectable Stage III (regional nodal or in-transit metastases), M1a (distant skin, subcutaneous, or nodal metastases), or M1b (lung metastases) versus M1c melanoma (all other visceral metastases or elevated serum LDH)]. The main efficacy outcome measure was progression-free survival (PFS) as assessed by the investigator. In addition, an independent radiology review committee (IRRC) assessed the following efficacy outcome measures in pre-specified supportive analyses: PFS, confirmed overall response rate (ORR), and duration of response (DoR).
The median age of patients in the BREAK-3 study was 52 years. The majority of the trial population was male (60%), white (99%), had an ECOG performance status of 0 (67%), M1c disease (66%), and normal LDH (62%). All patients had tumor tissue with mutations in BRAF V600E as determined by a clinical trial assay at a centralized testing site. Tumor samples from 243 patients (97%) were tested retrospectively, using an FDA-approved companion diagnostic test, THxID™-BRAF assay.
The median durations of follow-up prior to initiation of alternative treatment in patients randomized to receive TAFINLAR was 5.1 months and in the dacarbazine arm was 3.5 months. Twenty-eight (44%) patients crossed over from the dacarbazine arm at the time of disease progression to receive TAFINLAR.
The BREAK-3 study demonstrated a statistically significant increase in progression-free survival in the patients treated with TAFINLAR. Table 11 and Figure 1 summarize the PFS results.
Table 11. Investigator-Assessed Progression-Free Survival and Confirmed Overall Response Results in the BREAK-3 Study Abbreviations: CI, confidence interval; DoR, duration of response; HR, hazard ratio; NR, not reached.
aPike estimator, stratified by disease state.
bStratified log-rank test.Investigator-Assessed Endpoints TAFINLAR
N = 187Dacarbazine
N = 63Progression-Free Survival Number of Events (%) 78 (42%) 41 (65%) Progressive Disease 76 41 Death 2 0 Median, months (95% CI) 5.1 (4.9, 6.9) 2.7 (1.5, 3.2) HRa (95% CI) 0.33 (0.20, 0.54) P valueb < 0.0001 Confirmed Tumor Responses Overall Response Rate (95% CI) 52% (44%, 59%) 17% (9%, 29%) Complete Response, n (%) 6 (3%) 0 Partial Response, n (%) 91 (48%) 11 (17%) Duration of Response Median DoR, months (95% CI) 5.6 (5.4, NR) NR (5.0, NR) 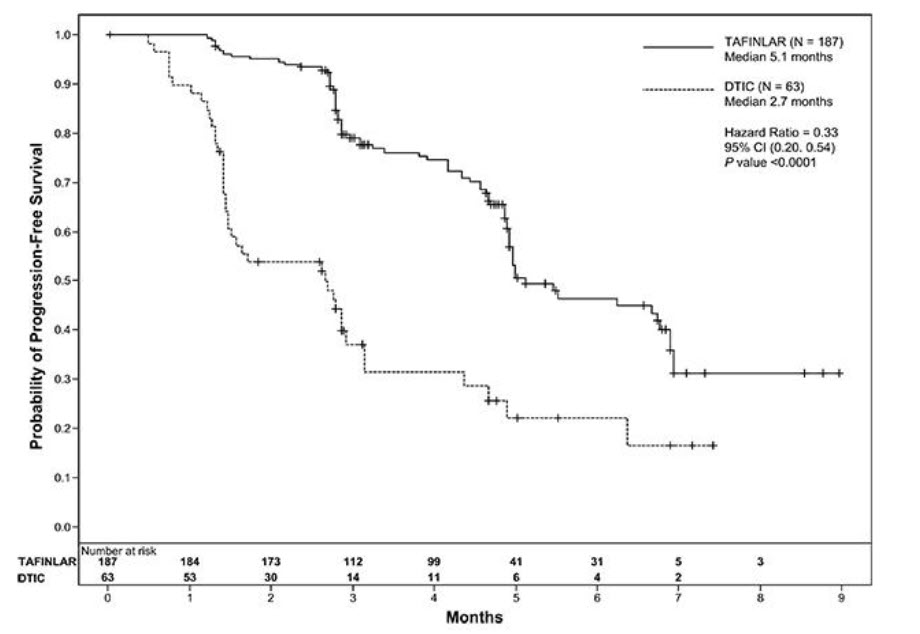
Figure 1. Kaplan-Meier Curves of Investigator-Assessed Progression-Free Survival in the BREAK-3 Study
In supportive analyses based on IRRC assessment and in an exploratory subgroup analysis of patients with retrospectively confirmed V600E mutation-positive melanoma with the THxID™-BRAF assay, the PFS results were consistent with those of the primary efficacy analysis.
