TYGACIL- tigecycline injection, powder, lyophilized, for solution
Tygacil by
Drug Labeling and Warnings
Tygacil by is a Prescription medication manufactured, distributed, or labeled by Wyeth Pharmaceuticals LLC, a subsidiary of Pfizer Inc., Patheon Italia S.p.A., Pfizer Ireland Pharmaceuticals, Pharmacia and Upjohn Company LLC, Wyeth Lederle SRL, Prime European Therapeuticals S.p.A dba Euticals S.p.A. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TYGACIL safely and effectively. See full prescribing information for TYGACIL.
TYGACIL® (tigecycline) for injection, for intravenous use
Initial U.S. Approval: 2005WARNING: ALL-CAUSE MORTALITY
See full prescribing information for complete boxed warning.
All-cause mortality was higher in patients treated with TYGACIL than comparators in a meta-analysis of clinical trials. The cause of this mortality risk difference of 0.6% (95% CI 0.1, 1.2) has not been established. TYGACIL should be reserved for use in situations when alternative treatments are not suitable (1.4, 5.1, 5.2, 6.1).
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
TYGACIL is a tetracycline class antibacterial indicated in patients 18 years of age and older for:
- Complicated skin and skin structure infections (1.1)
- Complicated intra-abdominal infections (1.2)
- Community-acquired bacterial pneumonia (1.3)
Limitations of Use: TYGACIL is not indicated for treatment of diabetic foot infection or hospital-acquired pneumonia, including ventilator-associated pneumonia (1.4).
To reduce the development of drug-resistant bacteria and maintain the effectiveness of TYGACIL and other antibacterial drugs, TYGACIL should be used only to treat infections that are proven or strongly suspected to be caused by bacteria.
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
For Injection: 50 mg, lyophilized powder for reconstitution in a single-dose 5 mL vial or 10 mL vial. (3)
CONTRAINDICATIONS
- Known hypersensitivity to tigecycline. (4)
WARNINGS AND PRECAUTIONS
- All-Cause Mortality: A meta-analysis of Phase 3 and 4 clinical trials demonstrated an increase in all-cause mortality in TYGACIL-treated patients compared to controls with a risk difference of 0.6% (95% CI 0.1, 1.2). The cause of this increase has not been established. An increase was also seen in a meta-analysis limited to the approved indications [0.6% (95% CI 0.0, 1.2)]. The greatest difference in mortality was seen in TYGACIL-treated patients with ventilator-associated pneumonia. (5.1, 5.2)
- Anaphylactic Reactions: have been reported with TYGACIL, and may be life-threatening. Avoid use in patients with known hypersensitivity to tetracyclines. (5.3)
- Hepatic Adverse Effects: have been reported with TYGACIL. Patients who develop abnormal liver function tests during TYGACIL therapy should be monitored for evidence of worsening hepatic function and evaluated for risk/benefit of continuing tigecycline therapy. (5.4)
- Pancreatitis, including fatalities, has been reported with TYGACIL. If pancreatitis is suspected, then consider stopping TYGACIL. (5.5)
- Tooth Discoloration and Enamel Hypoplasia: The use of TYGACIL during tooth development (last half of pregnancy, infancy, and childhood to the age of 8 years) may cause permanent discoloration of the teeth (yellow-gray-brown) and enamel hypoplasia. (5.6)
- Inhibition of Bone Growth: The use of TYGACIL during the second and third trimester of pregnancy, infancy, and childhood up to the age of 8 years may cause reversible inhibition of bone growth. (5.7)
- Clostridium difficile associated Diarrhea (CDAD): evaluate if diarrhea occurs. (5.8)
ADVERSE REACTIONS
The most common adverse reactions (incidence >5%) are nausea, vomiting, diarrhea, abdominal pain, headache, and increased SGPT. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc. at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Suitable anticoagulation test should be monitored if TYGACIL is administered to patients receiving warfarin. (7.1)
USE IN SPECIFIC POPULATIONS
- Lactation: Avoid breastfeeding for longer than 3 weeks while taking TYGACIL. A lactating woman may also pump and discard breast milk during treatment and for 9 days after the last dose of TYGACIL (8.2)
- Pediatrics: Use in patients under 18 years of age is not recommended. Pediatric trials were not conducted because of the higher risk of mortality seen in adult trials (8.4)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 1/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: ALL-CAUSE MORTALITY
1 INDICATIONS AND USAGE
1.1 Complicated Skin and Skin Structure Infections
1.2 Complicated Intra-abdominal Infections
1.3 Community-Acquired Bacterial Pneumonia
1.4 Limitations of Use
1.5 Usage
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Adult Dosage
2.2 Dosage in Patients With Hepatic Impairment
2.3 Dosage in Pediatric Patients
2.4 Preparation and Administration
2.5 Drug Compatibilities
2.6 Drug Incompatibilities
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 All-Cause Mortality
5.2 Mortality Imbalance and Lower Cure Rates in Hospital-Acquired Pneumonia
5.3 Anaphylactic Reactions
5.4 Hepatic Adverse Effects
5.5 Pancreatitis
5.6 Tooth Discoloration and Enamel Hypoplasia
5.7 Inhibition of Bone Growth
5.8 Clostridium difficile Associated Diarrhea
5.9 Sepsis/Septic Shock in Patients With Intestinal Perforation
5.10 Tetracycline-Class Adverse Effects
5.11 Development of Drug-Resistant Bacteria
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post-Marketing Experience
7 DRUG INTERACTIONS
7.1 Warfarin
7.2 Oral Contraceptives
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Complicated Skin and Skin Structure Infections
14.2 Complicated Intra-abdominal Infections
14.3 Community-Acquired Bacterial Pneumonia
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: ALL-CAUSE MORTALITY
An increase in all-cause mortality has been observed in a meta-analysis of Phase 3 and 4 clinical trials in TYGACIL-treated patients versus comparator. The cause of this mortality risk difference of 0.6% (95% CI 0.1, 1.2) has not been established. TYGACIL should be reserved for use in situations when alternative treatments are not suitable [see Indications and Usage (1.4), Warnings and Precautions (5.1, 5.2) and Adverse Reactions (6.1)].
-
1 INDICATIONS AND USAGE
1.1 Complicated Skin and Skin Structure Infections
Tigecycline for injection is indicated in patients 18 years of age and older for the treatment of complicated skin and skin structure infections caused by susceptible isolates of Escherichia coli, Enterococcus faecalis (vancomycin-susceptible isolates), Staphylococcus aureus (methicillin-susceptible and -resistant isolates), Streptococcus agalactiae, Streptococcus anginosus grp. (includes S. anginosus, S. intermedius, and S. constellatus), Streptococcus pyogenes, Enterobacter cloacae, Klebsiella pneumoniae, and Bacteroides fragilis.
1.2 Complicated Intra-abdominal Infections
Tigecycline for injection is indicated in patients 18 years of age and older for the treatment of complicated intra-abdominal infections caused by susceptible isolates of Citrobacter freundii, Enterobacter cloacae, Escherichia coli, Klebsiella oxytoca, Klebsiella pneumoniae, Enterococcus faecalis (vancomycin-susceptible isolates), Staphylococcus aureus (methicillin-susceptible and -resistant isolates), Streptococcus anginosus grp. (includes S. anginosus, S. intermedius, and S. constellatus), Bacteroides fragilis, Bacteroides thetaiotaomicron, Bacteroides uniformis, Bacteroides vulgatus, Clostridium perfringens, and Peptostreptococcus micros.
1.3 Community-Acquired Bacterial Pneumonia
Tigecycline for injection is indicated in patients 18 years of age and older for the treatment of community-acquired bacterial pneumonia caused by susceptible isolates of Streptococcus pneumoniae (penicillin-susceptible isolates), including cases with concurrent bacteremia, Haemophilus influenzae, and Legionella pneumophila.
