NUCALA- mepolizumab injection, powder, for solution NUCALA- mepolizumab injection, solution
Nucala by
Drug Labeling and Warnings
Nucala by is a Prescription medication manufactured, distributed, or labeled by GlaxoSmithKline LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use NUCALA safely and effectively. See full prescribing information for NUCALA.
NUCALA (mepolizumab) for injection, for subcutaneous use
NUCALA (mepolizumab) injection, for subcutaneous use
Initial U.S. Approval: 2015RECENT MAJOR CHANGES
Indications and Usage, Maintenance Treatment of Severe Asthma (1.1)
9/2019
Dosage and Administration, Severe Asthma (2.1)
9/2019
Dosage and Administration, Preparation and Administration of NUCALA for Injection Vial (2.3)
9/2019
Dosage and Administration, Preparation and Administration of NUCALA Injection Prefilled Autoinjector and Prefilled Syringe (2.4)
9/2019
INDICATIONS AND USAGE
NUCALA is an interleukin-5 (IL-5) antagonist monoclonal antibody (IgG1 kappa) indicated for:
- Add-on maintenance treatment of patients with severe asthma aged 6 years and older, and with an eosinophilic phenotype. (1.1)
- The treatment of adult patients with eosinophilic granulomatosis with polyangiitis (EGPA). (1.2)
Limitations of use: Not for relief of acute bronchospasm or status asthmaticus. (1.1)
DOSAGE AND ADMINISTRATION
- Severe asthma in patients aged 12 years and older: 100 mg administered subcutaneously once every 4 weeks. (2.1)
- Severe asthma in patients aged 6 to 11 years: 40 mg administered subcutaneously once every 4 weeks. (2.1)
- EGPA: 300 mg as 3 separate 100-mg injections administered subcutaneously once every 4 weeks. (2.2)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
History of hypersensitivity to mepolizumab or excipients in the formulation. (4)
WARNINGS AND PRECAUTIONS
- Hypersensitivity reactions (e.g., anaphylaxis, angioedema, bronchospasm, hypotension, urticaria, rash) have occurred after administration of NUCALA. Discontinue NUCALA in the event of a hypersensitivity reaction. (5.1)
- Do not use to treat acute bronchospasm or status asthmaticus. (5.2)
- Herpes zoster infections have occurred in patients receiving NUCALA. Consider vaccination if medically appropriate. (5.3)
- Do not discontinue systemic or inhaled corticosteroids abruptly upon initiation of therapy with NUCALA. Decrease corticosteroids gradually, if appropriate. (5.4)
- Treat patients with pre-existing helminth infections before therapy with NUCALA. If patients become infected while receiving treatment with NUCALA and do not respond to anti-helminth treatment, discontinue NUCALA until parasitic infection resolves. (5.5)
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥5%) include headache, injection site reaction, back pain, and fatigue. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact GlaxoSmithKline at 1-888-825-5249 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 9/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Maintenance Treatment of Severe Asthma
1.2 Eosinophilic Granulomatosis with Polyangiitis
2 DOSAGE AND ADMINISTRATION
2.1 Severe Asthma
2.2 Eosinophilic Granulomatosis with Polyangiitis
2.3 Preparation and Administration of NUCALA for Injection Vial
2.4 Preparation and Administration of NUCALA Injection Prefilled Autoinjector and Prefilled Syringe
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
5.2 Acute Asthma Symptoms or Deteriorating Disease
5.3 Opportunistic Infections: Herpes Zoster
5.4 Reduction of Corticosteroid Dosage
5.5 Parasitic (Helminth) Infection
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience in Severe Asthma
6.2 Clinical Trials Experience in Eosinophilic Granulomatosis with Polyangiitis
6.3 Immunogenicity
6.4 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Severe Asthma
14.2 Eosinophilic Granulomatosis with Polyangiitis
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Maintenance Treatment of Severe Asthma
NUCALA is indicated for the add-on maintenance treatment of patients with severe asthma aged 6 years and older, and with an eosinophilic phenotype [see Use in Specific Populations (8.4), Clinical Studies (14.1)].
Limitations of Use
NUCALA is not indicated for the relief of acute bronchospasm or status asthmaticus.
-
2 DOSAGE AND ADMINISTRATION
NUCALA is for subcutaneous (SC) use only.
2.1 Severe Asthma
Adults and Adolescents Aged 12 Years and Older
The recommended dosage of NUCALA in adults and adolescents aged 12 years and older is 100 mg administered once every 4 weeks by SC injection into the upper arm, thigh, or abdomen.
Pediatric Patients Aged 6 to 11 Years
The recommended dosage of NUCALA in children aged 6 to 11 years is 40 mg administered once every 4 weeks by SC injection into the upper arm, thigh, or abdomen.
2.2 Eosinophilic Granulomatosis with Polyangiitis
The recommended dosage of NUCALA is 300 mg administered once every 4 weeks by SC injection as 3 separate 100-mg injections into the upper arm, thigh, or abdomen. It is recommended that the individual 100-mg injections be administered at least 5 cm (approximately 2 inches) apart if more than 1 injection is administered at the same site. [See Dosage and Administration (2.3).]
Preparation of the 300-mg dose for treatment of EGPA requires the reconstitution of 3 separate 100-mg vials or administration of 3 prefilled autoinjectors or 3 prefilled syringes as described below [see Dosage and Administration (2.3, 2.4)].
2.3 Preparation and Administration of NUCALA for Injection Vial
NUCALA for injection should be reconstituted and administered by a healthcare professional. In line with clinical practice, monitoring of patients after administration of biologic agents is recommended [see Warnings and Precautions (5.1)].
Reconstitution Instructions
- 1. Reconstitute NUCALA for injection in the vial with 1.2 mL of Sterile Water for Injection, USP, preferably using a 2- or 3-mL syringe and a 21-gauge needle. The reconstituted solution will contain a concentration of 100 mg/mL mepolizumab. Do not mix with other medications.
- 2.
Direct the stream of Sterile Water for Injection vertically onto the center of the lyophilized powder, which may have a cake-like appearance. Gently swirl the vial for 10 seconds with a circular motion at 15-second intervals until the powder is dissolved.
Note: Do not shake the reconstituted solution during the procedure as this may lead to product foaming or precipitation. Reconstitution is typically complete within 5 minutes after the Sterile Water for Injection has been added, but it may take additional time. - 3. If a mechanical reconstitution device (swirler) is used to reconstitute NUCALA for injection, swirl at 450 rpm for no longer than 10 minutes. Alternatively, swirling at 1,000 rpm for no longer than 5 minutes is acceptable.
- 4. Visually inspect the reconstituted solution for particulate matter and clarity before use. The solution should be clear to opalescent and colorless to pale yellow or pale brown, essentially particle free. Small air bubbles, however, are expected and acceptable. If particulate matter remains in the solution or if the solution appears cloudy or milky, the solution must not be administered.
- 5.
If the reconstituted solution is not used immediately:
- store below 30°C (86°F),
- do not freeze, and
- discard if not used within 8 hours of reconstitution.
Administration of 100-mg Dose
- 1. For SC administration, preferably using a 1-mL polypropylene syringe fitted with a disposable 21- to 27-gauge x 0.5-inch (13-mm) needle.
- 2. Just before administration, remove 1 mL of reconstituted NUCALA for injection. Do not shake the reconstituted solution during the procedure as this could lead to product foaming or precipitation.
- 3. Administer the 1-mL injection (equivalent to 100 mg of mepolizumab) subcutaneously into the upper arm, thigh, or abdomen.
Administration of 40-mg Dose
- 1. For SC administration, preferably using a 1-mL polypropylene syringe fitted with a disposable 21- to 27-gauge x 0.5 inch (13-mm) needle.
- 2. Just before administration, remove 0.4 mL of reconstituted NUCALA. Do not shake the reconstituted solution during the procedure as this could lead to product foaming or precipitation.
- 3. Administer the 0.4-mL injection (equivalent to 40 mg of mepolizumab) subcutaneously into the upper arm, thigh, or abdomen.
Each vial of NUCALA should be used for a single patient, and any remainder of the contents should be discarded.