BREAK-MB Study
The activity of TAFINLAR for the treatment of BRAF V600E mutation-positive melanoma, metastatic to the brain was evaluated in a single-arm, open-label, two-cohort multicenter trial (the BREAK-MB study; NCT01266967). All patients received TAFINLAR 150 mg twice daily. Patients in Cohort A (n = 74) had received no prior local therapy for brain metastases, while patients in Cohort B (n = 65) had received at least one local therapy for brain metastases, including, but not limited to, surgical resection, whole brain radiotherapy, or stereotactic radiosurgery, such as gamma knife, linear-accelerated-based radiosurgery, or charged particles. In addition, patients in Cohort B were required to have evidence of disease progression in a previously treated lesion or an untreated lesion. Additional eligibility criteria were at least one measurable lesion of 0.5 cm or greater in largest diameter on contrast-enhanced MRI, stable or decreasing corticosteroid dose, and no more than two prior systemic regimens for treatment of metastatic disease. The major efficacy outcome measure was estimation of the overall intracranial response rate (OIRR) in each cohort.
The median age of patients in Cohort A was 50 years, 72% were male, 100% were white, 59% had a pre-treatment ECOG performance status of 0, and 57% had an elevated LDH value at baseline. The median age of patients in Cohort B was 51 years, 63% were male, 98% were white, 66% had a pre-treatment ECOG performance status of 0, and 54% had an elevated LDH value at baseline. The intracranial response rate as determined by an independent radiology review committee, masked to investigator response assessments, was 18% (95% CI: 10%, 28%) in Cohort A and 18% (95% CI: 10%, 30%) in Cohort B. The median duration of intracranial response was 4.6 months in both cohorts.
14.2 BRAF V600E or V600K Unresectable or Metastatic Melanoma – TAFINLAR With Trametinib
COMBI-d Study and COMBI-v Study
The safety and efficacy of TAFINLAR administered with trametinib were evaluated in two international, randomized, active-controlled trials: one double-blind trial (the COMBI-d study; NCT01584648) and one open-label trial (the COMBI-v study; NCT01597908).
The COMBI-d study compared TAFINLAR and trametinib to TAFINLAR and placebo as first-line therapy for patients with unresectable (Stage IIIC) or metastatic (Stage IV) BRAF V600E or V600K mutation-positive cutaneous melanoma. Patients were randomized (1:1) to receive TAFINLAR 150 mg twice daily and trametinib 2 mg once daily or TAFINLAR 150 mg twice daily plus matching placebo. Randomization was stratified by LDH level (> ULN vs. ≤ ULN) and BRAF mutation subtype (V600E vs. V600K). The major efficacy outcome was investigator-assessed progression-free survival (PFS) per RECIST v1.1 with additional efficacy outcome measures of overall survival (OS) and confirmed overall response rate (ORR).
The COMBI-v study compared TAFINLAR and trametinib to vemurafenib as first-line treatment therapy for patients with unresectable (Stage IIIC) or metastatic (Stage IV) BRAF V600E or V600K mutation-positive cutaneous melanoma. Patients were randomized (1:1) to receive TAFINLAR 150 mg twice daily and trametinib 2 mg once daily or vemurafenib 960 mg twice daily. Randomization was stratified by lactate dehydrogenase (LDH) level (> ULN vs. ≤ ULN) and BRAF mutation subtype (V600E vs. V600K). The major efficacy outcome measure was overall survival. Additional efficacy outcome measures were PFS and ORR as assessed by investigator per RECIST v1.1.
In the COMBI-d study, 423 patients were randomized to TAFINLAR plus trametinib (n = 211) or TAFINLAR plus placebo (n = 212). The median age was 56 years (range: 22 to 89 years), 53% were male, > 99% were white, 72% had ECOG performance status of 0, 4% had Stage IIIC, 66% had M1c disease, 65% had a normal LDH, and 2 patients had a history of brain metastases. All patients had tumor containing BRAF V600E or V600K mutations as determined by centralized testing, 85% with BRAF V600E mutations and 15% with BRAF V600K mutations.
In the COMBI-v study, 704 patients were randomized to TAFINLAR plus trametinib (n = 352) or single-agent vemurafenib (n = 352). The median age was 55 years (range: 18 to 91 years), 96% were white, and 55% were male, 6% percent of patients had Stage IIIC, 61% had M1c disease, 67% had a normal LDH, 70% had ECOG performance status of 0, 89% had BRAF V600E mutation-positive melanoma, and one patient had a history of brain metastases.
The COMBI-d and COMBI-v studies demonstrated statistically significant improvements in OS and PFS. Table 12 and Figures 2 and 3 summarize the efficacy results.
Table 12. Efficacy Results in Patients With BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanomaa Abbreviations: DoR, duration of response; ORR, overall response rate; CI, confidence interval; HR, hazard ratio; NR, not reached.
aP-value is comparing with the allocated alpha of 0.021 for the interim analysis based on 77% information.
