AREXVY- respiratory syncytial virus vaccine recombinant, adjuvanted kit
Arexvy by
Drug Labeling and Warnings
Arexvy by is a Other medication manufactured, distributed, or labeled by GlaxoSmithKline Biologicals SA. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use AREXVY safely and effectively. See full prescribing information for AREXVY.
AREXVY (Respiratory Syncytial Virus Vaccine, Adjuvanted) for injectable suspension, for intramuscular use
Initial U.S. Approval: 2023INDICATIONS AND USAGE
AREXVY is a vaccine indicated for active immunization for the prevention of lower respiratory tract disease (LRTD) caused by respiratory syncytial virus (RSV) in:
- individuals 60 years of age and older; (1)
- individuals 18 through 59 years of age who are at increased risk for LRTD caused by RSV. (1)
Limitations of Use
AREXVY is not for use in pregnant individuals. (1)
DOSAGE AND ADMINISTRATION
For intramuscular use.
After reconstitution, a single dose of AREXVY is either 0.5 mL (vial and vial presentation) or approximately 0.5 mL (vial and prefilled syringe presentation). (2.1)
AREXVY is supplied as two presentations:
- Vial and vial presentation: vial of Lyophilized Antigen Component to be reconstituted with the accompanying vial of Adjuvant Suspension Component. (2.2)
- Vial and prefilled syringe presentation: vial of Lyophilized Antigen Component to be reconstituted with the accompanying prefilled syringe of Adjuvant Suspension Component. (2.2)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
History of severe allergic reaction (e.g., anaphylaxis) to any component of the vaccine. (4)
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
- The most commonly reported adverse reactions (≥10%), in individuals 60 years of age and older, were injection site pain (60.9%), fatigue (33.6%), myalgia (28.9%), headache (27.2%), and arthralgia (18.1%). (6.1)
- The most commonly reported adverse reactions (≥10%), in individuals 50 through 59 years of age, were injection site pain (75.8%), fatigue (39.8%), myalgia (35.6%), headache (31.7%), arthralgia (23.4%), erythema (13.2%), and swelling (10.4%). (6.1)
- The most commonly reported adverse reactions (≥10%), in individuals 18 through 49 years of age, were injection site pain (76.0%), myalgia (59.9%), fatigue (59.6%), headache (43.6%), and arthralgia (28.3%). (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact GlaxoSmithKline at 1-888-825-5249 or VAERS at 1-800-822-7967 or www.vaers.hhs.gov.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 4/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dose and Schedule
2.2 Preparation
2.3 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Guillain-Barré Syndrome
5.2 Preventing and Managing Allergic Reactions
5.3 Syncope
5.4 Altered Immunocompetence
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Efficacy in Individuals 60 Years of Age and Older
14.2 Immunogenicity in Individuals 18 through 59 Years of Age at Increased Risk for LRTD caused by RSV
14.3 Concomitant Administration
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 Storage before Reconstitution
16.2 Storage after Reconstitution
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
AREXVY is a vaccine indicated for active immunization for the prevention of lower respiratory tract disease (LRTD) caused by respiratory syncytial virus (RSV) in:
- individuals 60 years of age and older;
- individuals 18 through 59 years of age who are at increased risk for LRTD caused by RSV.
Limitations of Use
AREXVY is not for use in pregnant individuals [see Use in Specific Populations (8.1)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Dose and Schedule
After reconstitution, a single dose of AREXVY is either 0.5 mL (vial and vial presentation) or approximately 0.5 mL (vial and prefilled syringe presentation) [see Dosage and Administration (2.2)].
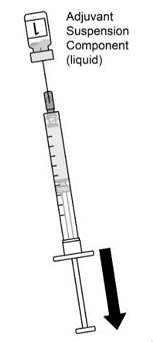
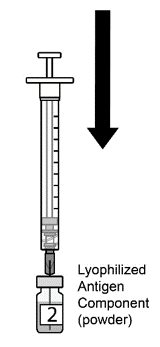
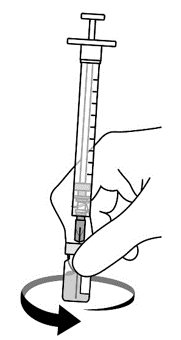
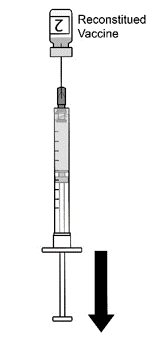
2.2 Preparation
AREXVY is supplied in two presentations as follows:
Vial and Vial Presentation
AREXVY is supplied in two vials that must be combined prior to administration. Prepare AREXVY by reconstituting the Lyophilized Antigen Component (a sterile white powder) with the accompanying Adjuvant Suspension Component (an opalescent, colorless to pale brownish sterile liquid), as described in the instructions below. Use only the supplied Adjuvant Suspension Component for reconstitution.
Vial and Prefilled Syringe Presentation
AREXVY is supplied in a vial of Lyophilized Antigen Component and a prefilled syringe of Adjuvant Suspension Component that must be combined prior to administration. Prepare AREXVY by reconstituting the Lyophilized Antigen Component (a sterile white powder) with the accompanying Adjuvant Suspension Component (an opalescent, colorless to pale brownish sterile liquid), as described in the instructions below. Use only the supplied Adjuvant Suspension Component for reconstitution.
2.3 Administration
For intramuscular use.
After reconstitution, AREXVY is an opalescent, colorless to pale brownish liquid. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. If either of these conditions exists, AREXVY should not be administered.
After reconstitution, administer AREXVY immediately or store protected from light in the refrigerator between 2°C and 8°C (36°F to 46°F) or at room temperature [up to 25°C (77°F)] and use within 4 hours. Discard reconstituted vaccine if not used within 4 hours.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Do not administer AREXVY to anyone with a history of a severe allergic reaction (e.g., anaphylaxis) to any component of AREXVY [see Description (11)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Guillain-Barré Syndrome
The results of a postmarketing observational study suggest an increased risk of Guillain-Barré syndrome during the 42 days following vaccination with AREXVY [see Adverse Reactions (6.2)].
5.2 Preventing and Managing Allergic Reactions
Appropriate medical treatment must be immediately available to manage potential anaphylactic reactions following administration of AREXVY.
-
6 ADVERSE REACTIONS
Table 1. Clinical Studies of AREXVY Study
Study Population
Number of Participants
Study 1
(NCT04886596)
Adults ≥60 years with or without medical conditions associated with an increased risk for LRTD caused by RSV
AREXVY: N = 12,467
Placebo: N = 12,499
Study 2
(NCT04732871)
Adults ≥60 years with or without medical conditions associated with an increased risk for LRTD caused by RSV
AREXVY: N = 1,653
Study 4
(NCT05590403)
Adults 50 through 59 years with or without medical conditions associated with an increased risk for LRTD caused by RSV
AREXVY: N = 769
Placebo: N = 383
Adults ≥60 years with or without medical conditions associated with an increased risk for LRTD caused by RSV
AREXVY: N = 381
Study 7
(NCT06389487)
Adults 18 through 49 years with medical conditions associated with an increased risk for LRTD caused by RSV
AREXVY: N = 1,029
Adults ≥60 years with or without medical conditions associated with an increased risk for LRTD caused by RSV
AREXVY: N = 429
Concomitant Administration Studies
Study 3
(NCT04841577)
Adults ≥60 years with or without medical conditions associated with an increased risk for LRTD caused by RSV
Concomitant administration of AREXVY with FLUARIX QUADRIVALENT: N = 442
Sequential administration of AREXVY and FLUARIX QUADRIVALENT: N = 443
Study 5
(NCT05559476)
Adults ≥65 years with or without medical conditions associated with an increased risk for LRTD caused by RSV
Concomitant administration of AREXVY with FLUZONE HIGH DOSE QUADRIVALENT: N = 516
Sequential administration of AREXVY and FLUZONE HIGH DOSE QUADRIVALENT: N = 513
Study 6 (NCT05568797)
Adults ≥65 years with or without medical conditions associated with an increased risk for LRTD caused by RSV
Concomitant administration of AREXVY with FLUAD QUADRIVALENT: N = 523
Sequential administration of AREXVY and FLUAD QUADRIVALENT: N = 522
In participants 60 years of age and older, the most commonly reported adverse reactions (≥10%) were injection site pain (60.9%), fatigue (33.6%), myalgia (28.9%), headache (27.2%), and arthralgia (18.1%).
In participants 50 through 59 years of age, the most commonly reported adverse reactions (≥10%) were injection site pain (75.8%), fatigue (39.8%), myalgia (35.6%), headache (31.7%), arthralgia (23.4%), erythema (13.2%), and swelling (10.4%).
In participants 18 through 49 years of age, the most commonly reported adverse reactions (≥10%) were injection site pain (76.0%), myalgia (59.9%), fatigue (59.6%), headache (43.6%), and arthralgia (28.3%).
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a vaccine cannot be directly compared with rates in the clinical trials of another vaccine and may not reflect the rates observed in practice.
