TRETINOIN Capsules
Tretinoin by
Drug Labeling and Warnings
Tretinoin by is a Prescription medication manufactured, distributed, or labeled by Bryant Ranch Prepack. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
TRETINOIN- tretinoin capsule, liquid filled
Bryant Ranch Prepack
----------
TRETINOIN Capsules
Rx only
WARNINGS
- Experienced Physician and Institution
Patients with acute promyelocytic leukemia (APL) are at high risk in general and can have severe adverse reactions to tretinoin. Tretinoin should therefore be administered only to patients with APL under the strict supervision of a physician who is experienced in the management of patients with acute leukemia and in a facility with laboratory and supportive services sufficient to monitor drug tolerance and protect and maintain a patient compromised by drug toxicity, including respiratory compromise. Use of tretinoin requires that the physician concludes that the possible benefit to the patient outweighs the following known adverse effects of the therapy.
2. Retinoic Acid-APL Syndrome
About 25% of patients with APL treated with tretinoin have experienced a syndrome called the retinoic-acid-APL (RA-APL) syndrome characterized by fever, dyspnea, acute respiratory distress, weight gain, radiographic pulmonary infiltrates, pleural and pericardial effusions, edema, and hepatic, renal, and multi-organ failure. This syndrome has occasionally been accompanied by impaired myocardial contractility and episodic hypotension. It has been observed with or without concomitant leukocytosis. Endotracheal intubation and mechanical ventilation have been required in some cases due to progressive hypoxemia, and several patients have expired with multi-organ failure. The syndrome generally occurs during the first month of treatment, with some cases reported following the first dose of tretinoin.
The management of the syndrome has not been defined rigorously, but high dose steroids given at the first suspicion of the RA-APL syndrome appear to reduce morbidity and mortality. At the first signs suggestive of the syndrome (unexplained fever, dyspnea and/or weight gain, abnormal chest auscultatory findings or radiographic abnormalities), high dose steroids (dexamethasone 10 mg intravenously administered every 12 hours for 3 days or until the resolution of symptoms) should be immediately initiated, irrespective of the leukocyte count. The majority of patients do not require termination of tretinoin therapy during treatment of the RA-APL syndrome. However, in cases of moderate and severe RA-APL syndrome, temporary interruption of tretinoin therapy should be considered.1
3. Leukocytosis at Presentation and Rapidly Evolving Leukocytosis During Tretinoin Treatment
During tretinoin treatment about 40% of patients will develop rapidly evolving leukocytosis. Patients who present with high WBC at diagnosis (> 5x109/L) have an increased risk of a further rapid increase in WBC counts. Rapidly evolving leukocytosis is associated with a higher risk of life threatening complications.
If signs and symptoms of the RA-APL syndrome are present together with leukocytosis, treatment with high dose steroids should be initiated immediately. Some investigators routinely add chemotherapy to tretinoin treatment in the case of patients presenting with a WBC count of > 5x109/L or in the case of a rapid increase in WBC count for patients leukopenic at start of treatment, and have reported a lower incidence of the RA-APL syndrome. Consideration could be given to adding full dose chemotherapy (including an anthracycline if not contraindicated) to the tretinoin therapy on day 1 or 2 for patients presenting with a WBC count of > 5x109/L, or immediately, for patients presenting with a WBC count of < 5x109/L, if the WBC count reaches ≥ 6x109/L by day 5, or ≥ 10x109/L by day 10, or ≥ 15x109/L by day 28.
4. Teratogenic Effects. Pregnancy Category D - see WARNINGS
There is a high risk that a severely deformed infant will result if tretinoin is administered during pregnancy. If, nonetheless, it is determined that tretinoin represents the best available treatment for a pregnant woman or a woman of childbearing potential, it must be assured that the patient has received full information and warnings of the risk to the fetus if she were to be pregnant and of the risk of possible contraception failure and has been instructed in the need to use two reliable forms of contraception simultaneously during therapy and for 1 month following discontinuation of therapy, and has acknowledged her understanding of the need for using dual contraception, unless abstinence is the chosen method.