bPFS and ORR were assessed by investigator.Endpoint COMBI-d Study COMBI-v Study TAFINLAR plus Trametinib
N = 211TAFINLAR
plus Placebo
N = 212TAFINLAR plus Trametinib
N = 352Vemurafenib
N = 352Overall Survival Number of Deaths (%) 99 (47%) 123 (58%) 100 (28%) 122 (35%) Median, months (95% CI) 25.1 (19.2, NR) 18.7 (15.2, 23.1) NR (18.3, NR) 17.2 (16.4, NR) HR (95% CI) 0.71 (0.55, 0.92) 0.69 (0.53, 0.89) P value (log-rank test) 0.01 0.005a Progression-Free Survivalb Number of Events (%) 102 (48%) 109 (51%) 166 (47%) 217 (62%) Median, months (95% CI) 9.3 (7.7, 11.1) 8.8 (5.9, 10.9) 11.4 (9.9, 14.9) 7.3 (5.8, 7.8) HR (95% CI) 0.75 (0.57, 0.99) 0.56 (0.46, 0.69) P value (log-rank test) 0.035 < 0.001 Overall Response Rateb ORR (95% CI) 66% (60%, 73%) 51% (44%, 58%) 64% (59%, 69%) 51% (46%, 56%) P value < 0.001 < 0.001 Complete Response 10% 8% 13% 8% Partial Response 56% 42% 51% 43% Median DoR, months
(95% CI)9.2
(7.4, NR)10.2
(7.5, NR)13.8
(11.0, NR)7.5
(7.3, 9.3)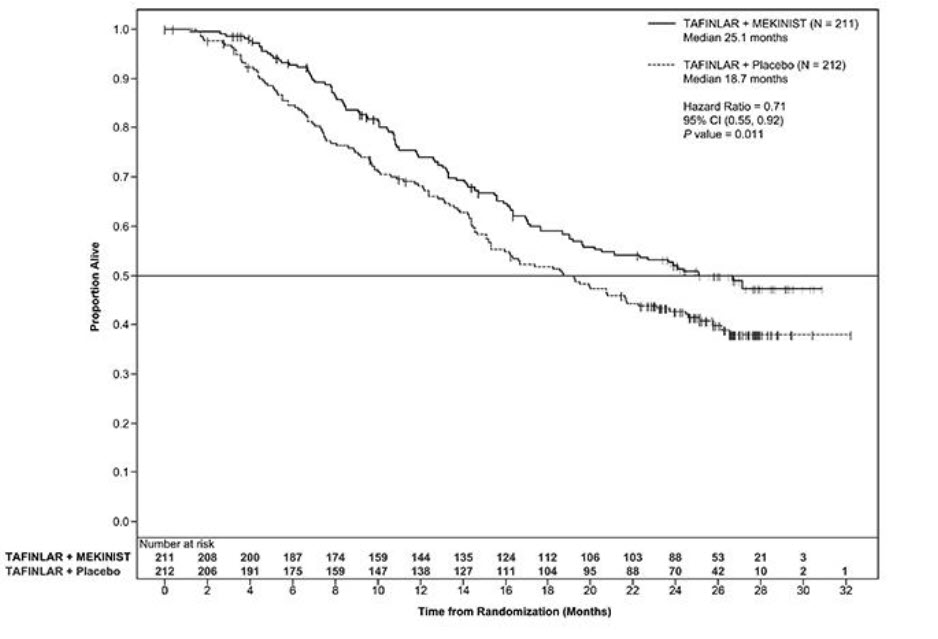
Figure 2. Kaplan-Meier Curves for Overall Survival in the COMBI-d Study
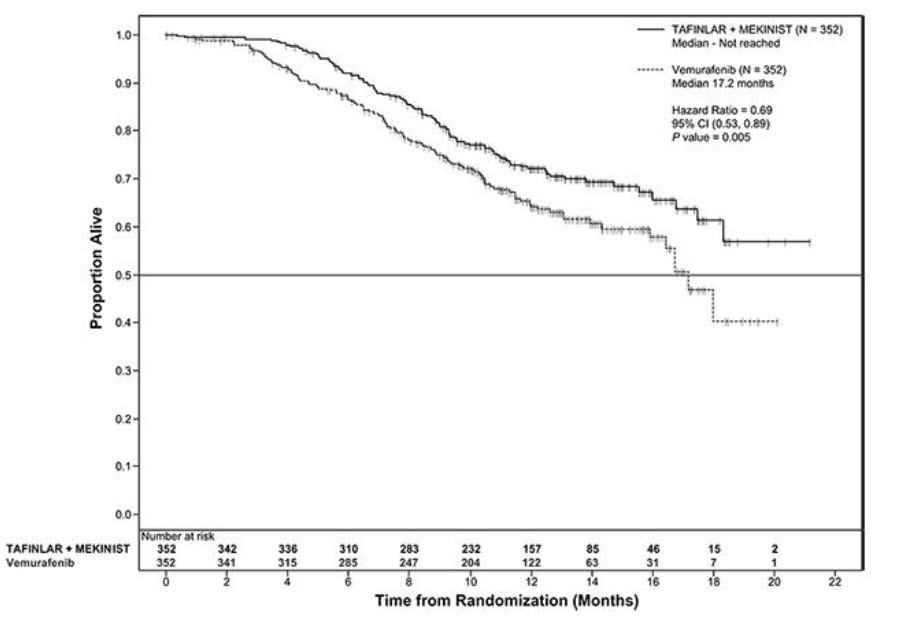
Figure 3. Kaplan-Meier Curves for Overall Survival in the COMBI-v Study
COMBI-MB Study
The activity of TAFINLAR with trametinib for the treatment of BRAF V600E or V600K mutation-positive melanoma, metastatic to the brain, was evaluated in a non-randomized, open-label, multi-center, multi-cohort trial (the COMBI-MB study; NCT02039947). Eligible patients were required to have at least one measurable intracranial lesion and to have no leptomeningeal disease, parenchymal brain metastasis greater than 4 cm in diameter, ocular melanoma, or primary mucosal melanoma. Patients received TAFINLAR 150 mg orally twice daily and trametinib 2 mg orally once daily until disease progression or unacceptable toxicity. The major efficacy outcome measure was intracranial response rate, defined as the percentage of patients with a confirmed intracranial response per RECIST v1.1, modified to allow up to five intracranial target lesions at least 5 mm in diameter, as assessed by independent review.