Individuals 60 Years of Age and Older
The safety of AREXVY was evaluated in 17,879 vaccine recipients.
Study 1 is a placebo‑controlled, Phase 3 clinical study conducted in Europe, North America, Asia, and the Southern Hemisphere (South Africa, Australia, and New Zealand), involving 24,966 participants, 60 years of age and older, who received AREXVY (N = 12,467) or saline placebo (N = 12,499). Study 2 is a non–placebo-controlled, open‑label, Phase 3 clinical study conducted in Europe, North America, and Asia, involving 1,653 participants, 60 years of age and older, who received AREXVY. Study 3 is a non–placebo-controlled, open‑label, Phase 3 clinical study conducted in New Zealand, Panama, and South Africa, involving participants 60 years of age and older who received 1 dose of AREXVY and FLUARIX QUADRIVALENT concomitantly (N = 442) or sequentially (N = 443). Study 5 is a non–placebo-controlled, open‑label, clinical study conducted in the United States involving participants 65 years of age and older who received 1 dose of AREXVY and FLUZONE HIGH DOSE QUADRIVALENT (influenza vaccine, high dose; manufactured by Sanofi Pasteur, Inc.) concomitantly (N = 516) or sequentially (N = 513). Study 6 is a non–placebo-controlled, open‑label, clinical study conducted in Belgium, Finland, France, Spain, and the United Kingdom involving participants 65 years of age and older who received 1 dose of AREXVY and FLUAD QUADRIVALENT (influenza vaccine, adjuvanted; manufactured by Seqirus, Inc.) concomitantly (N = 523) or sequentially (N = 522).
At the time of vaccination in study 1, the median age of the population was 69.0 years; 13,943 (55.8%) participants were 60 to 69 years of age, 8,978 (36.0%) participants were 70 to 79 years of age, and 2,045 (8.2%) participants were 80 years of age and older. The majority of participants were White (79.4%), followed by Black (8.7%), Asian (7.6%), and other racial/ethnic groups (4.3%); 5.5% were of Hispanic or Latino ethnicity; 51.7% were female. In study 2, the median age of the population at the time of vaccination was 69.0 years; 820 (49.6%) participants were 60 to 69 years of age, 621 (37.6%) participants were 70 to 79 years of age, and 212 (12.8%) participants were 80 years of age and older. In study 2, the majority of participants were White (67.8%), followed by Asian (30.0%), Black (2.0%), and other racial/ethnic groups (0.2%); 1.9% were of Hispanic or Latino ethnicity; 54.6% were female. In study 3, the median age of the population at the time of the vaccination was 67.0 years; 519 (58.6%) participants were 60 to 69 years of age, 288 (32.5%) participants were 70 to 79 years of age, and 78 (8.8%) participants were 80 years of age and older, respectively. In study 3, the majority of the participants were of mixed race (50.3%), followed by White (30.7%), and Black (16.0%); 34.7% were of Hispanic or Latino ethnicity; 51.5% were female. In study 5, the median age of the population was 70 years. The majority of the participants were White (68.7%), followed by other racial groups (15.8%), Black (13.6%), American Indian or Alaskan Native (1.0%), and Asian (0.8%); 32.1% were of Hispanic or Latino ethnicity; 49.6% were female. In study 6, the median age of the population was 71 years. The majority of the participants were White (99.3%), followed by American Indian or Alaska Native (0.3%), Unknown Race (0.1%), Black (0.1%), Asian (0.1%), and Mixed Race (0.1%); 1.8% were of Hispanic or Latino ethnicity; 49.3% were female.
Safety Data from Study 1
Solicited Adverse Reactions: In study 1, a subset of study participants (solicited safety set) was monitored for solicited adverse reactions using standardized paper diary cards during the 4 days (i.e., day of vaccination and the next 3 days) following a dose of AREXVY or placebo; 879 participants received AREXVY and 878 participants received placebo. The other study participants did not prospectively record solicited reactions on a diary card but may have reported them as unsolicited adverse reactions.
The reported frequencies of specific solicited local (administration site) and systemic adverse reactions (per participant) are presented in Table 2.
Table 2. Percentage of Participants with Solicited Local Adverse Reactions and Systemic Adverse Reactions within 4 Days of Vaccination in Individuals 60 Years of Age and Older (Solicited Safety Set with 4-Day Diary Card) N = Exposed set for solicited safety set included all participants with at least 1 documented dose and who completed their diary.
a Placebo was a saline solution.
b Any grade pain: Defined as any pain neither interfering with nor preventing normal everyday activities (Grade 1), painful when limb is moved and interferes with everyday activities (Grade 2), or significant pain at rest and prevents normal everyday activities (Grade 3).
c Any grade fatigue, myalgia, headache, arthralgia: Defined as event easily tolerated (Grade 1), interfering with normal activity (Grade 2), or preventing normal activity (Grade 3).
d Temperature taken by any route (oral, axillary, or tympanic).AREXVY
%
Placeboa
%
Local Adverse Reactions
N = 879
N = 874
Pain, Anyb
60.9
9.3
Pain, Grade 3b
1
0
Erythema, >20 mm
7.5
0.8
Erythema, >100 mm
0.2
0
Swelling, >20 mm
5.5
0.6
Swelling, >100 mm
0.2
0
Systemic Adverse Reactions
N = 879
N = 878
Fatigue, Anyc
33.6
16.1
Fatigue, Grade 3c
1.7
0.5
Myalgia, Anyc
28.9
8.2
Myalgia, Grade 3c
1.4
0.3
Headache, Anyc
27.2
12.6
Headache, Grade 3c
1.3
0
Arthralgia, Anyc
18.1
6.4
Arthralgia, Grade 3c
1.3
0.6
Fever, ≥38.0°C/100.4°Fd
2.0
0.3
Fever, >39.0°C/102.2°Fd
0.1
0.1
In the solicited safety set, the local administration site adverse reactions reported with AREXVY had a median duration of 2 days, and the systemic adverse reactions reported with AREXVY had a median duration ranging between 1 and 2 days.
Unsolicited Adverse Events: In all participants from study 1, unsolicited adverse events were monitored using paper diary cards during the 30-day period following vaccination (day of vaccination and the next 29 days).
Among participants in the solicited safety set, (AREXVY, N = 879 or placebo, N = 878), unsolicited adverse events occurring within 30 days after vaccination were reported in 14.9% and 14.6% of participants who received AREXVY and placebo, respectively.
In the exposed set, 24,966 participants, 60 years of age and older, received at least 1 dose of AREXVY (N = 12,467) or placebo (N = 12,499). Unsolicited adverse events occurring within 30 days of vaccination were reported in 33.0% and 17.8% of participants, respectively. The higher frequency of reported unsolicited adverse events among participants who received AREXVY, compared to participants who received placebo, was primarily attributed to events that are consistent with adverse reactions solicited among participants in the reactogenicity subset. Within 30 days after vaccination, atrial fibrillation was reported in 10 participants who received AREXVY and 4 participants who received placebo (of which 7 events in AREXVY arm and 1 event in placebo arm were serious); the onset of symptoms ranged from 1 to 30 days postvaccination. The currently available information on the atrial fibrillation is insufficient to determine a causal relationship to the vaccine. There were no other notable patterns or numerical imbalances between groups for specific categories of unsolicited adverse events.
Serious Adverse Events: In study 1, participants were monitored for all serious adverse events (SAEs) that occurred during the 6‑month period following administration of AREXVY (N = 12,467) or placebo (N = 12,499).
SAEs with onset within 6 months following vaccination were reported at similar rates in participants who received AREXVY (4.2%) or placebo (4.0%). Serious events of atrial fibrillation were reported in 13 participants who received AREXVY and 15 participants who received placebo within 6 months after vaccination.
Deaths: In study 1, with a median follow-up time of 12.2 months postvaccination, adverse events leading to death were reported for 110 participants (0.9%) who received a single dose of AREXVY (N = 12,469) and for 114 participants (0.9%) who received placebo (N = 12,503). Based on available information, there is no evidence of causal relationship to AREXVY. Causes of death among participants were consistent with those generally reported in adult and elderly populations.
Potential Immune‑Mediated Diseases: In study 1, participants were monitored for all potential immune‑mediated diseases (pIMDs) that occurred during the 6‑month period following administration of AREXVY (N = 12,467) or placebo (N = 12,499).
New onset pIMDs or exacerbation of existing pIMDs within 6 months following vaccination were reported for 0.3% of participants who received AREXVY and 0.3% of participants who received placebo. There were no notable imbalances between study groups in individual pIMDs reported.
Serious Adverse Events Reported from Other Studies
Study 2: Guillain-Barré syndrome beginning 9 days after AREXVY vaccination was reported in a participant enrolled in a study site in Japan.
Study 3: Acute disseminated encephalomyelitis (ADEM) was reported in 2 participants enrolled in a study site in South Africa; the onset of the symptoms was 7 and 22 days postvaccination, respectively. One event was fatal and the other non-fatal. These participants received AREXVY concomitantly with FLUARIX QUADRIVALENT. For both events, magnetic resonance imaging and cerebrospinal fluid analyses were not performed.
