ORKAMBI- lumacaftor and ivacaftor tablet, film coated ORKAMBI- lumacaftor and ivacaftor granule
ORKAMBI by
Drug Labeling and Warnings
ORKAMBI by is a Prescription medication manufactured, distributed, or labeled by Vertex Pharmaceuticals Incorporated. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ORKAMBI safely and effectively. See full prescribing information for ORKAMBI.
ORKAMBI® (lumacaftor/ivacaftor) tablets, for oral use
ORKAMBI® (lumacaftor/ivacaftor) oral granules
Initial U.S. Approval: 2015INDICATIONS AND USAGE
ORKAMBI is a combination of lumacaftor and ivacaftor, a cystic fibrosis transmembrane conductance regulator (CFTR) potentiator, indicated for the treatment of cystic fibrosis (CF) in patients age 2 years and older who are homozygous for the F508del mutation in the CFTR gene. If the patient's genotype is unknown, an FDA-cleared CF mutation test should be used to detect the presence of the F508del mutation on both alleles of the CFTR gene. (1)
Limitations of Use:
The efficacy and safety of ORKAMBI have not been established in patients with CF other than those homozygous for the F508del mutation. (1)
DOSAGE AND ADMINISTRATION
- Pediatric patients age 2 through 5 years and weighing less than 14 kg: one packet of granules (each containing lumacaftor 100 mg/ivacaftor 125 mg) mixed with 1 teaspoon (5 mL) of soft food or liquid and administered orally every 12 hours with fat-containing food. (2.1, 12.3)
- Pediatric patients age 2 through 5 years and weighing 14 kg or greater: one packet of granules (each containing lumacaftor 150 mg/ivacaftor 188 mg) mixed with 1 teaspoon (5 mL) of soft food or liquid and administered orally every 12 hours with fat-containing food. (2.1, 12.3)
- Pediatric patients age 6 through 11 years: two tablets (each containing lumacaftor 100 mg/ivacaftor 125 mg) taken orally every 12 hours with fat-containing food. (2.1, 12.3)
- Adults and pediatric patients age 12 years and older: two tablets (each containing lumacaftor 200 mg/ivacaftor 125 mg) taken orally every 12 hours with fat-containing food. (2.1, 12.3)
- Reduce dose in patients with moderate or severe hepatic impairment. (2.2, 8.6, 12.3)
- When initiating ORKAMBI in patients taking strong CYP3A inhibitors, reduce ORKAMBI dose for the first week of treatment. (2.3, 7.1, 12.3)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
- None. (4)
WARNINGS AND PRECAUTIONS
- Use in patients with advanced liver disease: ORKAMBI should be used with caution in these patients and only if the benefits are expected to outweigh the risks. If ORKAMBI is used in these patients, they should be closely monitored after the initiation of treatment and the dose should be reduced. Liver function decompensation, including liver failure leading to death, has been reported in CF patients with pre-existing cirrhosis with portal hypertension. (2.2, 5.1, 6.1)
- Liver-related events: Elevated transaminases (ALT/AST) have been observed in some cases associated with elevated bilirubin. Measure serum transaminases and bilirubin before initiating ORKAMBI, every 3 months during the first year of treatment, and annually thereafter. For patients with a history of ALT, AST, or bilirubin elevations, more frequent monitoring should be considered. Interrupt dosing in patients with ALT or AST >5 × upper limit of normal (ULN), or ALT or AST >3 × ULN with bilirubin >2 × ULN. Following resolution, consider the benefits and risks of resuming dosing. (5.2, 6.1)
- Respiratory events: Chest discomfort, dyspnea, and respiration abnormal were observed more commonly during initiation of ORKAMBI. Clinical experience in patients with percent predicted FEV1 (ppFEV1) <40 is limited, and additional monitoring of these patients is recommended during initiation of therapy. (5.3, 6.1)
- Blood pressure: Increased blood pressure has been observed in some patients. Periodically monitor blood pressure in all patients. (5.4, 6.1)
- Drug interactions: Use with sensitive CYP3A substrates or CYP3A substrates with a narrow therapeutic index may decrease systemic exposure of the medicinal products and co-administration is not recommended. Hormonal contraceptives should not be relied upon as an effective method of contraception and their use is associated with increased menstruation-related adverse reactions. Use with strong CYP3A inducers may diminish exposure of ivacaftor, which may diminish its effectiveness; therefore, co-administration is not recommended. (5.5, 6.1, 7, 12.3)
- Cataracts: Non-congenital lens opacities/cataracts have been reported in pediatric patients treated with ORKAMBI and ivacaftor, a component of ORKAMBI. Baseline and follow-up examinations are recommended in pediatric patients initiating ORKAMBI. (5.6)
ADVERSE REACTIONS
The most common adverse reactions to ORKAMBI (occurring in ≥5% of patients with CF homozygous for the F508del mutation in the CFTR gene) were dyspnea, nasopharyngitis, nausea, diarrhea, upper respiratory tract infection, fatigue, respiration abnormal, blood creatine phosphokinase increased, rash, flatulence, rhinorrhea, influenza. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Vertex Pharmaceuticals Incorporated at 1-877-634-8789 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 7/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information in Adults and Children Age 2 Years and Older
2.2 Dosage Adjustment for Patients with Hepatic Impairment
2.3 Dosage Adjustment for Patients Taking CYP3A Inhibitors
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Use in Patients with Advanced Liver Disease
5.2 Liver-related Events
5.3 Respiratory Events
5.4 Effect on Blood Pressure
5.5 Drug Interactions
5.6 Cataracts
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post-marketing Experience
7 DRUG INTERACTIONS
7.1 Inhibitors of CYP3A
7.2 Inducers of CYP3A
7.3 CYP3A Substrates
7.4 CYP2B6 and CYP2C Substrates
7.5 Digoxin and Other P-gp Substrates
7.6 Anti-allergics and Systemic Corticosteroids
7.7 Antibiotics
7.8 Antifungals
7.9 Anti-inflammatories
7.10 Antidepressants
7.11 Hormonal Contraceptives
7.12 Oral Hypoglycemics
7.13 Proton Pump Inhibitors, H2 Blockers, Antacids
7.14 Warfarin
7.15 Concomitant Drugs That Do Not Need Dose Adjustment
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
8.8 Patients with Severe Lung Dysfunction
8.9 Patients After Organ Transplantation
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
ORKAMBI is a combination of lumacaftor and ivacaftor indicated for the treatment of cystic fibrosis (CF) in patients age 2 years and older who are homozygous for the F508del mutation in the CFTR gene. If the patient's genotype is unknown, an FDA-cleared CF mutation test should be used to detect the presence of the F508del mutation on both alleles of the CFTR gene.
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information in Adults and Children Age 2 Years and Older
Table 1: Dosage of ORKAMBI in Patients Age 2 Years and Older Age ORKAMBI Dose Total Daily Dose 2 through 5 years and weighing less than 14 kg One lumacaftor 100 mg/ivacaftor 125 mg packet of granules every 12 hours with fat-containing food. lumacaftor 200 mg/ivacaftor 250 mg 2 through 5 years and weighing 14 kg or greater One lumacaftor 150 mg/ivacaftor 188 mg packet of granules every 12 hours with fat-containing food. lumacaftor 300 mg/ivacaftor 376 mg 6 through 11 years Take two lumacaftor 100 mg/ivacaftor 125 mg tablets every 12 hours with fat-containing food. lumacaftor 400 mg/ivacaftor 500 mg 12 years and older Take two lumacaftor 200 mg/ivacaftor 125 mg tablets every 12 hours with fat-containing food. lumacaftor 800 mg/ivacaftor 500 mg ORKAMBI oral granules
The entire content of each packet of oral granules should be mixed with one teaspoon (5 mL) of age-appropriate soft food or liquid and the mixture completely consumed. Some examples of soft foods include puréed fruits, flavored yogurt or pudding, and milk or juice. Food should be at room temperature or below. Each packet is for single use only. Once mixed, the product has been shown to be stable for one hour, and therefore should be ingested during this period.
A fat-containing meal or snack should be consumed just before or just after dosing for all formulations. Examples of appropriate fat-containing foods include eggs, avocados, nuts, butter, peanut butter, cheese pizza, whole-milk dairy products (such as whole milk, cheese, and yogurt), etc. If a patient misses a dose and remembers the missed dose within 6 hours, the patient should take the dose with fat-containing food. If more than 6 hours elapsed after the usual dosing time, the patient should skip that dose and resume the normal schedule for the following dose. A double dose should not be taken to make up for the forgotten dose [see Clinical Pharmacology (12.3) and Patient Counseling Information (17)].
2.2 Dosage Adjustment for Patients with Hepatic Impairment
For dose adjustment for patients with hepatic impairment, refer to Table 2.
Studies have not been conducted in patients with severe hepatic impairment (Child-Pugh Class C), but exposure is expected to be higher than in patients with moderate hepatic impairment. Therefore, use with caution at a maximum dose of 1 tablet in the morning and 1 tablet in the evening or less frequently, or 1 packet of oral granules once daily or less frequently in patients with severe hepatic impairment after weighing the risks and benefits of treatment [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3), and Patient Counseling Information (17)].
Table 2:Dosage Recommendations for Patients with Hepatic Impairment Age group Morning Evening - * or less frequently.
Mild (Child-Pugh Class A) 6 years and older No dose adjustment No dose adjustment 2 to 5 years old Moderate (Child-Pugh Class B) 6 years and older 2 tablets 1 tablet 2 to 5 years old 1 packet of oral granules 1 packet of oral granules every other day Severe (Child-Pugh Class C) 6 years and older 1 tablet* 1 tablet* 2 to 5 years old 1 packet of oral granules* No dose 2.3 Dosage Adjustment for Patients Taking CYP3A Inhibitors
No dose adjustment is necessary when CYP3A inhibitors are initiated in patients already taking ORKAMBI. However, when initiating ORKAMBI in patients currently taking strong CYP3A inhibitors (e.g., itraconazole), reduce ORKAMBI dose to 1 tablet daily or 1 packet of oral granules every other day for the first week of treatment. Following this period, continue with the recommended daily dose.
If ORKAMBI is interrupted for more than 1 week and then re-initiated while taking strong CYP3A inhibitors, patients should reduce ORKAMBI dose to 1 tablet daily or 1 packet of oral granules every other day for the first week of treatment re-initiation. Following this period, continue with the recommended daily dose.
-
3 DOSAGE FORMS AND STRENGTHS
Tablets: 100 mg lumacaftor and 125 mg ivacaftor; supplied as pink, oval-shaped, film-coated, fixed-dose combination tablets containing 100 mg of lumacaftor and 125 mg of ivacaftor. Each tablet is printed with the characters "1V125" in black ink on one side and plain on the other.
Tablets: 200 mg lumacaftor and 125 mg ivacaftor; supplied as pink, oval-shaped, film-coated, fixed-dose combination tablets containing 200 mg of lumacaftor and 125 mg of ivacaftor. Each tablet is printed with the characters "2V125" in black ink on one side and plain on the other.
