TAKHZYRO- lanadelumab-flyo injection, solution
TAKHZYRO by
Drug Labeling and Warnings
TAKHZYRO by is a Prescription medication manufactured, distributed, or labeled by Takeda Pharmaceuticals America, Inc., Rentschler Biopharma SE, Vetter Pharma-Fertigung GmbH & Co. KG, Catalent Indiana LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TAKHZYRO™ safely and effectively. See full prescribing information for TAKHZYRO™.
TAKHZYRO™ (lanadelumab-flyo) injection, for subcutaneous use.
Initial U.S. Approval: 2018.INDICATIONS AND USAGE
TAKHZYRO is a plasma kallikrein inhibitor (monoclonal antibody) indicated for prophylaxis to prevent attacks of hereditary angioedema (HAE) in patients 12 years and older. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Injection: 300 mg/2 mL (150 mg/mL) solution in a single-dose vial. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
Hypersensitivity reactions have been observed. In case of a severe hypersensitivity reaction, discontinue TAKHZYRO administration and institute appropriate treatment. (5.1)
ADVERSE REACTIONS
The most common adverse reactions are injection site reactions, upper respiratory infections, headache, rash, myalgia, dizziness, and diarrhea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Dyax Corp. (a wholly-owned, indirect subsidiary of Shire plc) at 1-800-828-2088 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 11/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosage
2.2 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
7 DRUG INTERACTIONS
7.1 Drug-Laboratory Test Interactions
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosage
The recommended starting dose is 300 mg every 2 weeks. A dosing interval of 300 mg every 4 weeks is also effective and may be considered if the patient is well-controlled (e.g., attack free) for more than 6 months.
2.2 Administration
TAKHZYRO is administered subcutaneously only.
TAKHZYRO is provided as a ready-to-use solution in a single-dose vial that does not require additional reconstitution or dilution for administration. TAKHZYRO is supplied as a clear to slightly opalescent, colorless to slightly yellow solution. Do not use the vial if it appears discolored or contains visible particles. Avoid vigorous agitation of the vial.
TAKHZYRO is intended for self-administration or administration by a caregiver. The patient or caregiver should be trained by a healthcare professional.
Take the TAKHZYRO vial out of the refrigerator 15 minutes before injecting to allow it to equilibrate to room temperature.
Using aseptic technique, withdraw the prescribed dose of TAKHZYRO from the vial using an 18-gauge needle. Change the needle on the syringe to a 27-gauge, ½-inch needle or other needle suitable for subcutaneous injection. Inject TAKHZYRO subcutaneously into the abdomen, thigh, or upper arm. Patients should inject the complete dose as prescribed by their physician. In clinical studies, the majority of patients self-administered TAKHZYRO over 10 to 60 seconds.
TAKHZYRO should be administered within 2 hours of preparing the dosing syringe. After the dosing syringe is prepared, it can be refrigerated at 36ºF to 46ºF (2°C to 8°C) and must be used within 8 hours.
Discard any unused portions of drug remaining in the vial and syringe.
For detailed instructions on the preparation and administration of TAKHZYRO see Instructions for Use.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
- 5 WARNINGS AND PRECAUTIONS
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of TAKHZYRO is primarily based on a 26-week, randomized, double-blind, parallel-group and placebo-controlled study (Trial 1) in 125 patients with Type I or II HAE. Eligible patients were also able to participate in an open-label extension study (Trial 2) up to 130 weeks. In Trial 1, a total of 84 patients with HAE aged 12 years and older received at least one dose of TAKHZYRO. Overall, 70% of patients were female and 90% of patients were Caucasian with a mean age of 41 years. The proportion of patients who discontinued study drug prematurely due to adverse events was 1.2% for TAKHZYRO-treated patients and 4.9% for placebo-treated patients. No deaths occurred in the trial.
The safety profile of TAKHZYRO was generally similar across all subgroups of patients, including analysis by age, sex, and geographic region.
Table 1 shows adverse reactions occurring in ≥10% of patients in any TAKHZYRO treatment group that also occurred at a higher rate than in the placebo treatment group in Trial 1.
Table 1 Adverse Reactions Observed in ≥10% of Patients Treated with TAKHZYRO in Trial 1 Adverse Reaction Placebo
(N=41)TAKHZYRO 150 mg q4wks
(N=28)300 mg q4wks
(N=29)300 mg q2wks
(N=27)Total
(N=84)n (%) n (%) n (%) n (%) n (%) N= number of patients; n =number of patients experiencing the event; q2wks = every 2 weeks; q4wks = every 4 weeks - * Injection site reactions include: pain, erythema, bruising, hematoma, hemorrhage, pruritus, swelling, induration, paraesthesia, reaction, warmth, edema and rash.
