CTEXLI- chenodiol tablet, film coated
CTEXLI by
Drug Labeling and Warnings
CTEXLI by is a Prescription medication manufactured, distributed, or labeled by Mirum Pharmaceuticals Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use CTEXLI safely and effectively. See full prescribing information for CTEXLI.
CTEXLI® (chenodiol) tablets, for oral use
Initial U.S. Approval: 1983INDICATIONS AND USAGE
CTEXLI is a bile acid indicated for treatment of cerebrotendinous xanthomatosis (CTX) in adults. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
CTEXLI tablets: 250 mg. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
Hepatotoxicity: Obtain baseline liver transaminase and total bilirubin levels in all patients and monitor yearly and as clinically indicated. Interrupt treatment until the levels have returned to baseline values. For persistent or recurrent liver test abnormalities, consider discontinuing CTEXLI. (5.1)
ADVERSE REACTIONS
The most common adverse reactions (incidence > 14%) are diarrhea, headache, abdominal pain, constipation, hypertension, muscular weakness, and upper respiratory tract infection. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Mirum Pharmaceuticals at 1-855-MRM-4YOU or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Recommendation Prior to CTEXLI Treatment Initiation
2.2 Recommended Dosage
2.3 Administration Modification and Monitoring
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on CTEXLI
7.2 Effect of CTEXLI on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Recommendation Prior to CTEXLI Treatment Initiation
Before initiating CTEXLI, obtain baseline liver transaminase (alanine aminotransferase [ALT] and aspartate aminotransferase [AST]) and total bilirubin levels in all patients [see Warnings and Precautions (5.1)].
2.2 Recommended Dosage
The recommended dosage of CTEXLI is 250 mg administered orally three times daily.
Administer CTEXLI with or without food. Swallow tablets whole.
Missed Dose
If a dose of CTEXLI is missed, advise the patient to skip the missed dose and to resume taking the prescribed dose at the next scheduled time. Patients should not take a double dose.
2.3 Administration Modification and Monitoring
If liver transaminase (ALT, AST) levels are elevated > 3 times the upper limit of normal (ULN) or total bilirubin level is >2 times ULN, interrupt treatment with CTEXLI until the levels have returned to baseline values. Monitor liver transaminase and total bilirubin levels yearly and as clinically indicated [see Warnings and Precautions (5.1)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
Chenodiol, including CTEXLI, has been associated with hepatotoxicity [see Adverse Reactions (6)]. In Trial 1, one CTEXLI-treated patient (7%) had increased ALT levels > 3 times ULN, which led to treatment interruption. Patients with pre-existing liver disease or bile duct abnormalities may be at higher risk for hepatotoxicity during treatment with CTEXLI. Published reports suggest patients who are poor sulfators of lithocholic acid are more likely to develop chenodiol-induced serum aminotransferase elevations [see Clinical Pharmacology (12.3)].
Obtain baseline liver transaminase (ALT, AST) and total bilirubin levels in all patients prior to treatment initiation with CTEXLI. If liver transaminase levels are elevated > 3 times ULN or total bilirubin level is >2 times ULN, interrupt treatment with CTEXLI until the levels have returned to baseline values. Monitor liver transaminase and total bilirubin levels yearly and as clinically indicated. For persistent or recurrent liver test abnormalities, consider discontinuing CTEXLI.
Inform the patient of the symptoms of hepatotoxicity (e.g., abdominal pain, bruising, dark-colored urine, fatigue, bleeding, jaundice, nausea, and pruritus). If clinical signs and symptoms consistent with hepatotoxicity occur, have the patient discontinue CTEXLI immediately.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reaction is described elsewhere in the labeling:
- Hepatotoxicity [see Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of CTEXLI was evaluated in a randomized, double blind, placebo-controlled, 2-period, 2-treatment crossover trial in 14 patients (16 to 55 years of age) with CTX (Trial 1). CTEXLI is not approved for use in pediatric patients. The dosage of CTEXLI was 250 mg orally three times a day [see Clinical Studies (14)]. The mean (SD) chenodiol exposure during Trial 1 was 139.1 (26.7) days.