Within 1 week prior to the institution of tretinoin therapy, the patient should have blood or urine collected for a serum or urine pregnancy test with a sensitivity of at least 50 mIU/mL. When possible, tretinoin therapy should be delayed until a negative result from this test is obtained. When a delay is not possible, the patient should be placed on two reliable forms of contraception. Pregnancy testing and contraception counseling should be repeated monthly throughout the period of tretinoin treatment.
DESCRIPTION
Tretinoin, USP is a retinoid that induces maturation of acute promyelocytic leukemia (APL) cells in culture. It is available in a 10 mg gelatin capsule for oral administration. Each capsule contains the following inactive ingredients: butylated hydroxyanisole, edetate disodium, gelatin, hydrogenated vegetable oil, polysorbate 80, soybean oil, vitamin E, and white wax (beeswax). The ingredients in the capsule shell include black iron oxide, red iron oxide, titanium dioxide and yellow iron oxide. The ingredients in the edible imprinting ink include D&C yellow no. 10 aluminum lake, FD&C blue no. 1 aluminum lake, FD&C blue no. 2 aluminum lake, FD&C red no. 40 aluminum lake, iron oxide black, propylene glycol and shellac glaze.
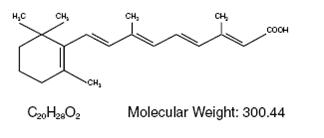
Chemically, tretinoin, USP is all-trans retinoic acid and is related to retinol (Vitamin A) and has the following chemical name: 3,7-Dimethyl-9-(2,6,6-trimethyl-1- cyclohexen-1-yl)-2,4,6,8-nonatetraenoic acid. It is a yellow to light orange crystalline powder, and has the following structural formula:
CLINICAL PHARMACOLOGY
Mechanism of Action
Tretinoin is not a cytolytic agent but instead induces cytodifferentiation and decreased proliferation of APL cells in culture and in vivo. In APL patients, tretinoin treatment produces an initial maturation of the primitive promyelocytes derived from the leukemic clone, followed by a repopulation of the bone marrow and peripheral blood by normal, polyclonal hematopoietic cells in patients achieving complete remission (CR). The exact mechanism of action of tretinoin in APL is unknown.
Pharmacokinetics
Tretinoin activity is primarily due to the parent drug. In human pharmacokinetics studies, orally administered drug was well absorbed into the systemic circulation, with approximately two-thirds of the administered radiolabel recovered in the urine. The terminal elimination half-life of tretinoin following initial dosing is 0.5 to 2 hours in patients with APL. There is evidence that tretinoin induces its own metabolism. Plasma tretinoin concentrations decrease on average to one-third of their day 1 values during 1 week of continuous therapy. Mean ± SD peak tretinoin concentrations decreased from 394 ± 89 to 138 ± 139 ng/mL, while area under the curve (AUC) values decreased from 537 ± 191 ngh/mL to 249 ± 185 ngh/mL during 45 mg/m2 daily dosing in 7 APL patients. Increasing the dose to “correct” for this change has not increased response.
Absorption
A single 45 mg/m2 (~ 80 mg) oral dose to APL patients resulted in a mean ± SD peak tretinoin concentration of 347 ± 266 ng/mL. Time to reach peak concentration was between 1 and 2 hours.
Distribution
The apparent volume of distribution of tretinoin has not been determined. Tretinoin is greater than 95% bound in plasma, predominately to albumin. Plasma protein binding remains constant over the concentration range of 10 to 500 ng/mL.
Metabolism
Tretinoin metabolites have been identified in plasma and urine. Cytochrome P450 enzymes have been implicated in the oxidative metabolism of tretinoin. Metabolites include 13- cis retinoic acid, 4-oxo trans retinoic acid, 4-oxo cis retinoic acid, and 4-oxo trans retinoic acid glucuronide. In APL patients, daily administration of a 45 mg/m2 dose of tretinoin resulted in an approximately tenfold increase in the urinary excretion of 4-oxo trans retinoic acid glucuronide after 2 to 6 weeks of continuous dosing, when compared to baseline values.