- † Includes upper respiratory infection, viral upper respiratory infection
- ‡ Includes headache, tension headache, sinus headache
- § Includes rash, rash maculopapular, rash erythematous
Injection site reactions* 14 (34) 16 (57) 13 (45) 15 (56) 44 (52) Upper respiratory infection† 13 (32) 3 (11) 9 (31) 12 (44) 24 (29) Headache‡ 9 (22) 3 (11) 6 (21) 9 (33) 18 (21) Rash§ 2(5) 2 (7) 3 (10) 1 (4) 6 (7) Myalgia 0 1 (4) 0 3 (11) 4 (5) Dizziness 0 1 (4) 3 (10) 1 (4) 5 (6) Diarrhea 2 (5) 3 (11) 0 1 (4) 4 (5) Injection site reactions primarily consisted mainly of pain, erythema, and bruising at the injection site. There was no meaningful difference in injection site reactions with self-administration.
Less Common Adverse Reactions
Other adverse reactions that occurred at a higher incidence in TAKHZYRO-treated patients compared to placebo include hypersensitivity (1% vs 0%), increased aspartate transaminase (2% vs 0%), and increased alanine transaminase (2% vs 0%).
Safety data from the ongoing open-label extension study, consisting of 109 rollover patients from Trial 1 and 103 non-rollover HAE patients, is consistent with controlled safety data from Trial 1.
Laboratory Abnormalities
Transaminase elevations
During the placebo-controlled treatment period in Trial 1, the number of TAKHZYRO-treated patients with maximum transaminase (ALT or AST) levels >8, >5, or >3 times the upper limit of normal (ULN) was 1 (1.2%), 0 (0%), or 3 (3.6%) respectively, compared to 0 in the placebo-treated patients. These transaminase elevations were asymptomatic and transient. No patients had elevated total bilirubin >2× ULN. One TAKHZYRO-treated patient permanently discontinued treatment due to elevated transaminases (4.1× ULN AST). None of the patients were reported to have serious adverse reactions of elevated transaminases.
6.2 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to lanadelumab-flyo in the study described below with the incidence of antibodies in other studies or to other products may be misleading.
In Trial 1, 10 (12%) lanadelumab-flyo-treated and 2 (5%) placebo-treated patients had at least 1 anti-drug antibody (ADA)-positive sample during the treatment period; antibody titers were low (range: 20 to 1280). The ADA response observed was transient in 2/10 lanadelumab-flyo and 1/2 placebo-treated patients. Pre-existing low titer antibodies were observed in 3 lanadelumab-flyo-treated patients and 1 placebo-treated patient with ADAs. Two patients receiving 150 mg q4wks had low titer antibodies classified as neutralizing.
The development of ADA including neutralizing antibodies against lanadelumab-flyo did not appear to adversely affect pharmacokinetics (PK), pharmacodynamics (PD), safety or clinical response.
-
7 DRUG INTERACTIONS
No dedicated drug interaction studies have been conducted [see Clinical Pharmacology (12.3)].
7.1 Drug-Laboratory Test Interactions
Coagulation tests
TAKHZYRO can increase activated partial thromboplastin time (aPTT) due to an interaction of TAKHZYRO with the aPTT assay. The reagents used in the aPTT laboratory test initiate intrinsic coagulation through the activation of plasma kallikrein in the contact system. Inhibition of plasma kallikrein by TAKHZYRO can increase aPTT in this assay. In Trial 1, prolongation of aPTT (>1× ULN) was observed at one or more time points in 3, 9, and 11 patients treated with TAKHZYRO 150 mg q4 wks, 300 mg q4 wks, and 300 mg q2 wks, respectively, compared to 5 placebo-treated patients. Only one patient in the 300 mg q2 wks treatment group experienced transient aPTT prolongation ≥1.5× ULN which was confounded by ongoing heparin therapy. None of the increases in aPTT in patients treated with TAKHZYRO were associated with abnormal bleeding adverse events. There were no differences in INR values between treatment groups.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on TAKHZYRO use in pregnant women to inform any drug associated risks. Monoclonal antibodies such as lanadelumab-flyo are transported across the placenta during the third trimester of pregnancy; therefore, potential effects on a fetus are likely to be greater during the third trimester of pregnancy. An enhanced pre-and postnatal development (ePPND) study conducted in pregnant monkeys at doses resulting in exposures of up to 33 times the exposure achieved (on an AUC basis) at the maximum recommended human dose (MRHD) revealed no evidence of harm to the developing fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
In the ePPND study, pregnant cynomolgus monkeys were administered lanadelumab-flyo once weekly at subcutaneous doses resulting in up to 33 times the exposure at the MRHD (on an AUC basis with maternal subcutaneous doses up to 50 mg/kg/week) from gestation day 20, at the beginning of organogenesis, through to parturition. There were no lanadelumab-flyo-related effects on maintenance of pregnancy or parturition. Maternal lanadelumab-flyo treatment had no effects on embryo-fetal development, survival, growth, or postnatal development of offspring through 3 months of age. Lanadelumab-flyo crossed the placenta in monkeys. Offspring were exposed to lanadelumab-flyo at approximately 50% of the maternal plasma concentration out to postnatal day 21 (PND 21). Lanadelumab-flyo concentrations were approximately equivalent in maternal and offspring plasma at PND 90.