Oral granules: Unit-dose packets containing lumacaftor 100 mg/ivacaftor 125 mg or lumacaftor 150 mg/ivacaftor 188 mg per packet; supplied as small, white to off- white granules in unit-dose packets.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Use in Patients with Advanced Liver Disease
Worsening of liver function, including hepatic encephalopathy, in patients with advanced liver disease has been reported. Liver function decompensation, including liver failure leading to death, has been reported in CF patients with pre-existing cirrhosis with portal hypertension while receiving ORKAMBI. Use ORKAMBI with caution in patients with advanced liver disease and only if the benefits are expected to outweigh the risks. If ORKAMBI is used in these patients, they should be closely monitored after the initiation of treatment and the dose should be reduced [see Dosage and Administration (2.2) and Adverse Reactions (6.1)].
5.2 Liver-related Events
Serious adverse reactions related to elevated transaminases have been reported in patients with CF receiving ORKAMBI. In some instances, these elevations have been associated with concomitant elevations in total serum bilirubin.
It is recommended that ALT, AST, and bilirubin be assessed prior to initiating ORKAMBI, every 3 months during the first year of treatment, and annually thereafter. For patients with a history of ALT, AST, or bilirubin elevations, more frequent monitoring should be considered. Patients who develop increased ALT, AST, or bilirubin should be closely monitored until the abnormalities resolve.
Dosing should be interrupted in patients with ALT or AST greater than 5 × upper limit of normal (ULN) when not associated with elevated bilirubin. Dosing should also be interrupted in patients with ALT or AST elevations greater than 3 × ULN when associated with bilirubin elevations greater than 2 × ULN. Following resolution of transaminase elevations, consider the benefits and risks of resuming dosing [see Adverse Reactions (6.1)].
5.3 Respiratory Events
Respiratory events (e.g., chest discomfort, dyspnea, and respiration abnormal) were observed more commonly in patients during initiation of ORKAMBI compared to those who received placebo. These events have led to drug discontinuation and can be serious, particularly in patients with advanced lung disease (percent predicted FEV1 <40). Clinical experience in patients with ppFEV1 <40 is limited, and additional monitoring of these patients is recommended during initiation of therapy [see Adverse Reactions (6.1)].
5.4 Effect on Blood Pressure
Increased blood pressure has been observed in some patients treated with ORKAMBI. Blood pressure should be monitored periodically in all patients being treated with ORKAMBI [see Adverse Reactions (6.1)].
5.5 Drug Interactions
Substrates of CYP3A
Lumacaftor is a strong inducer of CYP3A. Administration of ORKAMBI may decrease systemic exposure of medicinal products that are substrates of CYP3A, which may decrease therapeutic effect. Co-administration with sensitive CYP3A substrates or CYP3A substrates with a narrow therapeutic index is not recommended.
ORKAMBI may substantially decrease hormonal contraceptive exposure, reducing their effectiveness and increasing the incidence of menstruation-associated adverse reactions, e.g., amenorrhea, dysmenorrhea, menorrhagia, menstrual irregular (27% in women using hormonal contraceptives compared with 3% in women not using hormonal contraceptives). Hormonal contraceptives, including oral, injectable, transdermal, and implantable, should not be relied upon as an effective method of contraception when co-administered with ORKAMBI [see Adverse Reactions (6.1), Drug Interactions (7.3, 7.11), and Clinical Pharmacology (12.3)].
Strong CYP3A Inducers
Ivacaftor is a substrate of CYP3A4 and CYP3A5 isoenzymes. Use of ORKAMBI with strong CYP3A inducers, such as rifampin, significantly reduces ivacaftor exposure, which may reduce the therapeutic effectiveness of ORKAMBI. Therefore, co-administration with strong CYP3A inducers (e.g., rifampin, St. John's wort [Hypericum perforatum]) is not recommended [see Drug Interactions (7.2) and Clinical Pharmacology (12.3)].
5.6 Cataracts
Cases of non-congenital lens opacities have been reported in pediatric patients treated with ORKAMBI and ivacaftor, a component of ORKAMBI. Although other risk factors were present in some cases (such as corticosteroid use and exposure to radiation), a possible risk attributable to ivacaftor cannot be excluded [see Use in Specific Populations (8.4)]. Baseline and follow-up ophthalmological examinations are recommended in pediatric patients initiating ORKAMBI treatment.
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label:
- Use in Patients with Advanced Liver Disease [see Warnings and Precautions (5.1)]
- Liver-related Events [see Warnings and Precautions (5.2)]
- Respiratory Events [see Warnings and Precautions (5.3)]
- Effect on Blood Pressure [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The overall safety profile of ORKAMBI is based on the pooled data from 1108 patients with CF 12 years and older who are homozygous for the F508del mutation in the CFTR gene and who received at least one dose of study drug in 2 double-blind, placebo-controlled, Phase 3 clinical trials, each with 24 weeks of treatment (Trials 1 and 2).
In addition, the following clinical trials have been conducted:
- A 24-week open-label trial (Trial 3) in 58 patients with CF aged 6 through 11 years homozygous for the F508del-CFTR mutation.
- A 24-week, placebo-controlled trial (Trial 4) in 204 patients aged 6 through 11 years homozygous for the F508del-CFTR mutation.
- A 24-week, open label trial (Trial 5) in 46 patients aged 12 years and older homozygous for the F508del-CFTR mutation and with advanced lung disease (ppFEV1 <40).
- A 24-week, open-label trial (Trial 6) in 60 patients aged 2 through 5 years homozygous for the F508del-CFTR mutation.
Of the 1108 patients, in the pooled analyses of Trial 1 and Trial 2, 49% were female and 99% were Caucasian; 369 patients received ORKAMBI every 12 hours and 370 received placebo.
The proportion of patients who prematurely discontinued study drug due to adverse events was 5% for patients treated with ORKAMBI and 2% for patients who received placebo.
Serious adverse reactions, whether considered drug-related or not by the investigators, that occurred more frequently in patients treated with ORKAMBI included pneumonia, hemoptysis, cough, increased blood creatine phosphokinase, and transaminase elevations. These occurred in 1% or less of patients.
Table 3 shows adverse reactions occurring in ≥5% of patients with CF ages 12 years and older treated with ORKAMBI who are homozygous for the F508del mutation in the CFTR gene that also occurred at a higher rate than in patients who received placebo in the two double-blind, placebo-controlled trials.
Table 3: Incidence of Adverse Drug Reactions in ≥5% of ORKAMBI-Treated Patients Ages 12 Years and Older Who are Homozygous for the F508del Mutation in the CFTR Gene in 2 Placebo-Controlled Phase 3 Clinical Trials of 24 Weeks Duration Adverse Reaction
(Preferred Term)ORKAMBI
N=369
(%)Placebo
N=370
(%)Dyspnea 48 (13) 29 (8) Nasopharyngitis 48 (13) 40 (11) Nausea 46 (13) 28 (8) Diarrhea 45 (12) 31 (8) Upper respiratory tract infection 37 (10) 20 (5) Fatigue 34 (9) 29 (8) Respiration abnormal 32 (9) 22 (6) Blood creatine phosphokinase increased 27 (7) 20 (5) Rash 25 (7) 7 (2) Flatulence 24 (7) 11 (3) Rhinorrhea 21 (6) 15 (4) Influenza 19 (5) 8 (2) The safety profile from two pediatric trials in CF patients aged 6 through 11 years who are homozygous for the F508del-CFTR mutation, a 24-week, open-label, multicenter Phase 3 safety trial in 58 patients (Trial 3) and a 24-week, placebo-controlled, Phase 3 clinical trial (Trial 4) in 204 patients (103 received lumacaftor 200 mg/ivacaftor 250 mg every 12 hours and 101 received placebo), was similar to that observed in Trials 1 and 2. Adverse reactions that are not listed in Table 3, and that occurred in ≥5% of lumacaftor/ivacaftor-treated patients with an incidence of ≥3% higher than placebo included: productive cough (17.5% vs 5.9%), nasal congestion (16.5% vs 7.9%), headache (12.6% vs 8.9%), abdominal pain upper (12.6% vs 6.9%), and sputum increased (10.7% vs 2.0%).
In a 24-week, open-label, multicenter Phase 3 study in 60 patients aged 2 through 5 years with CF who are homozygous for the F508del-CFTR mutation (Trial 6) the safety profile was similar to that observed in studies in patients aged 6 years and older.
Additional information on selected adverse reactions from trials is detailed below.
Description of Selected Adverse Drug Reactions
Liver-related Adverse Reactions
In Trials 1 and 2, the incidence of maximum transaminase (ALT or AST) levels >8, >5, and >3 × ULN elevations was similar between patients treated with ORKAMBI and those who received placebo. Three patients who received ORKAMBI had liver-related serious adverse reactions, including 2 reported as transaminase elevations and 1 as hepatic encephalopathy, compared to none in the placebo group. Of these three, one had elevated transaminases (>3 × ULN) associated with bilirubin elevation >2 × ULN. Following discontinuation or interruption of ORKAMBI, transaminases decreased to <3 × ULN.
Among 6 patients with pre-existing cirrhosis and/or portal hypertension who received ORKAMBI, worsening liver function with increased ALT, AST, bilirubin, and hepatic encephalopathy was observed in one patient. The event occurred within 5 days of the start of dosing and resolved following discontinuation of ORKAMBI [see Warnings and Precautions (5.1, 5.2)].
During the 24-week, open-label Phase 3 clinical trial in 58 patients aged 6 through 11 years (Trial 3), the incidence of maximum transaminase (ALT or AST) levels >8, >5, and >3 × ULN was 5%, 9%, and 19%. No patients had total bilirubin levels > 2 × ULN. Lumacaftor/ivacaftor dosing was maintained or successfully resumed after interruption in all patients with transaminase elevations, except 1 patient who discontinued treatment permanently.
During the 24 week, placebo-controlled Phase 3 clinical trial in 204 patients aged 6 through 11 years (Trial 4), the incidence of maximum transaminase (ALT or AST) levels >8, >5, and >3 × ULN was 1%, 5%, and 13% in the lumacaftor/ivacaftor patients, and 2%, 3%, and 8% in the placebo treated patients. No patients had total bilirubin levels > 2 × ULN. Two patients in the lumacaftor/ivacaftor group and two patients in the placebo group discontinued treatment permanently due to transaminase elevations.
During the 24-week, open-label Phase 3 clinical study in 60 patients aged 2 through 5 years (Trial 6), the incidence of maximum transaminase (ALT or AST) levels >8, >5, and >3 × ULN was 8.3% (5/60), 11.7% (7/60), and 15.0% (9/60). No patients had total bilirubin levels > 2 × ULN. Three patients discontinued lumacaftor/ivacaftor treatment permanently due to transaminase elevations.
Respiratory Adverse Reactions
In Trials 1 and 2, the incidence of respiratory symptom-related adverse reactions (e.g., chest discomfort, dyspnea, and respiration abnormal) was more common in patients treated with ORKAMBI (22%) compared to patients who received placebo (14%). The incidence of these adverse reactions was more common in patients treated with ORKAMBI with lower pre-treatment FEV1. In patients treated with ORKAMBI, the majority of the events began during the first week of treatment [see Warnings and Precautions (5.3)].
During a 24-week, open label, Phase 3b clinical trial in 46 patients aged 12 years and older (Trial 5) with advanced lung disease (ppFEV1 <40) [mean ppFEV1 29.1 at baseline (range: 18.3 to 42.0)], the incidence of respiratory symptom-related adverse reactions was 65% [see Warnings and Precautions (5.3)].