Excretion
Studies with radiolabeled drug have demonstrated that after the oral administration of 2.75 and
50 mg doses of tretinoin, greater than 90% of the radioactivity was recovered in the urine and feces. Based upon data from 3 subjects, approximately 63% of radioactivity was recovered in the urine within 72 hours and 31% appeared in the feces within 6 days.
Special Populations
The pharmacokinetics of tretinoin have not been separately evaluated in women, in members of different ethnic groups, or in individuals with renal or hepatic insufficiency.
Drug-Drug Interactions
In 13 patients who had received daily doses of tretinoin for 4 consecutive weeks, administration of ketoconazole (400 to 1200 mg oral dose) 1 hour prior to the administration of the tretinoin dose on day 29 led to a 72% increase (218 ± 224 vs. 375 ± 285 ngh/mL) in tretinoin mean plasma AUC. The precise cytochrome P450 enzymes involved in these interactions have not been specified; CYP 3A4, 2C8 and 2E have been implicated in various preliminary reports.
Clinical Studies
Tretinoin has been investigated in 114 previously treated APL patients and in 67 previously untreated (“de novo”) patients in one open-label, uncontrolled single investigator clinical study (Memorial Sloan-Kettering Cancer Center [MSKCC]) and in two cohorts of compassionate cases treated by multiple investigators under the auspices of the National Cancer Institute (NCI). All patients received 45 mg/m2/day as a divided oral dose for up to 90 days or 30 days beyond the day that CR was reached. Results are shown in the following table:
| NR = Not Reached NA = Not Available |
||||||
|
|
||||||
|
MSKCC |
NCI Cohort 1 |
NCI Cohort 2 |
||||
|
Relapsed n=20 |
De Novo n=15 |
Relapsed* n=48 |
De Novo n=14 |
Relapsed n=46 |
De Novo† n=38 |
|
|
Complete Remission Median Survival (Mo) Median Follow-up (Mo) RA-APL Syndrome |
16 (80%) 10.8 9.9 4 (20%) |
11 (73%) NR 42.9 5 (33%) |
24 (50%) 5.8 5.6 10 (21%) |
5 (36%) 0.5 1.2 6 (43%) |
24 (52%) 8.8 8 NA |
26 (68%) NR 13.1 NA |
The median time to CR was between 40 and 50 days (range: 2 to 120 days). Most patients in these studies received cytotoxic chemotherapy during the remission phase. These results compare to the 30% to 50% CR rate and, ≤ 6 month median survival reported for cytotoxic chemotherapy of APL in the treatment of relapse.
Ten of 15 pediatric cases achieved CR (8 of 10 males and 2 of 5 females). There were insufficient patients of black, Hispanic or Asian derivation to estimate relative response rates in these groups, but responses were seen in each category.
Responses were seen in 3 of 4 patients for whom cytogenetic analysis failed to detect the t(15;17) translocation typically seen in APL. The t(15;17) translocation results in the PML/RARα gene, which appears necessary for this disease. Molecular genetic studies were not conducted in these cases, but it is likely they represent cases with a masked translocation giving rise to PML/RARα. Responses to tretinoin have not been observed in cases in which PML/RARα fusion has been shown to be absent.
INDICATIONS AND USAGE
Tretinoin Capsules are indicated for the induction of remission in patients with acute promyelocytic leukemia (APL), French-American-British (FAB) classification M3 (including the M3 variant), characterized by the presence of the t(15;17) translocation and/or the presence of the PML/RARα gene who are refractory to, or who have relapsed from, anthracycline chemotherapy, or for whom anthracycline based chemotherapy is contraindicated. Tretinoin is for the induction of remission only. The optimal consolidation or maintenance regimens have not been defined, but all patients should receive an accepted form of remission consolidation and/or maintenance therapy for APL after completion of induction therapy with tretinoin.