8.2 Lactation
Risk Summary
There are no data on the presence of lanadelumab-flyo in human milk, its effects on the breastfed infant, or its effects on milk production. Lanadelumab-flyo was detected in the milk of lactating cynomolgus monkeys at approximately 0.2% of the maternal plasma concentration. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for TAKHZYRO and any potential adverse effects on the breastfed infant from TAKHZYRO or from the underlying maternal condition.
8.4 Pediatric Use
The safety and efficacy of TAKHZYRO were evaluated in a subgroup of patients (N=10) aged 12 to <18 years in Trial 1. Results of the subgroup analysis by age were consistent with overall study results [see Adverse Reactions (6.1), Clinical Pharmacology (12.3) and Clinical Studies (14)]. An additional 13 adolescent patients aged 12 to <18 years were enrolled in the open-label extension study.
The safety and efficacy of TAKHZYRO in pediatric patients < 12 years of age have not been established.
8.5 Geriatric Use
The safety and efficacy of TAKHZYRO were evaluated in a subgroup of patients (N=5) aged ≥65 years in Trial 1. Results of the subgroup analysis by age were consistent with overall study results [see Adverse Reactions (6.1), Clinical Pharmacology (12.3) and Clinical Studies (14)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
Lanadelumab-flyo is non-plasma derived, recombinant, fully human, monoclonal antibody (IgG1/κ-light chain) produced in Chinese Hamster Ovary (CHO) cells. Based on the amino acid sequence, the molecular weight of the non-glycosylated lanadelumab-flyo is 146 kDa. The calculated molecular mass of the fully reduced light chain is 23 kDa. The calculated molecular mass of the fully reduced and non-glycosylated heavy chain is 49 kDa.
TAKHZYRO (lanadelumab-flyo) injection is a sterile, preservative-free, clear to slightly opalescent, colorless to slightly yellow solution for subcutaneous use.
Each mL of ready-to-use TAKHZYRO solution contains lanadelumab-flyo 150 mg, citric acid monohydrate (4.1 mg), L-histidine (7.8 mg), polysorbate 80 (0.1 mg), sodium chloride (5.3 mg), sodium phosphate dibasic dihydrate (5.3 mg), and Water for Injection, USP. The solution has a pH of approximately 6.0 and an osmolality of approximately 300 mOsm/kg.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Lanadelumab-flyo is a fully human monoclonal antibody (IgG1/κ-light chain) that binds plasma kallikrein and inhibits its proteolytic activity. Plasma kallikrein is a protease that cleaves high-molecular-weight-kininogen (HMWK) to generate cleaved HMWK (cHMWK) and bradykinin, a potent vasodilator that increases vascular permeability resulting in swelling and pain associated with HAE. In patients with HAE due to C1-inhibitor (C1-INH) deficiency or dysfunction, normal regulation of plasma kallikrein activity is not present, which leads to uncontrolled increases in plasma kallikrein activity and results in angioedema attacks. Lanadelumab-flyo decreases plasma kallikrein activity to control excess bradykinin generation in patients with HAE.
12.2 Pharmacodynamics
Concentration-dependent inhibition of plasma kallikrein, measured as reduction of cHMWK levels, was demonstrated after subcutaneous administration of TAKHZYRO 150 mg q4wks, 300 mg q4wks or 300 mg q2wks in patients with HAE.
TAKHZYRO did not prolong the QT/QTc interval.
12.3 Pharmacokinetics
Following subcutaneous administration, the pharmacokinetics of lanadelumab-flyo was approximately dose-proportional in the therapeutic dose range in patients with HAE (Table 2). The pharmacokinetic properties and exposure (steady state) of lanadelumab-flyo in HAE patients, following subcutaneous administration of 150 mg q4wks, 300 mg q4wks and 300 mg q2wks, are provided in Table 2. Following subcutaneous administration of TAKHZYRO, peak plasma concentrations are reached within 5 days, and terminal elimination half-life is ~2 weeks. The anticipated time to reach steady state concentration was approximately 70 days. At steady-state, the mean accumulation ratio is approximately 1.44, 1.42, and 2.43 for dosing regimen of 150 mg q4wks, 300 mg q4wks and 300 mg q2wks, respectively.
Table 2 Mean (SD) Pharmacokinetic Parameters of Lanadelumab-flyo Following Subcutaneous Administration (Trial 1) Pharmacokinetic Parameters Lanadelumab-flyo 150 mg q4wks
(N=28)300 mg q4wks
(N=29)300 mg q2wks
(N=27)CL/F: apparent clearance; Vc/F: apparent volume of distribution; AUCtau,ss: area under the curve over the dosing interval at steady-state; Cmax,ss: maximum concentration at steady-state; Cmin,ss: minimum concentration at steady state; Tmax: time to maximum concentration; t1/2 terminal elimination half-life. CL/F
(L/day)0.667 (0.162) 0.742 (0.239) 0.809 (0.370) Vc/F
(L)14.1 (2.93) 14.9 (4.45) 16.6 (4.79) AUCtau,ss
(µg*day/mL)233 (56.6) 441(137) 408 (138) Cmax,ss
(µg/mL)12.0 (3.01) 23.3 (7.94) 34.4 (11.2) Cmin,ss
(µg/mL)4.81 (1.40) 8.77 (2.80) 25.4 (9.18) tmax
(day)5.17 (1.09) 5.17 (1.12) 4.11 (0.377) t1/2
(day)14.9 (2.00) 14.2 (1.89) 15.0 (2.48) Specific Populations
Population pharmacokinetic analyses showed that age, gender and race did not meaningfully influence the pharmacokinetics of lanadelumab-flyo after correcting for body weight. Body weight was identified as an important covariate describing the variability of clearance and volume of distribution, resulting in higher exposure (AUC and Cmax) in lighter patients. However, this difference is not considered to be clinically relevant and no dose adjustments are recommended for any of these demographics.