The most common adverse reactions which occurred in two or more patients (>14%) during CTEXLI treatment (including the two 8-week open-label treatment periods) were diarrhea (36%), headache (21%), and abdominal pain (including abdominal pain upper) (14%), constipation (14%), hypertension (14%), muscular weakness (14%), and upper respiratory tract infection (14%).
In Trial 1, one CTEXLI-treated patient (7%) had increased ALT levels > 3x ULN, which led to treatment interruption.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of chenodiol. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Hepatobiliary Disorders: Hepatotoxicity [see Warnings and Precautions (5.1)]
- Immune System Disorders: Hypersensitivity reactions such as facial swelling, pruritus, rash, urticaria.
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on CTEXLI
Co-administration of bile acid sequestering agents, such as cholestyramine and colestipol, or aluminum-based antacids may decrease absorption of CTEXLI in the intestine and may result in decreased efficacy of CTEXLI. Avoid concomitant use of bile acid sequestering agents or aluminum-based antacids with CTEXLI.
7.2 Effect of CTEXLI on Other Drugs
Due to potential hepatotoxicity, CTEXLI may affect the pharmacodynamics of coumarin and its derivatives, causing unexpected prolongation of the prothrombin time and hemorrhage. If concomitant use of CTEXLI with coumarin or its derivatives is unavoidable, monitor prothrombin time. Adjust the dosage of coumarin or its derivatives in accordance with its approved product labeling.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from published case reports over decades of use with chenodiol during pregnancy have not identified an increased risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Serious hepatic, renal and adrenal lesions occurred in fetuses of female Rhesus monkeys treated at doses 1 to 2 times the recommended human dose based on body surface area (mg/m2). Hepatic lesions also occurred at doses comparable to the human dose based on body surface area in neonatal baboons born to mothers administered chenodiol during pregnancy (see Data). The animal study findings have not been demonstrated with human use.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the US general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
Hepatic lesions were reported in neonatal baboons whose mothers had received 18 to 38 mg/kg of chenodiol throughout pregnancy (0.6 to 1.4 times the recommended human dose based on body surface area). Serious hepatic, renal and adrenal lesions were also reported in fetuses of female Rhesus monkeys given 60 to 90 mg/kg/day from GD 21-45 of pregnancy (1 to 2 times the recommended human dose based on body surface area). Non-human primates form sulfate conjugates of the known hepatotoxic bacterial metabolite of chenodiol, lithocholic acid, to a lesser extent than reported in humans, which may exaggerate the toxicity of orally dosed chenodiol compared to humans. However, there is also evidence that the hepatobiliary toxicity is partly due to the parent drug, chenodiol.
8.2 Lactation
Risk Summary
There are no data on the presence of chenodiol in either human or animal milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for CTEXLI and any potential adverse effects on the breastfed infant from CTEXLI or the underlying maternal condition.
-
10 OVERDOSAGE
Cases of intentional overdose with chenodiol have been reported: one patient consumed 3 g to 4.5 g of chenodiol and another patient consumed 30 g of chenodiol. Clinical manifestations experienced by these patients included nausea, dizziness, and diarrhea.
In the event of an overdose, discontinue CTEXLI, monitor the patient, and institute general supportive measures if needed.
-
11 DESCRIPTION
CTEXLI (chenodiol) is a bile acid. Chenodiol is a bitter-tasting, white powder consisting of crystalline and amorphous particles that are freely soluble in methanol, acetone and acetic acid, and practically insoluble in water.