CONTRAINDICATIONS
Tretinoin is contraindicated in patients with a known hypersensitivity to tretinoin, any of its components, or other retinoids.
WARNINGS
Pregnancy Category D
See boxed WARNINGS
Tretinoin has teratogenic and embryotoxic effects in mice, rats, hamsters, rabbits and pigtail monkeys, and may be expected to cause fetal harm when administered to a pregnant woman. Tretinoin causes fetal resorptions and a decrease in live fetuses in all animals studied. Gross external, soft tissue and skeletal alterations occurred at doses higher than 0.7 mg/kg/day in mice, 2 mg/kg/day in rats, 7 mg/kg/day in hamsters, and at a dose of 10 mg/kg/day, the only dose tested, in pigtail monkeys (about 1/20, 1/4, and 1/2 and 4 times the human dose, respectively, on a mg/m2 basis).
There are no adequate and well-controlled studies in pregnant women. Although experience with humans administered tretinoin is extremely limited, increased spontaneous abortions and major human fetal abnormalities related to the use of other retinoids have been documented in humans. Reported defects include abnormalities of the CNS, musculoskeletal system, external ear, eye, thymus and great vessels; and facial dysmorphia, cleft palate, and parathyroid hormone deficiency. Some of these abnormalities were fatal. Cases of IQ scores less than 85, with or without obvious CNS abnormalities, have also been reported. All fetuses exposed during pregnancy can be affected and at the present time there is no antepartum means of determining which fetuses are and are not affected.
Effective contraception must be used by all females during tretinoin therapy and for 1 month following discontinuation of therapy. Contraception must be used even when there is a history of infertility or menopause, unless a hysterectomy has been performed. Whenever contraception is required, it is recommended that two reliable forms of contraception be used simultaneously, unless abstinence is the chosen method. If pregnancy does occur during treatment, the physician and patient should discuss the desirability of continuing or terminating the pregnancy.
Patients Without the t(15;17) Translocation
Initiation of therapy with tretinoin may be based on the morphological diagnosis of acute promyelocytic leukemia. Confirmation of the diagnosis of APL should be sought by detection of the t(15;17) genetic marker by cytogenetic studies. If these are negative, PML/RARα fusion should be sought using molecular diagnostic techniques. The response rate of other AML subtypes to tretinoin has not been demonstrated; therefore, patients who lack the genetic marker should be considered for alternative treatment.
Retinoic Acid-APL (RA-APL) Syndrome
In up to 25% of patients with APL treated with tretinoin, a syndrome occurs which can be fatal (see boxed WARNINGS and ADVERSE REACTIONS).
Leukocytosis at Presentation and Rapidly Evolving Leukocytosis During Tretinoin Treatment
See boxed WARNINGS.
Pseudotumor Cerebri
Retinoids, including tretinoin, have been associated with pseudotumor cerebri (benign intracranial hypertension), especially in pediatric patients. The concomitant use of other agents known to cause pseudotumor cerebri/intracranial hypertension, such as tetracyclines, might increase the risk of this condition (see PRECAUTIONS, Drug Interactions).2 Early signs and symptoms of pseudotumor cerebri include papilledema, headache, nausea and vomiting, and visual disturbances. Patients with these symptoms should be evaluated for pseudotumor cerebri, and, if present, appropriate care should be instituted in concert with neurological assessment.
Lipids
Up to 60% of patients experienced hypercholesterolemia and/or hypertriglyceridemia, which were reversible upon completion of treatment. The clinical consequences of temporary elevation of triglycerides and cholesterol are unknown, but venous thrombosis and myocardial infarction have been reported in patients who ordinarily are at low risk for such complications.
Elevated Liver Function Test Results
Elevated liver function test results occur in 50% to 60% of patients during treatment. Liver function test results should be carefully monitored during treatment and consideration be given to a temporary withdrawal of tretinoin if test results reach > 5 times the upper limit of normal values. However, the majority of these abnormalities resolve without interruption of tretinoin or after completion of treatment.