Pediatric Population
Based on population pharmacokinetics (PK) analyses, the mean lanadelumab-flyo (±SD) AUCss was 629 (204) µg*day/mL following SC administration of TAKHZYRO 300 mg every 2 weeks in pediatric patients 12 to less than 18 years of age. This is approximately 37% higher than the mean AUCss in adult patients (460 μg*day/mL) under the same dosing regimen, due to lower body weight in pediatric patients.
Renal Impairment
No dedicated studies have been conducted to evaluate the PK of lanadelumab-flyo in renal impairment patients. Based on population pharmacokinetic analysis, renal impairment (estimated GFR: 60 to 89 mL/min/1.73m2, [mild, N=98] and 30 to 59 mL/min/1.73m2, [moderate, N=9]) had no effect on the clearance or volume of distribution of lanadelumab-flyo.
Concomitant medications
The use of analgesic, antibacterial, antihistamine, anti-inflammatory and anti-rheumatic medications had no effect on clearance and volume of distribution of lanadelumab-flyo.
For breakthrough HAE attacks, use of rescue medications such as plasma-derived and recombinant C1-INH, icatibant or ecallantide had no effects on clearance and volume of distribution of lanadelumab-flyo.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Animal studies have not been conducted to evaluate the carcinogenic potential of lanadelumab-flyo. Published literature supports bradykinin, which is elevated in HAE, as a pro-tumorigenic molecule. However, the malignancy risk in humans from an antibody that inhibits plasma kallikrein activity, such as lanadelumab-flyo, which lowers bradykinin levels, is currently unknown.
Male and female fertility were unaffected based upon no observed adverse histopathological findings in the reproductive organs from sexually mature cynomolgus monkeys that received lanadelumab-flyo for 13 weeks at subcutaneous doses up to 50 mg/kg/week (resulting in approximately 22 times the exposure at the MRHD on an AUC basis).
-
14 CLINICAL STUDIES
Trial 1 (NCT02586805)
The efficacy of TAKHZYRO for the prevention of angioedema attacks in patients 12 years of age and older with Type I or II HAE was demonstrated in a multicenter, randomized, double-blind, placebo-controlled parallel-group study (Trial 1).
The study included 125 adult and adolescent patients with Type I or II HAE who experienced at least one investigator-confirmed attack per 4 weeks during the run-in period. Patients were randomized into 1 of 4 parallel treatment arms, stratified by baseline attack rate, in a 3:2:2:2 ratio (placebo, lanadelumab-flyo 150 mg q4wks, lanadelumab-flyo 300 mg q4wks, or lanadelumab-flyo 300 mg q2wks by subcutaneous injection) for the 26-week treatment period. Patients ≥18 years of age were required to discontinue other prophylactic HAE medications prior to entering the study; however, all patients were allowed to use rescue medications for treatment of breakthrough HAE attacks.
Overall, 90% of patients had Type I HAE. A history of laryngeal angioedema attacks was reported in 65% of patients and 56% were on prior long-term prophylaxis. During the study run-in period, attack rates of ≥3 attacks/month were observed in 52% of patients overall.
All TAKHZYRO treatment arms produced clinically meaningful and statistically significant reductions in the mean HAE attack rate compared to placebo across all primary and secondary endpoints in the Intent-to-Treat population (ITT) as shown in Table 3.
Table 3 Results of Primary and Secondary Efficacy Measures-ITT Population Endpoint Statistics Placebo
(N=41)TAKHZYRO 150 mg q4wks
(N=28)300 mg q4wks
(N=29)300 mg q2wks
(N=27)CI=confidence interval; SD=standard deviation; LS=least squares.
Note: Results are from a Poisson regression model accounting for over dispersion with fixed effects for treatment group (categorical) and normalized baseline attack rate (continuous), and the logarithm of time in days each patient was observed during the treatment period as an offset variable in the model.- * Primary efficacy endpoint.
- † Model-based treatment period HAE attack rate (attacks/4 weeks).
- ‡ Calculated as the ratio of the model-based treatment period HAE attack rates (lanadelumab/placebo) minus 1 multiplied by 100.
- § Adjusted p-values for multiple testing.