1.4 Limitations of Use
TYGACIL is not indicated for the treatment of diabetic foot infections. A clinical trial failed to demonstrate non-inferiority of TYGACIL for treatment of diabetic foot infections.
TYGACIL is not indicated for the treatment of hospital-acquired or ventilator-associated pneumonia. In a comparative clinical trial, greater mortality and decreased efficacy were reported in TYGACIL-treated patients [see Warnings and Precautions (5.2)].
1.5 Usage
To reduce the development of drug-resistant bacteria and maintain the effectiveness of TYGACIL and other antibacterial drugs, TYGACIL should be used only to treat infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
Appropriate specimens for bacteriological examination should be obtained in order to isolate and identify the causative organisms and to determine their susceptibility to tigecycline. TYGACIL may be initiated as empiric monotherapy before results of these tests are known.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Adult Dosage
The recommended dosage regimen for TYGACIL is an initial dose of 100 mg, followed by 50 mg every 12 hours. Intravenous infusions of TYGACIL should be administered over approximately 30 to 60 minutes every 12 hours.
The recommended duration of treatment with TYGACIL for complicated skin and skin structure infections or for complicated intra-abdominal infections is 5 to 14 days. The recommended duration of treatment with TYGACIL for community-acquired bacterial pneumonia is 7 to 14 days. The duration of therapy should be guided by the severity and site of the infection and the patient's clinical and bacteriological progress.
2.2 Dosage in Patients With Hepatic Impairment
No dosage adjustment is warranted in patients with mild to moderate hepatic impairment (Child Pugh A and Child Pugh B). In patients with severe hepatic impairment (Child Pugh C), the initial dose of TYGACIL should be 100 mg followed by a reduced maintenance dose of 25 mg every 12 hours. Patients with severe hepatic impairment (Child Pugh C) should be treated with caution and monitored for treatment response [see Clinical Pharmacology (12.3) and Use in Specific Populations (8.6)].
2.3 Dosage in Pediatric Patients
The safety and efficacy of the proposed pediatric dosing regimens have not been evaluated due to the observed increase in mortality associated with TYGACIL in adult patients. Avoid use of TYGACIL in pediatric patients unless no alternative antibacterial drugs are available. Under these circumstances, the following doses are suggested:
- Pediatric patients aged 8 to 11 years should receive 1.2 mg/kg of TYGACIL every 12 hours intravenously to a maximum dose of 50 mg of TYGACIL every 12 hours.
- Pediatric patients aged 12 to 17 years should receive 50 mg of TYGACIL every 12 hours.
The proposed pediatric doses of TYGACIL were chosen based on exposures observed in pharmacokinetic trials, which included small numbers of pediatric patients [see Use in Specific Populations (8.4) and Clinical Pharmacology (12.3)].
There are no data to provide dosing recommendations in pediatric patients with hepatic impairment.
2.4 Preparation and Administration
Each vial of TYGACIL should be reconstituted with 5.3 mL of 0.9% Sodium Chloride Injection, USP, 5% Dextrose Injection, USP, or Lactated Ringer's Injection, USP to achieve a concentration of 10 mg/mL of tigecycline. (Note: Each vial contains a 6% overage. Thus, 5 mL of reconstituted solution is equivalent to 50 mg of the drug.) The vial should be gently swirled until the drug dissolves. Reconstituted solution must be transferred and further diluted for intravenous infusion. Withdraw 5 mL of the reconstituted solution from the vial and add to a 100 mL intravenous bag for infusion (for a 100 mg dose, reconstitute two vials; for a 50 mg dose, reconstitute one vial). The maximum concentration in the intravenous bag should be 1 mg/mL. The reconstituted solution should be yellow to orange in color; if not, the solution should be discarded. Parenteral drug products should be inspected visually for particulate matter and discoloration (e.g., green or black) prior to administration. Once reconstituted, TYGACIL may be stored at room temperature (not to exceed 25°C/77°F) for up to 24 hours (up to 6 hours in the vial and the remaining time in the intravenous bag). If the storage conditions exceed 25°C (77°F) after reconstitution, tigecycline should be used immediately. Alternatively, TYGACIL mixed with 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP may be stored refrigerated at 2° to 8°C (36° to 46°F) for up to 48 hours following immediate transfer of the reconstituted solution into the intravenous bag.
TYGACIL may be administered intravenously through a dedicated line or through a Y-site. If the same intravenous line is used for sequential infusion of several drugs, the line should be flushed before and after infusion of TYGACIL with 0.9% Sodium Chloride Injection, USP, 5% Dextrose Injection, USP or Lactated Ringer's Injection, USP. Injection should be made with an infusion solution compatible with tigecycline and with any other drug(s) administered via this common line.
2.5 Drug Compatibilities
Compatible intravenous solutions include 0.9% Sodium Chloride Injection, USP, 5% Dextrose Injection, USP, and Lactated Ringer's Injection, USP. When administered through a Y-site, TYGACIL is compatible with the following drugs or diluents when used with either 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP: amikacin, dobutamine, dopamine HCl, gentamicin, haloperidol, Lactated Ringer's, lidocaine HCl, metoclopramide, morphine, norepinephrine, piperacillin/tazobactam (EDTA formulation), potassium chloride, propofol, ranitidine HCl, theophylline, and tobramycin.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
TYGACIL is contraindicated for use in patients who have known hypersensitivity to tigecycline. Reactions have included anaphylactic reactions [see Warnings and Precautions (5.3) and Adverse Reactions (6.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 All-Cause Mortality
An increase in all-cause mortality has been observed in a meta-analysis of Phase 3 and 4 clinical trials in TYGACIL-treated patients versus comparator-treated patients. In all 13 Phase 3 and 4 trials that included a comparator, death occurred in 4.0% (150/3788) of patients receiving TYGACIL and 3.0% (110/3646) of patients receiving comparator drugs. In a pooled analysis of these trials, based on a random effects model by trial weight, the adjusted risk difference of all-cause mortality was 0.6% (95% CI 0.1, 1.2) between TYGACIL and comparator-treated patients. An analysis of mortality in all trials conducted for approved indications (cSSSI, cIAI, and CABP), including post-market trials showed an adjusted mortality rate of 2.5% (66/2640) for tigecycline and 1.8% (48/2628) for comparator, respectively. The adjusted risk difference for mortality stratified by trial weight was 0.6% (95% CI 0.0, 1.2).
The cause of this mortality difference has not been established. Generally, deaths were the result of worsening infection, complications of infection or underlying co-morbidities. TYGACIL should be reserved for use in situations when alternative treatments are not suitable [see Boxed Warning, Indications and Usage (1.4), Warnings and Precautions (5.2) and Adverse Reactions (6.1)].
5.2 Mortality Imbalance and Lower Cure Rates in Hospital-Acquired Pneumonia
A trial of patients with hospital acquired, including ventilator-associated, pneumonia failed to demonstrate the efficacy of TYGACIL. In this trial, patients were randomized to receive TYGACIL (100 mg initially, then 50 mg every 12 hours) or a comparator. In addition, patients were allowed to receive specified adjunctive therapies. The sub-group of patients with ventilator-associated pneumonia who received TYGACIL had lower cure rates (47.9% versus 70.1% for the clinically evaluable population).
In this trial, greater mortality was seen in patients with ventilator-associated pneumonia who received TYGACIL (25/131 [19.1%] versus 15/122 [12.3%] in comparator-treated patients) [see Adverse Reactions (6.1)]. Particularly high mortality was seen among TYGACIL-treated patients with ventilator-associated pneumonia and bacteremia at baseline (9/18 [50.0%] versus 1/13 [7.7%] in comparator-treated patients).
5.3 Anaphylactic Reactions
Anaphylactic reactions have been reported with nearly all antibacterial agents, including TYGACIL, and may be life-threatening. TYGACIL is structurally similar to tetracycline-class antibiotics and should be avoided in patients with known hypersensitivity to tetracycline-class antibiotics.