PRECAUTIONS
General
Tretinoin has potentially significant toxic side effects in APL patients. Patients undergoing therapy should be closely observed for signs of respiratory compromise and/or leukocytosis (see boxed WARNINGS). Supportive care appropriate for APL patients, e.g., prophylaxis for bleeding, prompt therapy for infection, should be maintained during therapy with tretinoin.
There is a risk of thrombosis (both venous and arterial) which may involve any organ system, during the first month of treatment (see ADVERSE REACTIONS). Therefore, caution should be exercised when treating patients with the combination of tretinoin and anti-fibrinolytic agents, such as tranexamic acid, aminocaproic acid or aprotinin (see Drug Interactions).3,4
The ability to drive or operate machinery might be impaired in patients treated with tretinoin, particularly if they are experiencing dizziness or severe headache.
Microdosed progesterone preparations (“minipill”) may be an inadequate method of contraception during treatment with tretinoin.5
Laboratory Tests
The patient's hematologic profile, coagulation profile, liver function test results, and triglyceride and cholesterol levels should be monitored frequently.
Drug Interactions
Limited clinical data on potential drug interactions are available.
Drugs Metabolized By the Hepatic P450 System
As tretinoin is metabolized by the hepatic P450 system, there is a potential for alteration of pharmacokinetics parameters in patients administered concomitant medications that are also inducers or inhibitors of this system. Medications that generally induce hepatic P450 enzymes include rifampicin, glucocorticoids, phenobarbital and pentobarbital. Medications that generally inhibit hepatic P450 enzymes include ketoconazole, cimetidine, erythromycin, verapamil, diltiazem and cyclosporine. To date there are no data to suggest that co-use with these medications increases or decreases either efficacy or toxicity of tretinoin.
Agents Known to Cause Pseudotumor Cerebri/Intracranial Hypertension (Such as Tetracyclines)
Tretinoin may cause pseudotumor cerebri/intracranial hypertension. Concomitant administration of tretinoin and agents known to cause pseudotumor cerebri/intracranial hypertension as well might increase the risk of this condition (see WARNINGS).
Vitamin A
As with other retinoids, tretinoin must not be administered in combination with vitamin A because symptoms of hypervitaminosis A could be aggravated.
Anti-fibrinolytic Agents (Such as Tranexamic Acid, Aminocaproic Acid, or Aprotinin)
Cases of fatal thrombotic complications have been reported rarely in patients concomitantly treated with tretinoin and anti-fibrinolytic agents. Therefore, caution should be exercised when administering tretinoin concomitantly with these agents (see PRECAUTIONS, General).
Carcinogenesis, Mutagenesis and Impairment of Fertility
No long term carcinogenicity studies with tretinoin have been conducted. In short term carcinogenicity studies, tretinoin at a dose of 30 mg/kg/day (about 2 times the human dose on a mg/m2 basis) was shown to increase the rate of diethylnitrosamine (DEN)-induced mouse liver adenomas and carcinomas. Tretinoin was negative when tested in the Ames and Chinese hamster V79 cell HGPRT assays for mutagenicity. A twofold increase in the sister chromatid exchange (SCE) has been demonstrated in human diploid fibroblasts, but other chromosome aberration assays, including an in vitro assay in human peripheral lymphocytes and an in vivo mouse micronucleus assay, did not show a clastogenic or aneuploidogenic effect. Adverse effects on fertility and reproductive performance were not observed in studies conducted in rats at doses up to 5 mg/kg/day (about 2/3 the human dose on a mg/m2 basis). In a 6 week toxicology study in dogs, minimal to marked testicular degeneration, with increased numbers of immature spermatozoa, were observed at 10 mg/kg/day (about 4 times the equivalent human dose in mg/m2).
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, and because of the potential for serious adverse reactions from tretinoin in nursing infants, mothers should discontinue nursing prior to taking this drug.