Individuals 50 through 59 Years of Age
Study 4 was a Phase 3, observer‑blind, randomized, placebo‑controlled study conducted in Argentina, Canada, Germany, Japan, the Netherlands, Poland, Spain, and the U.S., in participants 50 through 59 years of age (N = 769 AREXVY; N = 383 saline placebo), including a subset of participants with stable chronic medical conditions associated with an increased risk for LRTD caused by RSV defined as chronic pulmonary disease, chronic cardiovascular disease, diabetes, chronic kidney or liver disease (N = 386 AREXVY; N = 191 saline placebo). The study also enrolled participants 60 years of age and older (N = 381 AREXVY) [see Clinical Studies (14.2)].
At the time of vaccination in study 4, the median age was 57 years for the entire study population and 55 years for the age group 50 through 59 years. The racial/ethnic and sex distribution of study participants were similar in the two age groups. The majority of participants were White (83.8%), followed by Asian (11.2%), Black (3.3%), and other racial/ethnic groups (1.7%); 14.3% were of Hispanic or Latino ethnicity; 52.1% were female.
In study 4, all participants were monitored for solicited adverse reactions during the 4 days following vaccination (i.e., day of vaccination and the next 3 days) and for unsolicited adverse events, during the 30-day period following vaccination (day of vaccination and the next 29 days), using standardized paper diary cards. Participants were monitored for all SAEs and for all pIMDs (serious and non-serious cases) that occurred during the 6-month period following vaccination. Among participants, 99.2% have completed at least 6 months of follow-up following vaccination.
Solicited Adverse Reactions: The reported frequencies of specific solicited, local (administration site), and systemic adverse reactions among participants 50 through 59 years of age are presented in Table 3.
Table 3. Percentage of Participants with Solicited Local Adverse Reactions and Systemic Adverse Reactions within 4 Days of Vaccination from Study 4 (Exposed Set) N = Exposed set included all participants with at least 1 documented dose and with completed diary card. a Placebo was a saline solution. b Any grade pain: Defined as any pain neither interfering with nor preventing normal everyday activities (Grade 1), painful when limb is moved and interferes with everyday activities (Grade 2), or significant pain at rest and prevents normal everyday activities (Grade 3). c Any grade fatigue, myalgia, headache, arthralgia: Defined as event easily tolerated (Grade 1), interfering with normal activity (Grade 2), or preventing normal activity (Grade 3). d Temperature taken by any route (oral or axillary). AREXVY
50 through 59 Years of Age
%
Placeboa
50 through 59 Years of Age
%
Local Adverse Reactions
N = 756
N = 379
Pain, Anyb
75.8
12.1
Pain, Grade 3b
3.4
0.3
Erythema, >20 mm
13.2
0.5
Erythema, >100 mm
0.5
0
Swelling, >20 mm
10.4
0.8
Swelling, >100 mm
0.1
0
Systemic Adverse Reactions
N = 756
N = 380
Fatigue, Anyc
39.8
18.2
Fatigue, Grade 3c
2.8
0.8
Myalgia, Anyc
35.6
9.7
Myalgia, Grade 3c
2.5
0.5
Headache, Anyc
31.7
16.8
Headache, Grade 3c
2.6
1.1
Arthralgia, Anyc
23.4
7.9
Arthralgia, Grade 3c
1.7
0.8
Fever, ≥38.0°C/100.4°Fd
3.2
1.1
Fever, >39.0°C/102.2°Fd
0.1
0.5
The rates of solicited local and systemic adverse reactions (Table 3) were similar in participants 50 through 59 years of age either with or without pre-defined, stable, chronic medical conditions associated with an increased risk for LRTD caused by RSV.
Overall, the median duration of solicited local adverse reactions and solicited systemic adverse reactions after AREXVY vaccination was 2-3 days and 1-2 days, respectively.
Unsolicited Adverse Events: Unsolicited adverse events within 30 days after vaccination were reported in 13.8% of participants, 50 through 59 years of age, who received AREXVY (N = 769) compared to 12.0% of participants who received placebo (N = 383). Within 30 days after vaccination, there were no cases of atrial fibrillation reported in participants 50 through 59 years of age.
Serious Adverse Events: In study 4, participants were monitored for all SAEs that occurred during the 6-month period following administration of AREXVY (N = 769) or placebo (N = 383). Among participants 50 through 59 years of age, SAEs with onset within 6 months postvaccination were reported in 2.3% of those who received AREXVY and 2.1% of those who received placebo.
Deaths: Among participants 50 through 59 years of age, adverse events leading to death within 12 months after vaccination were reported for 4 (0.5%) participants who received AREXVY (N = 769) and 1 (0.3%) participant who received placebo (N = 383). None of these deaths were considered causally related to AREXVY.
Potential Immune-Mediated Diseases: In study 4, participants were monitored for all pIMDs that occurred during the 6-month period following administration of AREXVY (N = 769) or placebo (N = 383). Among participants 50 through 59 years of age, new onset pIMDs or exacerbation of existing pIMDs with onset within 6 months postvaccination were reported in 0.5% of those who received AREXVY and 0.3% of those who received placebo. There were no notable imbalances between study groups in individual pIMDs reported.
Individuals 18 through 49 Years of Age
Study 7 was an open‑label Phase 3 study conducted in Australia, Canada, Germany, Japan, South Africa and the U.S., in participants 18 through 49 years of age with pre-specified, stable, chronic medical conditions leading to an increased risk for LRTD caused by RSV defined as chronic pulmonary disease, chronic cardiovascular disease, diabetes, chronic kidney or liver disease, or neurological or neuromuscular disease (N = 1,029). The study also enrolled participants 60 years of age and older (N = 429). All study participants received 1 dose of AREXVY [see Clinical Studies (14.2)].
At the time of vaccination in study 7, the median age was 40 years for the age group 18 through 49 years and 45 years for the entire study population. The racial/ethnic and sex distribution of study participants were similar in the two age groups. The majority of participants were White (57.0%), followed by Black (21.4%), Asian (11.9%), and other racial/ethnic groups (9.0%); 10.4% were of Hispanic or Latino ethnicity; 55.8% were female.
In study 7, all participants were monitored for solicited adverse reactions during the 4 days following vaccination (i.e., day of vaccination and the next 3 days), using standardized electronic diary (e‑diary) or as reported to an investigator. The unsolicited adverse events were collected during the 30‑day period following vaccination (i.e., day of vaccination and the next 29 days). Participants were monitored for all SAEs and for all pIMDs (serious and non‑serious cases) that occurred during the 6‑month period following vaccination. Among participants, 97.5% completed the study at month 6.
Solicited Adverse Reactions: The reported frequencies of specific solicited, local (administration site), and systemic adverse reactions among participants 18 through 49 years of age are presented in Table 4.
Table 4. Percentage of Participants with Solicited Local Adverse Reactions and Systemic Adverse Reactions within 4 Days of Vaccination from Study 7 (Exposed Set) N = Exposed set included all participants with at least 1 documented dose and with available data. a Any grade pain: Defined as any pain neither interfering with nor preventing normal everyday activities (Grade 1), painful when limb is moved and interferes with everyday activities (Grade 2), or significant pain at rest and prevents normal everyday activities (Grade 3). b Any grade fatigue, myalgia, headache, arthralgia: Defined as event easily tolerated (Grade 1), interfering with normal activity (Grade 2), or preventing normal activity (Grade 3). c Temperature taken by any route (oral or axillary). AREXVY
18 through 49 Years of Age
%
Local Adverse Reactions
N = 1026
Pain, Anya
76.0
Pain, Grade 3a
1.3
Erythema, >20 mm
8.6
Erythema, >100 mm
0.2
Swelling, >20 mm
7.0
Swelling, >100 mm
0.2
Systemic Adverse Reactions
N = 1027
Fatigue, Anyb
59.6
Fatigue, Grade 3b
3.0
Myalgia, Anyb
59.9
Myalgia, Grade 3b
2.1
Headache, Anyb
43.6
Headache, Grade 3b
2.1
Arthralgia, Anyb
28.3
Arthralgia, Grade 3b
1.2
Fever, ≥38.0°C/100.4°Fc
5.0
Fever, >39.0°C/102.2°Fc
1.1
Overall, the median duration of solicited local adverse reactions and solicited systemic adverse reactions after AREXVY vaccination was 2‑3 days and 1‑2 days, respectively.
Unsolicited Adverse Events: Unsolicited adverse events within 30 days after vaccination were reported in 18.5% of participants, 18 through 49 years of age, who received AREXVY (N = 1029). Within 30 days after vaccination, there was 1 case of non‑serious atrial fibrillation reported in a 45-year old participant with an onset of 6 days postvaccination. The participant had obesity (BMI ≥ 30kg/m2), had a medical history of paroxysmal atrial fibrillation, paroxysmal supraventricular tachycardia, hypercholesterolemia, insulin resistance and obstructive sleep apnea. The event did not result in changes to medication regimen taken for atrial fibrillation prior to study vaccination, was self-limited and resolved spontaneously 4 days after onset. The currently available information on atrial fibrillation is insufficient to determine a causal relationship to the vaccine.