The COMBI-MB study enrolled 121 patients with a BRAF V600E (85%) or V600K (15%) mutation. The median age was 54 years (range: 23 to 84 years), 58% were male, 100% were white, 8% were from the United States, 65% had a normal LDH value at baseline, and 97% had an ECOG performance status of 0 or 1. Intracranial metastases were asymptomatic in 87% and symptomatic in 13% of patients, 22% received prior local therapy for brain metastases, and 87% also had extracranial metastases.
The intracranial response rate was 50% (95% CI: 41, 60), with a complete response rate of 4.1% and a partial response rate of 46%. The median duration of intracranial response was 6.4 months (range: 1 to 31 months). Of the patients with an intracranial response, 9% had stable or progressive disease as their best overall response.
14.3 Adjuvant Treatment of BRAF V600E or V600K Mutation-Positive Melanoma
COMBI-AD (NCT 01682083) was an international, multi-center, randomized, double-blind, placebo-controlled trial that enrolled patients with Stage III melanoma with BRAF V600E or V600K mutations as detected by the THxID™-BRAF assay and pathologic involvement of regional lymph node(s). Patients were randomized (1:1) to receive TAFINLAR 150 mg twice daily and trametinib 2 mg once daily or two placebos for up to 1 year. Enrollment required complete resection of melanoma with complete lymphadenectomy within 12 weeks prior to randomization. The trial excluded patients with mucosal or ocular melanoma, unresectable in-transit metastases, distant metastatic disease, or prior systemic anticancer treatment, including radiotherapy. Randomization was stratified by BRAF mutation status (V600E or V600K) and American Joint Committee on Cancer (AJCC; 7th Edition) stage (IIIa, IIIb, or IIIc). The major efficacy outcome measure was relapse-free survival (RFS), defined as the time from randomization to disease recurrence (local, regional, or distant metastasis), new primary melanoma, or death from any cause, whichever occurred first as assessed by the investigator. Patients underwent imaging for tumor recurrence every 3 months for the first two years and every 6 months thereafter.
In COMBI-AD, a total of 870 patients were randomized: 438 to TAFINLAR administered with trametinib and 432 to placebo. Median age was 51 years (range 18-89), 55% were male, 99% were white, and 91% had an ECOG performance status of 0. Disease characteristics were AJCC Stage IIIa (18%), Stage IIIb (41%), Stage IIIc (40%), stage unknown (1%); BRAF V600E mutation (91%), BRAF V600K mutation (9%); macroscopic lymph nodes (65%); and tumor ulceration (41%). The median duration of follow-up (time from randomization to last contact or death) was 2.8 years.
COMBI-AD showed a statistically significant improvement in RFS in patients randomized to TAFINLAR administered with trametinib compared to those randomized to placebo. Efficacy results are presented in Table 13 and Figure 4.
Table 13. Efficacy Results in COMBI-AD in the Adjuvant Treatment of Melanoma Abbreviations: HR, hazard ratio; CI, confidence interval; NE, not estimable.
aPike estimator obtained from the stratified log-rank test estimator.