During the 24-week, open-label Phase 3 clinical trial (Trial 3) in 58 patients aged 6 through 11 years (mean baseline ppFEV1 was 91.4), the incidence of respiratory symptom-related adverse reactions was 3% (2/58).
During the 24 week, placebo-controlled Phase 3 clinical trial (Trial 4) in patients aged 6 through 11 years (mean ppFEV1 89.8 at baseline [range: 48.6 to 119.6]), the incidence of respiratory symptom-related adverse reactions was 11% in lumacaftor/ivacaftor patients and 9% in placebo patients. A decline in ppFEV1 at initiation of therapy was observed during serial post dose spirometry assessments. The absolute change from pre-dose at 4-6 hours post-dose was -7.7 on Day 1 and -1.3 on Day 15 in lumacaftor/ivacaftor patients. The post-dose decline was resolved by Week 16.
Menstrual Abnormalities
In Trials 1 and 2, the incidence of combined menstrual abnormality adverse reactions (e.g., amenorrhea, dysmenorrhea, menorrhagia, menstrual irregular) was more common in female patients treated with ORKAMBI (10%) compared to placebo (2%). These events occurred more frequently in the subset of female patients treated with ORKAMBI who were using hormonal contraceptives (27%) compared to those not using hormonal contraceptives (3%) [see Warnings and Precautions (5.5) and Drug Interactions (7.11)].
Increased Blood Pressure
In Trials 1 and 2, adverse reactions related to increases in blood pressure (e.g., hypertension, blood pressure increased) were reported in 1.1% (4/369) of patients treated with ORKAMBI and in no patients who received placebo.
The proportion of patients who experienced a systolic blood pressure value >140 mmHg or a diastolic blood pressure >90 mmHg on at least two occasions was 3.6% and 2.2% in patients treated with ORKAMBI, respectively, compared with 1.6% and 0.5% in patients who received placebo [see Warnings and Precautions (5.4)].
6.2 Post-marketing Experience
Because post-marketing reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Post-marketing cases of liver function decompensation including liver failure leading to death have been reported in CF patients with pre-existing cirrhosis with portal hypertension who were treated with ORKAMBI [see Warnings and Precautions (5.1)].
-
7 DRUG INTERACTIONS
7.1 Inhibitors of CYP3A
Co-administration of lumacaftor/ivacaftor with itraconazole, a strong CYP3A inhibitor, did not impact the exposure of lumacaftor, but increased ivacaftor exposure by 4.3-fold. Due to the induction effect of lumacaftor on CYP3A, at steady-state, the net exposure of ivacaftor is not expected to exceed that when given in the absence of lumacaftor at a dose of 150 mg every 12 hours (the approved dose of ivacaftor monotherapy). Therefore, no dose adjustment is necessary when CYP3A inhibitors are initiated in patients currently taking ORKAMBI. However, when initiating ORKAMBI in patients taking strong CYP3A inhibitors, reduce the ORKAMBI dose to 1 tablet daily or 1 packet of oral granules every other day (patients aged 2 through 5 years) for the first week of treatment to allow for the steady-state induction effect of lumacaftor. Following this period, continue with the recommended daily dose [see Dosage and Administration (2.3)].
Examples of strong CYP3A inhibitors include:
- ketoconazole, itraconazole, posaconazole, and voriconazole
- telithromycin, clarithromycin.
No dose adjustment is recommended when used with moderate or weak CYP3A inhibitors.
7.2 Inducers of CYP3A
Co-administration of lumacaftor/ivacaftor with rifampin, a strong CYP3A inducer, had minimal effect on the exposure of lumacaftor, but decreased ivacaftor exposure (AUC) by 57%. This may reduce the effectiveness of ORKAMBI. Therefore, co-administration with strong CYP3A inducers, such as rifampin, rifabutin, phenobarbital, carbamazepine, phenytoin, and St. John's wort (Hypericum perforatum), is not recommended [see Warnings and Precautions (5.5) and Clinical Pharmacology (12.3)].
No dose adjustment is recommended when used with moderate or weak CYP3A inducers.
7.3 CYP3A Substrates
Lumacaftor is a strong inducer of CYP3A. Co-administration of lumacaftor with ivacaftor, a sensitive CYP3A substrate, decreased ivacaftor exposure by approximately 80%. Administration of ORKAMBI may decrease systemic exposure of medicinal products which are substrates of CYP3A, thereby decreasing the therapeutic effect of the medicinal product.
Co-administration of ORKAMBI is not recommended with sensitive CYP3A substrates or CYP3A substrates with a narrow therapeutic index [see Warnings and Precautions (5.5) and Clinical Pharmacology (12.3)] such as:
- Benzodiazepines: midazolam, triazolam (consider an alternative to these benzodiazepines).
- Immunosuppressants: cyclosporine, everolimus, sirolimus, and tacrolimus (avoid the use of ORKAMBI).
7.4 CYP2B6 and CYP2C Substrates
In vitro studies suggest that lumacaftor has the potential to induce CYP2B6, CYP2C8, CYP2C9, and CYP2C19; inhibition of CYP2C8 and CYP2C9 has also been observed in vitro. Additionally, in vitro studies suggest that ivacaftor may inhibit CYP2C9. Therefore, concomitant use of ORKAMBI with CYP2B6, CYP2C8, CYP2C9, and CYP2C19 substrates may alter the exposure of these substrates.
7.5 Digoxin and Other P-gp Substrates
Based on in vitro results which showed P-gp inhibition and pregnane-X-receptor (PXR) activation, lumacaftor has the potential to both inhibit and induce P-gp. Additionally, a clinical study with ivacaftor monotherapy showed that ivacaftor is a weak inhibitor of P-gp. Therefore, concomitant use of ORKAMBI with P-gp substrates may alter the exposure of these substrates.
Monitor the serum concentration of digoxin and titrate the digoxin dose to obtain the desired clinical effect.
7.6 Anti-allergics and Systemic Corticosteroids
ORKAMBI may decrease the exposure of montelukast, which may reduce its efficacy. No dose adjustment for montelukast is recommended. Employ appropriate clinical monitoring, as is reasonable, when co-administered with ORKAMBI.
Concomitant use of ORKAMBI may reduce the exposure and effectiveness of prednisone and methylprednisolone. A higher dose of these systemic corticosteroids may be required to obtain the desired clinical effect.
7.7 Antibiotics
Concomitant use of ORKAMBI may decrease the exposure of clarithromycin, erythromycin, and telithromycin, which may reduce the effectiveness of these antibiotics. Consider an alternative to these antibiotics, such as ciprofloxacin, azithromycin, and levofloxacin.
7.8 Antifungals
Concomitant use of ORKAMBI may reduce the exposure and effectiveness of itraconazole, ketoconazole, posaconazole, and voriconazole. Concomitant use of ORKAMBI with these antifungals is not recommended. Monitor patients closely for breakthrough fungal infections if such drugs are necessary. Consider an alternative such as fluconazole.
7.9 Anti-inflammatories
Concomitant use of ORKAMBI may reduce the exposure and effectiveness of ibuprofen. A higher dose of ibuprofen may be required to obtain the desired clinical effect.
7.10 Antidepressants
Concomitant use of ORKAMBI may reduce the exposure and effectiveness of citalopram, escitalopram, and sertraline. A higher dose of these antidepressants may be required to obtain the desired clinical effect.
7.11 Hormonal Contraceptives
ORKAMBI may decrease hormonal contraceptive exposure, reducing the effectiveness. Hormonal contraceptives, including oral, injectable, transdermal, and implantable, should not be relied upon as an effective method of contraception when co-administered with ORKAMBI.
Concomitant use of ORKAMBI with hormonal contraceptives increased the menstrual abnormality events [see Adverse Reactions (6.1)]. Avoid concomitant use unless the benefit outweighs the risks.
7.12 Oral Hypoglycemics
Concomitant use of ORKAMBI may reduce the exposure and effectiveness of repaglinide, and may alter the exposure of sulfonylurea. A dose adjustment may be required to obtain the desired clinical effect. No dose adjustment is recommended for metformin.
7.13 Proton Pump Inhibitors, H2 Blockers, Antacids
ORKAMBI may reduce the exposure and effectiveness of proton pump inhibitors such as omeprazole, esomeprazole, and lansoprazole, and may alter the exposure of ranitidine. A dose adjustment may be required to obtain the desired clinical effect. No dose adjustment is recommended for calcium carbonate antacid.
7.14 Warfarin
ORKAMBI may alter the exposure of warfarin. Monitor the international normalized ratio (INR) when warfarin co-administration with ORKAMBI is required.
7.15 Concomitant Drugs That Do Not Need Dose Adjustment
No dosage adjustment of ORKAMBI or concomitant drug is recommended when ORKAMBI is given with the following: azithromycin, aztreonam, budesonide, ceftazidime, cetirizine, ciprofloxacin, colistimethate, colistin, dornase alfa, fluticasone, ipratropium, levofloxacin, pancreatin, pancrelipase, salbutamol, salmeterol, sulfamethoxazole and trimethoprim, tiotropium, and tobramycin. Based on the metabolism and route of elimination, ORKAMBI is not expected to impact the exposure of these drugs.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are limited and incomplete human data from clinical trials and post-marketing reports on use of ORKAMBI or its individual components, lumacaftor or ivacaftor, in pregnant women to inform a drug-associated risk. In animal reproduction studies, oral administration of lumacaftor to pregnant rats and rabbits during organogenesis demonstrated no teratogenicity or adverse effects on fetal development at doses that produced maternal exposures up to approximately 8 (rats) and 5 (rabbits) times the exposure at the maximum recommended human dose (MRHD). Oral administration of ivacaftor to pregnant rats and rabbits during organogenesis demonstrated no teratogenicity or adverse effects on fetal development at doses that produced maternal exposures up to approximately 7 (rats) and 45 (rabbits) times the exposure at the MRHD. No adverse developmental effects were observed after oral administration of either lumacaftor or ivacaftor to pregnant rats from organogenesis through lactation at doses that produced maternal exposures approximately 8 and 5 times the exposures at the MRHD, respectively (see Data). There are no animal reproduction studies with concomitant administration of lumacaftor and ivacaftor.
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Animal Data
Lumacaftor
In an embryo-fetal development study in pregnant rats dosed during the period of organogenesis from gestation days 7-17, lumacaftor was not teratogenic and did not affect fetal development or survival at exposures up to 8 times the MRHD (on an AUC basis at maternal oral doses up to 2000 mg/kg/day). In an embryo-fetal development study in pregnant rabbits dosed during the period of organogenesis from gestation days 7-19, lumacaftor was not teratogenic and did not affect fetal development or survival at exposures up to 5 times the MRHD (on an AUC basis at maternal oral doses up to 200 mg/kg/day). In a pre- and postnatal development study in pregnant female rats dosed from gestation day 6 through lactation day 20, lumacaftor had no effects on delivery or growth and development of offspring at exposures up to 8 times the MRHD (on an AUC basis at maternal oral doses up to 1000 mg/kg/day). Placental transfer of lumacaftor was observed in pregnant rats and rabbits.