Serious Adverse Events: In study 7, participants were monitored for all SAEs that occurred during the 6‑month period following the administration of AREXVY. Among participants 18 through 49 years of age (N = 1029), no SAEs related to vaccination with AREXVY with onset within 6 months postvaccination were reported.
Potential Immune-Mediated Diseases: In study 7, participants were monitored for all pIMDs that occurred during the 6‑month period following administration of AREXVY. Among participants 18 through 49 years of age (N = 1029), new onset pIMDs or exacerbation of existing pIMDs with onset within 6 months postvaccination were reported in 0.2% of those who received AREXVY. One non-serious case of hematoma that initially developed at an insulin injection site prior to vaccination and subsequently exacerbated as multiple hematomas at other body sites was reported in a 31-year old participant with an onset of 14 days postvaccination. The event resolved spontaneously 217 days after onset.
6.2 Postmarketing Experience
The following adverse reactions have been identified from spontaneous reports during postmarketing use of AREXVY. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to the vaccine.
Nervous System Disorders
Guillain-Barré Syndrome
Postmarketing Observational Study of the Risk of Guillain-Barré Syndrome following Vaccination with AREXVY
The association between vaccination with AREXVY and Guillain-Barré syndrome (GBS) was evaluated among Medicare beneficiaries 65 years of age and older. Using Medicare claims data, between May 2023 through July 2024, vaccinations with AREXVY were identified through Current Procedural Terminology (CPT)/Healthcare Common Procedure Coding System (HCPCS) codes and National Drug Codes, and potential cases of hospitalized GBS among recipients of AREXVY were identified through International Classification of Diseases (ICD) codes. GBS diagnoses in claims data were confirmed by medical record review when available.
The risk of GBS following vaccination with AREXVY was assessed in self-controlled case series analyses using a risk window of 1 to 42 days postvaccination and a control window of 43 to 90 days postvaccination. The analyses of all GBS cases based on claims data suggest an increased risk of GBS during the 42 days following vaccination with AREXVY, with an incidence rate ratio (GBS cases in the risk window/control window) of 2.46 (95% CI 1.19, 5.08) and an estimated 7 excess cases of GBS per million doses administered to individuals 65 years of age and older. The background risk of GBS in a study population influences the excess GBS case estimate and may differ between studies, precluding direct comparison to excess GBS case estimates from other vaccine studies or populations.
The analyses of GBS diagnoses in claims data were supported by analyses of GBS cases confirmed by medical record review and by analyses of GBS cases in individuals who received AREXVY alone, without other concomitantly administered vaccines. While the results of this observational study suggest an increased risk of GBS with AREXVY, available evidence is insufficient to establish a causal relationship.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
All pregnancies have a risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
There are no data from clinical trials on the use of AREXVY in pregnant individuals. In a clinical study that enrolled pregnant individuals who received an investigational unadjuvanted RSV vaccine that contained the same RSVPreF3 antigen as AREXVY, an increase in preterm births was observed compared with pregnant individuals who received placebo (sucrose reconstituted with saline). This observation raises uncertainty about the use of AREXVY in pregnant individuals [see Indications and Usage (1)].
A developmental and reproductive toxicology study was performed in female rabbits administered AREXVY prior to mating and during gestation (0.5 mL at each occasion). This study revealed no adverse effects on female fertility, embryo‑fetal, pre‑weaning and post‑natal development (see Data).
Available data are insufficient to determine a causal relationship between preterm birth and AREXVY.
Human Data: There are no data from clinical trials on the use of AREXVY in pregnant individuals. In a randomized controlled clinical trial that enrolled pregnant individuals in a 2:1 ratio, 3,557 received an investigational unadjuvanted RSV vaccine that contained the same RSVPreF3 antigen as AREXVY and 1,771 received placebo (sucrose reconstituted with saline) at 24 to 34 weeks gestation. Numerical imbalances between the vaccine and placebo groups were observed for preterm births (6.8% and 4.9%, respectively) and neonatal deaths (0.4% and 0.2%, respectively). The observed numerical imbalance in neonatal deaths was not considered as an independent safety signal but as a consequence of the identified risk of preterm birth (0.2% and 0 in the vaccine and placebo groups in preterm infants, respectively).
Animal Data: In a developmental toxicity study, female rabbits were administered AREXVY by intramuscular injection 28 and 14 days prior to mating, on gestation Days 3, 9, 16, and 24, and on lactation Day 7. The total dose was 0.5 mL on each occasion (a single human dose of AREXVY is 0.5 mL). No adverse effects on female fertility or pre‑weaning development up to post‑natal Day 35 were observed.
8.2 Lactation
Risk Summary
It is not known whether AREXVY is excreted in human milk. No human or animal data are available to assess the effects of AREXVY on the breastfed infant or on milk production/excretion.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for AREXVY and any potential adverse effects on the breastfed child from AREXVY or from the underlying maternal condition. For preventive vaccines, the underlying maternal condition is susceptibility to disease prevented by the vaccine.
-
11 DESCRIPTION
AREXVY (Respiratory Syncytial Virus Vaccine, Adjuvanted) is a sterile injectable suspension for intramuscular use. The vaccine is supplied as a vial of lyophilized recombinant respiratory syncytial virus glycoprotein F stabilized in pre‑fusion conformation (RSVPreF3) as the antigen component, which must be reconstituted at the time of use with the accompanying vial or prefilled syringe of AS01E adjuvant as the Adjuvant Suspension Component.
The RSVPreF3 antigen is expressed by culturing genetically engineered Chinese Hamster Ovary cells in media containing no antibiotics or animal‑derived proteins. The RSVPreF3 protein is purified by several chromatographic and filtration steps, formulated with excipients, filled into vials, and lyophilized.
The AS01E adjuvant is composed of 3‑O‑desacyl‑4’‑monophosphoryl lipid A (MPL) from Salmonella minnesota and QS‑21, a saponin purified from plant extract Quillaja saponaria Molina, combined in a liposomal formulation. The liposomes are composed of dioleoyl phosphatidylcholine (DOPC) and cholesterol in a phosphate‑buffered saline solution containing disodium phosphate anhydrous, potassium dihydrogen phosphate, sodium chloride, and water for injection.
After reconstitution of the vial and vial presentation, each 0.5 mL dose is formulated to contain 120 mcg of the recombinant RSVPreF3 antigen, 25 mcg of MPL, and 25 mcg of QS‑21. Each dose also contains 14.7 mg of Trehalose, 4.4 mg of sodium chloride, 0.83 mg of potassium dihydrogen phosphate, 0.26 mg of dipotassium phosphate, 0.18 mg of polysorbate 80, 0.15 mg of disodium phosphate anhydrous, 0.5 mg of DOPC, and 0.125 mg of cholesterol.
After reconstitution of the vial and prefilled syringe presentation, each dose (approximately 0.5 mL) is formulated to contain 120 mcg of the recombinant RSVPreF3 antigen, 25 mcg of MPL, and 25 mcg of QS‑21. Each dose also contains 16.3 mg of Trehalose, 4.4 mg of sodium chloride, 0.87 mg of potassium dihydrogen phosphate, 0.29 mg of dipotassium phosphate, 0.20 mg of polysorbate 80, 0.15 mg of disodium phosphate anhydrous, 0.5 mg of DOPC, and 0.125 mg of cholesterol.
AREXVY contains no preservative. Each dose may also contain residual amounts of host cell proteins (≤2.0%) and DNA (≤0.80 ng/mg) from the manufacturing process.
The stopper of the vial containing Lyophilized Antigen Component or Adjuvant Suspension Component and the tip cap and plunger stopper of the prefilled syringe containing Adjuvant Suspension Component are not made with natural rubber latex.
- 12 CLINICAL PHARMACOLOGY
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
14.1 Efficacy in Individuals 60 Years of Age and Older
Efficacy of AREXVY against RSV‑associated LRTD in individuals 60 years of age and older was evaluated for up to 3 RSV seasons in study 1, a Phase 3, randomized, placebo‑controlled, observer‑blind clinical study conducted in 17 countries from Northern and Southern Hemispheres.
The study excluded participants who were immunocompromised. Participants with pre‑existing, chronic, stable disease such as diabetes, hypertension, or cardiac disease were allowed to participate in the study if considered by the investigator as medically stable at the time of vaccination.
The primary population for efficacy analysis (referred to as the modified exposed set), included individuals 60 years of age and older receiving 1 dose of AREXVY or placebo and who did not report an RSV‑confirmed acute respiratory illness [ARI] prior to Day 15 after vaccination.