bLog-rank test stratified by disease stage (IIIA vs. IIIB vs. IIIC) and BRAF V600 mutation type (V600E vs. V600K).TAFINLAR plus Trametinib
N = 438Placebo
N = 432Relapse-Free Survival Number of Events (%) 166 (38) 248 (57) Median, months (95% CI) NE (44.5, NE) 16.6 (12.7, 22.1) HR (95% CI)a 0.47 (0.39, 0.58) P valueb < 0.0001 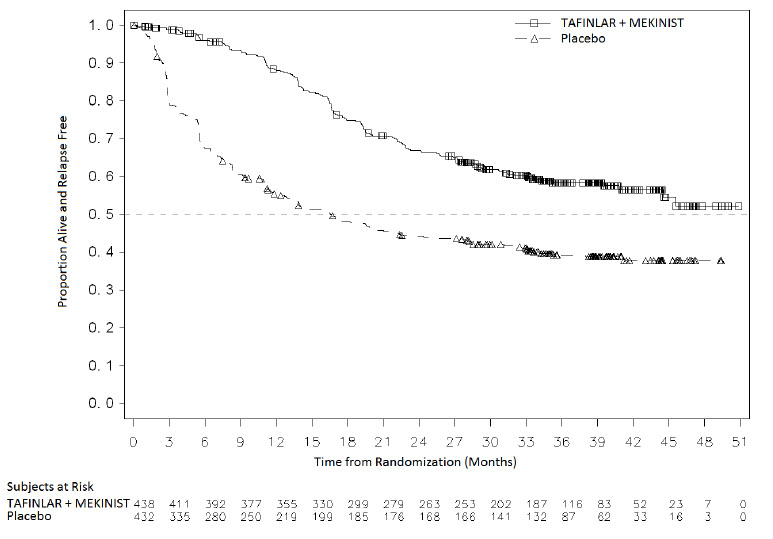
Figure 4. Kaplan-Meier Curves for Relapse-Free Survival in COMBI-AD in the Adjuvant Treatment of Melanoma
14.4 BRAF V600E Mutation-Positive Metastatic Non-Small Cell Lung Cancer
The safety and efficacy of TAFINLAR alone or administered with trametinib were evaluated in a multi-center, three-cohort, non-randomized, activity-estimating, open-label trial (Study BRF113928, NCT01336634). Key eligibility criteria were locally confirmed BRAF V600E mutation-positive metastatic NSCLC, no prior exposure to BRAF or MEK-inhibitor, and absence of EGFR mutation or ALK rearrangement (unless patients had progression on prior tyrosine kinase inhibitor therapy). Patients enrolled in Cohorts A and B were required to have received at least one previous platinum based chemotherapy regimen for NSCLC with demonstrated disease progression but no more than three prior systemic regimens. Patients enrolled in Cohort C could not have received prior systemic therapy for metastatic NSCLC. Patients in Cohort A received TAFINLAR 150 mg twice daily. Patients in Cohorts B and C received TAFINLAR 150 mg twice daily and trametinib 2 mg once daily. The major efficacy outcome measure was overall response rate (ORR) per RECIST v1.1 as assessed by independent review committee (IRC) and DoR.
There were a total of 171 patients enrolled which included 78 patients enrolled in Cohort A, 57 patients enrolled in Cohort B, and 36 patients enrolled in Cohort C. The characteristics of the study population were a median age of 66 years, 48% male; 81% white, 14% Asian, 3% black, and 2% Hispanic; 60% were former smokers, 32% were never smokers, and 8% current smokers; 27% had ECOG performance status (PS) 0, 63% had ECOG PS 1, and 11% had ECOG PS of 2; 99% had metastatic disease of which 6% had brain metastasis at baseline and 14% had liver metastasis at baseline; 11% had systemic anti-cancer therapy in the adjuvant setting and 58% of the 135 previously treated patients had only one line of prior systemic therapy for metastatic disease; and 98% had non-squamous histology.
Efficacy results are summarized in Table 14.
Table 14. Efficacy Results Based on Independent Review in Study BRF113928 Abbreviations: CI, confidence interval; DoR, duration of response; NE, not estimable. Treatment TAFINLAR TAFINLAR + Trametinib Population Previously Treated
N = 78Previously Treated
N = 57Treatment Naïve
N = 36Overall Response Rate ORR (95% CI) 27% (18%, 38%) 63% (49%, 76%) 61% (44%, 77%) Complete Response 1% 4% 3% Partial Response 26% 60% 58% Duration of Response n = 21 n = 36 n = 22 Median DoR, months (95% CI) 9.9 (4.2, NE) 12.6 (5.8, NE) NE (6.9, NE) % with DoR ≥ 6 months 52% 64% 59% In a subgroup analysis of patients with retrospectively, centrally confirmed BRAF V600E mutation-positive NSCLC with the Oncomine™ Dx Target Test, the ORR results were similar to those presented in Table 14.
14.5 BRAF V600E Mutation-Positive Locally Advanced or Metastatic Anaplastic Thyroid Cancer
The safety and efficacy of TAFINLAR administered with trametinib was evaluated in an activity estimating, nine-cohort, multi-center, non-randomized, open-label trial (Study BRF117019; NCT02034110) in patients with rare cancers with the BRAF V600E mutation, including locally advanced, unresectable, or metastatic ATC with no standard locoregional treatment options. Trial BRF117019 excluded patients who could not swallow or retain the medication; who received prior treatment with BRAF or MEK inhibitors; with symptomatic or untreated CNS metastases; or who had airway obstruction. Patients received TAFINLAR 150 mg twice daily and trametinib 2 mg once daily. The major efficacy outcome measure was overall response rate (ORR) per RECIST v1.1 as assessed by independent review committee (IRC) and DoR.
At the time of efficacy analysis, 23 patients were evaluable for response in the ATC cohort. Three additional patients were enrolled; however, there was insufficient time to assess response of these patients. Among the 26 patients enrolled, the median age was 70 years (range: 49-85); 50% were male, 50% white, 46% Asian; 100% had ECOG performance status of 0 or 1; and 54% had a prior history of differentiated thyroid cancer. Prior anti-cancer treatments included surgery (92%), external beam radiotherapy (81%), and systemic therapy (54%).