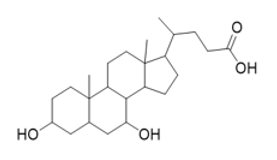
The chemical name of chenodiol is 3α,7α-dihydroxy-5-β-cholan-24-oic acid. The molecular formula is C24H40O4 and the molecular weight is 392.58 g/mol. The chemical structure is:
Each CTEXLI tablet contains 250 mg of chenodiol. Inactive ingredients are magnesium stearate, microcrystalline cellulose, pregelatinized starch, silicon dioxide, and sodium starch glycolate. The thin-film coating contains opadry YS 2 7035 (consisting of methylcellulose and glycerin) and sodium lauryl sulfate.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Endogenous chenodiol (chenodeoxycholic acid) is a primary bile acid, synthesized from cholesterol in the liver. In CTX, the major bile acid synthesis pathways are disrupted due to partial or total deficiency in sterol 27-hydroxylase encoded by the CYP27A1 gene.
CTEXLI may act to replace deficient levels of the endogenous bile acid chenodeoxycholic acid in patients with CTX. Increased chenodiol levels in the enterohepatic bile acid pool restore the activation of farnesoid X receptor (FXR) and downregulate CYP7A1 leading to suppression and reduction of atypical bile acids and bile alcohols including cholestanol and 23S-pentol.
12.2 Pharmacodynamics
In Trial 1, plasma cholestanol and urine 23S-pentol concentrations were elevated in patients with CTX. Treatment with CTEXLI resulted in reductions of plasma cholestanol and urine 23S-pentol concentrations in the 8-week run-in open label treatment period. Continued treatment with CTEXLI for 4 weeks in the double-blind treatment period resulted in the maintenance of low levels of urine 23S-pentol and additional reductions of plasma cholestanol [see Clinical Studies (14)].
12.3 Pharmacokinetics
In CTX patients, the geometric mean (%CV) maximum plasma concentration (Cmax), trough plasma concentration (Ctrough), and area under the plasma concentration-time curve (AUC0-8h) of chenodiol at steady state following the recommended dosage (250 mg administered orally three times daily) were 3.7 mcg/mL (60%), 0.7 mcg/mL (90%), and 12.5 mcg*h/mL (60%), respectively.
Absorption
The median (range) Tmax of chenodiol following an oral administration in CTX patients was 3 (0.5‑8) hours.
Distribution
Due to first-pass hepatic clearance, the body pool of chenodiol resides mainly in the enterohepatic circulation. The apparent volume of distribution of chenodiol at steady-state was 0.36 L/kg. The plasma protein binding of chenodiol was approximately 98%.
Elimination
The geometric mean total apparent clearance of chenodiol in CTX patients was 20 L/h.
Metabolism
Chenodiol is well absorbed from the small intestine and taken up by the liver where it is converted to its taurine and glycine conjugates and secreted into the bile along with other endogenous bile acids in the enterohepatic circulation. Chenodiol that escapes to the colon is converted by bacterial action to lithocholic acid. Humans have the capacity to form sulfate conjugates of lithocholic acid. About 80% of the lithocholate is excreted in the feces and the remainder is absorbed and converted in the liver to its poorly absorbed sulfolithocholyl conjugates.
Excretion
Conjugated chenodiol is either reabsorbed in the terminal ileum, deconjugated before excretion, or decomposed by bacteria to lithocholic acid.
Drug Interaction Studies
Based on in vitro studies, chenodiol and its glyco- and tauro- conjugates are not expected to inhibit CYPs 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, or 3A4, or induce CYPs 1A2 or 2B6 at the recommended dose of chenodiol of 250 mg TID. Chenodiol and its tauro- conjugate may upregulate CYP3A4 mRNA in vitro. The clinical significance of this upregulation is unknown.