Pediatric Use
There are limited clinical data on the pediatric use of tretinoin. Of 15 pediatric patients (age range: 1 to 16 years) treated with tretinoin, the incidence of complete remission was 67%. Safety and effectiveness in pediatric patients below the age of 1 year have not been established. Some pediatric patients experience severe headache and pseudotumor cerebri, requiring analgesic treatment and lumbar puncture for relief. Increased caution is recommended in the treatment of pediatric patients. Dose reduction may be considered for pediatric patients experiencing serious and/or intolerable toxicity; however, the efficacy and safety of tretinoin at doses lower than 45 mg/m2/day have not been evaluated in the pediatric population.
Geriatric Use
Of the total number of subjects in clinical studies of tretinoin, 21.4% were 60 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
ADVERSE REACTIONS
Virtually all patients experience some drug related toxicity, especially headache, fever, weakness, and fatigue. These adverse effects are seldom permanent or irreversible nor do they usually require interruption of therapy. Some of the adverse events are common in patients with APL, including hemorrhage, infections, gastrointestinal hemorrhage, disseminated intravascular coagulation, pneumonia, septicemia, and cerebral hemorrhage. The following describes the adverse events, regardless of drug relationship, that were observed in patients treated with tretinoin.
Typical Retinoid Toxicity
The most frequently reported adverse events were similar to those described in patients taking high doses of vitamin A and included headache (86%), fever (83%), skin/mucous membrane dryness (77%), bone pain (77%), nausea/vomiting (57%), rash (54%), mucositis (26%), pruritus (20%), increased sweating (20%), visual disturbances (17%), ocular disorders (17%), alopecia (14%), skin changes (14%), changed visual acuity (6%), bone inflammation (3%), visual field defects (3%).
RA-APL Syndrome
APL patients treated with tretinoin have experienced a potentially fatal syndrome characterized by fever, dyspnea, acute respiratory distress, weight gain, radiographic pulmonary infiltrates, pleural and pericardial effusions, edema, and hepatic, renal, and multi-organ failure. This syndrome has occasionally been accompanied by impaired myocardial contractility and episodic hypotension and has been observed with or without concomitant leukocytosis. Some patients have expired due to progressive hypoxemia and multi-organ failure. The syndrome generally occurs during the first month of treatment, with some cases reported following the first dose of tretinoin. The management of the syndrome has not been defined rigorously, but high dose steroids given at the first signs of the syndrome appear to reduce morbidity and mortality. Treatment with dexamethasone, 10 mg intravenously administered every 12 hours for 3 days or until resolution of symptoms, should be initiated without delay at the first suspicion of symptoms (one or more of the following: fever, dyspnea, weight gain, abnormal chest auscultatory findings or radiographic abnormalities). Sixty percent or more of patients treated with tretinoin may require high dose steroids because of these symptoms. The majority of patients do not require termination of tretinoin therapy during treatment of the syndrome.
Body as a Whole
General disorders related to tretinoin administration and/or associated with APL included malaise (66%), shivering (63%), hemorrhage (60%), infections (58%), peripheral edema (52%), pain (37%), chest discomfort (32%), edema (29%), disseminated intravascular coagulation (26%), weight increase (23%), injection site reactions (17%), anorexia (17%), weight decrease (17%), myalgia (14%), flank pain (9%), cellulitis (8%), face edema (6%), fluid imbalance (6%), pallor (6%), lymph disorders (6%), acidosis (3%), hypothermia (3%), ascites (3%).
Respiratory System Disorders
Respiratory system disorders were commonly reported in APL patients administered tretinoin. The majority of these events are symptoms of the RA-APL syndrome (see boxed WARNINGS). Respiratory system adverse events included upper respiratory tract disorders (63%), dyspnea (60%), respiratory insufficiency (26%), pleural effusion (20%), pneumonia (14%), rales (14%), expiratory wheezing (14%), lower respiratory tract disorders (9%), pulmonary infiltration (6%), bronchial asthma (3%), pulmonary edema (3%), larynx edema (3%), unspecified pulmonary disease (3%).