2.4 Preparation and Administration of NUCALA Injection Prefilled Autoinjector and Prefilled Syringe
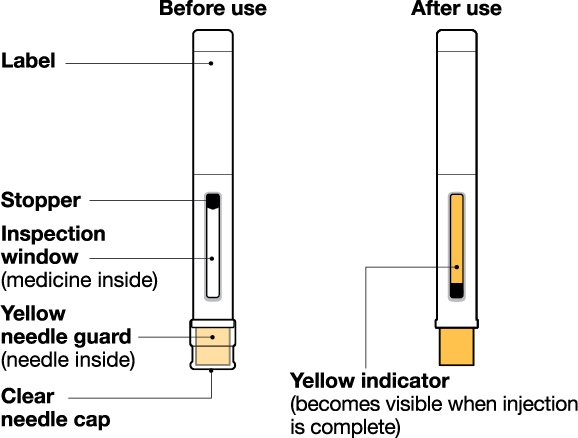
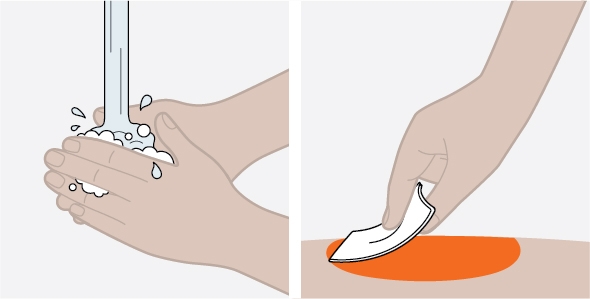
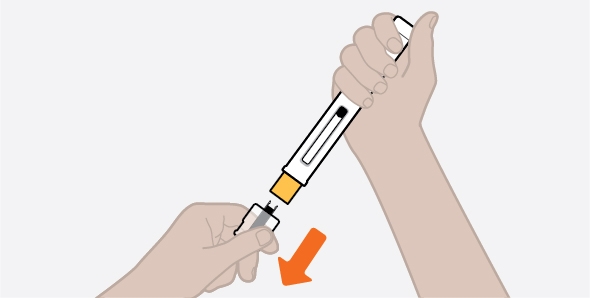
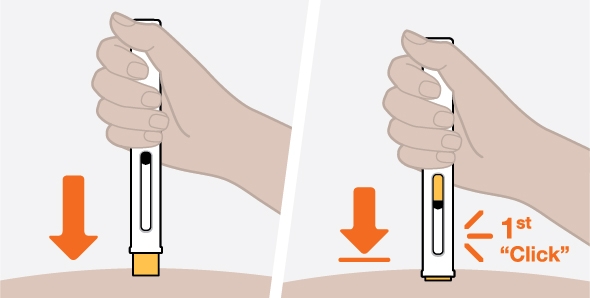
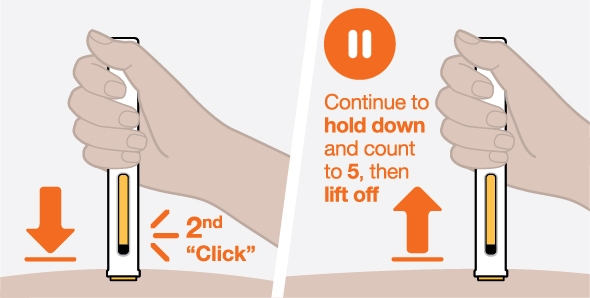
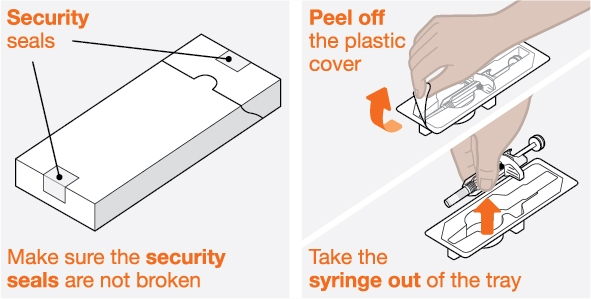
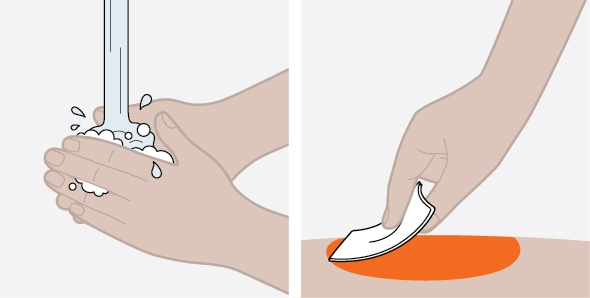
NUCALA injection is intended for use under the guidance of a healthcare provider. The NUCALA injection prefilled autoinjector and prefilled syringe are only for use in adults and adolescents aged 12 years and older. A patient may self-inject or the patient caregiver may administer NUCALA injection subcutaneously after the healthcare provider determines it is appropriate. Provide proper training in SC injection technique and on the preparation and administration of NUCALA injection prior to use according to the “Instructions for Use”.
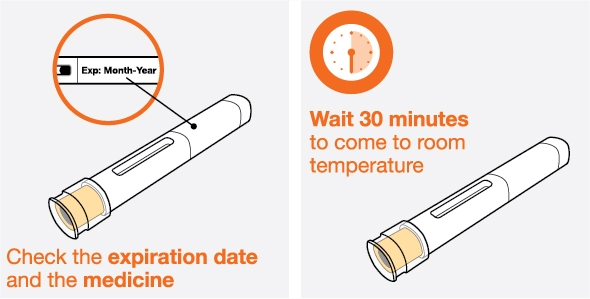
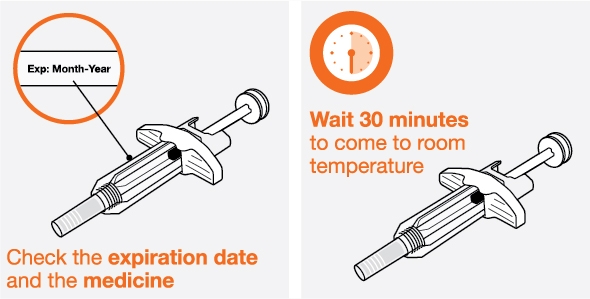
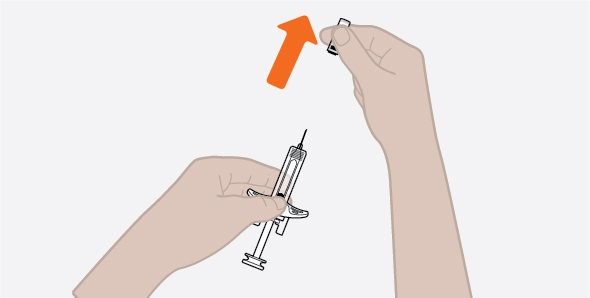
- 1. Remove the prefilled autoinjector or prefilled syringe from the refrigerator and allow it to sit at room temperature for 30 minutes prior to injection. Do not warm NUCALA in any other way.
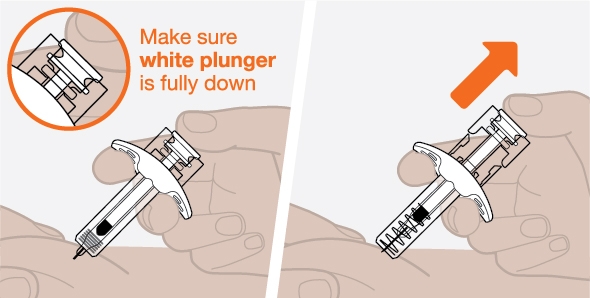
- 2. Prior to administration, visually inspect the window of the prefilled autoinjector or the prefilled syringe for particulate matter or discoloration. NUCALA injection should be clear to opalescent, colorless to pale yellow to pale brown in color. Do not use NUCALA injection if the product exhibits discoloration, cloudiness, or particulate matter. Do not use the NUCALA prefilled autoinjector or prefilled syringe if dropped on a hard surface.
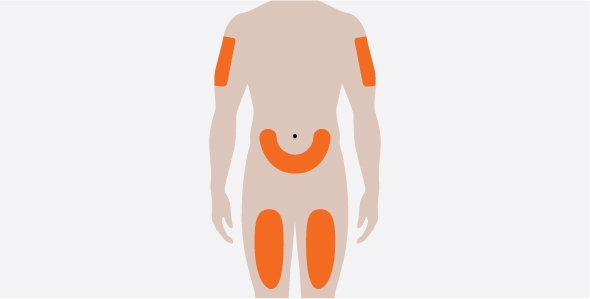
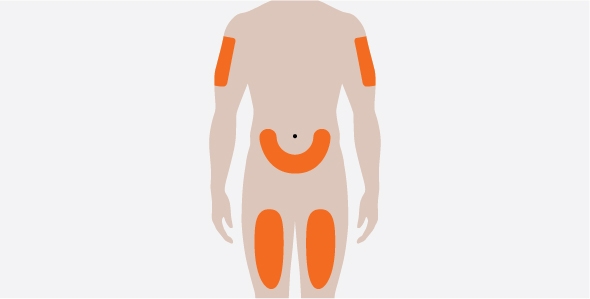
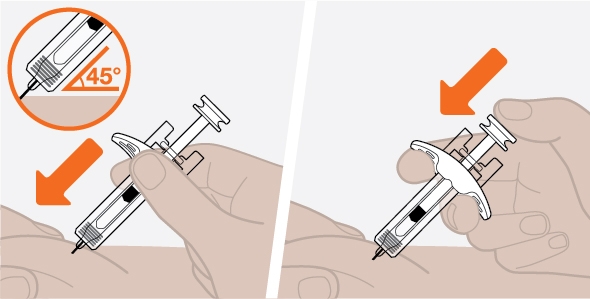
- 3. Administer the SC injection into the thigh or abdomen, avoiding the 5 cm (2 inches) around the navel. The upper arm can also be used if a caregiver administers the SC injection.
- 4. For use in EGPA, ensure injection sites for each SC injection are separated by at least 5 cm (2 inches).
- 5. Never give injections into areas where the skin is tender, bruised, red, or hard.
- 6. If a dose is missed, administer a dose as soon as possible. Thereafter, the patient can resume dosing on the usual day of administration. If the next dose is already due, then administer as planned.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
NUCALA should not be administered to patients with a history of hypersensitivity to mepolizumab or excipients in the formulation [see Description (11)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Hypersensitivity reactions (e.g., anaphylaxis, angioedema, bronchospasm, hypotension, urticaria, rash) have occurred following administration of NUCALA. These reactions generally occur within hours of administration, but in some instances can have a delayed onset (i.e., days). In the event of a hypersensitivity reaction, NUCALA should be discontinued [see Contraindications (4)].
5.2 Acute Asthma Symptoms or Deteriorating Disease
NUCALA should not be used to treat acute asthma symptoms or acute exacerbations. Do not use NUCALA to treat acute bronchospasm or status asthmaticus. Patients should seek medical advice if their asthma remains uncontrolled or worsens after initiation of treatment with NUCALA.
5.3 Opportunistic Infections: Herpes Zoster
Herpes zoster has occurred in subjects receiving NUCALA 100 mg in controlled clinical trials [see Adverse Reactions (6.1)]. Consider vaccination if medically appropriate.
5.4 Reduction of Corticosteroid Dosage
Do not discontinue systemic or inhaled corticosteroids (ICS) abruptly upon initiation of therapy with NUCALA. Reductions in corticosteroid dosage, if appropriate, should be gradual and performed under the direct supervision of a physician. Reduction in corticosteroid dosage may be associated with systemic withdrawal symptoms and/or unmask conditions previously suppressed by systemic corticosteroid therapy.
5.5 Parasitic (Helminth) Infection
Eosinophils may be involved in the immunological response to some helminth infections. Patients with known parasitic infections were excluded from participation in clinical trials. It is unknown if NUCALA will influence a patient’s response against parasitic infections. Treat patients with pre-existing helminth infections before initiating therapy with NUCALA. If patients become infected while receiving treatment with NUCALA and do not respond to anti-helminth treatment, discontinue treatment with NUCALA until infection resolves.