The primary efficacy analysis over the first RSV season included 24,960 participants randomized equally to receive 1 dose of AREXVY (N = 12,466) or placebo (N = 12,494).
The efficacy analysis up to the end of the second RSV season included participants who received a single dose of AREXVY before Season 1 (N = 12,469); due to valid informed consent obtained from some participants initially excluded from the primary efficacy analysis, there is a higher number of participants in the end of Season 2 analysis. Pre‑Season 2, participants who received AREXVY were re‑randomized to receive placebo (N = 4,991) or a second dose of AREXVY (N = 4,966). Participants who received placebo before Season 1 (N = 12,498) received a second dose of placebo before Season 2 (N = 10,031). The efficacy analysis up to the end of the third RSV season included participants who received a single dose of AREXVY before Season 1 (N = 12,468); duplicate data from one participant was excluded from end of Season 3 analysis.
The participants were followed up to the end of the third RSV season (median follow‑up time 30.6 months).
Among study 1 participants in the modified exposed set for the analysis of efficacy over the first RSV season, 51.7% were female; 79.4% were White, 8.7% were Black, 7.6% were Asian, and 4.3% were of other racial/ethnic groups including American Indian or Alaska Native, Native Hawaiian or Other Pacific Islander; 5.5% were of Hispanic or Latino ethnicity. The median age of participants was 69.0 years.
At baseline, 39.3% of participants had at least one comorbidity of interest; 19.7% of participants had an underlying cardiorespiratory condition (chronic obstructive pulmonary disease, asthma, any chronic respiratory/pulmonary disease, or chronic heart failure) and 25.8% of participants had endocrine and metabolic conditions (diabetes, advanced liver or renal disease). Among participants in the modified exposed set for the analysis of efficacy over 2 and over 3 RSV seasons, demographic and baseline characteristics were similar to those in the modified exposed set for the analysis of efficacy over the first RSV season.
In study 1, confirmed RSV cases were determined by quantitative Reverse Transcription Polymerase Chain Reaction (qRT‑PCR) on a nasopharyngeal swab during all ARI episodes. ARI was defined by the presence of at least 2 respiratory symptoms/signs for at least 24 hours (nasal congestion, sore throat, lower respiratory symptoms/signs, as described below), or at least 1 respiratory symptom/sign plus 1 systemic symptom/sign (fever or feverishness, fatigue, body aches, headache, decreased appetite) for at least 24 hours. LRTD was defined based on the following criteria: the participant must have experienced at least 2 lower respiratory symptoms/signs, including at least 1 lower respiratory sign for at least 24 hours, or experienced at least 3 lower respiratory symptoms for at least 24 hours. Lower respiratory symptoms included: new or increased sputum, new or increased cough, new or increased dyspnea (shortness of breath). Lower respiratory signs included: new or increased wheezing, crackles/rhonchi, respiratory rate ≥20 respirations/min, low or decreased oxygen saturation (O2 saturation <95% or ≤90% if baseline is <95%), need for oxygen supplementation.
In study 1, a severe RSV-associated LRTD was defined as an RT-PCR confirmed RSV-associated LRTD with at least 2 lower respiratory signs, or preventing normal, everyday activities or requiring supportive therapy.
Efficacy against Respiratory Syncytial Virus-Associated Lower Respiratory Tract Disease over the First Respiratory Syncytial Virus Season
The primary objective was to demonstrate the efficacy of AREXVY in the prevention of a first episode of confirmed RSV-A and/or B associated LRTD during the first season.
Compared with placebo, AREXVY significantly reduced the risk of developing RSV-associated LRTD by 82.6% (96.95% CI [57.9, 94.1]) in participants 60 years of age and older, which met the pre-specified success criterion for the primary study objective (Table 5). The median duration of efficacy follow-up was 6.7 months.
Vaccine efficacy analyses by age subgroup and for participants with at least one comorbidity of interest are presented in Table 5.
The vaccine efficacy against RSV‑A–associated LRTD and RSV‑B–associated LRTD was 84.6% (95% CI [32.1, 98.3]) and 80.9% (95% CI [49.4, 94.3]), respectively.
Table 5. Efficacy Analyses over the First Respiratory Syncytial Virus Season: First Respiratory Syncytial Virus-Associated Lower Respiratory Tract Disease Overall, by Age and Comorbidity Subgroups in Study 1a (Modified Exposed Set) Two-sided exact CI for vaccine efficacy is derived based on Poisson model adjusted by age categories and regions.
N = Number of participants included in each group.
n = Number of participants having first occurrence of RSV-confirmed LRTD occurring from Day 15 postvaccination.
a Study 1: NCT04886596.
b CI = Confidence Interval (96.95% for the overall ≥60 years and 95% for all subgroup analyses).c Vaccine Efficacy (VE) for ≥80 years cannot be reliably estimated due to the low number of cases accrued in this age group. Subgroup
AREXVY
Placebo
% Efficacy
(CI)b
N
n
Incidence Rate per 1,000 Person-Years
N
n
Incidence Rate per 1,000 Person-Years
Overall
(≥60 years)12,466
7
1.0
12,494
40
5.8
82.6 (57.9, 94.1)
60 to 69 years
6,963
4
1.0
6,979
21
5.5
81.0 (43.6, 95.3)
70 to 79 years
4,487
1
0.4
4,487
16
6.5
93.8 (60.2, 99.9)
≥80 years
1,016
2
3.6
1,028
3
5.4
33.8 (-477.7, 94.5)c
Participants with at least 1 comorbidity of interest
4,937
1
0.4
4,861
18
6.6
94.6 (65.9, 99.9)
Compared with placebo, AREXVY significantly reduced the risk of developing RSV-associated LRTD by 84.4% (95% CI [46.9, 97.0]) in participants 70 years of age and older.
Compared with placebo, AREXVY significantly reduced the risk of developing severe RSV-associated LRTD by 94.1% (95% CI [62.4, 99.9]) in participants 60 years of age and older. One case of severe RSV-associated LRTD in the group that received AREXVY and 17 cases in the group that received placebo were reported, amongst which 2 cases required supportive therapy i.e., oxygen supplementation.
Efficacy against Respiratory Syncytial Virus‑Associated Lower Respiratory Tract Disease over 2 and over 3 Respiratory Syncytial Virus Seasons
Participants 60 years of age and older who received 1 dose of AREXVY or placebo were followed over 3 RSV seasons (up to the end of the second and third seasons in the Northern Hemisphere), with a median follow‑up time of 17.8 months over 2 RSV seasons and 30.6 months over 3 RSV seasons. The vaccine efficacy against RSV‑associated LRTD over 2 RSV seasons was 67.2% (97.5% CI [48.2, 80.0]) and over 3 RSV seasons was 62.9% (97.5% CI [46.7, 74.8]).
The vaccine efficacy against RSV-A‑associated LRTD and RSV-B‑associated LRTD over 3 RSV seasons was 69.8% (97.5% CI [42.2, 85.7]) and 58.6% (97.5% CI [35.9, 74.1]), respectively.
Vaccine efficacy analyses by age subgroup and for participants with at least one comorbidity of interest are presented in Table 6.
Table 6. Efficacy Analyses over 2 and over 3 Respiratory Syncytial Virus Seasons: First Respiratory Syncytial Virus-Associated Lower Respiratory Tract Disease Overall, by Age and Comorbidity Subgroups in Study 1a (Modified Exposed Set) Two‑sided exact CI for vaccine efficacy is derived based on Poisson model adjusted by age categories, regions and season. N = Number of participants included in each group. n = Number of participants having first occurrence of RSV‑confirmed LRTD occurring from Day 15 postvaccination. a Study 1: NCT04886596. b Participants who received a second dose of AREXVY did not contribute to these efficacy analyses after receipt of Dose 2. c VE(%) Poisson method – adjusted by age, region, and season for overall (≥60 years) and participants with at least 1 comorbidity of interest; and adjusted by region and season for analysis by age category. d CI = Confidence Interval (97.5% for the overall ≥60 years and 95% for all subgroup analyses). e VE for ≥80 years cannot be reliably estimated due to the low number of cases accrued in this age group. Subgroup
AREXVYb
Placebo
% Efficacyc
(CI)d
N
n
Incidence Rate per 1,000 Person-Years
N
n
Incidence Rate per 1,000 Person-Years
Over 2 RSV Seasons
Overall
(≥60 years)12,469
30
2.0
12,498
139
8.0
67.2 (48.2, 80.0)
60 to 69 years
6,963
17
2.1
6,981
74
7.7
65.4 (40.4, 80.9)
70 to 79 years
4,489
9
1.7
4,489
55
8.8
74.9 (48.4, 89.2)
≥80 years
1,017
4
3.5
1,028
10
7.2
38.4 (-118.2, 86.1)e
Participants with at least 1 comorbidity of interest
4,983
16
2.7
4,919
72
10.6
66.7 (41.8, 82.0)
Over 3 RSV Seasons
Overall
(≥60 years)12,468
48
2.4
12,498
215
7.9
62.9 (46.7, 74.8)
60 to 69 years
6,962
28
2.5
6,981
117
7.6
60.3 (39.5, 74.8)
70 to 79 years
4,489
15
2.1
4,489
85
8.6
70.6 (48.4, 84.3)
≥80 years
1,017
5
3.3
1,028
13
6.0
36.2 (-94.0, 82.5)e
Participants with at least 1 comorbidity of interest
5,014
25
3.2
4,951
116
10.8
64.7 (45.1, 78.1)
In participants 70 years of age and older, over 2 RSV seasons and over 3 RSV seasons, the vaccine efficacy against RSV‑associated LRTD was 69.3% (95% CI [43.4, 84.6]) and 66.0% (95% CI [44.3, 80.2]), respectively.