5.4 Hepatic Adverse Effects
Increases in total bilirubin concentration, prothrombin time and transaminases have been seen in patients treated with tigecycline. Isolated cases of significant hepatic dysfunction and hepatic failure have been reported in patients being treated with tigecycline. Some of these patients were receiving multiple concomitant medications. Patients who develop abnormal liver function tests during tigecycline therapy should be monitored for evidence of worsening hepatic function and evaluated for risk/benefit of continuing tigecycline therapy. Hepatic dysfunction may occur after the drug has been discontinued.
5.5 Pancreatitis
Acute pancreatitis, including fatal cases, has occurred in association with tigecycline treatment. The diagnosis of acute pancreatitis should be considered in patients taking tigecycline who develop clinical symptoms, signs, or laboratory abnormalities suggestive of acute pancreatitis. Cases have been reported in patients without known risk factors for pancreatitis. Patients usually improve after tigecycline discontinuation. Consideration should be given to the cessation of the treatment with tigecycline in cases suspected of having developed pancreatitis [see Adverse Reactions (6.2)].
5.6 Tooth Discoloration and Enamel Hypoplasia
The use of TYGACIL during tooth development (last half of pregnancy, infancy, and childhood to the age of 8 years) may cause permanent discoloration of the teeth (yellow-gray-brown). This adverse reaction is more common during long-term use of tetracyclines, but it has been observed following repeated short-term courses. Enamel hypoplasia has also been reported. Advise the patient of the potential risk to the fetus if TYGACIL is used during the second or third trimester of pregnancy [see Use in Specific Populations (8.1, 8.4)].
5.7 Inhibition of Bone Growth
The use of TYGACIL during the second and third trimester of pregnancy, infancy and childhood up to the age of 8 years may cause reversible inhibition of bone growth. All tetracyclines form a stable calcium complex in any bone-forming tissue. A decrease in fibula growth rate has been observed in premature infants given oral tetracycline in doses of 25 mg/kg every 6 hours. This reaction was shown to be reversible when the tetracycline was discontinued. Advise the patient of the potential risk to the fetus if TYGACIL is used during the second or third trimester of pregnancy [see Use in Specific Populations (8.1, 8.4)].
5.8 Clostridium difficile Associated Diarrhea
Clostridium difficile associated diarrhea (CDAD) has been reported with use of nearly all antibacterial agents, including TYGACIL, and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon leading to overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibiotic use. Careful medical history is necessary since CDAD has been reported to occur over two months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibiotic use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibiotic treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated.
5.9 Sepsis/Septic Shock in Patients With Intestinal Perforation
Monotherapy with tigecycline should be avoided in patients with complicated intra-abdominal infections (cIAI) secondary to clinically apparent intestinal perforation. In cIAI studies (n=1642), 6 patients treated with TYGACIL and 2 patients treated with imipenem/cilastatin presented with intestinal perforations and developed sepsis/septic shock. The 6 patients treated with TYGACIL had higher APACHE II scores (median = 13) versus the 2 patients treated with imipenem/cilastatin (APACHE II scores = 4 and 6). Due to differences in baseline APACHE II scores between treatment groups and small overall numbers, the relationship of this outcome to treatment cannot be established.
5.10 Tetracycline-Class Adverse Effects
TYGACIL is structurally similar to tetracycline-class antibacterial drugs and may have similar adverse effects. Such effects may include: photosensitivity, pseudotumor cerebri, and anti-anabolic action (which has led to increased BUN, azotemia, acidosis, and hyperphosphatemia).
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described elsewhere in the labeling:
- All-Cause Mortality [see Boxed Warning and Warnings and Precautions (5.1)]
- Mortality Imbalance and Lower Cure Rates in Hospital-Acquired Pneumonia [see Warnings and Precautions (5.2)]
- Anaphylaxis [Warning and Precautions (5.3)]
- Hepatic Adverse Effects [Warnings and Precautions (5.4)]
- Pancreatitis [Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In clinical trials, 2514 patients were treated with TYGACIL. TYGACIL was discontinued due to adverse reactions in 7% of patients compared to 6% for all comparators. Table 1 shows the incidence of adverse reactions through test of cure reported in ≥2% of patients in these trials.
Table 1. Incidence (%) of Adverse Reactions Through Test of Cure Reported in ≥ 2% of Patients Treated in Clinical Studies Body System
Adverse ReactionsTYGACIL
(N=2514)Comparators*
(N=2307)- * Vancomycin/Aztreonam, Imipenem/Cilastatin, Levofloxacin, Linezolid.
- † LFT abnormalities in TYGACIL-treated patients were reported more frequently in the post therapy period than those in comparator-treated patients, which occurred more often on therapy.
Body as a Whole Abdominal pain 6 4 Abscess 2 2 Asthenia 3 2 Headache 6 7 Infection 7 5 Cardiovascular System Phlebitis 3 4 Digestive System Diarrhea 12 11 Dyspepsia 2 2 Nausea 26 13 Vomiting 18 9 Hemic and Lymphatic System Anemia 5 6 Metabolic and Nutritional Alkaline Phosphatase Increased 3 3 Amylase Increased 3 2 Bilirubinemia 2 1 BUN Increased 3 1 Healing Abnormal 3 2 Hyponatremia 2 1 Hypoproteinemia 5 3 SGOT Increased† 4 5 SGPT Increased† 5 5 Respiratory System Pneumonia 2 2 Nervous System Dizziness 3 3 Skin and Appendages Rash 3 4 In all 13 Phase 3 and 4 trials that included a comparator, death occurred in 4.0% (150/3788) of patients receiving TYGACIL and 3.0% (110/3646) of patients receiving comparator drugs. In a pooled analysis of these trials, based on a random effects model by trial weight, an adjusted risk difference of all-cause mortality was 0.6% (95% CI 0.1, 1.2) between TYGACIL and comparator-treated patients (see Table 2). The cause of the imbalance has not been established. Generally, deaths were the result of worsening infection, complications of infection or underlying co-morbidities.
Table 2. Patients with Outcome of Death by Infection Type TYGACIL Comparator Risk Difference* Infection Type n/N % n/N % % (95% CI) CAP = Community-acquired pneumonia; cIAI = Complicated intra-abdominal infections; cSSSI = Complicated skin and skin structure infections; HAP = Hospital-acquired pneumonia; VAP = Ventilator-associated pneumonia; RP = Resistant pathogens; DFI = Diabetic foot infections.
Note: The studies include 300, 305, 900 (cSSSI), 301, 306, 315, 316, 400 (cIAI), 308 and 313 (CAP), 311 (HAP), 307 [Resistant gram-positive pathogen study in patients with MRSA or Vancomycin-Resistant Enterococcus (VRE)], and 319 (DFI with and without osteomyelitis).- * The difference between the percentage of patients who died in TYGACIL and comparator treatment groups. The 95% CI for each infection type was calculated using the normal approximation method without continuity correction.
- † These are subgroups of the HAP population.
- ‡ Overall adjusted (random effects model by trial weight) risk difference estimate and 95% CI.
cSSSI 12/834 1.4 6/813 0.7 0.7 (-0.3, 1.7) cIAI 42/1382 3.0 31/1393 2.2 0.8 (-0.4, 2.0) CAP 12/424 2.8 11/422 2.6 0.2 (-2.0, 2.4) HAP 66/467 14.1 57/467 12.2 1.9 (-2.4, 6.3) Non-VAP† 41/336 12.2 42/345 12.2 0.0 (-4.9, 4.9) VAP† 25/131 19.1 15/122 12.3 6.8 (-2.1, 15.7) RP 11/128 8.6 2/43 4.7 3.9 (-4.0, 11.9) DFI 7/553 1.3 3/508 0.6 0.7 (-0.5, 1.8) Overall Adjusted 150/3788 4.0 110/3646 3.0 0.6 (0.1, 1.2)‡ An analysis of mortality in all trials conducted for approved indications - cSSSI, cIAI, and CABP, including post-market trials (one in cSSSI and two in cIAI) - showed an adjusted mortality rate of 2.5% (66/2640) for tigecycline and 1.8% (48/2628) for comparator, respectively. The adjusted risk difference for mortality stratified by trial weight was 0.6% (95% CI 0.0, 1.2).