The glyco- and tauro- conjugates of chenodiol are high affinity substrates for BSEP, and in vitro studies suggest that chenodiol may inhibit OATP1B1 and OATP1B3 at the recommended dose of 250 mg TID (clinical significance unknown), but chenodiol and its glyco- and tauro- conjugates are not predicted to inhibit P-gp, BCRP, OATP2B1, OAT1, OAT3, OCT1, OCT2, MATE1, or MATE2-K.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
A two-year oral study of chenodiol in rats did not show a carcinogenic potential at the tested levels of 15 to 60 mg/kg/day (0.2 to 0.6 times the recommended human dose based on body surface area). In additional long-term studies, chenodiol given at oral doses up to 600 mg/kg/day in rats (6 times the recommended human dose based on body surface area), and 1000 mg/kg/day in mice (5 times the recommended human dose based on body surface area) induced benign and malignant liver cell tumors in female rats and cholangiomas in female rats and male mice. Two-year studies of lithocholic acid (a major metabolite of chenodiol) in mice (125 to 250 mg/kg/day, equivalent to 0.7 to 1.4 times the recommended human dose based on body surface area) and rats (250 and 500 mg/kg/day, equivalent to 3 to 5 times the recommended human dose based on body surface area) found it not to be carcinogenic. The dietary administration of lithocholic acid to chickens is reported to cause hepatic adenomatous hyperplasia.
13.2 Animal Toxicology and/or Pharmacology
Chenodiol caused hepatobiliary toxicity (e.g., cholestasis) in many animal species, including rodents, non-rodents, and non-human primates at doses close to the human dose. Less efficient sulfation of the chenodiol metabolite, lithocholic acid, in non-human primates compared to humans is thought to cause the hepatobiliary toxicity of orally dosed chenodiol. However, there is evidence that the hepatobiliary toxicity is partly due to the parent drug, chenodiol.
-
14 CLINICAL STUDIES
The efficacy of CTEXLI for the treatment of patients with CTX was evaluated in Trial 1, which was a randomized, double-blind, placebo controlled, 2-period with 2-treatment crossover trial in patients ≥16 years of age (NCT 04270682).
In Trial 1, 14 patients were enrolled and 13 patients were randomized and treated in a crossover withdrawal design to receive either CTEXLI 250 mg or placebo orally three times daily for 4 weeks during 2 double-blind treatment periods. The study also included treatment with CTEXLI 250 mg three times daily during an 8 week run-in period and an 8-week open label period in between the 2 double-blind withdrawal periods. The total duration of study treatment was 24 weeks.
Of the 13 randomized patients, 62% were male and 39% were female. The baseline median age was 42 years (16-55) and median age at diagnosis was 35 years (15-55). CTEXLI is not approved for use in pediatric patients. The patient population consisted of 62% White, 15% Asian, and 23% Other (In the Other racial group, there was one patient who reported both White and Black). Ethnicity consisted of 15% Hispanic or Latino, 54% not Hispanic or Latino, and 31% unknown.
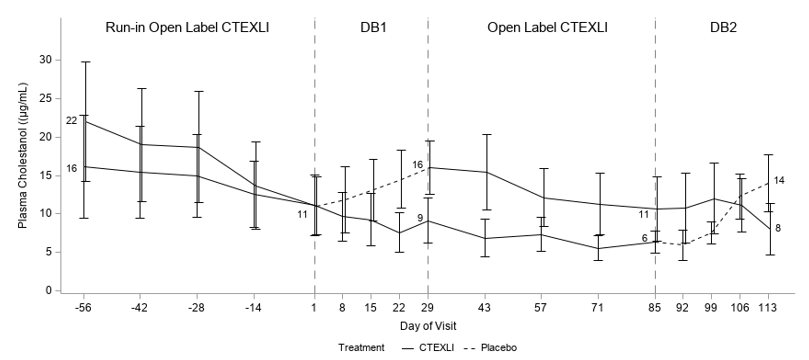
Plasma cholestanol and urine 23S-pentol were assessed at multiple time points as shown in Figure 1. For plasma cholestanol, the estimated mean change from baseline at day 29 was -2.3 µg/mL when patients continued CTEXLI treatment and 6.2 µg/mL when patients received placebo. The estimated treatment difference was -8.5 µg/mL (95% CI: -13.2, -3.9) (Table 1). For urine 23S-pentol, the estimated mean change from baseline at day 29 was 185 ng/mL when patients continued CTEXLI treatment and 29506 ng/mL when patients received placebo. The estimated treatment difference was -29321 ng/mL (95% CI: -45701, -12941).