The vaccine efficacy against severe RSV‑associated LRTD over 2 RSV seasons was 78.8% (95% CI [52.6, 92.0]) in participants 60 years of age and older (7 cases in participants who received 1 dose of AREXVY and 48 cases in participants who received placebo, amongst which supportive therapy (i.e., oxygen supplementation and positive airway pressure) was required for 1 case in participants who received 1 dose of AREXVY and 5 cases in participants who received placebo). The vaccine efficacy against severe RSV‑associated LRTD over 3 RSV seasons was 67.4% (95% CI [42.4, 82.7]) in participants 60 years of age and older (15 cases in participants who received 1 dose of AREXVY and 75 cases in participants who received placebo, amongst which supportive therapy (i.e., oxygen supplementation and positive airway pressure) was required for 2 cases in participants who received 1 dose of AREXVY and 5 cases in participants who received placebo).
Efficacy against Respiratory Syncytial Virus‑Associated Lower Respiratory Tract Disease over the Second Respiratory Syncytial Virus Season and over the Third Respiratory Syncytial Virus Season
The vaccine efficacy against RSV‑associated LRTD over the second RSV season with median follow‑up of 6.3 months was 56.1% (95% CI [28.2, 74.4]) in participants 60 years of age and older (20 cases among participants who received 1 dose of AREXVY and 91 cases among participants who received placebo).
The vaccine efficacy against RSV‑associated LRTD over the third RSV season with median follow‑up of 7 months was 48.0% (95% CI [8.7, 72.0]) in participants 60 years of age and older (16 cases among participants who received 1 dose of AREXVY and 61 cases among participants who received placebo).
14.2 Immunogenicity in Individuals 18 through 59 Years of Age at Increased Risk for LRTD caused by RSV
In study 4, individuals 50 through 59 years of age with an increased risk of LRTD caused by RSV due to certain chronic medical conditions (chronic pulmonary disease, chronic cardiovascular disease, diabetes, chronic kidney or liver disease) were randomized to receive AREXVY (N = 386) or saline placebo (N = 191). A comparator group of individuals 60 years and older also received AREXVY (N = 381) [see Adverse Reactions (6.1)].
In study 7, individuals 18 through 49 years of age with an increased risk of LRTD caused by RSV due to certain chronic medical conditions (participants may have reported more than 1 condition) were enrolled to receive AREXVY (N = 1,029; 426 of whom were part of the immunogenicity subset). These conditions included cardiopulmonary disorders (55.0%), diabetes mellitus (48.9%), chronic kidney disease (2.9%), chronic liver disease (6.2%), and neurologic or neuromuscular conditions (20.5%). A comparator group of individuals 60 years and older also received AREXVY (N = 429). All study participants received 1 dose of AREXVY [see Adverse Reactions (6.1)].
In both study 4 and study 7, effectiveness of AREXVY in individuals 50 through 59 years of age and individuals 18 through 49 years of age with chronic medical conditions was assessed by a comparison of RSV neutralizing antibody responses induced by AREXVY in these age groups to antibody responses of individuals 60 years of age and older. The neutralizing antibody responses to RSV‑A and RSV‑B subtypes in individuals 50 through 59 years of age and 18 through 49 years of age with chronic medical conditions met the criteria for immunobridging. The noninferiority criteria, as pre‑specified in the protocol, were met as the upper limit (UL) of the 2‑sided 95% CIs for the Geometric Mean Titer (GMT) ratios (GMT for individuals 60 years and older/GMT for individuals 50 through 59 years of age, and GMT for individuals 60 years and older/GMT for individuals 18 through 49 years of age) were ≤1.50 and the UL of the 2‑sided 95% CIs for seroresponse rate (SRR) differences (SRR for individuals 60 years and older minus SRR for individuals 50 through 59 years of age, and SRR for individuals 60 years and older minus SRR for individuals 18 through 49 years of age) were ≤10% for the RSV‑A and RSV‑B subtypes. For clarity, Table 7 presents the GMT ratio as GMT for individuals 50 through 59 years of age/GMT for individuals 60 years and older or GMT for individuals 18 through 49 years of age/GMT for individuals 60 years and older, for which the corresponding noninferiority criterion is a 2‑sided 95% CI lower limit (LL) ≥0.67. Similarly, Table 7 presents the SRR difference as SRR for individuals 50 through 59 years of age minus SRR for individuals 60 years and older or SRR for individuals 18 through 49 years of age minus SRR for individuals 60 years and older, for which the corresponding noninferiority criterion is a 2‑sided 95% CI LL ≥-10%.
Table 7. Adjusted Geometric Mean Titer (GMT) Values, Adjusted GMT Ratios, Seroresponse Rate (SRR) Values and SRR Difference for RSV-A and RSV-B Neutralizing Titers in Individuals 18 through 49 Years of Age (Study 7) and 50 through 59 Years of Age (Study 4) Compared to Individuals 60 Years of Age and Older (in the respective study) – Per Protocol Set ED60: Estimated Dilution 60; CI = Confidence Interval; GMT = Geometric Mean Titer; SRR = Seroresponse Rate (The SRR was defined as the percentage of participants having a fold increase in neutralization titers ≥4 at 1 month post‑study intervention administration over pre‑study intervention administration). N = Number of participants with both pre- and postvaccination results available; study-specific comparator group for 60 years of age and older. a Stable chronic medical conditions in 50 through 59 years of age were those associated with an increased risk for LRTD caused by RSV defined as chronic pulmonary disease, chronic cardiovascular disease, diabetes, chronic kidney or liver disease. b Stable chronic medical conditions in 18 through 49 years of age were those associated with an increased risk for LRTD caused by RSV defined as chronic pulmonary disease, chronic cardiovascular disease, diabetes, chronic kidney or liver disease, or neurological or neuromuscular disease. c Comparison is done using the group ratio of adjusted GMT (individuals 50 through 59 years of age with chronic medical conditions/individuals 60 years of age and older; and individuals 18 through 49 years of age with chronic medical conditions/individuals 60 years of age and older) (ANCOVA model applied to the logarithm 10 transformed titers). The ANCOVA model included the group as fixed effects and the pre‑dose logarithm 10 titer as covariate. d The GMT ratio was calculated by GMT for individuals 50 through 59 years of age with chronic medical conditions/GMT for individuals 60 years and older; and GMT for individuals 18 through 49 years of age with chronic medical conditions/GMT for individuals 60 years and older. e The SRR difference was calculated by SRR for individuals 50 through 59 years of age with chronic medical conditions minus SRR for individuals 60 years and older; and SRR for individuals 18 through 49 years of age with chronic medical conditions minus SRR for individuals 60 years and older. Individuals 50 through 59 Years of Age at Increased Risk for LRTD Caused by RSVa
RSV-A Neutralizing Titers (ED60)
Individuals 50 through 59 years of age with chronic medical conditions
(N = 343)
Individuals 60 years of age and older
(N = 342)
Adjusted GMTc
(95% CI)
8,922.7
(8,118.2, 9,806.9)
7,440.1
(6,768.4, 8,178.5)
Adjusted GMT Ratioc
(95% CI)d
1.20
(1.05, 1.37)
SRR %
(95% CI)
86.9
(82.8, 90.3)
80.4
(75.8, 84.5)
SRR Difference %
(95% CI)e
6.5
(0.9, 12.1)
RSV-B Neutralizing Titers (ED60)
Adjusted GMTc
(95% CI)
10,054.7
(9,225.4, 10,958.7)
8,062.8
(7,395.9, 8,789.9)
Adjusted GMT Ratioc
(95% CI)d
1.25
(1.10, 1.41)
SRR %
(95% CI)
81.6
(77.1, 85.6)
74.5
(69.5, 79.0)
SRR Difference %
(95% CI)e
7.2
(0.9, 13.3)
Individuals 18 through 49 Years of Age at Increased Risk for LRTD Caused by RSVb
RSV-A Neutralizing Titers (ED60)
Individuals 18 through 49 years of age with chronic medical conditions
(N = 394)
Individuals 60 years of age and older
(N = 417)
Adjusted GMTc
(95% CI)
11,914.6
(10,933.2, 12,984.2)
8,591.5
(7,902.7, 9,340.3)
Adjusted GMT Ratioc
(95% CI)d
1.39
(1.23, 1.56)
SRR %
(95% CI)
87.1
(83.3, 90.2)
77.7
(73.4, 81.6)
SRR Difference %
(95% CI)e
9.4
(4.1, 14.6)
RSV-B Neutralizing Titers (ED60)
Adjusted GMTc
(95% CI)
12,503.4
(11,490.5, 13,605.4)
9,087.6
(8,372.1, 9,864.2)
Adjusted GMT Ratioc
(95% CI)d
1.38
(1.22, 1.55)
SRR %
(95% CI)
87.3
(83.6, 90.4)
77.2
(72.9, 81.2)
SRR Difference %
(95% CI)e
10.1
(4.8, 15.3)
14.3 Concomitant Administration
Concomitant Administration with Influenza Vaccines
In 3 open-label clinical studies, participants were randomized 1:1 to receive 1 dose of AREXVY administered either concomitantly at Month 0 or sequentially 1 month apart (referred to as the control group) with FLUARIX QUADRIVALENT in study 3 (N = 885), FLUZONE HIGH DOSE QUADRIVALENT in study 5 (N = 1,029), or FLUAD QUADRIVALENT (N = 1,045) in study 6 [see Adverse Reactions (6)].