In comparative clinical studies, infection-related serious adverse reactions were more frequently reported for subjects treated with TYGACIL (7%) versus comparators (6%). Serious adverse reactions of sepsis/septic shock were more frequently reported for subjects treated with TYGACIL (2%) versus comparators (1%). Due to baseline differences between treatment groups in this subset of patients, the relationship of this outcome to treatment cannot be established [see Warnings and Precautions (5.9)].
The most common adverse reactions were nausea and vomiting which generally occurred during the first 1 – 2 days of therapy. The majority of cases of nausea and vomiting associated with TYGACIL and comparators were either mild or moderate in severity. In patients treated with TYGACIL, nausea incidence was 26% (17% mild, 8% moderate, 1% severe) and vomiting incidence was 18% (11% mild, 6% moderate, 1% severe).
In patients treated for complicated skin and skin structure infections (cSSSI), nausea incidence was 35% for TYGACIL and 9% for vancomycin/aztreonam; vomiting incidence was 20% for TYGACIL and 4% for vancomycin/aztreonam. In patients treated for complicated intra-abdominal infections (cIAI), nausea incidence was 25% for TYGACIL and 21% for imipenem/cilastatin; vomiting incidence was 20% for TYGACIL and 15% for imipenem/cilastatin. In patients treated for community-acquired bacterial pneumonia (CABP), nausea incidence was 24% for TYGACIL and 8% for levofloxacin; vomiting incidence was 16% for TYGACIL and 6% for levofloxacin.
Discontinuation from TYGACIL was most frequently associated with nausea (1%) and vomiting (1%). For comparators, discontinuation was most frequently associated with nausea (<1%).
The following adverse reactions were reported (<2%) in patients receiving TYGACIL in clinical studies:
Body as a Whole: injection site inflammation, injection site pain, injection site reaction, septic shock, allergic reaction, chills, injection site edema, injection site phlebitis
Cardiovascular System: thrombophlebitis
Digestive System: anorexia, jaundice, abnormal stools
Metabolic/Nutritional System: increased creatinine, hypocalcemia, hypoglycemia
Special Senses: taste perversion
Hemic and Lymphatic System: prolonged activated partial thromboplastin time (aPTT), prolonged prothrombin time (PT), eosinophilia, increased international normalized ratio (INR), thrombocytopenia
Skin and Appendages: pruritus
Urogenital System: vaginal moniliasis, vaginitis, leukorrhea
6.2 Post-Marketing Experience
The following adverse reactions have been identified during post-approval use of TYGACIL. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish causal relationship to drug exposure.
- anaphylactic reactions
- acute pancreatitis
- hepatic cholestasis, and jaundice
- severe skin reactions, including Stevens-Johnson Syndrome
- symptomatic hypoglycemia in patients with and without diabetes mellitus
-
7 DRUG INTERACTIONS
7.1 Warfarin
Prothrombin time or other suitable anticoagulation test should be monitored if TYGACIL is administered with warfarin [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
TYGACIL, like other tetracycline class antibacterial drugs, may cause permanent discoloration of deciduous teeth and reversible inhibition of bone growth when administered during the second and third trimesters of pregnancy [see Warnings and Precautions (5.6) (5.7), Data, and Use in Specific Populations (8.4)]. There are no available data on the risk of major birth defects or miscarriage following the use of TYGACIL during pregnancy. Administration of intravenous tigecycline in pregnant rats and rabbits during the period of organogenesis was associated with reduction in fetal weights and an increased incidence of skeletal anomalies (delays in bone ossification) at exposures of 5 and 1 times the human exposure at the recommended clinical dose in rats and rabbits, respectively. Advise the patient of the potential risk to the fetus if TYGACIL is used during the second or third trimester.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U. S. general population, the estimated background risk in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Human Data
The use of tetracycline-class antibacterial drugs, that includes TYGACIL, during tooth development (second and third trimester of pregnancy) may cause permanent discoloration of deciduous teeth. This adverse reaction is more common during long-term use of tetracyclines but has been observed following repeated short-term courses. TYGACIL may cause reversible inhibition of bone growth when administered during the second and third trimesters of pregnancy. A decrease in fibula growth rate has been observed in premature infants given oral tetracycline in doses of 25 mg/kg every 6 hours.
Animal Data
In embryo-fetal development studies, tigecycline was administered during the period of organogenesis at doses up to 12 mg/kg/day in rats and 4 mg/kg in rabbits or 5 and 1 times the systemic exposure at the recommended clinical dose, respectively. In the rat study, decreased fetal weight and fetal skeletal variations (reduced ossification of the pubic, ischial, and supraoccipital bones and increased incidences of rudimentary 14th rib) were observed in the presence of maternal toxicity at 12 mg/kg/day (5 times the recommended clinical dose based on systemic exposure). In rabbits, decreased fetal weights were observed in the presence of maternal toxicity at 4 mg/kg (equivalent to the human exposure at the recommended clinical dose).
In preclinical safety studies, 14C-labeled tigecycline crossed the placenta and was found in fetal tissues.
8.2 Lactation
Risk Summary
There are no data on the presence of tigecycline in human milk; however, tetracycline-class antibacterial drugs are present in breast milk. It is not known whether tigecycline has an effect on the breastfed infant or on milk production. Tigecycline has low oral bioavailability; therefore, infant exposure is expected to be low. Tigecycline is present in rat milk with little or no systemic exposure to tigecycline in nursing pups as a result of exposure via maternal milk. When a drug is present in animal milk, it is likely that the drug will be present in human milk.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for TYGACIL and any potential adverse effects on the breastfed child from TYGACIL or from the underlying maternal condition (see Clinical Considerations).
Clinical Considerations
Because of the theoretical risk of dental discoloration and inhibition of bone growth, avoid breastfeeding if taking TYGACIL for longer than three weeks. A lactating woman may also consider interrupting breastfeeding and pumping and discarding breastmilk during administration of TYGACIL and for 9 days (approximately 5 half-lives) after the last dose in order to minimize drug exposure to a breastfed infant.
8.4 Pediatric Use
Use in patients under 18 years of age is not recommended. Safety and effectiveness in pediatric patients below the age of 18 years have not been established. Because of the increased mortality observed in TYGACIL-treated adult patients in clinical trials, pediatric trials of TYGACIL to evaluate the safety and efficacy of TYGACIL were not conducted.
In situations where there are no other alternative antibacterial drugs, dosing has been proposed for pediatric patients 8 to 17 years of age based on data from pediatric pharmacokinetic studies [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
Because of effects on tooth development, use in patients under 8 years of age is not recommended [see Warnings and Precautions (5.7)].
8.5 Geriatric Use
Of the total number of subjects who received TYGACIL in Phase 3 clinical studies (n=2514), 664 were 65 and over, while 288 were 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, but greater sensitivity to adverse events of some older individuals cannot be ruled out.
No significant difference in tigecycline exposure was observed between healthy elderly subjects and younger subjects following a single 100 mg dose of tigecycline [see Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
No dosage adjustment is warranted in patients with mild to moderate hepatic impairment (Child Pugh A and Child Pugh B). In patients with severe hepatic impairment (Child Pugh C), the initial dose of tigecycline should be 100 mg followed by a reduced maintenance dose of 25 mg every 12 hours. Patients with severe hepatic impairment (Child Pugh C) should be treated with caution and monitored for treatment response [see Clinical Pharmacology (12.3) and Dosage and Administration (2.2)].