Ear Disorders
Ear disorders were consistently reported, with earache or feeling of fullness in the ears reported by 23% of the patients. Hearing loss and other unspecified auricular disorders were observed in 6% of patients, with infrequent (< 1%) reports of irreversible hearing loss.
Gastrointestinal Disorders
GI disorders included GI hemorrhage (34%), abdominal pain (31%), other gastrointestinal disorders (26%), diarrhea (23%), constipation (17%), dyspepsia (14%), abdominal distention (11%), hepatosplenomegaly (9%), hepatitis (3%), ulcer (3%), unspecified liver disorder (3%).
Cardiovascular and Heart Rate and Rhythm Disorders
Arrhythmias (23%), flushing (23%), hypotension (14%), hypertension (11%), phlebitis (11%), cardiac failure (6%) and for 3% of patients: cardiac arrest, myocardial infarction, enlarged heart, heart murmur, ischemia, stroke, myocarditis, pericarditis, pulmonary hypertension, secondary cardiomyopathy.
Central and Peripheral Nervous System Disorders and Psychiatric
Dizziness (20%), paresthesias (17%), anxiety (17%), insomnia (14%), depression (14%), confusion (11%), cerebral hemorrhage (9%), intracranial hypertension (9%), agitation (9%), hallucination (6%) and for 3% of patients: abnormal gait, agnosia, aphasia, asterixis, cerebellar edema, cerebellar disorders, convulsions, coma, CNS depression, dysarthria, encephalopathy, facial paralysis, hemiplegia, hyporeflexia, hypotaxia, no light reflex, neurologic reaction, spinal cord disorder, tremor, leg weakness, unconsciousness, dementia, forgetfulness, somnolence, slow speech.
Urinary System Disorders
Renal insufficiency (11%), dysuria (9%), acute renal failure (3%), micturition frequency (3%), renal tubular necrosis (3%), enlarged prostate (3%).
Miscellaneous Adverse Events
Isolated cases of erythema nodosum, basophilia and hyperhistaminemia, Sweet's syndrome, organomegaly, hypercalcemia, pancreatitis and myositis have been reported.
Additional Adverse Reactions Reported With Tretinoin
Cardiovascular
Cases of thrombosis (both venous and arterial) involving various sites (e.g., cerebrovascular accident, myocardial infarction, renal infarct) have been reported rarely (see PRECAUTIONS, General).
OVERDOSAGE
In case of overdose with tretinoin, reversible signs of hypervitaminosis A (headache, nausea, vomiting, mucocutaneous symptoms) can appear. The maximal tolerated dose in patients with myelodysplastic syndrome or solid tumors was 195 mg/m2/day. The maximal tolerated dose in pediatric patients was lower at 60 mg/m2/day. Overdosage with other retinoids has been associated with transient headache, facial flushing, cheilosis, abdominal pain, dizziness and ataxia. These symptoms have quickly resolved without apparent residual effects.
There is no specific treatment in the case of an overdose; however, it is important that the patient be treated in a special hematological unit.
DOSAGE AND ADMINISTRATION
The recommended dose is 45 mg/m2/day administered as two evenly divided doses until complete remission is documented. Therapy should be discontinued 30 days after achievement of complete remission or after 90 days of treatment, whichever occurs first.
If after initiation of treatment of tretinoin the presence of the t(15;17) translocation is not confirmed by cytogenetics and/or by polymerase chain reaction studies and the patient has not responded to tretinoin, alternative therapy appropriate for acute myelogenous leukemia should be considered.
Tretinoin is for the induction of remission only. Optimal consolidation or maintenance regimens have not been determined. All patients should, therefore, receive a standard consolidation and/or maintenance chemotherapy regimen for APL after induction therapy with tretinoin, unless otherwise contraindicated.