Number of HAE Attacks from Day 0 to 182* LS Mean (95% CI) monthly attack rate† 1.97
(1.64, 2.36)0.48
(0.31, 0.73)0.53
(0.36, 0.77)0.26
(0.14, 0.46)% Reduction relative to placebo (95% CI)‡ 76
(61, 85)73
(59, 82)87
(76, 93)Adjusted p-values§ <0.001 <0.001 <0.001 Number of HAE Attacks Requiring Acute Treatment from Day 0 to 182 LS Mean (95% CI) monthly attack rate† 1.64
(1.34, 2.00)0.31
(0.18, 0.53)0.42
(0.28, 0.65)0.21
(0.11, 0.40)% Reduction relative to placebo (95% CI)‡ 81
(66, 89)74
(59, 84)87
(75, 93)Adjusted p-values§ <0.001 <0.001 <0.001 Number of Moderate or Severe HAE Attacks from Day 0 to 182 LS Mean (95% CI) monthly attack rate† 1.22
(0.97, 1.52)0.36
(0.22, 0.58)0.32
(0.20, 0.53)0.20
(0.11, 0.39)% Reduction relative to placebo (95% CI)‡ 70
(50, 83)73
(54, 84)83
(67, 92)Adjusted p-values§ <0.001 <0.001 <0.001 The mean reduction in HAE attack rate was consistently higher across the TAKHZYRO treatment arms compared to placebo regardless of the baseline history of prior long-term prophylaxis, laryngeal attacks, or attack rate during the run-in period.
Additional pre-defined exploratory endpoints included the percentage of patients who were attack free for the entire 26-week treatment period and the percentage of patients achieving threshold (≥50%, ≥70%, ≥90%) reductions in HAE attack rates compared to run-in during the 26-week treatment period. A ≥50% reduction in HAE attack rate was observed in 100% of patients on 300 mg q2wks or q4wks and 89% on 150 mg q4wks compared to 32% of placebo patients. A ≥70% reduction in HAE attack rates was observed in 89%, 76%, and 79% of patients on 300 mg q2wks, 300 mg q4wks, and 150 mg q4wks, respectively, compared to 10% of placebo patients. A ≥90% reduction in HAE attack rates was observed 67%, 55%, and 64% of patients on 300 mg q2wks, 300 mg q4wks, and 150 mg q4wks, respectively, compared to 5% of placebo patients.
The percentage of attack-free patients for the entire 26-week treatment period was 44%, 31%, and 39% in the TAKHZYRO 300 mg q2wks, 300 mg q4wks, and 150 mg q4wks groups respectively, compared to 2% of placebo patients.
Trial 2 (NCT02741596)
Patients who completed Trial 1 were eligible to rollover into an open-label extension study. Rollover patients, regardless of randomization group in Trial 1, received a single dose of TAKHZYRO 300 mg at study entry and were followed until the first HAE attack occurred. All efficacy endpoints were exploratory in this uncontrolled, unblinded study. At week 4 post-dose, approximately 80% of patients who had been in the 300 mg q2wks treatment group (N=25) in Trial 1 remained attack-free. After the first HAE attack, all patients received open-label treatment with TAKHYZRO 300 mg q2wks.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
- TAKHZYRO (lanadelumab-flyo) injection is a ready-to-use, clear to slightly opalescent, colorless to slightly yellow solution supplied in a carton containing one single-dose glass vial with chlorobutyl rubber stopper, aluminum crimp seal and polypropylene flip-off cap.
- NDC: 47783-644-01: 300 mg/2 mL (150 mg/mL) vial.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use). Inform patients of the risks and benefits of TAKHZYRO before prescribing or administering to the patient.
Hypersensitivity
Advise patients to seek immediate medical attention if they experience any symptoms of serious hypersensitivity reactions [see Warnings and Precautions (5.1)].
Self-administration:
- Ensure that the patient/caregiver receives clear instructions and training on subcutaneous administration and has demonstrated the ability to perform a subcutaneous injection.
- Instruct patients or caregivers in the technique of proper syringe and needle disposal, and advise them not to reuse these items. Instruct patients to dispose needles and syringes in a puncture-resistant container.
-
SPL UNCLASSIFIED SECTION
For more information, visit www.TAKHZYRO.com
Manufactured by:
Dyax Corp.
300 Shire Way
Lexington, MA 02421U.S. License No. 1789
TAKHZYRO™ is a trademark or registered trademark of Dyax Corp., a wholly-owned, indirect subsidiary of Shire plc. SHIRE and the Shire Logo are trademarks or registered trademarks of Shire Pharmaceutical Holdings Ireland Limited or its affiliates.
©2018 Shire. All rights reserved.
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration Issued: 11/2018 PATIENT INFORMATION
TAKHZYRO™ (tak-ZYE-roe)
(lanadelumab-flyo)
injection, for subcutaneous useRead this Patient Information before you start using TAKHZYRO and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or your treatment. What is TAKHZYRO?
TAKHZYRO is a prescription medicine used to prevent attacks of Hereditary Angioedema (HAE) in people 12 years of age and older. It is not known if TAKHZYRO is safe and effective in children under 12 years of age.Before you use TAKHZYRO, tell your healthcare provider about all of your medical conditions, including if you: - are pregnant or planning to become pregnant. It is not known if TAKHZYRO can harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if TAKHZYRO passes into your breastmilk. Talk to your healthcare provider about the best way to feed your baby while using TAKHZYRO.