In studies 3 and 5, the prespecified criteria for noninferiority of the immune responses in the concomitant administration group versus the control group were met for the RSV‑A neutralizing titers (studies 3 and 5), the RSV‑B neutralizing titers (study 5), and for the haemagglutinin inhibition (HI) titers against influenza strains A/Hong Kong/H3N2, A/Victoria/H1N1, B/Phuket/Yamagata and B/Washington/Victoria (study 3) and influenza strains A/Victoria/H1N1, A/Darwin/H3N2, B/Austria/Victoria and B/Phuket/Yamagata (study 5). The prespecified criterion for noninferiority was an upper limit of the 2-sided 95% confidence interval on the GMT ratio (control group/concomitant group) ≤1.5, which corresponds to a lower limit of the 2-sided 95% confidence interval on the GMT ratio (concomitant group/control group) ≥0.67.
In study 6, the prespecified criteria for noninferiority of the immune responses in the concomitant administration group versus the control group were met for the RSV‑A neutralizing titers and for HI titers against 3 of 4 influenza vaccine strains (A/Victoria/H1N1, B/Austria/Victoria, and B/Phuket/Yamagata). The prespecified criterion for noninferiority was an upper limit of the 2-sided 95% confidence interval on the GMT ratio (control group/concomitant group) ≤1.5, which corresponds to a lower limit of the 2-sided 95% confidence interval on the GMT ratio (concomitant group/control group) ≥0.67. The noninferiority criterion was not met for HI titers against A/Darwin/H3N2 influenza strain, with the lower limit of the 95% CI of the GMT ratio of 0.65. In a post‑hoc descriptive analysis, the GMT ratio [concomitant group/control group] was 0.86 [2‑sided 95% CI (0.77, 0.97)] for RSV‑B neutralizing titers.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
AREXVY is supplied in two presentations, as follows:
Vial and Vial Presentation:
In this presentation, AREXVY is supplied as 2 components: A single‑dose vial of Lyophilized Antigen Component (powder) and a single‑dose vial of Adjuvant Suspension Component (liquid) (packaged without syringes or needles).
Table 8. Vial and Vial Presentation for AREXVY Presentation
Carton NDC Number
Components
Adjuvant Suspension Component (liquid)
Lyophilized Antigen Component (powder)
Outer carton of 10 doses
58160-848-11
10 vials
NDC: 58160-744-03
10 vials
NDC: 58160-723-03
Vial and Prefilled Syringe Presentation:
In this presentation, AREXVY is supplied as 2 components: A single-dose vial of Lyophilized Antigen Component (powder) and a prefilled, ungraduated TIP-LOK syringe (Luer Lock syringe) of Adjuvant Suspension Component (liquid) (packaged without needles). TIP-LOK syringes are to be used with Luer Lock compatible needles.
Table 9. Vial and Prefilled Syringe Presentation for AREXVY Presentation
Carton NDC Number
Components
Adjuvant Suspension Component (liquid)
Lyophilized Antigen Component (powder)
Outer carton of 1 dose
58160-882-12
1 prefilled syringe
NDC: 58160-749-07
1 vial
NDC: 58160-719-03
Outer carton of 10 doses
58160-894-15
10 prefilled syringes
NDC: 58160-749-07
10 vials
NDC: 58160-719-03
16.1 Storage before Reconstitution
Adjuvant Suspension Component vials and prefilled syringes: Store refrigerated between 2°C and 8°C (36°F and 46°F). Store in the original package in order to protect vials and prefilled syringes from light. Do not freeze. Discard if the Adjuvant Suspension Component has been frozen.
Lyophilized Antigen Component vials: Store refrigerated between 2°C and 8°C (36°F and 46°F). Store in the original package in order to protect vials from light. Do not freeze. Discard if the Lyophilized Antigen Component has been frozen.
16.2 Storage after Reconstitution
- Administer immediately or store in the refrigerator between 2°C and 8°C (36°F to 46°F) or at room temperature [up to 25°C (77°F)] for up to 4 hours prior to use.
- Protect reconstituted vaccine from light.
- Discard reconstituted vaccine if not used within 4 hours.
- Do not freeze. Discard if the vaccine has been frozen.
-
17 PATIENT COUNSELING INFORMATION
- Inform vaccine recipients that AREXVY is not for use in pregnant individuals.
- Inform vaccine recipients of the potential benefits and risks of vaccination with AREXVY.
- Inform vaccine recipients about the potential for adverse reactions that have been observed following administration of AREXVY.
- Provide the Vaccine Information Statements, which are available free of charge at the Centers for Disease Control and Prevention (CDC) website (www.cdc.gov/vaccines).
AREXVY, FLUARIX QUADRIVALENT, and TIP-LOK are trademarks owned by or licensed to the GSK group of companies. The other brands listed are trademarks owned by or licensed to their respective owners and are not owned by or licensed to the GSK group of companies. The makers of these brands are not affiliated with and do not endorse the GSK group of companies or its products.
Manufactured by GlaxoSmithKline Biologicals
Rixensart, Belgium, U.S. License 1617, and
Distributed by GlaxoSmithKline
Durham, NC 27701©2026 GSK group of companies or its licensor.
ARV:8PI
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
NDC: 58160-848-11
AREXVY
Respiratory Syncytial Virus Vaccine, Adjuvanted
For 60 Years of Age and Older
For 18 through 59 Years at Increased Risk for Lower Respiratory Tract Disease caused by RSV
Contents (10 doses of AREXVY):
10 Vials containing Adjuvant Suspension Component
10 Vials containing Lyophilized Antigen Component
After reconstitution, a single dose of AREXVY is 0.5 mL
GSK
Adjuvant Suspension Component Made in Belgium
Lyophilized Antigen Component Made in Belgium
©2026 GSK group of companies or its licensor.
Rev. 03/26
527405
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL (Antigen)
NDC: 58160-723-03
Lyophilized Antigen Component
Vial 2 of 2
NOT TO BE USED ALONE
Reconstitute with Adjuvant Suspension Component to form Respiratory Syncytial Virus Vaccine, Adjuvanted (AREXVY)
0.5-mL Single Dose After Reconstitution
Mfd. By GlaxoSmithKline Biologicals
Rev 04/23
513499
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL (Antigen)
NDC: 58160-744-03
Adjuvant Suspension Component
Vial 1 of 2
NOT TO BE USED ALONE
Add to 1 vial of Lyophilized Antigen Component to form Arexvy
Mft. By GlaxoSmithKline Biologicals
Rev 04/23
513531
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
NDC: 58160-882-12
AREXVY
Respiratory Syncytial Virus Vaccine, Adjuvanted
For 60 Years of Age and Older
For 18 through 59 Years at Increased Risk for Lower Respiratory Tract Disease caused by RSV
RSV
Rx only
NOTICE: Combine one vial of Lyophilized Antigen Component and one prefilled syringe of Adjuvant Suspension Component before use.
Contents (1 dose of AREXVY):
1 Vial containing Lyophilized Antigen Component
1 Syringe containing Adjuvant Suspension Component
After reconstitution, a single dose of AREXVY is approximately 0.5 mL
For intramuscular use
TIP-LOK syringe to be used with Luer Lock compatible needle
NEEDLE NOT INCLUDED
GSK
AREXVY
Adjuvant Suspension Component Made in Belgium
Lyophilized Antigen Component Made in Belgium
©2026 GSK group of companies or its licensor.