Figure 1: Mean (SE) of Observed Plasma Cholestanol by Treatment Sequence (All Randomized Patients)
DB = double-blind. Solid line represents treatment with CTEXLI and dashed line represents treatment with placebo. The mean values of plasma cholestanol at baselines and end of each DB period are annotated in the figure.
Table 1: Summary Results for Plasma Cholestanol and Urine 23S Pentol For each study treatment (placebo or CTEXLI), the mean value at Baseline was calculated as the mean of the measurements obtained prior to receiving the study treatment during the double-blind study duration; and the mean value at Day 29 was calculated as the mean of the measurements at Day 29 at the end of the study treatment.
For each patient at each visit, the measurement of urine 23S-pentol was calculated as the geometric mean of first 3 morning void urine samples collected within 5 days prior to the visit.Plasma Cholestanol (µg/mL)
Mean (SD)
CTEXLI
(N = 13)Placebo
(N = 13)
Baseline
10.8 (10.0)
8.8 (7.8)
Day 29
8.5 (7.0)
15.1 (8.8)
Change from Baseline at Day 29
-2.3 (3.9)
6.2 (5.6)
Treatment Difference
-8.5 (95% CI: -13.2, -3.9)
Urine 23S- Pentol (ng/mL)
Baseline
1811 (1693)
1773 (1940)
Day 29
1996 (1341)
31279 (27595)
Change from Baseline at Day 29
185 (1479)
29506 (27257)
Treatment Difference
-29321 (95% CI: -45701, -12941)
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
CTEXLI (chenodiol) tablets are supplied as 250 mg white film-coated tablets imprinted with “MP” on one side and "250" on the other side.
NDC: 79378-310-90: 100 count bottle
Storage and Handling
Store CTEXLI at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
-
17 PATIENT COUNSELING INFORMATION
Hepatotoxicity
Inform the patient of the symptoms of hepatotoxicity (e.g., abdominal pain, bruising, dark-colored urine, fatigue, bleeding, jaundice, nausea, and pruritus). Instruct the patient to discontinue CTEXLI immediately and seek medical care should symptoms occur [see Warnings and Precautions (5.1)].
Rx only
Manufactured for:
Mirum Pharmaceuticals, Inc.
Foster City, CA 94404© 2025 Mirum Pharmaceuticals, Inc.
CTEXLI® is a registered trademark of Mirum Pharmaceuticals, Inc. -
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
QR 2D;
NDC: 79378-310-90;
GTIN: XXXXXXXXXXXXXX;
LOT: XXXXXX;
EXP: YYYY-MM-DD;NDC: 79378-310-90;
Ctexli; (chenodiol); tablets; 250 mg; For oral use; 100 Tablets; Rx only;
Recommended Dosage: see Prescribing Information.
Store at 20°C to 25°C (68°F to 77°F), excursions permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
KEEP THIS AND ALL MEDICATION OUT OF THE REACH OF CHILDREN.
Manufactured for
Mirum Pharmaceuticals, Inc.
Foster City, CA 94404 -
INGREDIENTS AND APPEARANCE
CTEXLI
chenodiol tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 79378-310 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CHENODIOL (UNII: 0GEI24LG0J) (CHENODIOL - UNII:0GEI24LG0J) CHENODIOL 250 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) MAGNESIUM STEARATE (UNII: 70097M6I30) METHYLCELLULOSE (100 MPA.S) (UNII: 4GFU244C4J) GLYCERIN (UNII: PDC6A3C0OX) SODIUM LAURYL SULFATE (UNII: 368GB5141J) STARCH, CORN (UNII: O8232NY3SJ) Product Characteristics Color WHITE Score no score Shape ROUND Size 10mm Flavor Imprint Code MP;250 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 79378-310-90 100 in 1 BOTTLE; Type 0: Not a Combination Product 04/04/2025 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA219488 04/04/2025 Labeler - Mirum Pharmaceuticals Inc. (116902386)

Trademark Results [CTEXLI]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 CTEXLI 98499814 not registered Live/Pending |
Mirum Pharmaceuticals, Inc. 2024-04-15 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.