HOW SUPPLIED
Tretinoin Capsules are available as:
| 10 mg: | Two-piece hard gelatin capsule with brown opaque cap and dark yellow opaque body, filled with yellow viscous oily suspension. Imprinted in black ink with stylized barr 808. Available in bottles of 100 capsules (NDC: 63629-8752-1). |
Store at 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature].
Dispense in a tight, light-resistant container as defined in the USP, with a child-resistant closure (as required).
KEEP THIS AND ALL MEDICATIONS OUT OF THE REACH OF CHILDREN.
PROTECT FROM LIGHT
ANNOTATIONS
- Tallman MS, et al. Clinical description of 44 patients with acute promyelocytic leukemia who developed retinoic acid syndrome. Blood. 2000; 95(1): 90-95.
- Longauer M. Drug Safety Report No. 1007960: Concomitant use of tetracyclines. March 27, 2002.
- Longauer M. Drug Safety Report No. 1008342: Thromboses involving any sites. March 27, 2002.
- Longauer M. Issue Work-Up: Safety of concomitant administration of Tretinoin and anti-fibrinolytic agents. May 4, 2001.
- Longauer M. Drug Safety Report No. 1007959: Interaction with low-dose progestogens (“minipill”). March 27, 2002.
- Longauer M. Issue Work-Up: Thrombocytosis. December 18, 2000.
- Longauer M. Safety Evaluation Update: Genital Ulcerations. Provided in Periodic Safety Update Report on Tretinoin Oral Period: April 1, 1998-March 31, 1991 (Research Report No. B- 170’967, May 31, 1999).
- Longauer M. Issue Work-Up: Vasculitis. September 26, 2000.
TEVA PHARMACEUTICALS USA, INC.
North Wales, PA 19454
Rev. B 10/2015
INFORMATION FOR THE PATIENT
WARNING TO FEMALE PATIENTS
YOU MUST NOT BECOME PREGNANT DURING TRETINOIN TREATMENT. There is an extremely high risk that a deformed baby will result if you become pregnant while taking tretinoin, in any amount, for even short periods of time. Potentially any exposed fetuses can be affected. There is also an increased risk of miscarriage. Premature births may also occur.
Effective contraception (birth control) should be discussed with your doctor. Two forms of reliable contraception must be used during therapy, and must be continued for one month after tretinoin treatment has stopped. If directed by your doctor, two forms of reliable contraception must also be used simultaneously for at least one month before beginning therapy. It is recommended that you either abstain from sexual intercourse or use two reliable kinds of birth control at the same time. Birth control must be used even if you think you cannot become pregnant, unless you have had a hysterectomy.
If you are pregnant or become pregnant while on tretinoin therapy or during the month after treatment has stopped, immediately contact your doctor to discuss the desirability of continuing the pregnancy.
YOU MUST NOT TAKE TRETINOIN IF YOU ARE A NURSING MOTHER.
Tretinoin should not be taken by nursing mothers since it is not known whether it is excreted in human milk. Since many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from tretinoin, mothers should discontinue nursing prior to taking this drug.
General guidelines for taking your medication...
- Call your doctor if you have any questions or experience any severe or troubling symptoms.
- Tretinoin does not need to be refrigerated. However, do not expose the capsules to sunlight or excessive heat.
- Be sure to take your medication as prescribed by your doctor. Read the prescription label on the package carefully. If there is anything you don’t understand, ask your doctor or pharmacist to explain it to you.
- Keep tretinoin and all medications out of the reach of children.
Store at 20° to 25°C (68° to 77°F).
PROTECT FROM LIGHT
TEVA PHARMACEUTICALS USA, INC.
North Wales, PA 19454
Rev. B 10/2015

| TRETINOIN
tretinoin capsule, liquid filled |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| Labeler - Bryant Ranch Prepack (171714327) |
| Registrant - Bryant Ranch Prepack (171714327) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Bryant Ranch Prepack | 171714327 | REPACK(63629-8752) , RELABEL(63629-8752) | |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.