Efficacy results are summarized in Table 15.
Table 15. Efficacy Results in the ATC Cohort Based on Independent Review of Study BRF117019 Abbreviations: ATC, anaplastic thyroid cancer; DoR, duration of response; CI, confidence interval. ATC Cohort Population (evaluable for response) N = 23 Overall Response Rate ORR (95% CI) 61% (39%, 80%) Complete Response 4% Partial Response 57% Duration of Response % with DoR ≥ 6 months 64% -
16
HOW SUPPLIED/STORAGE AND HANDLING
50 mg capsules: Dark red capsule imprinted with ‘GS TEW’ and ‘50 mg’ available in bottles of 120 (NDC: 0078-0682-66). Each bottle contains a silica gel desiccant.
75 mg capsules: Dark pink capsule imprinted with ‘GS LHF’ and ‘75 mg’ available in bottles of 120 (NDC: 0078-0681-66). Each bottle contains a silica gel desiccant.
Store at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C and 30°C (59°F and 86°F) [see USP Controlled Room Temperature].
-
17
PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
New Cutaneous and Non-cutaneous Malignancies
Advise patients that TAFINLAR increases the risk of developing new primary cutaneous and non-cutaneous malignancies. Advise patients to contact their healthcare provider immediately for any new lesions, changes to existing lesions on their skin, or signs and symptoms of other malignancies [see Warnings and Precautions (5.1)].
Hemorrhage
Advise patients that TAFINLAR when administered with trametinib increases the risk of intracranial and gastrointestinal hemorrhage and to contact their healthcare provider to seek immediate medical attention for signs or symptoms of unusual bleeding or hemorrhage [see Warnings and Precautions (5.3)].
Cardiomyopathy
Advise patients that TAFINLAR can cause cardiomyopathy and to immediately report any signs or symptoms of heart failure to their healthcare provider [see Warnings and Precautions (5.4)].
Uveitis
Advise patients that TAFINLAR can cause uveitis, including iritis and iridocyclitis and to contact their healthcare provider if they experience any changes in their vision [see Warnings and Precautions (5.5)].
Serious Febrile Reactions
Advise patients that TAFINLAR can cause pyrexia, including serious febrile reactions. Inform patients that the incidence and severity of pyrexia are increased when TAFINLAR is given administered with trametinib. Instruct patients to contact their healthcare provider if they develop fever while taking TAFINLAR [see Warnings and Precautions (5.6)].
Serious Skin Toxicities
Advise patients that TAFINLAR can cause serious skin toxicities and to contact their healthcare provider for progressive or intolerable rash. Advise patients to contact their healthcare provider immediately if they develop signs and symptoms of a severe skin reaction [see Warnings and Precautions (5.7)].
Hyperglycemia
Advise patients that TAFINLAR can impair glucose control in diabetic patients resulting in the need for more intensive hypoglycemic treatment and to contact their healthcare provider to report symptoms of severe hyperglycemia [see Warnings and Precautions (5.8)].
Glucose-6-phosphate Dehydrogenase (G6PD) Deficiency
Advise patients that TAFINLAR may cause hemolytic anemia in patients with G6PD deficiency. Advise patients with known G6PD deficiency to contact their healthcare provider to report signs or symptoms of anemia or hemolysis [see Warnings and Precautions (5.9)].
Embryo-Fetal Toxicity
- Advise pregnant women and females of reproductive potential of the potential risk to a fetus [see Warnings and Precautions (5.10), Use in Specific Populations (8.1, 8.3)].
- Advise females to contact their healthcare provider of a known or suspected pregnancy.
- Advise females of reproductive potential to use effective non-hormonal contraception during treatment and for 2 weeks after discontinuation of treatment with TAFINLAR.
- Advise male patients with female partners of reproductive potential to use condoms during treatment with TAFINLAR and for at least 2 weeks after the last dose.
Infertility
Advise males and females of reproductive potential of the potential risk for impaired fertility with TAFINLAR [see Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment with TAFINLAR and for 2 weeks after the last dose of TAFINLAR [see Use in Specific Populations (8.2)].
Administration
Instruct patients to take TAFINLAR at least 1 hour before or at least 2 hours after a meal [see Dosage and Administration (2.6)].
THxID™ is a trademark of bioMérieux.
Oncomine™ Dx Target Test is a trademark of Life Technologies Corporation, a part of Thermo Fisher Scientific Inc.
Distributed by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936© Novartis
T2020-45
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: April 2020 MEDICATION GUIDE
TAFINLAR® (TAFF-in-lar)
(dabrafenib)
capsulesImportant information: If your healthcare provider prescribes TAFINLAR for you to be taken with trametinib, also read the Patient Information leaflet that comes with trametinib. What is the most important information I should know about TAFINLAR?
TAFINLAR may cause serious side effects, including:
Risk of new cancers. TAFINLAR, when used alone or with trametinib, may cause skin cancers, called cutaneous squamous cell carcinoma (cuSCC), keratoacanthoma, basal cell carcinoma, or melanoma.
Talk with your healthcare provider about your risk for these cancers.