-
6 ADVERSE REACTIONS
The following adverse reactions are described in greater detail in other sections:
- Hypersensitivity reactions [see Warnings and Precautions (5.1)]
- Opportunistic infections: herpes zoster [see Warnings and Precautions (5.3)]
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
6.1 Clinical Trials Experience in Severe Asthma
Adult and Adolescent Subjects Aged 12 Years and Older
A total of 1,327 subjects with severe asthma were evaluated in 3 randomized, placebo-controlled, multicenter trials of 24 to 52 weeks’ duration (Trial 1, NCT #01000506; Trial 2, NCT #01691521; and Trial 3, NCT #01691508). Of these, 1,192 had a history of 2 or more exacerbations in the year prior to enrollment despite regular use of high-dose ICS plus additional controller(s) (Trials 1 and 2), and 135 subjects required daily oral corticosteroids (OCS) in addition to regular use of high-dose ICS plus additional controller(s) to maintain asthma control (Trial 3). All subjects had markers of eosinophilic airway inflammation [see Clinical Studies (14.1)]. Of the subjects enrolled, 59% were female, 85% were white, and ages ranged from 12 to 82 years. Mepolizumab was administered subcutaneously or intravenously once every 4 weeks; 263 subjects received NUCALA (mepolizumab 100 mg SC) for at least 24 weeks. Serious adverse events that occurred in more than 1 subject and in a greater percentage of subjects receiving NUCALA 100 mg (n = 263) than placebo (n = 257) included 1 event, herpes zoster (2 subjects vs. 0 subjects, respectively). Approximately 2% of subjects receiving NUCALA 100 mg withdrew from clinical trials due to adverse events compared with 3% of subjects receiving placebo.
The incidence of adverse reactions in the first 24 weeks of treatment in the 2 confirmatory efficacy and safety trials (Trials 2 and 3) with NUCALA 100 mg is shown in Table 1.
Table 1. Adverse Reactions with NUCALA with ≥3% Incidence and More Common than Placebo in Subjects with Severe Asthma (Trials 2 and 3) Adverse Reaction
NUCALA
(Mepolizumab 100 mg Subcutaneous)
(n = 263)
%
Placebo
(n = 257)
%
Headache
19
18
Injection site reaction
8
3
Back pain
5
4
Fatigue
5
4
Influenza
3
2
Urinary tract infection
3
2
Abdominal pain upper
3
2
Pruritus
3
2
Eczema
3
<1
Muscle spasms
3
<1
52-Week Trial: Adverse reactions from Trial 1 with 52 weeks of treatment with mepolizumab 75 mg intravenous (IV) (n = 153) or placebo (n = 155) and with ≥3% incidence and more common than placebo and not shown in Table 1 were: abdominal pain, allergic rhinitis, asthenia, bronchitis, cystitis, dizziness, dyspnea, ear infection, gastroenteritis, lower respiratory tract infection, musculoskeletal pain, nasal congestion, nasopharyngitis, nausea, pharyngitis, pyrexia, rash, toothache, viral infection, viral respiratory tract infection, and vomiting. In addition, 3 cases of herpes zoster occurred in subjects receiving mepolizumab 75 mg IV compared with 2 subjects in the placebo group.
Systemic Reactions, including Hypersensitivity Reactions: In Trials 1, 2, and 3 described above, the percentage of subjects who experienced systemic (allergic and non-allergic) reactions was 5% in the placebo group and 3% in the group receiving NUCALA 100 mg. Systemic allergic/hypersensitivity reactions were reported by 2% of subjects in the placebo group and 1% of subjects in the group receiving NUCALA 100 mg. The most commonly reported manifestations of systemic allergic/hypersensitivity reactions reported in the group receiving NUCALA 100 mg included rash, pruritus, headache, and myalgia. Systemic non-allergic reactions were reported by 2% of subjects in the group receiving NUCALA 100 mg and 3% of subjects in the placebo group. The most commonly reported manifestations of systemic non-allergic reactions reported in the group receiving NUCALA 100 mg included rash, flushing, and myalgia. A majority of the systemic reactions in subjects receiving NUCALA 100 mg (5/7) were experienced on the day of dosing.
Injection Site Reactions: Injection site reactions (e.g., pain, erythema, swelling, itching, burning sensation) occurred at a rate of 8% in subjects receiving NUCALA 100 mg compared with 3% in subjects receiving placebo.
Long-term Safety: Nine hundred ninety-eight subjects received NUCALA 100 mg in ongoing open-label extension studies, during which additional cases of herpes zoster were reported. The overall adverse event profile has been similar to the severe asthma trials described above.
Pediatric Subjects Aged 6 to 11 Years
The safety data for NUCALA is based upon 1 open-label clinical trial that enrolled 36 subjects with severe asthma aged 6 to 11 years. Subjects received 40 mg (for those weighing <40 kg) or 100 mg (for those weighing ≥40 kg) of NUCALA administered subcutaneously once every 4 weeks. Subjects received NUCALA for 12 weeks (initial short phase). After a treatment interruption of 8 weeks, 30 subjects received NUCALA for a further 52 weeks (long phase). The adverse reaction profile for subjects aged 6 to 11 years was similar to that observed in subjects aged 12 years and older.
6.2 Clinical Trials Experience in Eosinophilic Granulomatosis with Polyangiitis
A total of 136 subjects with EGPA were evaluated in 1 randomized, placebo-controlled, multicenter, 52-week treatment trial. Subjects received 300 mg of NUCALA or placebo subcutaneously once every 4 weeks. Subjects enrolled had a diagnosis of EGPA for at least 6 months prior to enrollment with a history of relapsing or refractory disease and were on a stable dosage of oral prednisolone or prednisone of greater than or equal to 7.5 mg/day (but not greater than 50 mg/day) for at least 4 weeks prior to enrollment [see Clinical Studies (14.2)]. Of the subjects enrolled, 59% were female, 92% were white, and ages ranged from 20 to 71 years. No additional adverse reactions were identified to those reported in the severe asthma trials.
Systemic Reactions, including Hypersensitivity Reactions
In the 52-week trial, the percentage of subjects who experienced systemic (allergic and non‑allergic) reactions was 1% in the placebo group and 6% in the group receiving 300 mg of NUCALA. Systemic allergic/hypersensitivity reactions were reported by 1% of subjects in the placebo group and 4% of subjects in the group receiving 300 mg of NUCALA. The manifestations of systemic allergic/hypersensitivity reactions reported in the group receiving 300 mg of NUCALA included rash, pruritus, flushing, fatigue, hypertension, warm sensation in trunk and neck, cold extremities, dyspnea, and stridor. Systemic non-allergic reactions were reported by 1 (1%) subject in the group receiving 300 mg of NUCALA and no subjects in the placebo group. The reported manifestation of systemic non-allergic reactions reported in the group receiving 300 mg of NUCALA was angioedema. Half of the systemic reactions in subjects receiving 300 mg of NUCALA (2/4) were experienced on the day of dosing.
Injection Site Reactions
Injection site reactions (e.g., pain, erythema, swelling) occurred at a rate of 15% in subjects receiving NUCALA compared with 13% in subjects receiving placebo.
6.3 Immunogenicity
In adult and adolescent subjects with severe asthma receiving NUCALA 100 mg, 15/260 (6%) had detectable anti-mepolizumab antibodies. Neutralizing antibodies were detected in 1 subject with asthma receiving NUCALA 100 mg. Anti-mepolizumab antibodies slightly increased (approximately 20%) the clearance of mepolizumab. There was no evidence of a correlation between anti-mepolizumab antibody titers and change in eosinophil level. The clinical relevance of the presence of anti-mepolizumab antibodies is not known. In the clinical trial of children aged 6 to 11 years with severe asthma receiving NUCALA 40 or 100 mg, 2/35 (6%) had detectable anti‑mepolizumab antibodies during the initial short phase of the trial. No children had detectable anti‑mepolizumab antibodies during the long phase of the trial.
In subjects with EGPA receiving 300 mg of NUCALA, 1/68 (<2%) had detectable anti-mepolizumab antibodies. No neutralizing antibodies were detected in any subjects with EGPA.
The reported frequency of anti-mepolizumab antibodies may underestimate the actual frequency due to lower assay sensitivity in the presence of high drug concentration. The data reflect the percentage of patients whose test results were positive for antibodies to mepolizumab in specific assays. The observed incidence of antibody positivity in an assay is highly dependent on several factors, including assay sensitivity and specificity, assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease.
6.4 Postmarketing Experience
In addition to adverse reactions reported from clinical trials, the following adverse reactions have been identified during postapproval use of NUCALA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. These events have been chosen for inclusion due to either their seriousness, frequency of reporting, or causal connection to NUCALA or a combination of these factors.
Immune System Disorders
Hypersensitivity reactions, including anaphylaxis.
- 7 DRUG INTERACTIONS
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women with asthma exposed to NUCALA during pregnancy. Healthcare providers can enroll patients or encourage patients to enroll themselves by calling 1-877-311-8972 or visiting www.mothertobaby.org/asthma.
Risk Summary
The data on pregnancy exposure are insufficient to inform on drug-associated risk. Monoclonal antibodies, such as mepolizumab, are transported across the placenta in a linear fashion as pregnancy progresses; therefore, potential effects on a fetus are likely to be greater during the second and third trimester of pregnancy. In a prenatal and postnatal development study conducted in cynomolgus monkeys, there was no evidence of fetal harm with IV administration of mepolizumab throughout pregnancy at doses that produced exposures up to approximately 9 times the exposure at the maximum recommended human dose (MRHD) of 300 mg SC (see Data).
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryofetal Risk: In women with poorly or moderately controlled asthma, evidence demonstrates that there is an increased risk of preeclampsia in the mother and prematurity, low birth weight, and small for gestational age in the neonate. The level of asthma control should be closely monitored in pregnant women and treatment adjusted as necessary to maintain optimal control.