How should I use TAKHZYRO? - See the detailed "Instructions for Use" that comes with this patient information leaflet about the right way to prepare and inject TAKHZYRO.
- Use TAKHZYRO exactly as your healthcare provider tells you to use it.
- TAKHZYRO is given as an injection under your skin (subcutaneous) by you or a caregiver.
- Your healthcare provider should show you or your caregiver how to prepare and inject your dose of TAKHZYRO before you inject yourself for the first time.
- Do not try to inject TAKHZYRO unless you have been trained by your healthcare provider.
What are the possible side effects of TAKHZYRO?
TAKHZYRO may cause serious side effects, including allergic reactions. Allergic reactions may happen with TAKHZYRO. Call your healthcare provider or get emergency help right away if you have any of the following symptoms:- wheezing
- difficulty breathing
- chest tightness
- fast heartbeat
- faintness
- rash
- hives
The most common side effects of TAKHZYRO are: - injection site reactions (pain, redness, and bruising)
- upper respiratory infections
- headache
Call your doctor for medical advice about side effects. You may report side effects to the FDA at 1-800-FDA-1088.General information about the safe and effective use of TAKHZYRO.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use TAKHZYRO for a condition for which it is not prescribed. Do not give TAKHZYRO to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about TAKHZYRO that is written for health professionals.What are the ingredients in TAKHZYRO?
Active ingredient: lanadelumab
Inactive ingredients: citric acid monohydrate, L-histidine, sodium chloride, sodium phosphate dibasic dihydrate and water for injection.
Manufactured by:
Dyax Corp
300 Shire Way
Lexington, MA 02421
U.S. License No. 1789
TAKHZYRO™ is a trademark or registered trademark of Dyax Corp., a wholly-owned, indirect subsidiary of Shire plc. SHIRE and the Shire Logo are trademarks or registered trademarks of Shire Pharmaceutical Holdings Ireland Limited or its affiliates.
©2018 Shire. All rights reserved.
For more information, visit www.TAKHZYRO.com or call 1-800-828-2088.
-
INSTRUCTIONS FOR USETAKHZYRO™ (tak-ZYE-roe)(lanadelumab-flyo)injection, for subcutaneous use
Be sure that you read, understand, and follow the Instructions for Use before injecting TAKHZYRO. A healthcare provider should show you how to prepare and inject TAKHZYRO properly before you use it for the first time. Contact your healthcare provider if you have any questions.
Important information:
- TAKHZYRO is a ready-to-use solution for injection under the skin (subcutaneous). It is supplied in a single-dose, glass vial.
- Your healthcare provider will prescribe the dose that you should take.
- Only use the syringes, transfer needles, and injection needles that your healthcare provider prescribes.
- Only use the syringes, transfer needles and injection needles 1 time. Throw away (dispose of) any used syringes and needles.
Storing TAKHZYRO:
- Store TAKHZYRO in the refrigerator at 36°F to 46°F (2°C to 8°C). Do not freeze.
- Store TAKHZYRO in the original carton to protect the vial from light.
- Do not shake TAKHZYRO.
- Keep TAKHZYRO and all medicines out of the reach of children.
Supplies needed for your TAKHZYRO Injection
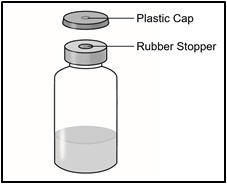
1 Vial containing TAKHZYRO 
TAKHZYRO Instructions for Use 
2 Alcohol wipes 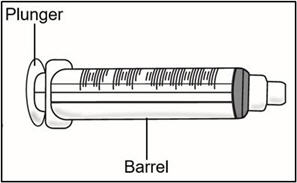
1 Empty 3 mL syringe 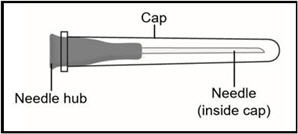
18G Transfer needle (longer needle)
Do not use the transfer needle to inject TAKHZYRO.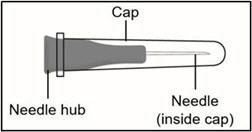
27G ½-inch Injection needle (shorter needle).
Do not use the injection needle to withdraw TAKHYZRO from the vial.
Sharps disposal container. See "STEP 6" at the end of this Instructions for Use. STEP 1: Prepare for your injection
- Gather all supplies and place them on a well-lighted flat work surface.
- Take the vial out of the refrigerator 15 minutes before use and allow it to reach room temperature before preparing an injection.
- Check the expiration date on the box and vial label of TAKHZYRO. Do not use if the expiration date has passed.
- Check the supplies for damage. Do not use if they appear damaged.
- Clean your work area and wash your hands before preparing your dose. Do not touch any surface or body part, especially your face, after washing your hands before injection.
- Remove the vial from the packaging. Do not use the vial if the plastic cap covering is missing.
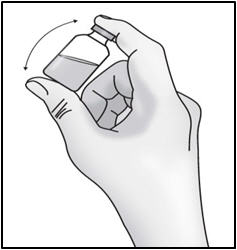
- Gently turn the vial upside down (invert) 3 to 5 times to mix the medicine. Do not shake to avoid foaming.