Ivacaftor
In an embryo-fetal development study in pregnant rats dosed during the period of organogenesis from gestation days 7-17, ivacaftor was not teratogenic and did not affect fetal survival at exposures up to 7 times the MRHD (based on summed AUCs for ivacaftor and its metabolites at maternal oral doses up to 200 mg/kg/day). In an embryo-fetal development study in pregnant rabbits dosed during the period of organogenesis from gestation days 7-19, ivacaftor was not teratogenic and did not affect fetal development or survival at exposures up to 45 times the MRHD (on an ivacaftor AUC basis at maternal oral doses up to 100 mg/kg/day). In a pre- and postnatal development study in pregnant female rats dosed from gestation day 7 through lactation day 20, ivacaftor had no effects on delivery or growth and development of offspring at exposures up to 5 times the MRHD (based on summed AUCs for ivacaftor and its metabolites at maternal oral doses up to 100 mg/kg/day). Decreased fetal body weights were observed at a maternally toxic dose that produced exposures 7 times the MRHD (based on summed AUCs for ivacaftor and its metabolites at a maternal oral dose of 200 mg/kg/day). Placental transfer of ivacaftor was observed in pregnant rats and rabbits.
8.2 Lactation
Risk Summary
There is no information regarding the presence of lumacaftor or ivacaftor in human milk, the effects on the breastfed infant, or the effects on milk production. Both lumacaftor and ivacaftor are excreted into the milk of lactating rats; however, due to species-specific differences in lactation physiology, animal lactation data may not reliably predict levels in human milk (see Data). The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ORKAMBI and any potential adverse effects on the breastfed child from ORKAMBI or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
ORKAMBI may decrease hormonal contraceptive exposure, reducing the effectiveness. Hormonal contraceptives, including oral, injectable, transdermal, and implantable, should not be relied upon as an effective method of contraception when co-administered with ORKAMBI [see Warnings and Precautions (5.5) and Drug Interactions (7.11)].
8.4 Pediatric Use
The efficacy of ORKAMBI in children ages 2 through 11 years is extrapolated from efficacy in patients ages 12 years and older homozygous for the F508del mutation in the CFTR gene with support from population pharmacokinetic analyses showing similar drug exposure levels in patients ages 12 years and older and in children ages 2 through 11 years [see Clinical Pharmacology (12.3)].
Safety data were obtained from a 24-week, open-label, Phase 3 clinical trial in 58 patients aged 6 through 11 years, mean age 9 years (Trial 3) and a 24-week, placebo-controlled, Phase 3 clinical trial in 204 patients aged 6 through 11 years (Trial 4). Trial 3 evaluated subjects with a screening ppFEV1 ≥40 [mean ppFEV1 91.4 at baseline (range: 55 to 122.7)]. Trial 4 evaluated subjects with a screening ppFEV1 ≥ 70 (mean ppFEV1 89.8 at baseline [range: 48.6 to 119.6]). The safety profile of ORKAMBI in children 6 through 11 years of age was similar to that in patients 12 years and older [see Adverse Reactions (6.1)].
In Trial 3, spirometry (ppFEV1) was assessed as a planned safety endpoint. The within-group LS mean absolute change from baseline in ppFEV1 at Week 24 was 2.5 percentage points. At the Week 26 safety follow-up visit (following a planned discontinuation) ppFEV1 was also assessed. The within-group LS mean absolute change in ppFEV1 from Week 24 at Week 26 was -3.2 percentage points.
Additional safety data were obtained from Trial 6, a 24-week, open-label, Phase 3 clinical trial in 60 patients aged 2 through 5 years at screening (mean age at baseline 3.7 years). The safety profile in Trial 6 was similar to that in patients aged 6 years and older [see Adverse Reactions (6.1)].
The safety and efficacy of ORKAMBI in patients with CF younger than age 2 years have not been established. Cases of non-congenital lens opacities have been reported in pediatric patients treated with ORKAMBI and ivacaftor, a component of ORKAMBI. Although other risk factors were present in some cases (such as corticosteroid use and exposure to radiation), a possible risk attributable to ivacaftor cannot be excluded [see Warnings and Precautions (5.6)].
Juvenile Animal Toxicity Data
In a juvenile toxicology study in which ivacaftor was administered to rats from postnatal days 7 to 35, cataracts were observed at all dose levels, ranging from 0.3 to 2 times the MRHD (based on summed AUCs for ivacaftor and its metabolites at oral doses of 10-50 mg/kg/day). This finding has not been observed in older animals.
8.5 Geriatric Use
CF is largely a disease of children and young adults. Clinical trials of ORKAMBI did not include sufficient numbers of patients 65 years of age and over to determine whether they respond differently from younger patients.
8.6 Hepatic Impairment
No dose adjustment is necessary for patients with mild hepatic impairment (Child-Pugh Class A). A dose reduction to 2 tablets in the morning and 1 tablet in the evening is recommended for patients 6 years and older with moderate hepatic impairment (Child-Pugh Class B). A dose reduction to 1 packet of oral granules in the morning daily and 1 packet of oral granules in the evening every other day is recommended for patients 2 to 5 years old with moderate hepatic impairment (Child-Pugh Class B).
Studies have not been conducted in patients with severe hepatic impairment (Child-Pugh Class C), but exposure is expected to be higher than in patients with moderate hepatic impairment. Therefore, use with caution at a maximum dose of 1 tablet in the morning and 1 tablet in the evening or less frequently, or 1 packet of oral granules once daily or less frequently in patients with severe hepatic impairment after weighing the risks and benefits of treatment [see Warnings and Precautions (5.1), Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Patient Counseling Information (17)].
8.7 Renal Impairment
ORKAMBI has not been studied in patients with mild, moderate, or severe renal impairment or in patients with end-stage renal disease. No dose adjustment is necessary for patients with mild to moderate renal impairment. Caution is recommended while using ORKAMBI in patients with severe renal impairment (creatinine clearance less than or equal to 30 mL/min) or end-stage renal disease.
8.8 Patients with Severe Lung Dysfunction
The Phase 3 trials (Trials 1 and 2) included 29 patients receiving ORKAMBI with ppFEV1 <40 at baseline. The treatment effect in this subgroup was comparable to that observed in patients with ppFEV1 ≥40.
8.9 Patients After Organ Transplantation
ORKAMBI has not been studied in patients with CF who have undergone organ transplantation. Use in transplanted patients is not recommended due to potential drug-drug interactions [see Drug Interactions (7.3)].
-
10 OVERDOSAGE
There have been no reports of overdose with ORKAMBI.
The highest repeated dose was lumacaftor 1000 mg once daily/ivacaftor 450 mg q12h administered to 49 healthy subjects for 7 days in a trial evaluating the effect of ORKAMBI on electrocardiograms (ECGs). Adverse events reported at an increased incidence of ≥5% compared to the lumacaftor 600 mg/ivacaftor 250 mg dosing period and placebo included: headache (29%), transaminase increased (18%), and generalized rash (10%).
No specific antidote is available for overdose with ORKAMBI. Treatment of overdose consists of general supportive measures including monitoring of vital signs and observation of the clinical status of the patient.
-
11 DESCRIPTION
The active ingredients in ORKAMBI tablets are lumacaftor, which has the following chemical name: 3-[6-({[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropyl]carbonyl}amino)-3-methylpyridin-2-yl]benzoic acid, and ivacaftor, a CFTR potentiator, which has the following chemical name: N-(2,4-di-tert-butyl-5-hydroxyphenyl)-1,4-dihydro-4-oxoquinoline-3-carboxamide. The molecular formula for lumacaftor is C24H18F2N2O5 and for ivacaftor is C24H28N2O3. The molecular weights for lumacaftor and ivacaftor are 452.41 and 392.49, respectively. The structural formulas are:
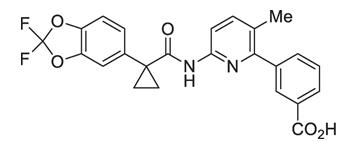
lumacaftor
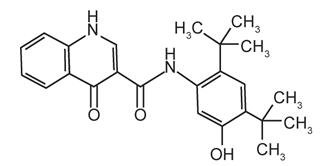
ivacaftor
Lumacaftor is a white to off-white powder that is practically insoluble in water (0.02 mg/mL). Ivacaftor is a white to off-white powder that is practically insoluble in water (<0.05 microgram/mL).
ORKAMBI is available as a pink, oval-shaped, film-coated tablet for oral administration containing 200 mg of lumacaftor and 125 mg of ivacaftor. Each ORKAMBI tablet contains 200 mg of lumacaftor and 125 mg of ivacaftor, and the following inactive ingredients: cellulose, microcrystalline; croscarmellose sodium; hypromellose acetate succinate; magnesium stearate; povidone; and sodium lauryl sulfate. The tablet film coat contains carmine, FD&C Blue #1, FD&C Blue #2, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide. The printing ink contains ammonium hydroxide, iron oxide black, propylene glycol, and shellac.
ORKAMBI is also available as a pink, oval-shaped, film-coated tablet for oral administration containing 100 mg of lumacaftor and 125 mg of ivacaftor. Each ORKAMBI tablet contains 100 mg of lumacaftor and 125 mg of ivacaftor, and the following inactive ingredients: cellulose, microcrystalline; croscarmellose sodium; hypromellose acetate succinate; magnesium stearate; povidone; and sodium lauryl sulfate. The tablet film coat contains carmine, FD&C Blue #1, FD&C Blue #2, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide. The printing ink contains ammonium hydroxide, iron oxide black, propylene glycol, and shellac.
ORKAMBI is also available as white to off-white granules for oral administration and enclosed in a unit-dose packet containing lumacaftor 100 mg/ivacaftor 125 mg or lumacaftor 150 mg/ivacaftor 188 mg per packet. Each unit-dose packet of ORKAMBI oral granules contains lumacaftor 100 mg/ivacaftor 125 mg or lumacaftor 150 mg/ivacaftor 188 mg per packet and the following inactive ingredients: cellulose, microcrystalline; croscarmellose sodium; hypromellose acetate succinate; povidone; sodium lauryl sulfate.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The CFTR protein is a chloride channel present at the surface of epithelial cells in multiple organs. The F508del mutation results in protein misfolding, causing a defect in cellular processing and trafficking that targets the protein for degradation and therefore reduces the quantity of CFTR at the cell surface. The small amount of F508del-CFTR that reaches the cell surface is less stable and has low channel-open probability (defective gating activity) compared to wild-type CFTR protein.
Lumacaftor improves the conformational stability of F508del-CFTR, resulting in increased processing and trafficking of mature protein to the cell surface. Ivacaftor is a CFTR potentiator that facilitates increased chloride transport by potentiating the channel-open probability (or gating) of the CFTR protein at the cell surface. In vitro studies have demonstrated that both lumacaftor and ivacaftor act directly on the CFTR protein in primary human bronchial epithelial cultures and other cell lines harboring the F508del-CFTR mutation to increase the quantity, stability, and function of F508del-CFTR at the cell surface, resulting in increased chloride ion transport. In vitro responses do not necessarily correspond to in vivo pharmacodynamic response or clinical benefit.