Data
Animal Data: In a prenatal and postnatal development study, pregnant cynomolgus monkeys received mepolizumab from gestation Days 20 to 140 at doses that produced exposures up to approximately 9 times that achieved with the MRHD (on an AUC basis with maternal IV doses up to 100 mg/kg once every 4 weeks). Mepolizumab did not elicit adverse effects on fetal or neonatal growth (including immune function) up to 9 months after birth. Examinations for internal or skeletal malformations were not performed. Mepolizumab crossed the placenta in cynomolgus monkeys. Concentrations of mepolizumab were approximately 2.4 times higher in infants than in mothers up to Day 178 postpartum. Levels of mepolizumab in milk were ≤0.5% of maternal serum concentration.
In a fertility, early embryonic, and embryofetal development study, pregnant CD-1 mice received an analogous antibody, which inhibits the activity of murine interleukin-5 (IL-5), at an IV dose of 50 mg/kg once per week throughout gestation. The analogous antibody was not teratogenic in mice. Embryofetal development of IL-5–deficient mice has been reported to be generally unaffected relative to wild-type mice.
8.2 Lactation
Risk Summary
There is no information regarding the presence of mepolizumab in human milk, the effects on the breastfed infant, or the effects on milk production. However, mepolizumab is a humanized monoclonal antibody (IgG1 kappa), and immunoglobulin G (IgG) is present in human milk in small amounts. Mepolizumab was present in the milk of cynomolgus monkeys postpartum following dosing during pregnancy [see Use in Specific Populations (8.1)]. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for NUCALA and any potential adverse effects on the breastfed infant from mepolizumab or from the underlying maternal condition.
8.4 Pediatric Use
Severe Asthma
The safety and efficacy of NUCALA for severe asthma, and with an eosinophilic phenotype, have been established in pediatric patients aged 6 years and older.
Use of NUCALA in adolescents aged 12 to 17 years is supported by evidence from adequate and well-controlled trials in adults and adolescents. A total of 28 adolescents aged 12 to 17 years with severe asthma were enrolled in the Phase 3 asthma trials. Of these, 25 were enrolled in the 32-week exacerbation trial (Trial 2, NCT #01691521) and had a mean age of 14.8 years. Subjects had a history of 2 or more exacerbations in the previous year despite regular use of medium- or high-dose ICS plus additional controller(s) with or without OCS and had blood eosinophils of ≥150 cells/mcL at screening or ≥300 cells/mcL within 12 months prior to enrollment. [See Clinical Studies (14.1).] Subjects had a reduction in the rate of exacerbations that trended in favor of mepolizumab. Of the 19 adolescents who received mepolizumab, 9 received 100 mg and the mean apparent clearance in these subjects was 35% less than that of adults. The safety profile observed in adolescents was generally similar to that of the overall population in the Phase 3 studies [see Adverse Reactions (6.1)].
Use of NUCALA in children aged 6 to 11 years with severe asthma, and with an eosinophilic phenotype, is supported by evidence from adequate and well-controlled trials in adults and adolescents with additional pharmacokinetic, pharmacodynamic, and safety data in children aged 6 to 11 years. A single, open-label clinical trial (NCT #02377427) was conducted in 36 children aged 6 to 11 years (mean age: 8.6 years, 31% female) with severe asthma. Enrollment criteria were the same as for adolescents in the 32-week exacerbation trial (Trial 2). Based upon the pharmacokinetic data from this trial, a dose of 40 mg SC every 4 weeks was determined to have similar exposure to adults and adolescents administered a dose of 100 mg SC [see Clinical Pharmacology (12.3)].
The efficacy of NUCALA in children aged 6 to 11 years is extrapolated from efficacy in adults and adolescents with support from pharmacokinetic analyses showing similar drug exposure levels for 40 mg administered subcutaneously every 4 weeks in children aged 6 to 11 years compared with adults and adolescents [see Clinical Pharmacology (12.3)]. The safety profile and pharmacodynamic response observed in this trial for children aged 6 to 11 years were similar to that seen in adults and adolescents [see Adverse Reactions (6.1), Clinical Pharmacology (12.2)].
The safety and efficacy in pediatric patients aged younger than 6 years with severe asthma have not been established.
Eosinophilic Granulomatosis with Polyangiitis
The safety and efficacy in patients aged younger than 18 years with EGPA have not been established.
8.5 Geriatric Use
Clinical trials of NUCALA did not include sufficient numbers of subjects aged 65 years and older that received NUCALA (n = 46) to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function and of concomitant disease or other drug therapy. Based on available data, no adjustment of the dosage of NUCALA in geriatric patients is necessary, but greater sensitivity in some older individuals cannot be ruled out.
-
10 OVERDOSAGE
Single doses of up to 1,500 mg have been administered intravenously to adult subjects in a clinical trial with eosinophilic disease without evidence of dose-related toxicities.
There is no specific treatment for an overdose with mepolizumab. If overdose occurs, the patient should be treated supportively with appropriate monitoring as necessary.
-
11 DESCRIPTION
Mepolizumab is a humanized IL-5 antagonist monoclonal antibody. Mepolizumab is produced by recombinant DNA technology in Chinese hamster ovary cells. Mepolizumab has a molecular weight of approximately 149 kDa.
NUCALA (mepolizumab) for injection is a sterile, preservative-free, white to off-white, lyophilized powder in a single-dose vial for SC injection after reconstitution. Upon reconstitution with 1.2 mL of Sterile Water for Injection, USP, the resulting concentration is 100 mg/mL and delivers 1 mL [see Dosage and Administration (2.3)]. Each vial delivers 100 mg of mepolizumab, polysorbate 80 (0.67 mg), sodium phosphate dibasic heptahydrate (7.14 mg), and sucrose (160 mg), with a pH of 7.0.
The vial stopper is not made with natural rubber latex.
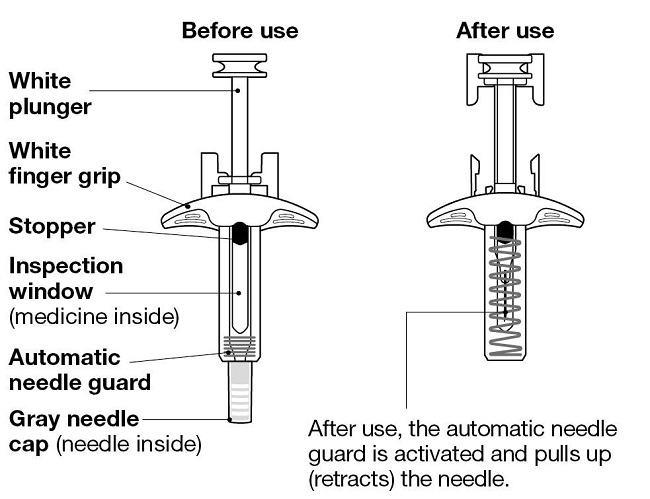
NUCALA injection is a sterile, preservative-free, clear to opalescent, colorless to pale yellow to pale brown solution for SC use. It is supplied in a single-dose, 1-mL, prefilled autoinjector with a fixed 29-gauge, half-inch needle or in a single-dose, 1-mL, prefilled syringe with a fixed 29-gauge, half-inch needle with a needle guard. Each 1 mL delivers 100 mg mepolizumab, citric acid monohydrate (0.95 mg), EDTA disodium dihydrate (0.019 mg), polysorbate 80 (0.20 mg), sodium phosphate dibasic heptahydrate (4.16 mg), and sucrose (120 mg), with a pH of 6.3.
The prefilled autoinjector and prefilled syringe are not made with natural rubber latex.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Mepolizumab is an IL-5 antagonist (IgG1 kappa). IL-5 is the major cytokine responsible for the growth and differentiation, recruitment, activation, and survival of eosinophils. Mepolizumab binds to IL-5 with a dissociation constant of 100 pM, inhibiting the bioactivity of IL-5 by blocking its binding to the alpha chain of the IL-5 receptor complex expressed on the eosinophil cell surface. Inflammation is an important component in the pathogenesis of asthma and EGPA. Multiple cell types (e.g., mast cells, eosinophils, neutrophils, macrophages, lymphocytes) and mediators (e.g., histamine, eicosanoids, leukotrienes, cytokines) are involved in inflammation. Mepolizumab, by inhibiting IL-5 signaling, reduces the production and survival of eosinophils; however, the mechanism of mepolizumab action in asthma and EGPA has not been definitively established.
12.2 Pharmacodynamics
The pharmacodynamic response (blood eosinophil reduction) following repeat doses of mepolizumab administered subcutaneously or intravenously was evaluated in adult subjects with asthma and blood eosinophil levels >200 cells/mcL. Subjects received 1 of 4 mepolizumab treatments (administered every 28 days for a total of 3 doses): 12.5 mg SC, 125 mg SC, 250 mg SC, or 75 mg IV. Sixty-six of the 70 randomized subjects completed the trial. Compared with baseline levels, blood eosinophils decreased in a dose-dependent manner. A reduction in blood eosinophil levels was observed in all treatment groups by Day 3 (48 hours post-dose). On Day 84 (4 weeks post-last dose), the observed geometric mean reduction from baseline in blood eosinophils was 64%, 78%, 84%, and 90% in the 12.5-mg SC, 75-mg IV, 125-mg SC, and 250‑mg SC treatment groups, respectively. The model-predicted SC doses providing 50% and 90% of maximal reduction of blood eosinophils at Day 84 were estimated to be 11 and 99 mg, respectively. These results, along with the clinical efficacy data from the dose-ranging exacerbation trial in adult and adolescent subjects with severe asthma (Trial 1) supported the evaluation of mepolizumab 75 mg IV and 100 mg SC in the confirmatory severe asthma trials [see Clinical Studies (14.1)]. Following SC administration of mepolizumab 100 mg every 4 weeks for 32 weeks in adult and adolescent subjects with severe asthma (Trial 2), blood eosinophils were reduced to a geometric mean count of 40 cells/mcL, which corresponds to a geometric mean reduction of 84% compared with placebo.