- Look at the medicine in the vial for visible particles or a change in the color. Medicine should be colorless to slightly yellow. Do not use if you see particles or a change in color.
Important: Do not shake. 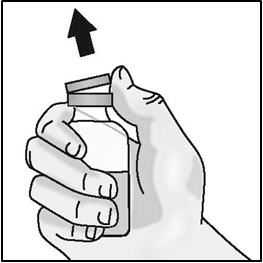
- Remove the plastic cap from the medicine vial. Do not remove the medicine vial rubber stopper.
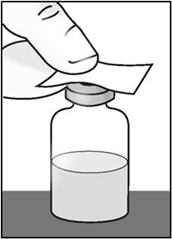
- Place the vial on a flat surface. Clean the medicine vial rubber stopper with an alcohol wipe and allow it to dry.
STEP 2: Attach transfer needle to syringe
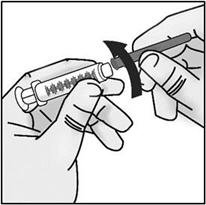
- Screw the 18G transfer needle to the 3mL syringe.
Important: Do not remove the transfer needle cap from the needle when attaching to the syringe. 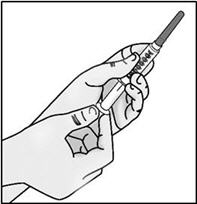
- Pull back the plunger to fill the syringe with air equal to the amount of medicine in the vial.
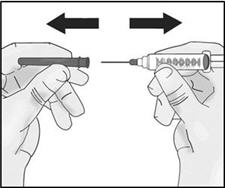
- Hold the syringe by the barrel with one hand and the transfer needle cap with the other hand.
- Pull off the transfer needle cap straight away from the syringe and away from your body. Do not pull on the plunger. Place the transfer needle cap down on a clean flat surface.
- Do not touch the needle tip.
STEP 3: Transfer TAKHZYRO into syringe and switch to the injection needle
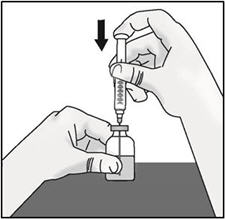
- Keep the vial on the flat surface and insert the transfer needle into the center of the rubber stopper.
- Push the plunger down to inject air into the vial and hold the plunger down.
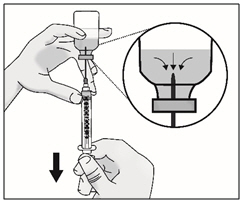
- Slowly turn the vial upside down with transfer needle and syringe attached. Pull back on the plunger to withdraw the full dose in the vial.
Important: Be sure to keep the tip of the transfer needle in the medicine to avoid drawing air in as you pull back the plunger. 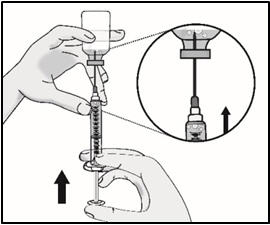
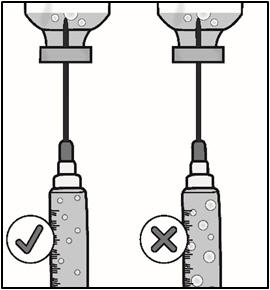
- Remove large air bubbles by gently tapping on the syringe barrel with your fingers until the bubbles rise to the top of the syringe.
- Slowly push the plunger, allowing air to go back into the vial, until the medicine reaches the top of the syringe.
- Repeat these steps until large air bubbles are removed.
Important: Check again to make sure you have the right amount of medicine in your syringe. If you do not have enough medicine, pull back on the plunger again while keeping the needle in the medicine to get your full dose. 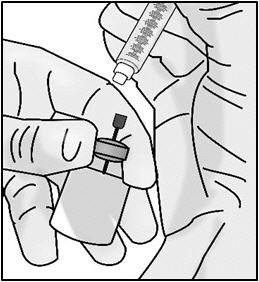
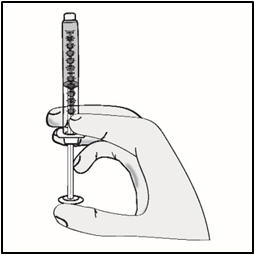
- Return the vial to an upright position.
- Without removing the needle from the vial, unscrew the syringe by holding the needle hub and turning the syringe counter clockwise.
- Return the syringe to an upright position.
- Throw away the 18G transfer needle and the vial in a sharps disposal container (See Step 6).
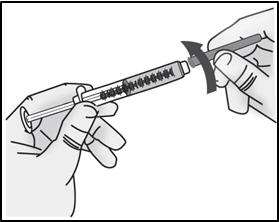
- Screw the 27G ½-inch injection needle to the syringe.
Important: Do not remove the needle cap from the injection needle when attaching to the syringe.
Do not use the transfer needle to inject TAKHZYRO as this may cause harm such as pain and bleeding.STEP 4: Select and prepare injection site
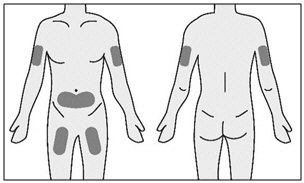
- TAKHZYRO can be self-injected in your stomach (abdomen) or thigh. If given by a caregiver, TAKHZYRO may also be injected in the upper arm.