Check your skin and tell your healthcare provider right away about any skin changes, including a:
- new wart
- skin sore or reddish bump that bleeds or does not heal
- change in size or color of a mole
Your healthcare provider should also check for cancers that may not occur on the skin. Tell your healthcare provider about any new symptoms that develop during treatment with TAFINLAR.
See "What are the possible side effects of TAFINLAR?" for more information about side effects.What is TAFINLAR?
TAFINLAR is a prescription medicine used:
- alone or in combination with a medicine called trametinib to treat a type of skin cancer called melanoma:
○ that has spread to other parts of the body or cannot be removed by surgery, and
○ that has a certain type of abnormal “BRAF” gene. - in combination with trametinib, to help prevent melanoma that has a certain type of abnormal “BRAF” gene from coming back after the cancer has been removed by surgery.
- in combination with trametinib to treat a type of lung cancer called non-small cell lung cancer (NSCLC):
○ that has spread to other parts of the body, and
○ that has a certain type of abnormal “BRAF” gene. - in combination with trametinib to treat a type of thyroid cancer called anaplastic thyroid cancer (ATC):
○ that has spread to other parts of the body and you have no satisfactory treatment options and
○ that has a certain type of abnormal “BRAF” gene
Your healthcare provider will perform a test to make sure that TAFINLAR is right for you.
It is not known if TAFINLAR alone or TAFINLAR with trametinib is safe and effective in children.Before you take TAFINLAR, tell your healthcare provider about all of your medical conditions, including if you:
- have had bleeding problems
- have heart problems
- have eye problems
- have liver or kidney problems
- have diabetes
- plan to have surgery, dental, or other medical procedures
- have a deficiency of the glucose-6-phosphate dehydrogenase (G6PD) enzyme
- are a male (including one who has had a vasectomy) with a female partner of reproductive potential
○ Males (including those who have had a vasectomy) should use condoms during sexual intercourse during treatment with TAFINLAR and for at least 2 weeks after the last dose of TAFINLAR - are pregnant or plan to become pregnant. TAFINLAR can harm your unborn baby.
○ Females who are able to become pregnant should use effective birth control (contraception) during treatment with TAFINLAR and for 2 weeks after the last dose of TAFINLAR.
○ Birth control methods that contain hormones (such as birth control pills, injections, or transdermal systems) may not work as well during treatment with TAFINLAR. You should use another effective method of birth control during treatment with TAFINLAR.
○ Talk to your healthcare provider about birth control methods that may be right for you during this time.
○ Tell your healthcare provider right away if you become pregnant or think you might be pregnant during treatment with TAFINLAR.
- are breastfeeding or plan to breastfeed. It is not known if TAFINLAR passes into your breast milk.
○ Do not breastfeed during treatment and for 2 weeks after your last dose of TAFINLAR. Talk to your healthcare provider about the best way to feed your baby during this time.
How should I take TAFINLAR? - Take TAFINLAR exactly as your healthcare provider tells you. Do not change your dose or stop TAFINLAR unless your healthcare provider tells you.
- Your healthcare provider may change your dose of TAFINLAR, temporarily stop, or completely stop your treatment with TAFINLAR if you develop certain side effects.
- Take TAFINLAR 2 times a day, about 12 hours apart.
- Take TAFINLAR at least 1 hour before or 2 hours after a meal.
- Do not open, crush, or break TAFINLAR capsules.
- If you miss a dose of TAFINLAR, take it as soon as you remember. If it is within 6 hours of your next scheduled dose, just take your next dose at your regular time. Do not make up for the missed dose.
What are the possible side effects of TAFINLAR?
TAFINLAR may cause serious side effects, including:
-
See “What is the most important information I should know about TAFINLAR?”
-
bleeding problems. TAFINLAR, when taken with trametinib, can cause serious bleeding problems, especially in your brain or stomach, that can lead to death. Call your healthcare provider and get medical help right away if you have any signs of bleeding, including:
○ headaches, dizziness, or feeling weak
○ cough up blood or blood clots
○ vomit blood or your vomit looks like “coffee grounds”
○ red or black stool that looks like tar
-
heart problems, including heart failure. Your healthcare provider should check your heart function before and during treatment with TAFINLAR. Call your healthcare provider right away if you have any of the following signs and symptoms of a heart problem:
○ feeling like your heart is pounding or racing
○ shortness of breath
○ swelling of your ankles or feet
○ feeling lightheaded
-
eye problems. TAFINLAR can cause severe eye problems that can lead to blindness. Call your healthcare provider right away if you get these symptoms of eye problems:
○ blurred vision, loss of vision, or other vision changes
○ see color dots
○ halo (see blurred outline around objects)
○ eye pain, swelling, or redness
-
fever. Fever is common during treatment with TAFINLAR, but may also be serious. When taking TAFINLAR with trametinib, fever may happen more often or may be more severe. In some cases, chills or shaking chills, too much fluid loss (dehydration), low blood pressure, dizziness, or kidney problems may happen with the fever. Call your healthcare provider right away if you get a fever during treatment with TAFINLAR.