The pharmacodynamic response (blood eosinophil reduction) was also evaluated in children aged 6 to 11 years with severe asthma. Following SC administration of mepolizumab 40 mg every 4 weeks for 52 weeks, blood eosinophils were reduced to a geometric mean count of 48 cells/mcL. This corresponds to a geometric mean reduction from baseline of 85%.
The magnitude of reduction in adults, adolescents, and children was observed within 4 weeks of treatment and was maintained throughout the treatment periods.
For adults with EGPA, following SC administration of mepolizumab 300 mg every 4 weeks for 52 weeks, blood eosinophils were reduced to a geometric mean count of 38 cells/mcL. There was a geometric mean reduction of 83% compared with placebo and this magnitude of reduction was observed within 4 weeks of treatment [see Clinical Studies (14.2)].
12.3 Pharmacokinetics
Following SC dosing in adult subjects with asthma, mepolizumab exhibited approximately dose‑proportional pharmacokinetics over a dose range of 12.5 to 250 mg. The pharmacokinetic properties of mepolizumab observed in adult subjects with EGPA were similar to the pharmacokinetic properties observed in adult and adolescent subjects with severe asthma.
Systemic exposure following administration of mepolizumab 300 mg subcutaneously in adult subjects with EGPA was approximately 3 times that of mepolizumab 100 mg administered subcutaneously in adult and adolescent subjects with severe asthma (Trial 2).
Absorption
Following 100-mg SC administration in the upper arm of adult and adolescent subjects with asthma, the bioavailability of mepolizumab was estimated to be approximately 80%.
Following repeat SC administration once every 4 weeks, there was approximately a 2-fold accumulation at steady state.
Distribution
The population central volume of distribution of mepolizumab in adult subjects with asthma is estimated to be 3.6 L for a 70-kg individual.
Metabolism
Mepolizumab is a humanized IgG1 monoclonal antibody that is degraded by proteolytic enzymes widely distributed in the body and not restricted to hepatic tissue.
Elimination
Following SC administration of mepolizumab in adult subjects with asthma, the mean terminal half‑life (t1/2) ranged from 16 to 22 days. The population apparent systemic clearance of mepolizumab in adult and adolescent subjects with asthma is estimated to be 0.28 L/day for a 70‑kg individual.
Specific Populations
Racial Groups and Male and Female Patients: Population pharmacokinetics analyses indicated there was no significant effect of race and gender on mepolizumab clearance.
Age: Population pharmacokinetics analyses indicated there was no significant effect of age on mepolizumab clearance.
Pediatric Patients: Mepolizumab pharmacokinetics following SC administration in subjects aged 6 to 11 years with severe asthma was investigated in the initial 12-week treatment phase of an open-label clinical trial. Exposures (AUC) following Sc administration of either 40 mg (for children weighing <40 kg) or 100 mg (for children weighing ≥40 kg) were 1.32 and 1.97 times higher, respectively, compared with that observed in adults and adolescents receiving 100 mg. Based on these results, simulation of a 40-mg SC dose every 4 weeks in children aged 6 to 11 years, irrespective of body weight, resulted in predicted exposures similar to that observed in adults and adolescents.
Patients with Renal Impairment: No clinical trials have been conducted to investigate the effect of renal impairment on the pharmacokinetics of mepolizumab. Based on population pharmacokinetic analyses, mepolizumab clearance was comparable between subjects with creatinine clearance values between 50 and 80 mL/min and patients with normal renal function. There are limited data available in subjects with creatinine clearance values <50 mL/min; however, mepolizumab is not cleared renally.
Patients with Hepatic Impairment: No clinical trials have been conducted to investigate the effect of hepatic impairment on the pharmacokinetics of mepolizumab. Since mepolizumab is degraded by widely distributed proteolytic enzymes, not restricted to hepatic tissue, changes in hepatic function are unlikely to have any effect on the elimination of mepolizumab.
Drug Interaction Studies
No formal drug interaction studies have been conducted with NUCALA. In population pharmacokinetics analyses of Phase 3 studies, there was no evidence of an effect of commonly coadministered small molecule drugs on mepolizumab exposure.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal studies have not been performed to evaluate the carcinogenic potential of mepolizumab. Published literature using animal models suggests that IL-5 and eosinophils are part of an early inflammatory reaction at the site of tumorigenesis and can promote tumor rejection. However, other reports indicate that eosinophil infiltration into tumors can promote tumor growth. Therefore, the malignancy risk in humans from an antibody to IL-5 such as mepolizumab is unknown.
Male and female fertility were unaffected based upon no adverse histopathological findings in the reproductive organs from cynomolgus monkeys receiving mepolizumab for 6 months at IV dosages up to 100 mg/kg once every 4 weeks (approximately 20 times the MRHD of 300 mg on an AUC basis). Mating and reproductive performance were unaffected in male and female CD-1 mice receiving an analogous antibody, which inhibits the activity of murine IL-5, at an IV dosage of 50 mg/kg once per week.
-
14 CLINICAL STUDIES
14.1 Severe Asthma
The asthma development program for NUCALA in subjects aged 12 years and older included 3 double-blind, randomized, placebo‑controlled trials: 1 dose-ranging and exacerbation trial (Trial 1, NCT #01000506) and 2 confirmatory trials (Trial 2, NCT #01691521 and Trial 3, NCT #01691508). Mepolizumab was administered every 4 weeks in all 3 trials as add-on to background treatment. All subjects continued their background asthma therapy throughout the duration of the trials.
Dose-Ranging and Exacerbation Trial
Trial 1 was a 52-week dose-ranging and exacerbation-reduction trial in subjects with severe asthma with a history of 2 or more exacerbations in the previous year despite regular use of high-dose ICS plus additional controller(s) with or without OCS. Subjects enrolled in this trial were required to have at least 1 of the following 4 pre-specified criteria in the previous 12 months: blood eosinophil count ≥300 cells/mcL, sputum eosinophil count ≥3%, exhaled nitric oxide concentration ≥50 ppb, or deterioration of asthma control after ≤25% reduction in regular maintenance ICS/OCS. Three IV dosages of mepolizumab (75, 250, and 750 mg) administered once every 4 weeks were evaluated compared with placebo. Results from this trial and the pharmacodynamic study supported the evaluation of mepolizumab 75 mg IV and 100 mg SC in the subsequent trials [see Clinical Pharmacology (12.2)]. NUCALA is not indicated for IV use and should only be administered by the SC route.
Confirmatory Trials
A total of 711 subjects with severe asthma were studied in the 2 confirmatory trials (Trials 2 and 3). In these 2 trials subjects were required to have blood eosinophils of ≥150 cells/mcL at screening (within 6 weeks of dosing) or blood eosinophils of ≥300 cells/mcL within 12 months of enrollment. The screening blood eosinophils of ≥150 cells/mcL criterion was derived from exploratory analyses of data from Trial 1. Trial 2 was a 32-week placebo- and active-controlled trial in subjects with severe asthma with a history of 2 or more exacerbations in the previous year despite regular use of high-dose ICS plus additional controller(s) with or without OCS. Subjects received mepolizumab 75 mg IV (n = 191), NUCALA 100 mg (n = 194), or placebo (n = 191) once every 4 weeks for 32 weeks.
Trial 3 was a 24-week OCS-reduction trial in subjects with severe asthma who required daily OCS in addition to regular use of high-dose ICS plus additional controller(s) to maintain asthma control. Subjects in Trial 3 were not required to have a history of exacerbations in the previous year. Subjects received NUCALA 100 mg (n = 69) or placebo (n = 66) once every 4 weeks for 24 weeks. The baseline mean OCS use was similar in the 2 treatment groups: 13.2 mg in the placebo group and 12.4 mg in the group receiving NUCALA 100 mg.
The demographics and baseline characteristics of these 3 trials are provided in Table 2.
Table 2. Demographics and Baseline Characteristics of Severe Asthma Trials FEV1 = forced expiratory volume in 1 second, SABA = short-acting beta2-agonist, FVC = forced vital capacity. Trial 1
(N = 616)
Trial 2
(N = 576)
Trial 3
(N = 135)
Mean age (y)
49
50
50
Female, n (%)
387 (63)
328 (57)
74 (55)
White, n (%)
554 (90)
450 (78)
128 (95)
Duration of asthma, mean (y)
19
20
19
Never smoked, n (%)
483 (78)
417 (72)
82 (61)
Baseline FEV1, L
1.88
1.82
1.95
Baseline % predicted FEV1
60
61
59
Baseline % reversibility
25
27
26
Baseline post-SABA FEV1/FVC
0.67
0.66
0.66
Geometric mean eosinophil count at baseline, cells/mcL
250
290
240
Mean number of exacerbations in previous year
3.6
3.6
3.1
Exacerbations
The primary endpoint for Trials 1 and 2 was the frequency of exacerbations defined as worsening of asthma requiring use of oral/systemic corticosteroids and/or hospitalization and/or emergency department visits. For subjects on maintenance OCS, an exacerbation requiring OCS was defined as the use of oral/systemic corticosteroids at least double the existing dose for at least 3 days. Compared with placebo, subjects receiving NUCALA 100 mg or mepolizumab 75 mg IV experienced significantly fewer exacerbations. Additionally, compared with placebo, there were fewer exacerbations requiring hospitalization and/or emergency department visits and exacerbations requiring only in-patient hospitalization with NUCALA 100 mg (Table 3).