12.2 Pharmacodynamics
Sweat Chloride Evaluation
Changes in sweat chloride in response to relevant doses of lumacaftor alone or in combination with ivacaftor were evaluated in a double-blind, placebo-controlled, Phase 2 clinical trial in patients with CF 18 years of age and older either homozygous or heterozygous for the F508del mutation. In that trial, 10 patients (homozygous for F508del) completed dosing with lumacaftor alone 400 mg q12h for 28 days followed by the addition of ivacaftor 250 mg q12h for an additional 28 days and 25 patients (homozygous or heterozygous for F508del) completed dosing with placebo. The treatment difference between lumacaftor 400 mg q12h alone and placebo evaluated as mean change in sweat chloride from baseline to Day 28 compared to placebo was -8.2 mmol/L (95% CI -14, -2). The treatment difference between the combination of lumacaftor 400 mg/ivacaftor 250 mg q12h and placebo evaluated as mean change in sweat chloride from baseline to Day 56 compared to placebo was -11 mmol/L (95% CI -18, -4).
Changes in sweat chloride in response to lumacaftor/ivacaftor were also evaluated in a 24-week, open-label Phase 3 clinical trial (Trial 3) in 58 patients with CF, aged 6 through 11 years (homozygous for F508del) who received lumacaftor 200 mg/ivacaftor 250 mg q12h for 24 weeks. Patients treated with lumacaftor/ivacaftor had a reduction in sweat chloride at Day 15 that was sustained through Week 24. The within-group LS mean absolute change from baseline in sweat chloride was -20.4 mmol/L at Day 15 and -24.8 mmol/L at Week 24. In addition, sweat chloride was also assessed after a 2-week washout period to evaluate the off-drug response. The within-group LS mean absolute change in sweat chloride from Week 24 at Week 26 following the 2-week washout period was 21.3 mmol/L.
Changes in sweat chloride in response to lumacaftor/ivacaftor were also evaluated in a 24-week, open-label Phase 3 clinical study (Trial 6) in 60 patients with CF, aged 2 through 5 years (homozygous for F508del) who received either lumacaftor 100 mg/ivacaftor 125 mg every 12 hours or lumacaftor 150 mg/ivacaftor 188 mg every 12 hours for 24 weeks. Treatment with lumacaftor/ivacaftor demonstrated a reduction in sweat chloride at Week 4 that was sustained through Week 24. The mean absolute change from baseline in sweat chloride was –31.7 mmol/L (95% CI: -35.7, -27.6) at Week 24. In addition, sweat chloride was also assessed after a 2-week washout period to evaluate the off-drug response. The mean absolute change in sweat chloride from Week 24 at Week 26 following the 2-week washout period was an increase of 33.0 mmol/L (95% CI: 28.9, 37.1; P<0.0001).
There was no direct correlation between decrease in sweat chloride levels and improvement in lung function (ppFEV1).
Cardiac Electrophysiology
The effect of multiple doses of lumacaftor 600 mg once daily/ivacaftor 250 mg q12h and lumacaftor 1000 mg once daily/ivacaftor 450 mg q12h on QTc interval was evaluated in a randomized, placebo- and active-controlled (400 mg moxifloxacin), parallel, thorough QT study in 168 healthy subjects. No meaningful changes in QTc interval were observed with either lumacaftor 600 mg once daily/ivacaftor 250 mg q12h and lumacaftor 1000 mg once daily/ivacaftor 450 mg q12h dose groups. A maximum decrease in mean heart rate of up to 8 beats per minute (bpm) from baseline was observed with lumacaftor/ivacaftor treatment. In Trials 1 and 2, a similar decrease in heart rate was observed in patients during initiation of ORKAMBI (lumacaftor 400 mg/ivacaftor 250 mg q12h).
12.3 Pharmacokinetics
The exposure (AUC) of lumacaftor is approximately 2-fold higher in healthy adult volunteers compared to exposure in patients with CF. The exposure of ivacaftor is similar between healthy adult volunteers and patients with CF. After twice-daily dosing, steady-state plasma concentrations of lumacaftor and ivacaftor in healthy subjects were generally reached after approximately 7 days of treatment, with an accumulation ratio of approximately 1.9 for lumacaftor. The steady-state exposure of ivacaftor is lower than that of Day 1 due to the CYP3A induction effect of lumacaftor.
Table 4: Mean (SD) Pharmacokinetic Parameters of Lumacaftor and Ivacaftor at Steady State in Subjects with CF Drug Cmax
(μg/mL)t½*
(h)AUC0-12h
(μg∙h/mL)- * Based on lumacaftor 200 mg q12h/ivacaftor 250 mg q12h studied in healthy subjects
Lumacaftor 400 mg q12h/ Lumacaftor 25.0 (7.96) 25.2 (9.94) 198 (64.8) Ivacaftor 250 mg q12h Ivacaftor 0.602 (0.304) 9.34 (3.81) 3.66 (2.25) Absorption
When a single dose of lumacaftor/ivacaftor was administered with fat-containing foods, lumacaftor exposure was approximately 2 times higher and ivacaftor exposure was approximately 3 times higher than when taken in a fasting state.
Following multiple oral dose administration of lumacaftor in combination with ivacaftor, the exposure of lumacaftor generally increased proportional to dose over the range of 200 mg every 24 hours to 400 mg every 12 hours. The median (range) tmax of lumacaftor is approximately 4.0 hours (2.0; 9.0) in the fed state.
Following multiple oral dose administration of ivacaftor in combination with lumacaftor, the exposure of ivacaftor generally increased with dose from 150 mg every 12 hours to 250 mg every 12 hours. The median (range) tmax of ivacaftor is approximately 4.0 hours (2.0; 6.0) in the fed state.
Distribution
Lumacaftor is approximately 99% bound to plasma proteins, primarily to albumin. After oral administration of 200 mg every 24 hours for 28 days to patients with CF in a fed state, the mean (±SD) for apparent volumes of distribution was 86.0 (69.8) L.
Ivacaftor is approximately 99% bound to plasma proteins, primarily to alpha 1-acid glycoprotein and albumin.
Elimination
The half-life of lumacaftor is approximately 26 hours in patients with CF. The typical apparent clearance, CL/F (CV), of lumacaftor was estimated to be 2.38 L/hr (29.4%) for patients with CF. The half-life of ivacaftor when given with lumacaftor is approximately 9 hours in healthy subjects. The typical CL/F (CV) of ivacaftor when given in combination with lumacaftor was estimated to be 25.1 L/hr (40.5%) for patients with CF.
Metabolism
Lumacaftor is not extensively metabolized in humans with the majority of lumacaftor excreted unchanged in the feces. In vitro and in vivo data indicate that lumacaftor is mainly metabolized via oxidation and glucuronidation.
Ivacaftor is extensively metabolized in humans. In vitro and in vivo data indicate that ivacaftor is primarily metabolized by CYP3A. M1 and M6 are the two major metabolites of ivacaftor in humans.
Excretion
Following oral administration of lumacaftor, the majority of lumacaftor (51%) is excreted unchanged in the feces. There was minimal elimination of lumacaftor and its metabolites in urine (only 8.6% of total radioactivity was recovered in the urine with 0.18% as unchanged parent).
Following oral administration of ivacaftor alone, the majority of ivacaftor (87.8%) is eliminated in the feces after metabolic conversion. There was minimal elimination of ivacaftor and its metabolites in urine (only 6.6% of total radioactivity was recovered in the urine).
Specific Populations
Age: Pediatric Population
The following conclusions about exposures between adults and the pediatric population are based on population pharmacokinetics (PK) analyses:
Pediatric patients 2 through 5 years of age
Following oral administration of ORKAMBI oral granules, lumacaftor 100 mg/ivacaftor 125 mg every 12 hours, the mean lumacaftor (±SD) AUCss was 180 (45.5) µg/mL*h and is comparable to the mean AUCss in patients 12 years and older administered ORKAMBI tablets. The mean ivacaftor (±SD) AUCss was 5.92 (4.61) µg/mL*h and is comparable to the mean AUCss in patients 12 years and older administered ORKAMBI tablets. Following oral administration of ORKAMBI oral granules, lumacaftor 150 mg/ivacaftor 188 mg every 12 hours, the mean lumacaftor (±SD) AUCss was 217 (48.6) µg/mL*h and is comparable to the mean AUCss in patients 12 years and older administered ORKAMBI tablets. The mean ivacaftor (±SD) AUCss was 5.90 (1.93) µg/mL*h and is comparable to the mean AUCss in patients 12 years and older administered ORKAMBI tablets [see Use in Specific Populations (8.4)].
Pediatric patients 6 through 11 years of age
Following oral administration of ORKAMBI tablets, lumacaftor 200 mg/ivacaftor 250 mg every 12 hours, the mean lumacaftor (±SD) AUCss was 203 (57.4) µg/mL*h and is comparable to the mean AUCss in patients 12 years and older administered ORKAMBI tablets, lumacaftor 400 mg/ivacaftor 250 mg every 12 hours. The mean ivacaftor (±SD) AUCss was 5.26 (3.08) µg/mL*h and is comparable to the mean AUCss in patients 12 years and older administered ORKAMBI tablets, lumacaftor 400 mg/ivacaftor 250 mg every 12 hours [see Use in Specific Populations (8.4)].
Pediatric patients 12 to less than 18 years of age
Following oral administration of ORKAMBI tablets, lumacaftor 400 mg/ivacaftor 250 mg every 12 hours, the mean lumacaftor (±SD) AUCss was 241 (61.4) µg/mL*h and is comparable to the mean AUCss in adult patients administered ORKAMBI tablets, lumacaftor 400 mg/ivacaftor 250 mg every 12 hours. The mean ivacaftor (±SD) AUCss was 3.90 (1.56) µg/mL*h and is comparable to the mean AUCss in adult patients administered ORKAMBI tablets, lumacaftor 400 mg/ivacaftor 250 mg every 12 hours [see Use in Specific Populations (8.4)].
Sex
The pharmacokinetics of ORKAMBI was evaluated using a population PK analysis of data from clinical studies of lumacaftor given in combination with ivacaftor. Results indicate no clinically relevant difference in pharmacokinetic parameters for lumacaftor and ivacaftor between males and females.
Renal Impairment
Pharmacokinetic studies have not been performed with ORKAMBI in patients with renal impairment [see Use in Specific Populations (8.7)].
Hepatic Impairment
Following multiple doses of lumacaftor/ivacaftor for 10 days, subjects with moderately impaired hepatic function (Child-Pugh Class B, score 7 to 9) had approximately 50% higher exposures (AUC0-12h) and approximately 30% higher Cmax for both lumacaftor and ivacaftor compared with healthy subjects matched for demographics. Pharmacokinetic studies have not been conducted in patients with mild (Child-Pugh Class A, score 5 to 6) or severe hepatic impairment (Child-Pugh Class C, score 10 to 15) receiving ORKAMBI [see Dosage and Administration (2.2), Warnings and Precautions (5.1), Adverse Reactions (6), and Use in Specific Populations (8.6)].
Drug Interaction Studies
Drug interaction studies were performed with lumacaftor/ivacaftor and other drugs likely to be co-administered or drugs commonly used as probes for pharmacokinetic interaction studies [see Drug Interactions (7)].