- Clean your injection site with an alcohol wipe and allow it to dry completely.
Important: - You should use a different injection site each time you receive an injection to keep your skin healthy.
- The area you choose for injection should be at least 2 inches (5 cm) away from any scars or your belly button (navel). Do not choose an area that is bruised, swollen, or painful.
- TAKHZYRO should be given within 2 hours of preparing the dosing syringe at room temperature. After the dosing syringe is prepared, it can be refrigerated at 36°F to 46°F (2°C to 8°C) and must be used within 8 hours of preparation. Take the dosing syringe out of the refrigerator 15 minutes before use and allow it to reach room temperature prior to injecting.
STEP 5: Inject TAKHZYRO
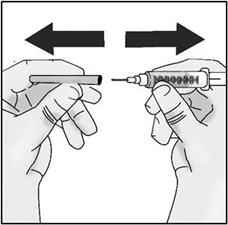
- Hold the syringe by the barrel with one hand and the injection needle cap with the other hand.
- Pull off the injection needle cap straight away from the syringe and away from your body. Do not pull on the plunger. Do not touch the needle tip or allow it to touch any other surface.
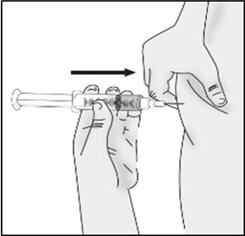
- Gently pinch about 1 inch of skin at your cleaned chosen injection site and insert the needle.
Important: Be sure to inject under the skin which is not too shallow (skin layer) or too deep (muscle). 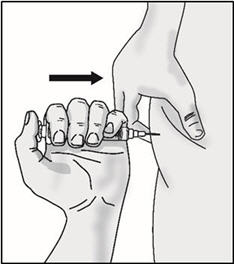
- Push the plunger slowly until no medicine remains in the syringe. Release the skin fold and gently remove the needle. Do not recap the needle.
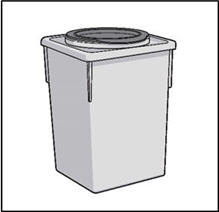
STEP 6: THROW AWAY (DISPOSE OF) NEEDLE AND SYRINGE
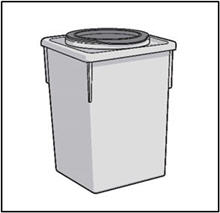
- Place the 27G ½-inch Injection needle and the syringe in a sharps container.
- If you do not have a FDA-cleared sharps disposal, you may use a household container that is:
- Made of heavy-duty plastic,
- Can be closed with a tight fitting, puncture-resistant lid, without sharps being able to come out,
- Upright and stable during use,
- Leak-resistant, and
- Properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps in the state you live in, go to the FDA's website at:
http://www.fda.gov/safesharpsdisposal.
Important: Always keep the sharps disposal container out of the reach of children.
For more information, visit www.TAKHZYRO.com
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured by:
Dyax Corp.
300 Shire Way
Lexington, MA 02421U.S. License No: 1789
TAKHZYRO™ is a trademark or registered trademark of Dyax Corp., a wholly-owned, indirect subsidiary of Shire plc. SHIRE and the Shire Logo are trademarks or registered trademarks of Shire Pharmaceutical Holdings Ireland Limited or its affiliates.
©2018 Shire. All rights reserved.
Approved: 11/2018
-
PRINCIPAL DISPLAY PANEL - 2 mL Vial Carton
Rx Only
NDC: 47783-644-01
TAKHZYRO™
(lanadelumab-flyo)
injection300 mg/2 mL (150 mg/mL)
Single-dose Vial, for Subcutaneous use only.
2 mL
1 single-dose vial.Shire

-
INGREDIENTS AND APPEARANCE
TAKHZYRO
lanadelumab-flyo injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 47783-644 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength lanadelumab (UNII: 2372V1TKXK) (lanadelumab - UNII:2372V1TKXK) lanadelumab 300 mg in 2 mL Inactive Ingredients Ingredient Name Strength SODIUM PHOSPHATE, DIBASIC, DIHYDRATE (UNII: 94255I6E2T) CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) HISTIDINE (UNII: 4QD397987E) SODIUM CHLORIDE (UNII: 451W47IQ8X) POLYSORBATE 80 (UNII: 6OZP39ZG8H) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 47783-644-01 1 in 1 CARTON 08/24/2018 1 5 mL in 1 VIAL, GLASS; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761090 08/24/2018 Labeler - Dyax Corp. (609869318) Establishment Name Address ID/FEI Business Operations Rentschler Biopharma SE 313166479 API MANUFACTURE(47783-644) Establishment Name Address ID/FEI Business Operations Catalent Indiana, LLC 172209277 MANUFACTURE(47783-644)
Trademark Results [TAKHZYRO]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 TAKHZYRO 87738368 not registered Live/Pending |
Dyax Corp. 2017-12-29 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.