Table 3. Rate of Exacerbations in Severe Asthma Trials 1 and 2 (Intent-to-Treat Population) IV = intravenous, SC = subcutaneous. Trial
Treatment
Exacerbations per Year
Rate
Difference
Rate Ratio
(95% CI)
All exacerbations
Trial 1
Placebo (n = 155)
2.40
Mepolizumab 75 mg IV (n = 153)
1.24
1.16
0.52
(0.39, 0.69)
Trial 2
Placebo (n = 191)
1.74
Mepolizumab 75 mg IV (n = 191)
0.93
0.81
0.53
(0.40, 0.72)
NUCALA 100 mg SC (n = 194)
0.83
0.91
0.47
(0.35, 0.64)
Exacerbations requiring hospitalization/emergency room visit
Trial 1
Placebo (n = 155)
0.43
Mepolizumab 75 mg IV (n = 153)
0.17
0.26
0.40
(0.19, 0.81)
Trial 2
Placebo (n = 191)
0.20
Mepolizumab 75 mg IV (n = 191)
0.14
0.06
0.68
(0.33, 1.41)
NUCALA 100 mg SC (n = 194)
0.08
0.12
0.39
(0.18, 0.83)
Exacerbations requiring hospitalization
Trial 1
Placebo (n = 155)
0.18
Mepolizumab 75 mg IV (n = 153)
0.11
0.07
0.61
(0.28, 1.33)
Trial 2
Placebo (n = 191)
0.10
Mepolizumab 75 mg IV (n = 191)
0.06
0.04
0.61
(0.23, 1.66)
NUCALA 100 mg SC (n = 194)
0.03
0.07
0.31
(0.11, 0.91)
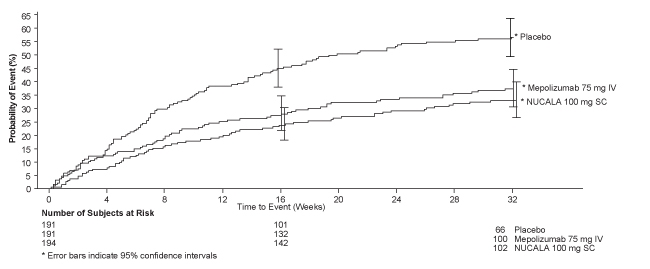
The time to first exacerbation was longer for the groups receiving NUCALA 100 mg and mepolizumab 75 mg IV compared with placebo in Trial 2 (Figure 1).
Figure 1. Kaplan-Meier Cumulative Incidence Curve for Time to First Exacerbation (Severe Asthma Trial 2)
IV = intravenous, SC = subcutaneous.
Trial 1 data were explored to determine criteria that could identify subjects likely to benefit from treatment with NUCALA. The exploratory analysis suggested that baseline blood eosinophil count of ≥150 cells/mcL was a potential predictor of treatment benefit. Exploratory analysis of Trial 2 data also suggested that baseline blood eosinophil count (obtained within 6 weeks of initiation of dosing) of ≥150 cells/mcL was a potential predictor of efficacy and showed a trend of greater exacerbation benefit with increasing blood eosinophil count. In Trial 2, subjects enrolled solely on the basis of the historical blood eosinophil count of ≥300 cells/mcL in the previous 12 months, but who had a baseline blood eosinophil count <150 cells/mcL, had virtually no exacerbation benefit following treatment with NUCALA 100 mg compared with placebo.
The Asthma Control Questionnaire-5 (ACQ-5) was assessed in Trials 1 and 2, and the St. George’s Respiratory Questionnaire (SGRQ) was assessed in Trial 2. In Trial 1, the ACQ-5 responder rate (defined as a decrease in score of 0.5 or more as threshold) for the 75-mg IV mepolizumab arm was 47% compared with 50% for placebo with an odds ratio (OR) of 1.1 (95% CI: 0.7, 1.7). In Trial 2, the ACQ-5 responder rate for the treatment arm for NUCALA 100 mg was 57% compared with 45% for placebo with an OR of 1.8 (95% CI: 1.2, 2.8). In Trial 2, the SGRQ responder rate (defined as a decrease in score of 4 or more as threshold) for the treatment arm for NUCALA 100 mg was 71% compared with 55% for placebo with an OR of 2.1 (95% CI: 1.3, 3.2).
Oral Corticosteroid Reduction
Trial 3 evaluated the effect of NUCALA 100 mg on reducing the use of maintenance OCS. The primary endpoint was the percent reduction of OCS dose during Weeks 20 to 24 compared with baseline dose, while maintaining asthma control. Subjects were classified according to their change in OCS use during the trial with the following categories: 90% to 100% decrease, 75% to <90% decrease, 50% to <75% decrease, >0% to <50% decrease, and no improvement (i.e., no change or any increase or lack of asthma control or withdrawal of treatment). Compared with placebo, subjects receiving NUCALA 100 mg achieved greater reductions in daily maintenance OCS dose, while maintaining asthma control. Sixteen (23%) subjects in the group receiving NUCALA 100 mg versus 7 (11%) in the placebo group had a 90% to 100% reduction in their OCS dose. Twenty-five (36%) subjects in the group receiving NUCALA 100 mg versus 37 (56%) in the placebo group were classified as having no improvement for OCS dose. Additionally, 54% of subjects receiving NUCALA 100 mg achieved at least a 50% reduction in the daily prednisone dose compared with 33% of subjects receiving placebo (95% CI for difference: 4%, 37%). An exploratory analysis was also performed on the subgroup of 29 subjects in Trial 3 who had an average baseline and screening blood eosinophil count <150 cells/mcL. Five (29%) subjects in the group receiving NUCALA 100 mg versus 0 (0%) in the placebo group had a 90% to 100% reduction in their dose. Four (24%) subjects in the group receiving NUCALA 100 mg versus 8 (67%) in the placebo group were classified as having no improvement for OCS dose. The ACQ and SGRQ were also assessed in Trial 3 and showed results similar to those in Trial 2.
Lung Function
Change from baseline in mean forced expiratory volume in 1 second (FEV1) was measured in all 3 trials and is presented in Table 4. Compared with placebo, NUCALA 100 mg did not provide consistent improvements in mean change from baseline in FEV1.
Table 4. Change from Baseline in FEV1 (mL) in Severe Asthma Trials a Dose = 75 mg intravenous.
b Forced expiratory volume in 1 second (FEV1) at Week 52.
c Dose = 100 mg subcutaneous.
d FEV1 at Week 32.Trial
Difference from Placebo in Mean Change from
Baseline FEV1 (mL) (95% CI)Week 12
Week 24
Weeks 32/52
1a
10 (-87, 108)
5 (-98, 108)
61 (-39, 161)b
2c
52 (-30, 134)
76 (-6, 159)
98 (11, 184)d
3c
56 (-91, 203)
114 (-42, 271)
NA
The effect of mepolizumab on lung function was also studied in a 12-week, placebo-controlled trial enrolling patients with asthma on a moderate dose of ICS with evidence of symptoms and lung function impairment. Enrollment was not dependent on a history of exacerbations or a pre-specified eosinophil count. Change from baseline in FEV1 at Week 12 was numerically lower in the mepolizumab treatment groups than the placebo group.
14.2 Eosinophilic Granulomatosis with Polyangiitis
A total of 136 adult subjects with EGPA were evaluated in a randomized, placebo-controlled, multicenter, 52-week trial (NCT #02020889). Subjects received 300 mg of NUCALA or placebo administered subcutaneously once every 4 weeks while continuing their stable OCS therapy. Starting at Week 4, OCS was tapered during the treatment period at the discretion of the investigator. The co-primary endpoints were the total accrued duration of remission over the 52‑week treatment period, defined as Birmingham Vasculitis Activity Score (BVAS) = 0 (no active vasculitis) plus prednisolone or prednisone dose less than or equal to 4 mg/day, and the proportion of subjects in remission at both Week 36 and Week 48 of treatment. The BVAS is a clinician-completed tool to assess clinically active vasculitis that would likely require treatment, after exclusion of other causes.
The demographics and baseline characteristics of subjects in this trial are provided in Table 5.
Table 5. Demographics and Baseline Characteristics in EGPA a Prednisone or prednisolone equivalent.
b e.g., Azathioprine, methotrexate, mycophenolic acid.
EGPA = eosinophilic granulomatosis with polyangiitis, SD = standard deviation.N = 136
Mean age (y)
48.5
Female, n (%)
80 (59)
White, n (%)
125 (92)
Duration (y) of EGPA, mean (SD)
5.5 (4.63)
History of >1 confirmed relapse in past 2 years, n (%)
100 (74)
Refractory disease, n (%)
74 (54)
Recurrence of EGPA symptoms, n (%)
68 (50)
Failed induction treatment, n (%)
6 (4)
Baseline oral corticosteroida daily dose (mg), median (range)
12 (7.5-50)
Receiving immunosuppressive therapyb, n (%)
72 (53)
Remission
Subjects receiving 300 mg of NUCALA achieved a significantly greater accrued time in remission compared with placebo. A significantly higher proportion of subjects receiving 300 mg of NUCALA achieved remission at both Week 36 and Week 48 compared with placebo (Table 6). Results of the components of remission are also shown in Table 6. In addition, significantly more subjects receiving 300 mg of NUCALA achieved remission within the first 24 weeks and remained in remission for the remainder of the 52-week study treatment period compared with placebo (19% for 300 mg of NUCALA versus 1% for placebo; OR 19.7; 95% CI: 2.3, 167.9).
Table 6. Remission and Components of Remission in EGPA a An odds ratio >1 favors mepolizumab.
EGPA = eosinophilic granulomatosis with polyangiitis, OCS = oral corticosteroid, BVAS = Birmingham Vasculitis Activity Score.Remission
(OCS ≤4 mg/day + BVAS = 0)
OCS ≤4 mg/day
BVAS = 0
Placebo
n = 68
NUCALA 300 mg
n = 68
Placebo
n = 68
NUCALA 300 mg
n = 68
Placebo
n = 68
NUCALA 300 mg
n = 68
Accrued duration over 52 weeks, n (%)
0
55 (81)
32 (47)
46 (68)
27 (40)
6 (9)
3 (4)
>0 to <12 weeks
8 (12)
8 (12)
12 (18)
5 (7)
15 (22)
13 (19)
12 to <24 weeks
3 (4)
9 (13)
6 (9)
12 (18)
11 (16)
5 (7)
24 to <36 weeks
0
10 (15)
2 (3)
10 (15)
17 (25)
2 (3)
≥36 weeks
2 (3)
9 (13)
2 (3)
14 (21)
19 (28)
45 (66)
Odds ratio (mepolizumab/placebo)a
(95% CI)
5.9
(2.7, 13.0)
5.1
(2.5, 10.4)
3.7
(1.8, 7.6)
Proportion of subjects at both Weeks 36 and 48
Subjects, n (%)
2 (3)
22 (32)
7 (10)
28 (41)
23 (34)
34 (50)
Odds ratio (mepolizumab/placebo)a
(95% CI)
16.7
(3.6, 77.6)
6.6
(2.6, 17.1)
1.9
(0.9, 4.2)
Additionally, a statistically significant benefit for these endpoints was demonstrated using remission defined as BVAS = 0 plus prednisolone/prednisone ≤7.5 mg/day.