-
10 OVERDOSAGE
No specific information is available on the treatment of overdosage with tigecycline. Intravenous administration of TYGACIL at a single dose of 300 mg over 60 minutes in healthy volunteers resulted in an increased incidence of nausea and vomiting. Tigecycline is not removed in significant quantities by hemodialysis.
-
11 DESCRIPTION
TYGACIL (tigecycline) is a tetracycline class antibacterial for intravenous infusion. The chemical name of tigecycline is (4S,4aS,5aR,12aS)-9-[2-(tert-butylamino)acetamido]-4,7-bis(dimethylamino)-1,4,4a,5,5a,6,11,12a-octahydro-3,10,12,12a-tetrahydroxy-1,11-dioxo-2-naphthacenecarboxamide. The empirical formula is C29H39N5O8 and the molecular weight is 585.65.
The following represents the chemical structure of tigecycline:
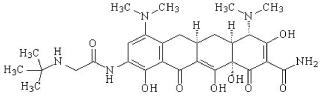
Figure 1: Structure of Tigecycline TYGACIL is an orange lyophilized powder or cake. Each TYGACIL single-dose 5 mL or 10 mL vial contains 50 mg tigecycline lyophilized powder for reconstitution for intravenous infusion and 100 mg of lactose monohydrate. The pH is adjusted with hydrochloric acid, and if necessary sodium hydroxide. The product does not contain preservatives.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tigecycline is a tetracycline class antibacterial [see Microbiology (12.4)].
12.3 Pharmacokinetics
The mean pharmacokinetic parameters of tigecycline after single and multiple intravenous doses based on pooled data from clinical pharmacology studies are summarized in Table 3. Intravenous infusions of tigecycline were administered over approximately 30 to 60 minutes.
Table 3. Mean (CV%) Pharmacokinetic Parameters of Tigecycline Single Dose
100 mg
(N=224)Multiple Dose*
50 mg every 12h
(N=103)- * 100 mg initially, followed by 50 mg every 12 hours
- † 30-minute infusion
- ‡ 60-minute infusion
Cmax (mcg/mL)† 1.45 (22%) 0.87 (27%) Cmax (mcg/mL)‡ 0.90 (30%) 0.63 (15%) AUC (mcg∙h/mL) 5.19 (36%) - - AUC0–24h (mcg∙h/mL) - - 4.70 (36%) Cmin (mcg/mL) - - 0.13 (59%) t½ (h) 27.1 (53%) 42.4 (83%) CL (L/h) 21.8 (40%) 23.8 (33%) CLr (mL/min) 38.0 (82%) 51.0 (58%) Vss (L) 568 (43%) 639 (48%) Distribution
The in vitro plasma protein binding of tigecycline ranges from approximately 71% to 89% at concentrations observed in clinical studies (0.1 to 1.0 mcg/mL). The steady-state volume of distribution of tigecycline averaged 500 to 700 L (7 to 9 L/kg), indicating tigecycline is extensively distributed beyond the plasma volume and into the tissues.
Following the administration of tigecycline 100 mg followed by 50 mg every 12 hours to 33 healthy volunteers, the tigecycline AUC0–12h (134 mcg∙h/mL) in alveolar cells was approximately 78-fold higher than the AUC0–12h in the serum, and the AUC0–12h (2.28 mcg∙h/mL) in epithelial lining fluid was approximately 32% higher than the AUC0–12h in serum. The AUC0–12h (1.61 mcg∙h/mL) of tigecycline in skin blister fluid was approximately 26% lower than the AUC0–12h in the serum of 10 healthy subjects.
In a single-dose study, tigecycline 100 mg was administered to subjects prior to undergoing elective surgery or medical procedure for tissue extraction. Concentrations at 4 hours after tigecycline administration were higher in gallbladder (38-fold, n=6), lung (3.7-fold, n=5), and colon (2.3-fold, n=6), and lower in synovial fluid (0.58-fold, n=5), and bone (0.35-fold, n=6) relative to serum. The concentration of tigecycline in these tissues after multiple doses has not been studied.
Elimination
Metabolism
Tigecycline is not extensively metabolized. In vitro studies with tigecycline using human liver microsomes, liver slices, and hepatocytes led to the formation of only trace amounts of metabolites. In healthy male volunteers receiving 14C-tigecycline, tigecycline was the primary 14C-labeled material recovered in urine and feces, but a glucuronide, an N-acetyl metabolite, and a tigecycline epimer (each at no more than 10% of the administered dose) were also present.
Tigecycline is a substrate of P-glycoprotein (P-gp) based on an in vitro study using a cell line overexpressing P-gp. The potential contribution of P-gp-mediated transport to the in vivo disposition of tigecycline is not known.
Excretion
The recovery of total radioactivity in feces and urine following administration of 14C-tigecycline indicates that 59% of the dose is eliminated by biliary/fecal excretion, and 33% is excreted in urine. Approximately 22% of the total dose is excreted as unchanged tigecycline in urine. Overall, the primary route of elimination for tigecycline is biliary excretion of unchanged tigecycline and its metabolites. Glucuronidation and renal excretion of unchanged tigecycline are secondary routes.
Specific Populations
Hepatic Impairment
In a study comparing 10 patients with mild hepatic impairment (Child Pugh A), 10 patients with moderate hepatic impairment (Child Pugh B), and 5 patients with severe hepatic impairment (Child Pugh C) to 23 age and weight matched healthy control subjects, the single-dose pharmacokinetic disposition of tigecycline was not altered in patients with mild hepatic impairment. However, systemic clearance of tigecycline was reduced by 25% and the half-life of tigecycline was prolonged by 23% in patients with moderate hepatic impairment (Child Pugh B). Systemic clearance of tigecycline was reduced by 55%, and the half-life of tigecycline was prolonged by 43% in patients with severe hepatic impairment (Child Pugh C). Dosage adjustment is necessary in patients with severe hepatic impairment (Child Pugh C) [see Use in Specific Populations (8.6) and Dosage and Administration (2.2)].
Renal Impairment
A single dose study compared 6 subjects with severe renal impairment (creatinine clearance <30 mL/min), 4 end stage renal disease (ESRD) patients receiving tigecycline 2 hours before hemodialysis, 4 ESRD patients receiving tigecycline 1 hour after hemodialysis, and 6 healthy control subjects. The pharmacokinetic profile of tigecycline was not significantly altered in any of the renally impaired patient groups, nor was tigecycline removed by hemodialysis. No dosage adjustment of TYGACIL is necessary in patients with renal impairment or in patients undergoing hemodialysis.
Geriatric Patients
No significant differences in pharmacokinetics were observed between healthy elderly subjects (n=15, age 65–75; n=13, age >75) and younger subjects (n=18) receiving a single 100-mg dose of TYGACIL. Therefore, no dosage adjustment is necessary based on age [see Use in Specific Populations (8.5)].
Pediatric Patients
A single-dose safety, tolerability, and pharmacokinetic study of tigecycline in pediatric patients aged 8–16 years who recently recovered from infections was conducted. The doses administered were 0.5, 1, or 2 mg/kg. The study showed that for children aged 12–16 years (n = 16) a dosage of 50 mg twice daily would likely result in exposures comparable to those observed in adults with the approved dosing regimen. Large variability observed in children aged 8 to 11 years of age (n = 8) required additional study to determine the appropriate dosage.
A subsequent tigecycline dose-finding study was conducted in 8–11 year old patients with cIAI, cSSSI, or CABP. The doses of tigecycline studied were 0.75 mg/kg (n = 17), 1 mg/kg (n = 21), and 1.25 mg/kg (n=20). This study showed that for children aged 8–11 years, a 1.2 mg/kg dose would likely result in exposures comparable to those observed in adults resulting with the approved dosing regimen [see Dosage and Administration (2.3)].