Potential for Lumacaftor/Ivacaftor to Affect Other Drugs
Lumacaftor is a strong inducer of CYP3A. Co-administration of lumacaftor with ivacaftor, a sensitive CYP3A substrate, decreased ivacaftor exposure by 80%. Ivacaftor is a weak inhibitor of CYP3A when given as monotherapy. The net effect of lumacaftor/ivacaftor therapy is strong CYP3A induction [see Drug Interactions (7.3)].
Based on in vitro results which showed P-gp inhibition and PXR activation, lumacaftor has the potential to both inhibit and induce P-gp. A clinical study with ivacaftor monotherapy showed that ivacaftor is a weak inhibitor of P-gp. Therefore, concomitant use of ORKAMBI with P-gp substrates may alter the exposure of these substrates [see Drug Interactions (7.5)].
In vitro studies suggest that lumacaftor has the potential to induce CYP2B6, CYP2C8, CYP2C9, and CYP2C19; inhibition of CYP2C8 and CYP2C9 has also been observed in vitro. In vitro studies suggest that ivacaftor may inhibit CYP2C9. Therefore, concomitant use of ORKAMBI with CYP2B6, CYP2C8, CYP2C9, and CYP2C19 substrates may alter the exposure of these substrates [see Drug Interactions (7.4)].
Potential for Other Drugs to Affect Lumacaftor/Ivacaftor
Lumacaftor exposure is not affected by concomitant CYP3A inducers or inhibitors. Exposure of ivacaftor when given in combination with lumacaftor is reduced by concomitant CYP3A inducers and increased by concomitant CYP3A inhibitors [see Dosage and Administration (2.3), Warnings and Precautions (5.5), and Drug Interactions (7)].
The effects of co-administered drugs on the exposure of lumacaftor and ivacaftor are shown in Table 5 [see Dosage and Administration (2.3), Warnings and Precautions (5.5), and Drug Interactions (7)].
Table 5: Impact of Other Drugs on Lumacaftor 200 mg q12h/Ivacaftor 250 mg q12h Co-administered Drug Dose of Co-administered Drug Effect on PK* Mean Ratio (90% CI) of Lumacaftor and Ivacaftor
No Effect=1.0AUC Cmax CI = Confidence interval; PK = Pharmacokinetics - * ↑ = increase, ↓ = decrease, ↔ = no change.
- † The net exposure of ivacaftor is not expected to exceed that when given in the absence of lumacaftor at a dose of 150 mg every 12 hours, the approved dose of ivacaftor monotherapy.
CYP3A inhibitor:
itraconazole200 mg once daily ↔ Lumacaftor 0.97
(0.91, 1.02)0.99
(0.92, 1.05)↑ Ivacaftor 4.30†
(3.78, 4.88)3.64†
(3.19, 4.17)CYP3A inducer:
rifampin600 mg once daily ↔ Lumacaftor 0.87
(0.81, 0.93)0.96
(0.87, 1.05)↓ Ivacaftor 0.43
(0.38, 0.49)0.50
(0.43, 0.58)Other:
ciprofloxacin750 mg q12h ↔ Lumacaftor 0.86
(0.79, 0.95)0.88
(0.80, 0.97)↔ Ivacaftor 1.29
(1.12, 1.48)1.29
(1.11, 1.49) -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No studies of carcinogenicity, mutagenicity, or impairment of fertility were conducted with ORKAMBI; however, studies are available for individual components, lumacaftor and ivacaftor, as described below.
Lumacaftor
A two-year study in Sprague-Dawley rats and a 26-week study in transgenic Tg.rasH2 mice were conducted to assess carcinogenic potential of lumacaftor. No evidence of tumorigenicity was observed in rats at lumacaftor oral doses up to 1000 mg/kg/day (approximately 5 and 13 times the MRHD on a lumacaftor AUC basis in males and females, respectively). No evidence of tumorigenicity was observed in Tg.rasH2 mice at lumacaftor oral doses up to 1500 and 2000 mg/kg/day in female and male mice, respectively. Lumacaftor was negative for genotoxicity in the following assays: Ames test for bacterial gene mutation, in vitro chromosomal aberration assay in Chinese hamster ovary cells, and in vivo mouse micronucleus test.
Lumacaftor had no effects on fertility and reproductive performance indices in male and female rats at an oral dose of 1000 mg/kg/day (approximately 3 and 8 times, respectively, the MRHD on a lumacaftor AUC basis).
Ivacaftor
Two-year studies were conducted in mice and rats to assess carcinogenic potential of ivacaftor. No evidence of tumorigenicity was observed in mice and rats at ivacaftor oral doses up to 200 mg/kg/day and 50 mg/kg/day, respectively (approximately equivalent to 3 and 10 times the MRHD based on summed AUCs of ivacaftor and its metabolites).
Ivacaftor was negative for genotoxicity in the following assays: Ames test for bacterial gene mutation, in vitro chromosomal aberration assay in Chinese hamster ovary cells, and in vivo mouse micronucleus test.
Ivacaftor impaired fertility and reproductive performance indices in male and female rats at an oral dose of 200 mg/kg/day (approximately 15 and 7 times the MRHD based on summed AUCs of ivacaftor and its metabolites). Increases in prolonged diestrus were observed in females at 200 mg/kg/day. Ivacaftor also increased the number of females with all nonviable embryos and decreased corpora lutea, implantations, and viable embryos in rats at 200 mg/kg/day (approximately 7 times the MRHD based on summed AUCs of ivacaftor and its metabolites) when dams were dosed prior to and during early pregnancy. These impairments of fertility and reproductive performance in male and female rats at 200 mg/kg/day were attributed to severe toxicity. No effects on male or female fertility and reproductive performance indices were observed at an oral dose of ≤100 mg/kg/day (approximately 8 and 5 times the MRHD based on summed AUCs of ivacaftor and its metabolites).
-
14 CLINICAL STUDIES
Dose Ranging
Dose ranging for the clinical program consisted primarily of one double-blind, placebo-controlled, multiple-cohort trial which included 97 Caucasian patients with CF (homozygous for the F508del mutation) 18 years of age and older with a screening ppFEV1 ≥40. In the trial, 76 patients (homozygous for the F508del mutation) were randomized to receive lumacaftor alone at once-daily doses of 200 mg, 400 mg, or 600 mg or 400 mg q12h for 28 days followed by the addition of ivacaftor 250 mg q12h and 27 patients (homozygous or heterozygous for the F508del mutation) received placebo. During the initial 28-day lumacaftor monotherapy period, treatment with lumacaftor demonstrated a dose-dependent decrease in ppFEV1 compared to placebo. Changes from Day 1 at Day 28 in ppFEV1 compared to placebo were 0.24, -1.4, -2.7, and -4.6 for the 200 mg once daily, 400 mg once daily, 600 mg once daily, and 400 mg q12h lumacaftor doses, respectively. Following the addition of ivacaftor 250 mg q12h, the changes from Day 1 at Day 56 in ppFEV1 compared to placebo were 3.8, 2.7, 5.6, and 4.2, respectively.
Sweat chloride was also assessed in this trial. Following the initial 28 days of lumacaftor monotherapy, the changes from Day 1 at Day 28 in sweat chloride compared to placebo were -4.9, -8.3, -6.1, and -8.2 mmol/L for the 200 mg once daily, 400 mg once daily, 600 mg once daily, and 400 mg q12h lumacaftor doses, respectively. Following the addition of ivacaftor 250 mg q12h, the changes from Day 1 at Day 56 in sweat chloride compared to placebo were -5.0, -9.8, -9.5, and -11 mmol/L, respectively.
These data supported the evaluation of lumacaftor 400 mg/ivacaftor 250 mg q12h (ORKAMBI) and lumacaftor 600 mg once daily/ivacaftor 250 mg q12h in the confirmatory trials.
Confirmatory
The efficacy of ORKAMBI in patients with CF who are homozygous for the F508del mutation in the CFTR gene was evaluated in two randomized, double-blind, placebo-controlled, 24-week clinical trials (Trials 1 and 2) in 1108 clinically stable patients with CF of whom 369 patients received ORKAMBI twice daily.
Trial 1 evaluated 549 patients with CF who were aged 12 years and older (mean age 25.1 years) with ppFEV1 at screening between 40-90 [mean ppFEV1 60.7 at baseline (range: 31.1 to 94.0)]. Trial 2 evaluated 559 patients aged 12 years and older (mean age 25.0 years) with ppFEV1 at screening between 40-90 [mean ppFEV1 60.5 at baseline (range: 31.3 to 99.8)]. Patients with a history of colonization with organisms such as Burkholderia cenocepacia, Burkholderia dolosa, or Mycobacterium abscessus, or who had 3 or more abnormal liver function tests (ALT, AST, AP, GGT ≥3 × the ULN or total bilirubin ≥2 × the ULN) were excluded.
Patients in both trials were randomized 1:1:1 to receive either ORKAMBI (lumacaftor 400 mg q12h/ivacaftor 250 mg q12h; or lumacaftor 600 mg once daily/ivacaftor 250 mg q12h) or placebo. Patients took the study drug with fat-containing food for 24 weeks in addition to their prescribed CF therapies (e.g., bronchodilators, inhaled antibiotics, dornase alfa, and hypertonic saline).
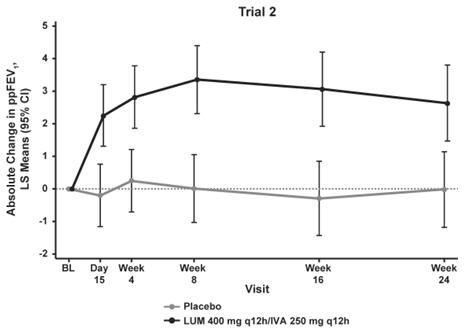
The primary efficacy endpoint in both trials was change in lung function as determined by absolute change from baseline in ppFEV1 at Week 24, assessed as the average of the treatment effects at Week 16 and at Week 24. In both trials, treatment with ORKAMBI resulted in a statistically significant improvement in ppFEV1. The treatment difference between ORKAMBI and placebo for the mean absolute change in ppFEV1 from baseline at Week 24 (assessed as the average of the treatment effects at Week 16 and at Week 24) was 2.6 percentage points [95% CI (1.2, 4.0)] in Trial 1 (P=0.0003) and 3.0 percentage points [95% CI (1.6, 4.4)] in Trial 2 (P<0.0001). These changes persisted throughout the 24-week treatment period (Figure 1). Improvements in ppFEV1 were observed regardless of age, disease severity, sex, and geographic region.
Figure 1. Absolute Change From Baseline at Each Visit in Percent Predicted FEV1 in Trial 1 and Trial 2. 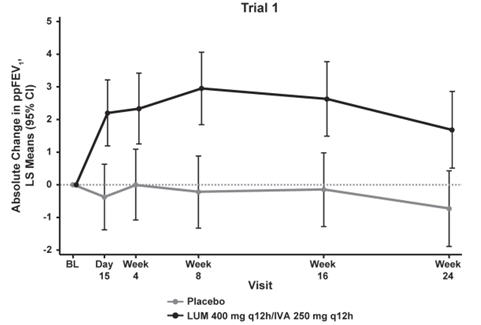
LS = least squares; q12h = every 12 hours Key secondary efficacy variables included relative change from baseline in ppFEV1 at Week 24, assessed as the average of the treatment effects at Week 16 and at Week 24; absolute change from baseline in BMI at Week 24; absolute change from baseline in Cystic Fibrosis Questionnaire-Revised (CFQ-R) Respiratory Domain score at Week 24, a measure of respiratory symptoms relevant to patients with CF such as cough, sputum production, and difficulty breathing; proportion of patients achieving ≥5% relative change from baseline in ppFEV1 using the average of Week 16 and Week 24; and number of pulmonary exacerbations through Week 24. For the purposes of these trials, a pulmonary exacerbation was defined as a change in antibiotic therapy (IV, inhaled, or oral) as a result of 4 or more of 12 pre-specified sino-pulmonary signs/symptoms.