Relapse
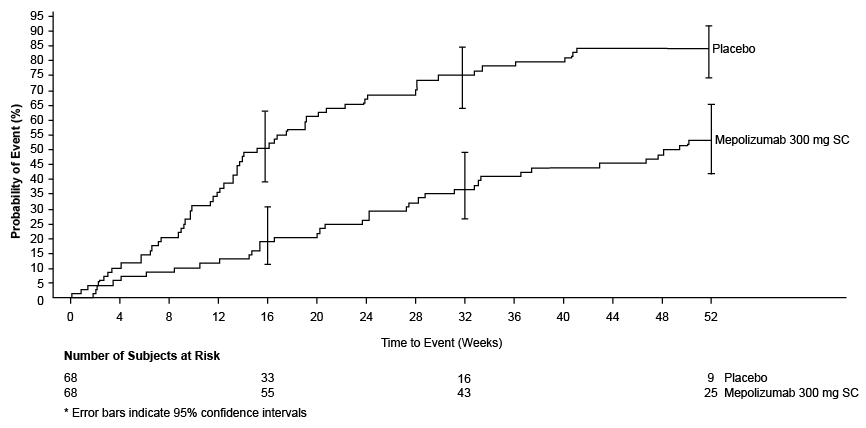
The time to first relapse (defined as worsening related to vasculitis, asthma, or sino-nasal symptoms requiring an increase in dose of corticosteroids or immunosuppressive therapy or hospitalization) was significantly longer for subjects receiving 300 mg of NUCALA compared with placebo with a hazard ratio of 0.32 (95% CI: 0.21, 0.5) (Figure 2). Additionally, subjects receiving 300 mg of NUCALA had a reduction in rate of relapse compared with subjects receiving placebo (rate ratio 0.50; 95% CI: 0.36, 0.70 for 300 mg of NUCALA compared with placebo). The incidence and number of relapse types (vasculitis, asthma, sino‑nasal) were numerically lower with mepolizumab compared with placebo.
Figure 2. Kaplan-Meier Plot of Time to First Relapse in EGPA
EGPA = eosinophilic granulomatosis with polyangiitis, SC = subcutaneous.
Corticosteroid Reduction
Subjects receiving 300 mg of NUCALA had a significantly greater reduction in average daily OCS dose compared with subjects receiving placebo during Weeks 48 to 52 (Table 7).
Table 7. Average Daily Oral Corticosteroid Dose during Weeks 48 to 52 in EGPA a Analyzed using a proportional odds model with covariates of treatment group, baseline oral corticosteroid daily dose, baseline Birmingham Vasculitis Activity Score, and region.
b An odds ratio <1 favors mepolizumab.
EGPA = eosinophilic granulomatosis with polyangiitis.Number (%) of Subjects
Placebo
n = 68
NUCALA 300 mg
n = 68
0
2 (3)
12 (18)
>0 to ≤4.0 mg
3 (4)
18 (26)
>4.0 to ≤7.5 mg
18 (26)
10 (15)
>7.5 mg
45 (66)
28 (41)
Comparison: mepolizumab/placeboa
Odds ratiob
---
0.20
95% CI
---
0.09, 0.41
Asthma Control Questionnaire-6 (ACQ-6)
The ACQ-6, a 6-item questionnaire completed by the subject, was developed to measure the adequacy of asthma control and change in asthma control. The on-treatment ACQ-6 responder rate during Weeks 48 to 52 (defined as a decrease in score of 0.5 or more compared with baseline) was 22% for 300 mg of NUCALA and 16% for placebo (OR 1.56; 95% CI: 0.63, 3.88 for 300 mg of NUCALA compared with placebo).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
NUCALA for Injection
NUCALA (mepolizumab) for injection is a sterile, preservative-free, white to off-white, lyophilized powder for reconstitution and SC injection in a single-dose glass vial with a flip-off seal. The vial stopper is not made with natural rubber latex.
NUCALA for injection is supplied as:
100-mg single-dose vials in cartons of 1 (NDC: 0173-0881-01).
Store vials below 77°F (25°C). Do not freeze. Store in the original carton to protect from light.
NUCALA Injection
NUCALA (mepolizumab) injection is a sterile, preservative-free, clear to opalescent, colorless to pale yellow to pale brown solution for SC use. Each single-dose prefilled autoinjector or single-dose prefilled syringe is designed to deliver 100 mg of mepolizumab in 1 mL of solution. The autoinjectors and syringes are not made with natural rubber latex.
NUCALA injection is supplied as:
100 mg/mL, single-dose, prefilled autoinjector with attached 29-gauge, half-inch needle in cartons of 1 (NDC: 0173-0892-01).
100 mg/mL, single-dose, prefilled glass syringe with attached 29-gauge, half-inch needle in cartons of 1 (NDC: 0173-0892-42).
Prior to Dispensing: Refrigerate prefilled autoinjectors and prefilled syringes at 36°F to 46°F (2°C to 8°C). Keep the product in the original carton to protect from light. Do not freeze. Do not shake. Avoid exposure to heat.
Following Dispensing: Refrigerate prefilled autoinjectors and prefilled syringes at 36°F to 46°F (2°C to 8°C). Keep the product in the original carton to protect from light until the time of use. Do not freeze. Do not shake. Avoid exposure to heat.
If necessary, an unopened carton can be stored outside the refrigerator at up to 86°F (30°C) for up to 7 days. Discard if left out of the refrigerator for more than 7 days.
NUCALA injection must be administered within 8 hours after removal from the carton. Discard if not administered within 8 hours.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Hypersensitivity Reactions
Inform patients that hypersensitivity reactions (e.g., anaphylaxis, angioedema, bronchospasm, hypotension, urticaria, rash) have occurred after administration of NUCALA. Instruct patients to contact their physicians if such reactions occur.
Not for Acute Symptoms or Deteriorating Disease
Inform patients that NUCALA does not treat acute asthma symptoms or acute exacerbations. Inform patients to seek medical advice if their asthma remains uncontrolled or worsens after initiation of treatment with NUCALA.
Opportunistic Infections: Herpes Zoster
Inform patients that herpes zoster infections have occurred in patients receiving NUCALA and where medically appropriate, inform patients that vaccination should be considered.
Reduction of Corticosteroid Dosage
Inform patients to not discontinue systemic or inhaled corticosteroids except under the direct supervision of a physician. Inform patients that reduction in corticosteroid dose may be associated with systemic withdrawal symptoms and/or unmask conditions previously suppressed by systemic corticosteroid therapy.
Pregnancy Exposure Registry
Inform women there is a pregnancy exposure registry that monitors pregnancy outcomes in women with asthma exposed to NUCALA during pregnancy and that they can enroll in the Pregnancy Exposure Registry by calling 1-877-311-8972 or by visiting www.mothertobaby.org/asthma [see Use in Specific Populations (8.1)].
Trademarks are owned by or licensed to the GSK group of companies.
Manufactured by
GlaxoSmithKline LLC
Philadelphia, PA 19112
U.S. License Number 1727
Distributed by
GlaxoSmithKline
Research Triangle Park, NC 27709
©2019 GSK group of companies or its licensor.
NCL:5PI
-
PATIENT PACKAGE INSERT
Patient Information
NUCALA (new KAH la)
(mepolizumab)
for injection, for subcutaneous use
NUCALA (new KAH la)
(mepolizumab)
injection, for subcutaneous use
What is NUCALA?
-
NUCALA is a prescription medicine used with other medicines:
- o for add-on maintenance treatment of severe asthma in people 6 years of age and older whose asthma is not controlled with their current asthma medicines. NUCALA helps prevent severe asthma attacks (exacerbations).
- o for the treatment of adults with eosinophilic granulomatosis with polyangiitis (EGPA). NUCALA helps reduce symptoms and flares, and it may allow your healthcare provider to reduce your oral corticosteroid medicine.
- Medicines such as NUCALA reduce blood eosinophils. Eosinophils are a type of white blood cells that may contribute to your disease.
- NUCALA is not used to treat sudden breathing problems that occur with asthma.
It is not known if NUCALA is safe and effective in children with severe asthma under 12 years of age.
It is not known if NUCALA is safe and effective in children and adolescents with EGPA under 18 years of age.
Do not use NUCALA if you are allergic to mepolizumab or any of the ingredients in NUCALA. See the end of this leaflet for a complete list of ingredients in NUCALA.
Before receiving NUCALA, tell your healthcare provider about all of your medical conditions, including if you:
- have a parasitic (helminth) infection.
- are taking oral or inhaled corticosteroid medicines. Do not stop taking your corticosteroid medicines unless instructed by your healthcare provider. This may cause other symptoms that were controlled by the corticosteroid medicine to come back.
-
are pregnant or plan to become pregnant. It is not known if NUCALA may harm your unborn baby.
- o Pregnancy Registry. There is a pregnancy registry for women with asthma who receive NUCALA while pregnant. The purpose of the registry is to collect information about the health of you and your baby. You can talk to your healthcare provider about how to take part in this registry or you can get more information and register by calling 1-877-311-8972 or go to www.mothertobaby.org/asthma.
- are breastfeeding or plan to breastfeed. You and your healthcare provider should decide if you will use NUCALA and breastfeed. You should not do both without talking with your healthcare provider first.
- Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
- Do not stop taking your other medicines unless instructed to do so by your healthcare provider.
How will I receive NUCALA?
Your healthcare provider will prescribe the dose that is right for you depending on what you are being treated for.
When injection is given by a healthcare provider:
- A healthcare provider will inject NUCALA under your skin (subcutaneously) every 4 weeks.