Gender
In a pooled analysis of 38 women and 298 men participating in clinical pharmacology studies, there was no significant difference in the mean (±SD) tigecycline clearance between women (20.7±6.5 L/h) and men (22.8±8.7 L/h). Therefore, no dosage adjustment is necessary based on gender.
Race
In a pooled analysis of 73 Asian subjects, 53 Black subjects, 15 Hispanic subjects, 190 White subjects, and 3 subjects classified as "other" participating in clinical pharmacology studies, there was no significant difference in the mean (±SD) tigecycline clearance among the Asian subjects (28.8±8.8 L/h), Black subjects (23.0±7.8 L/h), Hispanic subjects (24.3±6.5 L/h), White subjects (22.1±8.9 L/h), and "other" subjects (25.0±4.8 L/h). Therefore, no dosage adjustment is necessary based on race.
Drug Interaction Studies
Digoxin
TYGACIL (100 mg followed by 50 mg every 12 hours) and digoxin (0.5 mg followed by 0.25 mg, orally, every 24 hours) were co-administered to healthy subjects in a drug interaction study. Tigecycline slightly decreased the Cmax of digoxin by 13%, but did not affect the AUC or clearance of digoxin. This small change in Cmax did not affect the steady-state pharmacodynamic effects of digoxin as measured by changes in ECG intervals. In addition, digoxin did not affect the pharmacokinetic profile of tigecycline. Therefore, no dosage adjustment of either drug is necessary when TYGACIL is administered with digoxin.
Warfarin
Concomitant administration of TYGACIL (100 mg followed by 50 mg every 12 hours) and warfarin (25 mg single-dose) to healthy subjects resulted in a decrease in clearance of R-warfarin and S-warfarin by 40% and 23%, an increase in Cmax by 38% and 43% and an increase in AUC by 68% and 29%, respectively. Tigecycline did not significantly alter the effects of warfarin on INR. In addition, warfarin did not affect the pharmacokinetic profile of tigecycline. However, prothrombin time or other suitable anticoagulation test should be monitored if tigecycline is administered with warfarin.
In vitro studies in human liver microsomes indicate that tigecycline does not inhibit metabolism mediated by any of the following 6 cytochrome P450 (CYP) isoforms: 1A2, 2C8, 2C9, 2C19, 2D6, and 3A4. Therefore, TYGACIL is not expected to alter the metabolism of drugs metabolized by these enzymes. In addition, because tigecycline is not extensively metabolized, clearance of tigecycline is not expected to be affected by drugs that inhibit or induce the activity of these CYP450 isoforms.
In vitro studies using Caco-2 cells indicate that tigecycline does not inhibit digoxin flux, suggesting that tigecycline is not a P-glycoprotein (P-gp) inhibitor. This in vitro information is consistent with the lack of effect of tigecycline on digoxin clearance noted in the in vivo drug interaction study described above.
Tigecycline is a substrate of P-gp based on an in vitro study using a cell line overexpressing P-gp. The potential contribution of P-gp-mediated transport to the in vivo disposition of tigecycline is not known. Coadministration of P-gp inhibitors (e.g., ketoconazole or cyclosporine) or P-gp inducers (e.g., rifampicin) could affect the pharmacokinetics of tigecycline.
12.4 Microbiology
Mechanism of Action
Tigecycline inhibits protein translation in bacteria by binding to the 30S ribosomal subunit and blocking entry of amino-acyl tRNA molecules into the A site of the ribosome. This prevents incorporation of amino acid residues into elongating peptide chains. In general, tigecycline is considered bacteriostatic; however, TYGACIL has demonstrated bactericidal activity against isolates of S. pneumoniae and L. pneumophila.
Resistance
To date there has been no cross-resistance observed between tigecycline and other antibacterials. Tigecycline is less affected by the two major tetracycline-resistance mechanisms, ribosomal protection and efflux. Additionally, tigecycline is not affected by resistance mechanisms such as beta-lactamases (including extended spectrum beta-lactamases), target-site modifications, macrolide efflux pumps or enzyme target changes (e.g. gyrase/topoisomerases). However, some ESBL-producing isolates may confer resistance to tigecycline via other resistance mechanisms. Tigecycline resistance in some bacteria (e.g. Acinetobacter calcoaceticus-Acinetobacter baumannii complex) is associated with multi-drug resistant (MDR) efflux pumps.
Interaction with Other Antimicrobials
In vitro studies have not demonstrated antagonism between tigecycline and other commonly used antibacterials.
Antimicrobial Activity
Tigecycline has been shown to be active against most of the following microorganisms, both in vitro and in clinical infections [see Indications and Usage (1)].
Gram-positive bacteria
Enterococcus faecalis (vancomycin-susceptible isolates)
Staphylococcus aureus (methicillin-susceptible and -resistant isolates)
Streptococcus agalactiae
Streptococcus anginosus group (includes S. anginosus, S. intermedius, and S. constellatus)
Streptococcus pneumoniae (penicillin-susceptible isolates)
Streptococcus pyogenesGram-negative bacteria
Citrobacter freundii
Enterobacter cloacae
Escherichia coli
Haemophilus influenzae
Klebsiella oxytoca
Klebsiella pneumoniae
Legionella pneumophilaAnaerobic bacteria
Bacteroides fragilis
Bacteroides thetaiotaomicron
Bacteroides uniformis
Bacteroides vulgatus
Clostridium perfringens
Peptostreptococcus microsThe following in vitro data are available, but their clinical significance is unknown. At least 90 percent of the following bacteria exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for tigecycline against isolates of similar genus or organism group. However, the efficacy of tigecycline in treating clinical infections caused by these bacteria has not been established in adequate and well-controlled clinical trials.
Gram-positive bacteria
Enterococcus avium
Enterococcus casseliflavus
Enterococcus faecalis (vancomycin-resistant isolates)
Enterococcus faecium (vancomycin-susceptible and -resistant isolates)
Enterococcus gallinarum
Listeria monocytogenes
Staphylococcus epidermidis (methicillin-susceptible and -resistant isolates)
Staphylococcus haemolyticusGram-negative bacteria
Acinetobacter baumannii1
Aeromonas hydrophila
Citrobacter koseri
Enterobacter aerogenes
Haemophilus influenzae (ampicillin-resistant)
Haemophilus parainfluenzae
Pasteurella multocida
Serratia marcescens
Stenotrophomonas maltophiliaAnaerobic bacteria
Bacteroides distasonis
Bacteroides ovatus
Peptostreptococcus spp.
Porphyromonas spp.
Prevotella spp.Other bacteria
Mycobacterium abscessus
Mycobacterium fortuitum
- 1 There have been reports of the development of tigecycline resistance in Acinetobacter infections seen during the course of standard treatment. Such resistance appears to be attributable to an MDR efflux pump mechanism. While monitoring for relapse of infection is important for all infected patients, more frequent monitoring in this case is suggested. If relapse is suspected, blood and other specimens should be obtained and cultured for the presence of bacteria. All bacterial isolates should be identified and tested for susceptibility to tigecycline and other appropriate antimicrobials.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Lifetime studies in animals have not been performed to evaluate the carcinogenic potential of tigecycline. No mutagenic or clastogenic potential was found in a battery of tests, including in vitro chromosome aberration assay in Chinese hamster ovary (CHO) cells, in vitro forward mutation assay in CHO cells (HGRPT locus), in vitro forward mutation assays in mouse lymphoma cells, and in vivo mouse micronucleus assay. Tigecycline did not affect mating or fertility in rats at exposures up to 5 times the human daily dose based on AUC (28 mcg∙hr/mL at 12 mg/kg/day). In female rats, there were no compound-related effects on ovaries or estrous cycles at exposures up to 5 times the human daily dose based on AUC.