-
serious skin reactions. Skin rash is a common side effect of TAFINLAR. TAFINLAR can also cause other skin reactions. In some cases these rashes and other skin reactions can be severe or serious, and may need to be treated in a hospital or lead to death.
Tell your healthcare provider if you get a skin rash or acne that bothers you or worsens.
Tell your healthcare provider right away if you develop any of the following signs or symptoms of a severe skin reaction, including:
○ blisters or peeling of your skin
○ mouth sores○ blisters on your lips, or around your mouth or eyes
○ high fever or flu-like symptoms
○ enlarged lymph nodes-
increased blood sugar (hyperglycemia). Some people may develop high blood sugar or worsening diabetes during treatment with TAFINLAR. If you are diabetic, your healthcare provider should check your blood sugar levels closely during treatment with TAFINLAR. Your diabetes medicine may need to be changed. Tell your healthcare provider if you have any of the following symptoms of severe high blood sugar:
○ increased thirst
○ urinating more often than normal, or urinating an increased amount of urine
- TAFINLAR may cause healthy red blood cells to break down too early in people with G6PD deficiency. This may lead to a type of anemia called hemolytic anemia where the body does not have enough healthy red blood cells. Tell your healthcare provider if you have any of the following signs or symptoms:
○ yellow skin (jaundice)
○ weakness or dizziness
○ shortness of breath
thickening of the outer layers of the skin warts headache hair loss fever redness, swelling, peeling, or tenderness of hands or feet joint aches The most common side effects of TAFINLAR when taken with trametinib in people with melanoma that has spread to other parts of the body or cannot be removed by surgery include: fever chills rash joint aches headache cough The most common side effects of TAFINLAR when taken with trametinib to help prevent melanoma from coming back after the cancer has been removed by surgery include: fever chills fatigue diarrhea nausea vomiting headache joint aches rash muscle aches The most common side effects of TAFINLAR when taken with trametinib in people with NSCLC include: fever rash fatigue swelling of face, arms, and legs nausea chills vomiting bleeding diarrhea cough dry skin shortness of breath decreased appetite TAFINLAR may cause fertility problems in females. This could affect your ability to become pregnant. Talk to your healthcare provider if this is a concern for you.
TAFINLAR may cause lower sperm counts in males. This could affect the ability to father a child. Talk to your healthcare provider if this is a concern for you.
These are not all of the possible side effects of TAFINLAR.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
You may also report side effects to Novartis Pharmaceuticals Corporation at 1-888-669-6682.How should I store TAFINLAR?
- Store TAFINLAR at room temperature between 68°F to 77°F (20°C to 25°C).
General information about the safe and effective use of TAFINLAR
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use TAFINLAR for a condition for which it was not prescribed. Do not give TAFINLAR to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about TAFINLAR that is written for health professionals.What are the ingredients in TAFINLAR?
Active ingredient: dabrafenib
Inactive ingredients: colloidal silicon dioxide, magnesium stearate, microcrystalline cellulose
Capsule shells: hypromellose, red iron oxide (E172), titanium dioxide (E171).
Distributed by: Novartis Pharmaceuticals Corporation, East Hanover, New Jersey 07936
For more information, go to www.TAFINLAR.com or call 1-888-669-6682.
T2020-46 - new wart
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
NDC: 0078-0682-66
Tafinlar® (Dabrafenib) Capsules
50 mg
Rx only
120 Capsules
Dispense with Medication Guide attached or provided separately.
NOVARTIS
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
NDC: 0078-0681-66
Tafinlar® (Dabrafenib) Capsules
75 mg
Rx only
120 Capsules
Dispense with Medication Guide attached or provided separately.
NOVARTIS

-
INGREDIENTS AND APPEARANCE
TAFINLAR
dabrafenib capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-0682 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DABRAFENIB MESYLATE (UNII: B6DC89I63E) (DABRAFENIB - UNII:QGP4HA4G1B) DABRAFENIB 50 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) HYPROMELLOSES (UNII: 3NXW29V3WO) FERRIC OXIDE RED (UNII: 1K09F3G675) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color RED Score no score Shape OVAL Size 18mm Flavor Imprint Code GSTEW;50mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0682-66 120 in 1 BOTTLE; Type 0: Not a Combination Product 04/12/2016 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA202806 04/12/2016 TAFINLAR
dabrafenib capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-0681 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DABRAFENIB MESYLATE (UNII: B6DC89I63E) (DABRAFENIB - UNII:QGP4HA4G1B) DABRAFENIB 75 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) HYPROMELLOSES (UNII: 3NXW29V3WO) FERRIC OXIDE RED (UNII: 1K09F3G675) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color RED Score no score Shape OVAL Size 20mm Flavor Imprint Code GSLHF;75mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0681-66 120 in 1 BOTTLE; Type 0: Not a Combination Product 04/01/2016 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA202806 04/01/2016 Labeler - Novartis Pharmaceuticals Corporation (002147023)
Trademark Results [Tafinlar]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 TAFINLAR 85094477 3976920 Live/Registered |
NOVARTIS PHARMA AG 2010-07-28 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.