Table 6: Summary of Other Efficacy Endpoints in Trials 1 and 2* Trial 1 Trial 2 Placebo
(n=184)ORKAMBI
LUM 400 mg q12h/IVA 250 mg q12h
(n=182)Placebo
(n=187)ORKAMBI
LUM 400 mg q12h/IVA 250 mg q12h
(n=187)- * In each trial, a hierarchical testing procedure was performed within each active treatment arm for primary and secondary endpoints vs. placebo; at each step, P≤0.0250 and all previous tests also meeting this level of significance was required for statistical significance.
- † Assessed as the average of the treatment effects at Week 16 and Week 24.
- ‡ Indicates statistical significance confirmed in the hierarchical testing procedure. Other efficacy measures considered not statistically significant.
Relative change in ppFEV1 at Week 24† (%) Treatment difference
(95% CI)– 4.3
(1.9, 6.8)
P=0.0006‡– 5.3
(2.7, 7.8)
P<0.0001‡Absolute change in BMI at Week 24 (kg/m2) Treatment difference
(95% CI)– 0.1
(-0.1, 0.3)– 0.4
(0.2, 0.5)
P=0.0001‡Absolute change in CFQ-R Respiratory Domain Score (Points) at Week 24 Treatment difference
(95% CI)– 1.5
(-1.7, 4.7)– 2.9
(-0.3, 6.0)Proportion of patients with ≥5% relative change in ppFEV1 at Week 24† % 22% 37% 23% 41% Odds ratio
(95% CI)– 2.1
(1.3, 3.3)– 2.4
(1.5, 3.7)Number of pulmonary exacerbations through Week 24 # of events (rate per 48 weeks) 112 (1.1) 73 (0.7) 139 (1.2) 79 (0.7) Rate ratio
(95% CI)– 0.7
(0.5, 0.9)– 0.6
(0.4, 0.8) -
16 HOW SUPPLIED/STORAGE AND HANDLING
ORKAMBI (lumacaftor 200 mg/ivacaftor 125 mg) is supplied as pink, oval-shaped tablets; each tablet contains 200 mg of lumacaftor and 125 mg of ivacaftor, printed with "2V125" in black ink on one side and plain on the other, and is packaged as follows:
112–count tablet box containing a 4-week supply (4 weekly cartons of 7 daily blister strips with 4 tablets per strip). NDC 51167-809-01 ORKAMBI (lumacaftor 100 mg/ivacaftor 125 mg) is supplied as pink, oval-shaped tablets; each tablet contains 100 mg of lumacaftor and 125 mg of ivacaftor, printed with "1V125" in black ink on one side and plain on the other, and is packaged as follows:
112–count tablet box containing a 4-week supply (4 weekly cartons of 7 daily blister strips with 4 tablets per strip). NDC 51167-700-02 ORKAMBI (lumacaftor/ivacaftor) oral granules are supplied as small white to off-white granules and enclosed in unit dose packets as follows:
56-count carton (contains 56 unit-dose packets of lumacaftor 100 mg/ivacaftor 125 mg per packet) NDC 51167-900-01 56-count carton (contains 56 unit-dose packets of lumacaftor 150 mg/ivacaftor 188 mg per packet) NDC 51167-500-02 -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Advanced Liver Disease
Inform patients that worsening of liver function, including hepatic encephalopathy, in patients with advanced liver disease occurred in some patients treated with ORKAMBI. Liver function decompensation, including liver failure leading to death, has been reported in CF patients with pre-existing cirrhosis with portal hypertension while receiving ORKAMBI. If ORKAMBI is used in these patients, they should be closely monitored after the initiation of treatment and the dose should be reduced [see Dosage and Administration (2.2) and Warnings and Precautions (5.1)].
Abnormalities in Liver Function and Testing
Inform patients that abnormalities in liver function have occurred in patients treated with ORKAMBI. Blood tests to measure transaminases (ALT and AST) and bilirubin will be performed prior to initiating ORKAMBI, every 3 months during the first year of therapy, and annually thereafter [see Warnings and Precautions (5.2)].
Respiratory Events
Inform patients that chest discomfort, dyspnea, and respiration abnormal were more common during initiation of ORKAMBI therapy, especially in patients with advanced lung disease. Additional monitoring of patients with ppFEV1 <40 is recommended during initiation of therapy [see Warnings and Precautions (5.3)].
Effect on Blood Pressure
Inform patients that increased blood pressure has been observed in some patients treated with ORKAMBI and that periodic monitoring of their blood pressure during treatment is recommended [see Warnings and Precautions (5.4)].
Drug Interactions with CYP3A Inhibitors and Inducers
Ask patients to tell you all the medications they are taking, including any herbal supplements or vitamins. Co–administration with sensitive CYP3A substrates or CYP3A substrates with a narrow therapeutic index is not recommended [see Warnings and Precautions (5.5), Drug Interactions (7), and Clinical Pharmacology (12.3)].
Instruct patients on alternative methods of birth control because hormonal contraceptives should not be relied upon as an effective method of contraception and there is an increased incidence of menstruation-related adverse reactions when co-administered with ORKAMBI [see Warnings and Precautions (5.5), Adverse Reactions (6.1), and Drug Interactions (7.11)].
When initiating ORKAMBI in patients taking strong CYP3A inhibitors (e.g., itraconazole), instruct the patient to reduce the dose of ORKAMBI to 1 tablet daily or 1 packet of oral granules every other day for the first week of treatment. Following this period, continue with the recommended daily dose [see Dosage and Administration (2.3), Drug Interactions (7.1), and Clinical Pharmacology (12.3)].
Patients should be instructed to tell their doctor if they stop ORKAMBI for more than 1 week while they are also taking a strong CYP3A inhibitor because the dose of ORKAMBI would need to be reduced upon re-initiation. The dose of ORKAMBI should be reduced to 1 tablet daily or 1 packet of oral granules every other day for the first week upon treatment re-initiation. Following this period, continue with the recommended daily dose [see Dosage and Administration (2.3), Drug Interactions (7.1), and Clinical Pharmacology (12.3)].
Use in Patients with Hepatic Impairment
Inform patients with moderate hepatic impairment (Child-Pugh Class B) to reduce the dose of ORKAMBI as follows:
Tablets - 2 tablets in the morning and 1 tablet in the evening.
Granules - alternate daily between 2 packets of oral granules per day (1 in the morning and 1 in the evening) and 1 packet per day.If initiating ORKAMBI in a patient with severe hepatic impairment, after weighing the risks and benefits of treatment, instruct the patient to take a maximum dose of 1 tablet every 12 hours or less frequently, or 1 packet of oral granules once daily or less frequently [see Dosage and Administration (2.2), Warnings and Precautions (5.1), Adverse Reactions (6.1), and Clinical Pharmacology (12.3)].
Administration
Inform patients that ORKAMBI is best absorbed by the body when taken with fat-containing food. A typical CF diet will satisfy this requirement. Examples of fat-containing foods include eggs, avocados, nuts, butter, peanut butter, cheese pizza, whole-milk dairy products (such as whole milk, cheese, and yogurt), etc. [see Dosage and Administration (2.1) and Clinical Pharmacology (12.3)].
Inform patients and caregivers that ORKAMBI oral granules should be mixed with one teaspoon (5 mL) of age-appropriate soft food or liquid and completely consumed to ensure delivery of the entire dose. Food or liquid should be at or below room temperature. Once mixed, the product has been shown to be stable for one hour, and therefore should be consumed during this period. Some examples of appropriate soft foods or liquids may include puréed fruits, flavored yogurt or pudding, and milk or juice.
Inform patients that if a dose is missed and they remember the missed dose within 6 hours, the patients should take the dose with fat-containing food. If more than 6 hours elapsed after the usual dosing time, the patients should skip that dose and resume the normal schedule for the following dose. Patients should be informed not to take a double dose to make up for the forgotten dose [see Dosage and Administration (2.1)].
Cataracts
Inform patients that abnormalities of the eye lens (cataract) have been noted in some children and adolescents receiving ORKAMBI and ivacaftor, a component of ORKAMBI. Baseline and follow-up ophthalmological examinations are recommended in pediatric patients initiating ORKAMBI treatment [see Warnings and Precautions (5.6)].
-
SPL UNCLASSIFIED SECTION
Manufactured for
Vertex Pharmaceuticals Incorporated
50 Northern Avenue
Boston, MA 02210ORKAMBI, the ORKAMBI logo, VERTEX, and the VERTEX triangle logo are registered trademarks of Vertex Pharmaceuticals Incorporated.
All other trademarks referenced herein are the property of their respective owners.
©2019 Vertex Pharmaceuticals Incorporated
ALL RIGHTS RESERVED -
PATIENT PACKAGE INSERT
PATIENT INFORMATION
ORKAMBI (or-KAM-bee)
(Lumacaftor/Ivacaftor)
Tablets and Oral GranulesThis Patient Information has been approved by the U.S. Food and Drug Administration. June 2019 What is ORKAMBI?
ORKAMBI is a prescription medicine used for the treatment of cystic fibrosis (CF) in patients age 2 years and older who have two copies of the F508del mutation (F508del/F508del) in their CFTR gene.
ORKAMBI should not be used in patients other than those who have two copies of the F508del mutation in their CFTR gene.
It is not known if ORKAMBI is safe and effective in children under 2 years of age.
Do not take ORKAMBI if you take certain medicines or herbal supplements such as:
- antibiotics: rifampin (RIFAMATE®, RIFATER®) or rifabutin (MYCOBUTIN®)
- seizure medications: phenobarbital, carbamazepine (TEGRETOL®, CARBATROL®, and EQUETRO®), or phenytoin (DILANTIN®, PHENYTEK®)
- sedatives and anti-anxiety medicines: triazolam (HALCION®) or midazolam (DORMICUM®, HYPNOVEL®, and VERSED®)
- immunosuppressant medicines: cyclosporine, everolimus (ZORTRESS®), sirolimus (RAPAMUNE®), or tacrolimus (ASTAGRAF XL®, ENVARSUS® XR, PROGRAF®, PROTOPIC®)
- St. John's wort (Hypericum perforatum)
Talk to your doctor before taking ORKAMBI if you take any of the medicines or supplements listed above.
Before taking ORKAMBI, tell your healthcare provider about all of your medical conditions, including if you:
- have or have had liver problems
- have had an organ transplant
- have kidney problems
- are using birth control (hormonal contraceptives, including oral, injectable, transdermal, or implantable forms). Hormonal contraceptives should not be used as a method of birth control when taking ORKAMBI. Talk to your doctor about the best birth control method you should use while taking ORKAMBI.