13.2 Animal Toxicology and/or Pharmacology
In two week studies, decreased erythrocytes, reticulocytes, leukocytes, and platelets, in association with bone marrow hypocellularity, have been seen with tigecycline at exposures of 8 times and 10 times the human daily dose based on AUC in rats and dogs, (AUC of approximately 50 and 60 mcg∙hr/mL at doses of 30 and 12 mg/kg/day) respectively. These alterations were shown to be reversible after two weeks of dosing.
-
14 CLINICAL STUDIES
14.1 Complicated Skin and Skin Structure Infections
TYGACIL was evaluated in adults for the treatment of complicated skin and skin structure infections (cSSSI) in two randomized, double-blind, active-controlled, multinational, multicenter studies (Studies 1 and 2). These studies compared TYGACIL (100 mg intravenous initial dose followed by 50 mg every 12 hours) with vancomycin (1 g intravenous every 12 hours)/aztreonam (2 g intravenous every 12 hours) for 5 to 14 days. Patients with complicated deep soft tissue infections including wound infections and cellulitis (≥10 cm, requiring surgery/drainage or with complicated underlying disease), major abscesses, infected ulcers, and burns were enrolled in the studies. The primary efficacy endpoint was the clinical response at the test of cure (TOC) visit in the co-primary populations of the clinically evaluable (CE) and clinical modified intent-to-treat (c-mITT) patients. See Table 4. Clinical cure rates at TOC by pathogen in the microbiologically evaluable patients are presented in Table 5.
Table 4. Clinical Cure Rates from Two Studies in Complicated Skin and Skin Structure Infections after 5 to 14 Days of Therapy TYGACIL*
n/N (%)Vancomycin/Aztreonam†
n/N (%)- * 100 mg initially, followed by 50 mg every 12 hours
- † Vancomycin (1 g every 12 hours)/Aztreonam (2 g every 12 hours)
Study 1 CE 165/199 (82.9) 163/198 (82.3) c-mITT 209/277 (75.5) 200/260 (76.9) Study 2 CE 200/223 (89.7) 201/213 (94.4) c-mITT 220/261 (84.3) 225/259 (86.9) Table 5. Clinical Cure Rates By Infecting Pathogen in Microbiologically Evaluable Patients with Complicated Skin and Skin Structure Infections* Pathogen TYGACIL
n/N (%)Vancomycin/Aztreonam
n/N (%)- * Two cSSSI pivotal studies and two Resistant Pathogen studies
- † Includes Streptococcus anginosus, Streptococcus intermedius, and Streptococcus constellatus
Escherichia coli 29/36 (80.6) 26/30 (86.7) Enterobacter cloacae 10/12 (83.3) 15/15 (100) Enterococcus faecalis (vancomycin-susceptible only) 15/21 (71.4) 19/24 (79.2) Klebsiella pneumoniae 12/14 (85.7) 15/16 (93.8) Methicillin-susceptible Staphylococcus aureus (MSSA) 124/137 (90.5) 113/120 (94.2) Methicillin-resistant Staphylococcus aureus (MRSA) 79/95 (83.2) 46/57 (80.7) Streptococcus agalactiae 8/8 (100) 11/14 (78.6) Streptococcus anginosus grp.† 17/21 (81.0) 9/10 (90.0) Streptococcus pyogenes 31/32 (96.9) 24/27 (88.9) Bacteroides fragilis 7/9 (77.8) 4/5 (80.0) 14.2 Complicated Intra-abdominal Infections
TYGACIL was evaluated in adults for the treatment of complicated intra-abdominal infections (cIAI) in two randomized, double-blind, active-controlled, multinational, multicenter studies (Studies 1 and 2). These studies compared TYGACIL (100 mg intravenous initial dose followed by 50 mg every 12 hours) with imipenem/cilastatin (500 mg intravenous every 6 hours) for 5 to 14 days. Patients with complicated diagnoses including appendicitis, cholecystitis, diverticulitis, gastric/duodenal perforation, intra-abdominal abscess, perforation of intestine, and peritonitis were enrolled in the studies. The primary efficacy endpoint was the clinical response at the TOC visit for the co-primary populations of the microbiologically evaluable (ME) and the microbiologic modified intent-to-treat (m-mITT) patients. See Table 6. Clinical cure rates at TOC by pathogen in the microbiologically evaluable patients are presented in Table 7.
Table 6. Clinical Cure Rates from Two Studies in Complicated Intra-abdominal Infections after 5 to 14 Days of Therapy TYGACIL*
n/N (%)Imipenem/Cilastatin†
n/N (%)- * 100 mg initially, followed by 50 mg every 12 hours
- † Imipenem/Cilastatin (500 mg every 6 hours)
Study 1 ME 199/247 (80.6) 210/255 (82.4) m-mITT 227/309 (73.5) 244/312 (78.2) Study 2 ME 242/265 (91.3) 232/258 (89.9) m-mITT 279/322 (86.6) 270/319 (84.6) Table 7. Clinical Cure Rates By Infecting Pathogen in Microbiologically Evaluable Patients with Complicated Intra-abdominal Infections* Pathogen TYGACIL
n/N (%)Imipenem/Cilastatin
n/N (%)- * Two cIAI pivotal studies and two Resistant Pathogen studies
- † Includes Streptococcus anginosus, Streptococcus intermedius, and Streptococcus constellatus
Citrobacter freundii 12/16 (75.0) 3/4 (75.0) Enterobacter cloacae 15/17 (88.2) 16/17 (94.1) Escherichia coli 284/336 (84.5) 297/342 (86.8) Klebsiella oxytoca 19/20 (95.0) 17/19 (89.5) Klebsiella pneumoniae 42/47 (89.4) 46/53 (86.8) Enterococcus faecalis 29/38 (76.3) 35/47 (74.5) Methicillin-susceptible Staphylococcus aureus (MSSA) 26/28 (92.9) 22/24 (91.7) Methicillin-resistant Staphylococcus aureus (MRSA) 16/18 (88.9) 1/3 (33.3) Streptococcus anginosus grp.† 101/119 (84.9) 60/79 (75.9) Bacteroides fragilis 68/88 (77.3) 59/73 (80.8) Bacteroides thetaiotaomicron 36/41 (87.8) 31/36 (86.1) Bacteroides uniformis 12/17 (70.6) 14/16 (87.5) Bacteroides vulgatus 14/16 (87.5) 4/6 (66.7) Clostridium perfringens 18/19 (94.7) 20/22 (90.9) Peptostreptococcus micros 13/17 (76.5) 8/11 (72.7) 14.3 Community-Acquired Bacterial Pneumonia
TYGACIL was evaluated in adults for the treatment of community-acquired bacterial pneumonia (CABP) in two randomized, double-blind, active-controlled, multinational, multicenter studies (Studies 1 and 2). These studies compared TYGACIL (100 mg intravenous initial dose followed by 50 mg every 12 hours) with levofloxacin (500 mg intravenous every 12 or 24 hours). In Study 1, after at least 3 days of intravenous therapy, a switch to oral levofloxacin (500 mg daily) was permitted for both treatment arms. Total therapy was 7 to 14 days. Patients with community-acquired bacterial pneumonia who required hospitalization and intravenous therapy were enrolled in the studies. The primary efficacy endpoint was the clinical response at the test of cure (TOC) visit in the co-primary populations of the clinically evaluable (CE) and clinical modified intent-to-treat (c-mITT) patients. See Table 8. Clinical cure rates at TOC by pathogen in the microbiologically evaluable patients are presented in Table 9.
Table 8. Clinical Cure Rates from Two Studies in Community-Acquired Bacterial Pneumonia after 7 to 14 Days of Total Therapy TYGACIL*
n/N (%)Levofloxacin†
n/N (%)95% CI‡ - * 100 mg initially, followed by 50 mg every 12 hours
- † Levofloxacin (500 mg intravenous every 12 or 24 hours)
- ‡ 95% confidence interval for the treatment difference
- § After at least 3 days of intravenous therapy, a switch to oral levofloxacin (500 mg daily) was permitted for both treatment arms in Study 1.