When injection is given by a patient or patient caregiver with a prefilled syringe or prefilled autoinjector:
- Use NUCALA every 4 weeks exactly as your healthcare provider tells you to.
- Read the Instructions for Use that comes with NUCALA for instructions about the right way to give your injections at home.
- NUCALA may be prescribed as a single-dose prefilled autoinjector or as a single-dose prefilled syringe for people 12 years of age and older.
- Before you use NUCALA, your healthcare provider will show you or your caregiver how to give the injections.
- You should inject NUCALA under your skin (subcutaneously) into your thigh or stomach (abdomen). Also, a caregiver may give the injection in your upper arm.
- If you miss a dose, inject a dose as soon as possible. Then continue (resume) your injection on your regular dosing schedule. If you do not notice that you have missed a dose until it is time for your next scheduled dose, then inject the next scheduled dose as planned. If you are not sure when to inject NUCALA, call your healthcare provider.
What are the possible side effects of NUCALA?
NUCALA can cause serious side effects, including:
- allergic (hypersensitivity) reactions, including anaphylaxis. Serious allergic reactions can happen after you get your NUCALA injection. Allergic reactions can sometimes happen hours or days after you get a dose of NUCALA. Tell your healthcare provider or get emergency help right away if you have any of the following symptoms of an allergic reaction:
ο swelling of your face, mouth, and tongue ο breathing problems
ο fainting, dizziness, feeling lightheaded (low blood pressure) ο rash
ο hives
- herpes zoster infections. Herpes zoster infections that can cause shingles have happened in people who received NUCALA.
The most common side effects of NUCALA include: headache, injection site reactions (pain, redness, swelling, itching, or a burning feeling at the injection site), back pain, and weakness (fatigue).
These are not all the possible side effects of NUCALA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store NUCALA?
- Store prefilled autoinjectors and prefilled syringes in the refrigerator between 36°F to 46°F (2°C to 8°C).
- Keep prefilled autoinjectors and prefilled syringes in the original carton until time of use to protect from light.
- Do not freeze. Do not shake. Keep away from heat.
- If necessary, an unopened carton can be stored outside the refrigerator at up to 86°F (30°C) for up to 7 days.
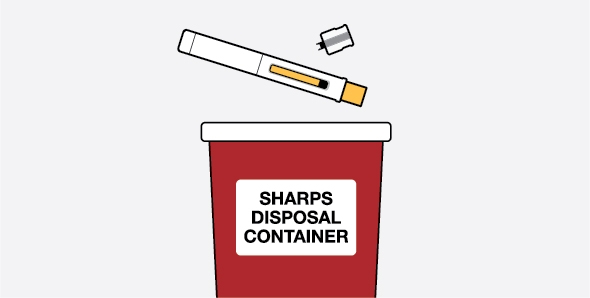
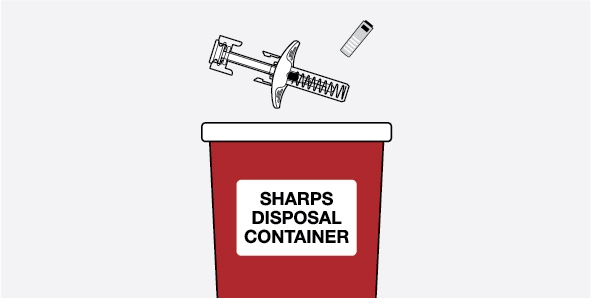
- Safely throw away prefilled autoinjectors and prefilled syringes if the unopened carton is left out of the refrigerator for more than 7 days.
- Prefilled autoinjectors and prefilled syringes must be used within 8 hours after you take them out of the carton. Safely throw away if not used within 8 hours.
- Safely throw away medicine that is out of date or no longer needed.
Keep NUCALA and all medicines out of the reach of children.
General information about the safe and effective use of NUCALA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information Leaflet. Do not give NUCALA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about NUCALA that is written for health professionals.
What are the ingredients in NUCALA?
Active Ingredient: mepolizumab.
Inactive Ingredients (vials): polysorbate 80, sodium phosphate dibasic heptahydrate, and sucrose.
Inactive Ingredients (prefilled autoinjectors and prefilled syringes): citric acid monohydrate, EDTA disodium dihydrate, polysorbate 80, sodium phosphate dibasic heptahydrate, and sucrose.
For more information about NUCALA, call 1-888-825-5249 or visit our website at www.NUCALA.com.
Trademarks are owned by or licensed to the GSK group of companies.
Manufactured by:
GlaxoSmithKline LLC, Philadelphia, PA 19112, U.S. License No. 1727
Distributed by:
GlaxoSmithKline, Research Triangle Park, NC 27709
©2019 GSK group of companies or its licensor.
NCL:5PIL
- This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: September 2019
-
NUCALA is a prescription medicine used with other medicines:
-
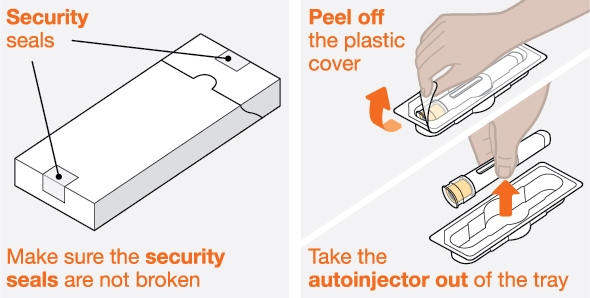
INSTRUCTIONS FOR USE
-
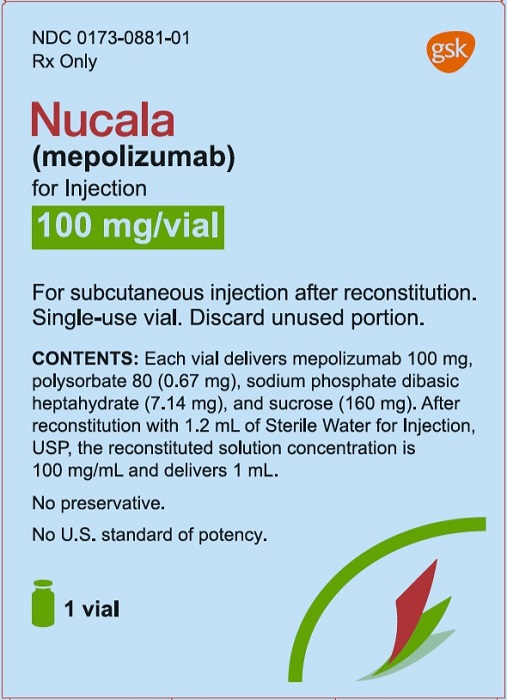
Package/Label Display Panel
PRINCIPAL DISPLAY PANEL
NDC: 0173-0881-01
Nucala
(mepolizumab)
for Injection
100 mg/vial
Rx Only
For subcutaneous injection after reconstitution.
Reconstituted solution contains 100mg/mL.
Single-dose vial. Discard unused portion.
Contents: Each vial delivers 100 mg of mepolizumab, polysorbate 80 (0.67 mg), sodium phosphate dibasic heptahydrate (7.14 mg), and sucrose (160 mg).
No preservative.
No U.S. standard of potency.
1 vial
Do not accept if plastic overseal is missing or not securely fitted.
©2019 GSK group of companies or its licensor.
- Rev. 9/19
- 62000000043659
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
NDC: 0173-0892-01
Nucala
(mepolizumab)
100 mg/mL
Rx Only
Prefilled Autoinjector
For subcutaneous use only.
Contents:
-1 Single-Dose 1-mL. Prefilled Autoinjector
-Instructions for Use
-Prescribing Information
-Patient Information
©2019 GSK group of companies or its licensor.
Do not accept if security seal is missing or broken.
- 62000000038009 Rev. 4/19
-
INGREDIENTS AND APPEARANCE
NUCALA
mepolizumab injection, powder, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0173-0881 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength MEPOLIZUMAB (UNII: 90Z2UF0E52) (MEPOLIZUMAB - UNII:90Z2UF0E52) MEPOLIZUMAB 100 mg in 1 mL Inactive Ingredients Ingredient Name Strength POLYSORBATE 80 (UNII: 6OZP39ZG8H) SODIUM PHOSPHATE, DIBASIC, HEPTAHYDRATE (UNII: 70WT22SF4B) SUCROSE (UNII: C151H8M554) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0173-0881-01 1 in 1 CARTON 11/04/2015 1 1 mL in 1 VIAL; Type 0: Not a Combination Product 2 NDC: 0173-0881-61 1 in 1 CARTON 06/17/2016 2 1 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125526 11/04/2015 NUCALA
mepolizumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0173-0892 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength MEPOLIZUMAB (UNII: 90Z2UF0E52) (MEPOLIZUMAB - UNII:90Z2UF0E52) MEPOLIZUMAB 100 mg in 1 mL Inactive Ingredients Ingredient Name Strength CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) EDETATE DISODIUM (UNII: 7FLD91C86K) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SODIUM PHOSPHATE, DIBASIC, HEPTAHYDRATE (UNII: 70WT22SF4B) SUCROSE (UNII: C151H8M554) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0173-0892-01 1 in 1 CARTON 06/06/2019 1 1 mL in 1 SYRINGE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 2 NDC: 0173-0892-42 1 in 1 CARTON 06/06/2019 2 1 mL in 1 SYRINGE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 3 NDC: 0173-0892-61 1 in 1 CARTON 06/06/2019 3 1 mL in 1 SYRINGE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 4 NDC: 0173-0892-63 1 in 1 CARTON 06/06/2019 4 1 mL in 1 SYRINGE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761122 06/06/2019 Labeler - GlaxoSmithKline LLC (167380711)

Trademark Results [Nucala]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 NUCALA 97086351 not registered Live/Pending |
Xie, Wenfu 2021-10-21 |
 NUCALA 86293514 4900162 Live/Registered |
Glaxo Group Limited 2014-05-28 |
 NUCALA 85371627 4714559 Live/Registered |
Glaxo Group Limited 2011-07-14 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.