Rev. 4/26
MCK-166310-01
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL (Antigen)
NDC: 58160-719-03
Lyophilized Antigen Component
NOT TO BE USED ALONE
Reconstitute with Adjuvant Suspension Component to form Respiratory Syncytial Virus Vaccine, Adjuvanted (AREXVY)
Mfd. By GlaxoSmithKline Biologicals
Single dose 0.5-mL after reconstitution
Rx Only
Rev 10/25
MCK-166311-01
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL (Antigen)
NDC: 58160-749-07
Adjuvant Suspension Component
NOT TO BE USED ALONE
Add to 1 vial of Lyophilized Antigen Component to form Respiratory Syncytial Virus Vaccine, Adjuvanted (AREXVY)
Mft. By GlaxoSmithKline Biologicals
Rx Only
Rev 9/25
MCK-166312-01
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
NDC: 58160-894-15
AREXVY
Respiratory Syncytial Virus Vaccine, Adjuvanted
RSV
Rx only
NOTICE: Combine one vial of Lyophilized Antigen Component and one prefilled syringe of Adjuvant Suspension Component before use.
For 60 Years of Age and Older
For 18 through 59 Years at Increased Risk for Lower Respiratory Tract Disease caused by RSV
Contents (10 dose of AREXVY):
10 VialS containing Lyophilized Antigen Component
10 SyringeS containing Adjuvant Suspension Component
After reconstitution, a single dose of AREXVY is approximately 0.5 mL
For intramuscular use
TIP-LOK syringe to be used with Luer Lock compatible needle
NEEDLE NOT INCLUDED
GSK
AREXVY
Adjuvant Suspension Component Made in Belgium
Lyophilized Antigen Component Made in Belgium
©2026 GSK group of companies or its licensor.
Rev. 4/26
MCK-166243-01
-
INGREDIENTS AND APPEARANCE
AREXVY
respiratory syncytial virus vaccine recombinant, adjuvanted kitProduct Information Product Type VACCINE Item Code (Source) NDC: 58160-848 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 58160-848-11 1 in 1 CARTON; Type 0: Not a Combination Product Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 10 VIAL 5 mL Part 2 10 VIAL 5 mL Part 1 of 2 ANTIGEN
respiratory syncytial virus glycoprotein f injection, powder, lyophilized, for suspensionProduct Information Item Code (Source) NDC: 58160-723 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RECOMBINANT RESPIRATORY SYNCYTIAL VIRUS PRE-FUSION F PROTEIN ANTIGEN (UNII: M739EB7427) (RECOMBINANT RESPIRATORY SYNCYTIAL VIRUS PRE-FUSION F PROTEIN ANTIGEN - UNII:M739EB7427) RECOMBINANT RESPIRATORY SYNCYTIAL VIRUS PRE-FUSION F PROTEIN ANTIGEN 120 ug in 0.5 mL Inactive Ingredients Ingredient Name Strength TREHALOSE DIHYDRATE (UNII: 7YIN7J07X4) POLYSORBATE 80 (UNII: 6OZP39ZG8H) MONOBASIC POTASSIUM PHOSPHATE (UNII: 4J9FJ0HL51) DIBASIC POTASSIUM PHOSPHATE (UNII: CI71S98N1Z) Product Characteristics Color WHITE Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 58160-723-03 0.5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125775 05/03/2023 Part 2 of 2 ADJUVANT
as01 inhalantProduct Information Item Code (Source) NDC: 58160-744 Route of Administration INTRAMUSCULAR Inactive Ingredients Ingredient Name Strength MONOPHOSPHORYL LIPID A TRIETHYLAMINE (UNII: UNY5G9WB4K) QUILLAJA SAPONIN (QS-21) (UNII: 61H83WZX3U) 1,2-DIOLEOYL-SN-GLYCERO-3-PHOSPHOCHOLINE (UNII: H026DM5V6U) CHOLESTEROL (UNII: 97C5T2UQ7J) SODIUM CHLORIDE (UNII: 451W47IQ8X) SODIUM PHOSPHATE, DIBASIC, ANHYDROUS (UNII: 22ADO53M6F) MONOBASIC POTASSIUM PHOSPHATE (UNII: 4J9FJ0HL51) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 58160-744-03 0.5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125775 05/03/2023 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125775 05/03/2023 AREXVY
respiratory syncytial virus vaccine recombinant, adjuvanted kitProduct Information Product Type VACCINE Item Code (Source) NDC: 58160-894 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 58160-894-15 1 in 1 CARTON; Type 0: Not a Combination Product Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 10 VIAL 5 mL Part 2 10 SYRINGE 5 mL Part 1 of 2 ANTIGEN
respiratory syncytial virus glycoprotein f injection, powder, lyophilized, for suspensionProduct Information Item Code (Source) NDC: 58160-719 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RECOMBINANT RESPIRATORY SYNCYTIAL VIRUS PRE-FUSION F PROTEIN ANTIGEN (UNII: M739EB7427) (RECOMBINANT RESPIRATORY SYNCYTIAL VIRUS PRE-FUSION F PROTEIN ANTIGEN - UNII:M739EB7427) RECOMBINANT RESPIRATORY SYNCYTIAL VIRUS PRE-FUSION F PROTEIN ANTIGEN 120 ug in 0.5 mL Inactive Ingredients Ingredient Name Strength TREHALOSE DIHYDRATE (UNII: 7YIN7J07X4) POLYSORBATE 80 (UNII: 6OZP39ZG8H) MONOBASIC POTASSIUM PHOSPHATE (UNII: 4J9FJ0HL51) DIBASIC POTASSIUM PHOSPHATE (UNII: CI71S98N1Z) Product Characteristics Color WHITE Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 58160-719-03 0.5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125775 04/30/2026 Part 2 of 2 ADJUVANT
as01 inhalantProduct Information Item Code (Source) NDC: 58160-749 Route of Administration INTRAMUSCULAR Inactive Ingredients Ingredient Name Strength MONOPHOSPHORYL LIPID A TRIETHYLAMINE (UNII: UNY5G9WB4K) QUILLAJA SAPONIN (QS-21) (UNII: 61H83WZX3U) 1,2-DIOLEOYL-SN-GLYCERO-3-PHOSPHOCHOLINE (UNII: H026DM5V6U) CHOLESTEROL (UNII: 97C5T2UQ7J) SODIUM CHLORIDE (UNII: 451W47IQ8X) SODIUM PHOSPHATE, DIBASIC, ANHYDROUS (UNII: 22ADO53M6F) MONOBASIC POTASSIUM PHOSPHATE (UNII: 4J9FJ0HL51) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 58160-749-07 0.5 mL in 1 SYRINGE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125775 04/30/2026 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125775 04/30/2026 AREXVY
respiratory syncytial virus vaccine recombinant, adjuvanted kitProduct Information Product Type VACCINE Item Code (Source) NDC: 58160-882 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 58160-882-12 1 in 1 CARTON; Type 0: Not a Combination Product Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 10 VIAL 5 mL Part 2 10 SYRINGE 5 mL Part 1 of 2 ANTIGEN
respiratory syncytial virus glycoprotein f injection, powder, lyophilized, for suspensionProduct Information Item Code (Source) NDC: 58160-719 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RECOMBINANT RESPIRATORY SYNCYTIAL VIRUS PRE-FUSION F PROTEIN ANTIGEN (UNII: M739EB7427) (RECOMBINANT RESPIRATORY SYNCYTIAL VIRUS PRE-FUSION F PROTEIN ANTIGEN - UNII:M739EB7427) RECOMBINANT RESPIRATORY SYNCYTIAL VIRUS PRE-FUSION F PROTEIN ANTIGEN 120 ug in 0.5 mL Inactive Ingredients Ingredient Name Strength TREHALOSE DIHYDRATE (UNII: 7YIN7J07X4) POLYSORBATE 80 (UNII: 6OZP39ZG8H) MONOBASIC POTASSIUM PHOSPHATE (UNII: 4J9FJ0HL51) DIBASIC POTASSIUM PHOSPHATE (UNII: CI71S98N1Z) Product Characteristics Color WHITE Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 58160-719-03 0.5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125775 04/30/2026 Part 2 of 2 ADJUVANT
as01 inhalantProduct Information Item Code (Source) NDC: 58160-749 Route of Administration INTRAMUSCULAR Inactive Ingredients Ingredient Name Strength MONOPHOSPHORYL LIPID A TRIETHYLAMINE (UNII: UNY5G9WB4K) QUILLAJA SAPONIN (QS-21) (UNII: 61H83WZX3U) 1,2-DIOLEOYL-SN-GLYCERO-3-PHOSPHOCHOLINE (UNII: H026DM5V6U) CHOLESTEROL (UNII: 97C5T2UQ7J) SODIUM CHLORIDE (UNII: 451W47IQ8X) SODIUM PHOSPHATE, DIBASIC, ANHYDROUS (UNII: 22ADO53M6F) MONOBASIC POTASSIUM PHOSPHATE (UNII: 4J9FJ0HL51) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 58160-749-07 0.5 mL in 1 SYRINGE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125775 04/30/2026 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125775 04/30/2026 Labeler - GlaxoSmithKline Biologicals SA (372748392)











Trademark Results [Arexvy]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 AREXVY 97052261 not registered Live/Pending |
GlaxoSmithKline Biologicals SA 2021-09-29 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.