Study 1§ CE 125/138 (90.6) 136/156 (87.2) (-4.4, 11.2) c-mITT 149/191 (78) 158/203 (77.8) (-8.5, 8.9) Study 2 CE 128/144 (88.9) 116/136 (85.3) (-5.0, 12.2) c-mITT 170/203 (83.7) 163/200 (81.5) (-5.6, 10.1) Table 9. Clinical Cure Rates By Infecting Pathogen in Microbiologically Evaluable Patients with Community-Acquired Bacterial Pneumonia* Pathogen TYGACIL
n/N (%)Levofloxacin
n/N
(%)- * Two CABP studies
- † Includes cases of concurrent bacteremia [cure rates of 20/22 (90.9%) versus 13/18 (72.2%) for TYGACIL and levofloxacin respectively]
Haemophilus influenzae 14/17 (82.4) 13/16 (81.3) Legionella pneumophila 10/10 (100.0) 6/6 (100.0) Streptococcus pneumoniae (penicillin-susceptible only)† 44/46 (95.7) 39/44 (88.6) To further evaluate the treatment effect of tigecycline, a post-hoc analysis was conducted in CABP patients with a higher risk of mortality, for whom the treatment effect of antibiotics is supported by historical evidence. The higher-risk group included CABP patients from the two studies with any of the following factors:
- Age ≥50 years
- PSI score ≥3
- Streptococcus pneumoniae bacteremia
The results of this analysis are shown in Table 10. Age ≥50 was the most common risk factor in the higher-risk group.
Table 10. Post-hoc Analysis of Clinical Cure Rates in Patients with Community-Acquired Bacterial Pneumonia Based on Risk of Mortality* TYGACIL
n/N (%)Levofloxacin
n/N
(%)95% CI† - * Patients at higher risk of death include patients with any one of the following: ≥50 year of age; PSI score ≥3; or bacteremia due to Streptococcus pneumoniae
- † 95% confidence interval for the treatment difference
- ‡ After at least 3 days of intravenous therapy, a switch to oral levofloxacin (500 mg daily) was permitted for both treatment arms in Study 1.
Study 1‡ CE Higher risk Yes 93/103 (90.3) 84/102 (82.4) (-2.3, 18.2) No 32/35 (91.4) 52/54 (96.3) (-20.8, 7.1) c-mITT Higher risk Yes 111/142 (78.2) 100/134 (74.6) (-6.9, 14) No 38/49 (77.6) 58/69 (84.1) (-22.8, 8.7) Study 2 CE Higher risk Yes 95/107 (88.8) 68/85 (80) (-2.2, 20.3) No 33/37 (89.2) 48/51 (94.1) (-21.1, 8.6) c-mITT Higher risk Yes 112/134 (83.6) 93/120 (77.5) (-4.2, 16.4) No 58/69 (84.1) 70/80 (87.5) (-16.2, 8.8) -
16 HOW SUPPLIED/STORAGE AND HANDLING
TYGACIL (tigecycline) for injection is supplied in a single-dose 5 mL glass vial or 10 mL glass vial, each containing 50 mg tigecycline lyophilized powder for reconstitution.
Supplied:
5 mL - 10 vials/box. NDC: 0008-4990-02
10 mL - 10 vials/box. NDC: 0008-4990-20
Prior to reconstitution, TYGACIL should be stored at 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30°C (59° to 86°F). [See USP Controlled Room Temperature.] The reconstituted solution of TYGACIL may be stored at room temperature (not to exceed 25°C/77°F) for up to 24 hours (up to 6 hours in the vial and the remaining time in the intravenous bag) [see Dosage and Administration (2.1)].
-
17 PATIENT COUNSELING INFORMATION
Tooth Discoloration and Inhibition of Bone Growth
Advise pregnant women that TYGACIL may cause permanent discoloration of deciduous teeth and reversible inhibition of bone growth when administered during the second and third trimesters of pregnancy [see Warnings and Precautions (5.6, 5.7) and Use in Specific Populations (8.1, 8.4)].
Lactation
Advise a woman not to breastfeed for longer than 3 weeks while taking TYGACIL because of the lack of data on effects due to prolonged breastfeeding, and the theoretical risk of dental discoloration and inhibition of bone growth. Women may also consider reducing infant exposure through pumping and discarding breastmilk during and for 9 days after the last dose of tigecycline [see Use in Specific Populations (8.2)].
Diarrhea
Advise patients, their families, or caregivers that diarrhea is a common problem caused by antibacterial drugs, including TYGACIL. Sometimes, frequent watery or bloody diarrhea may occur and may be a sign of a more serious intestinal infection. If severe watery or bloody diarrhea develops, advise patients to contact his or her healthcare provider [see Warnings and Precautions (5.8)].
Development of Resistance
Patients should be counseled that antibacterial drugs including TYGACIL should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When TYGACIL is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by TYGACIL or other antibacterial drugs in the future.
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL - 50 mg Vial Label
NDC: 0008-4990-19
Single Use Vial
Tygacil®
(tigecycline)
for injection50 mg
tigecycline per vial
For I.V. Infusion Only
-
PRINCIPAL DISPLAY PANEL - 50 mg Vial Carton
10 Single Use Vials
NDC: 0008-4990-20
Contains 10 of NDC: 0008-4990-19Rx only
Tygacil®
(tigecycline)
for injection50 mg
tigecycline per vial
Pfizer Injectables
RECONSTITUTED SOLUTION
MUST BE FURTHER DILUTED
FOR I.V. INFUSION
-
INGREDIENTS AND APPEARANCE
TYGACIL
tigecycline injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0008-4990 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TIGECYCLINE (UNII: 70JE2N95KR) (TIGECYCLINE - UNII:70JE2N95KR) TIGECYCLINE 50 mg in 5 mL Inactive Ingredients Ingredient Name Strength HYDROCHLORIC ACID (UNII: QTT17582CB) SODIUM HYDROXIDE (UNII: 55X04QC32I) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) 100 mg in 5 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0008-4990-20 10 in 1 CARTON 06/01/2005 1 NDC: 0008-4990-19 50 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product 2 NDC: 0008-4990-02 10 in 1 CARTON 06/01/2005 2 NDC: 0008-4990-01 50 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021821 06/01/2005 Labeler - Wyeth Pharmaceuticals LLC, a subsidiary of Pfizer Inc. (113008515) Establishment Name Address ID/FEI Business Operations Patheon Italia S.p.A. 338336589 ANALYSIS(0008-4990) , MANUFACTURE(0008-4990) Establishment Name Address ID/FEI Business Operations Pfizer Ireland Pharmaceuticals 985586408 ANALYSIS(0008-4990) Establishment Name Address ID/FEI Business Operations Pharmacia and Upjohn Company LLC 618054084 LABEL(0008-4990) , PACK(0008-4990) Establishment Name Address ID/FEI Business Operations Wyeth Lederle SRL 542812040 ANALYSIS(0008-4990) , LABEL(0008-4990) , MANUFACTURE(0008-4990) , PACK(0008-4990) Establishment Name Address ID/FEI Business Operations Pfizer Ireland Pharmaceuticals 985104227 ANALYSIS(0008-4990) , API MANUFACTURE(0008-4990) Establishment Name Address ID/FEI Business Operations Prime European Therapeuticals S.p.A dba Euticals S.p.A 429351620 ANALYSIS(0008-4990) , API MANUFACTURE(0008-4990)

Trademark Results [Tygacil]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 TYGACIL 78546429 3493859 Dead/Cancelled |
WYETH LLC 2005-01-12 |
 TYGACIL 78530600 3455234 Dead/Cancelled |
WYETH LLC 2004-12-10 |
 TYGACIL 78295227 2993202 Live/Registered |
WYETH LLC 2003-09-03 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.