- are pregnant or plan to become pregnant. It is not known if ORKAMBI will harm your unborn baby. You and your doctor should decide if you will take ORKAMBI while you are pregnant.
- are breastfeeding or planning to breastfeed. It is not known if ORKAMBI passes into your breast milk. You and your doctor should decide if you will take ORKAMBI while you are breastfeeding.
ORKAMBI may affect the way other medicines work, and other medicines may affect how ORKAMBI works.
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements, because the dose of ORKAMBI may need to be adjusted when taken with certain medications.
Ask your doctor or pharmacist for a list of these medicines if you are not sure.
Especially tell your doctor if you take:
- antifungal medications including ketoconazole (such as NIZORAL®), itraconazole (such as SPORANOX®), posaconazole (such as NOXAFIL®), or voriconazole (such as VFEND®)
- antibiotics including telithromycin (such as KETEK®), clarithromycin (such as BIAXIN®), or erythromycin (such as ERY-TAB®)
Know the medicines you take. Keep a list of them to show your doctor and pharmacist when you get a new medicine.
How should I take ORKAMBI?
- Take ORKAMBI exactly as your doctor tells you to take it.
- Always take ORKAMBI tablets or granules with foods that contain fat. Examples of fat containing foods include eggs, avocados, nuts, butter, peanut butter, cheese pizza, whole-milk dairy products (such as whole milk, cheese, and yogurt).
- Take your doses of ORKAMBI 12 hours apart.
- If you miss a dose within 6 hours of when you usually take it, take your dose with fat-containing food as soon as possible.
- If you miss a dose and it is more than 6 hours after the time you usually take it, skip that dose only and take the next dose when you usually take it. Do not take 2 doses at the same time to make up for your missed dose.
- Tell your doctor if you stop ORKAMBI for more than 1 week. Your doctor may need to change your dose of ORKAMBI or other medicines you take.
- Each ORKAMBI box contains 4 weekly cartons.
- Each carton contains 7 daily blister strips.
- Each blister strip contains 4 tablets so you can take 2 tablets for the morning and 2 tablets for the evening.
- You may cut along the dotted line to separate your morning dose from your evening dose.
- To take your morning dose, unpeel the paper backing from a blister strip (do not push tablet through backing) to remove 2 ORKAMBI tablets and take them with fat-containing food.
- 12 hours after your previous dose, open another blister strip (do not push tablet through backing) to remove 2 ORKAMBI tablets and take them with fat-containing food.
- Hold the packet with the cut line on top.
- Shake the packet gently to settle the ORKAMBI granules.
- Tear or cut packet open along cut line.
- Carefully pour all of the ORKAMBI granules in the packet into 1 teaspoon (5 mL) of soft food or liquid in a small container (like an empty bowl).
- The food or liquid should be at or below room temperature.
- Examples of soft foods or liquids include puréed fruits, flavored yogurt or pudding, and milk or juice.
- Mix the ORKAMBI granules with food or liquid.
- After mixing, give ORKAMBI within 1 hour. Make sure all medicine is taken.
- Give a child fat-containing food just before or just after the ORKAMBI granules dose (see examples above).
What are the possible side effects of ORKAMBI?
ORKAMBI can cause serious side effects, including:
-
Worsening of liver function in people with severe liver disease. The worsening of liver function can be serious or cause death. Talk to your doctor if you have been told you have liver disease as your doctor may need to adjust the dose of ORKAMBI.
-
High liver enzymes in the blood, which can be a sign of liver injury in people receiving ORKAMBI. Your doctor will do blood tests to check your liver:
- before you start ORKAMBI
- every 3 months during your first year of taking ORKAMBI
- every year while you are taking ORKAMBI
Call your doctor right away if you have any of the following symptoms of liver problems:
- pain or discomfort in the upper right stomach (abdominal) area
- loss of appetite
- dark, amber-colored urine
- yellowing of your skin or the white part of your eyes
- nausea or vomiting
- confusion
- Breathing problems such as shortness of breath or chest tightness in patients when starting ORKAMBI, especially in patients who have poor lung function. If you have poor lung function, your doctor may monitor you more closely when you start ORKAMBI.
- An increase in blood pressure in some people receiving ORKAMBI. Your doctor should monitor your blood pressure during treatment with ORKAMBI.
- Abnormality of the eye lens (cataract) in some children and adolescents receiving ORKAMBI. If you are a child or adolescent, your doctor should perform eye examinations before and during treatment with ORKAMBI to look for cataracts.
- breathing problems such as shortness of breath and chest tightness
- nausea
- diarrhea
- fatigue
- increase in a certain blood enzyme called creatine phosphokinase
- rash
- gas
- common cold, including sore throat, stuffy or runny nose
- flu or flu-like symptoms
- irregular, missed, or abnormal periods (menses) and increase in the amount of menstrual bleeding
Additional side effects in children
Side effects seen in children are similar to those seen in adults and adolescents. Additional common side effects seen in children include:
- cough with sputum
- stuffy nose
- headache
- stomach pain
- increase in sputum
These are not all the possible side effects of ORKAMBI.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store ORKAMBI?
- Store ORKAMBI at room temperature between 68°F to 77°F (20°C to 25°C).
- Do not use ORKAMBI after the expiration date on the package.
Keep ORKAMBI and all medicines out of the reach of children.
General information about the safe and effective use of ORKAMBI.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use ORKAMBI for a condition for which it was not prescribed. Do not give ORKAMBI to other people, even if they have the same symptoms you have. It may harm them.
You can ask your pharmacist or doctor for information about ORKAMBI that is written for health professionals.
What are the ingredients in ORKAMBI?
ORKAMBI Tablets
Active ingredients: lumacaftor and ivacaftor
Inactive ingredients: cellulose, microcrystalline; croscarmellose sodium; hypromellose acetate succinate; magnesium stearate; povidone; and sodium lauryl sulfate.The tablet film coat contains: carmine, FD&C Blue #1, FD&C Blue #2, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
The printing ink contains: ammonium hydroxide, iron oxide black, propylene glycol, and shellac.
ORKAMBI Oral Granules
Active ingredients: lumacaftor and ivacaftor
Inactive ingredients: cellulose, microcrystalline; croscarmellose sodium; hypromellose acetate succinate; povidone; and sodium lauryl sulfate.VERTEX
Manufactured for: Vertex Pharmaceuticals Incorporated; 50 Northern Avenue, Boston, MA 02210
For more information, go to www.orkambi.com or call 1 877 752 5933.
ORKAMBI, the ORKAMBI logo, VERTEX, and the VERTEX triangle logo are registered trademarks of Vertex Pharmaceuticals Incorporated.
All other trademarks referenced herein are the property of their respective owners.
©2019 Vertex Pharmaceuticals Incorporated -
PRINCIPAL DISPLAY PANEL - 200 mg/125 mg tablet Box
Rx only
NDC: 51167-809-01ORKAMBI®
(Lumacaftor/Ivacaftor) tablets
200 mg/125 mg per tablet112 tablets

-
PRINCIPAL DISPLAY PANEL - 100 mg/125 mg tablet Box
Rx only
NDC: 51167-700-02ORKAMBI®
(Lumacaftor/Ivacaftor) tablets
100 mg/125 mg per tablet112 tablets

-
PRINCIPAL DISPLAY PANEL - 100 mg/125 mg packet Carton
Rx only
NDC: 51167-900-01ORKAMBI®
(Lumacaftor/Ivacaftor)100 mg
125 mgOral Granules
100 mg/125 mg per packet56 packets
Carton contains: 4 individual wallets
with 14 packets per walletLift here to open
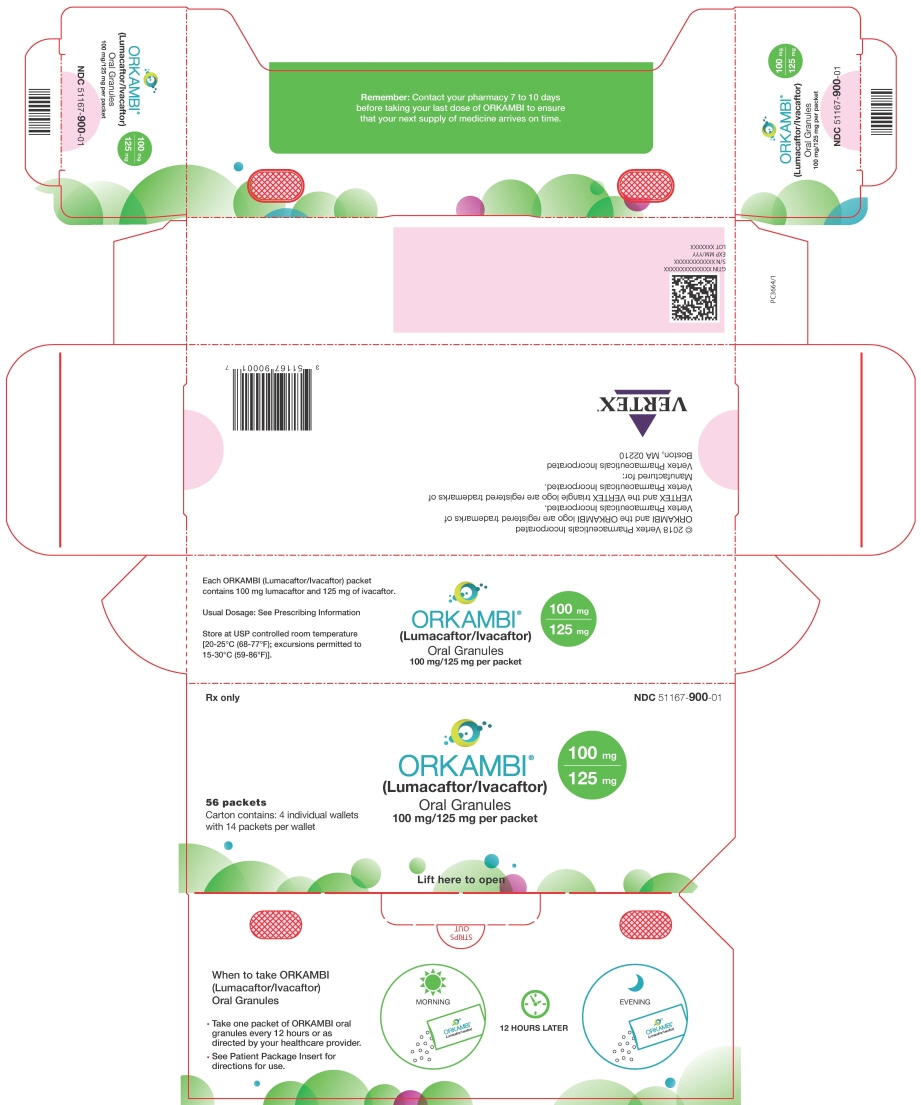
-
PRINCIPAL DISPLAY PANEL - 150 mg/188 mg packet Carton
Rx only
NDC: 51167-500-02
ORKAMBI®
(Lumacaftor/Ivacaftor)150 mg
188 mgOral Granules
150 mg/188 mg per packet56 packets
Carton contains: 4 individual wallets
with 14 packets per walletLift here to open
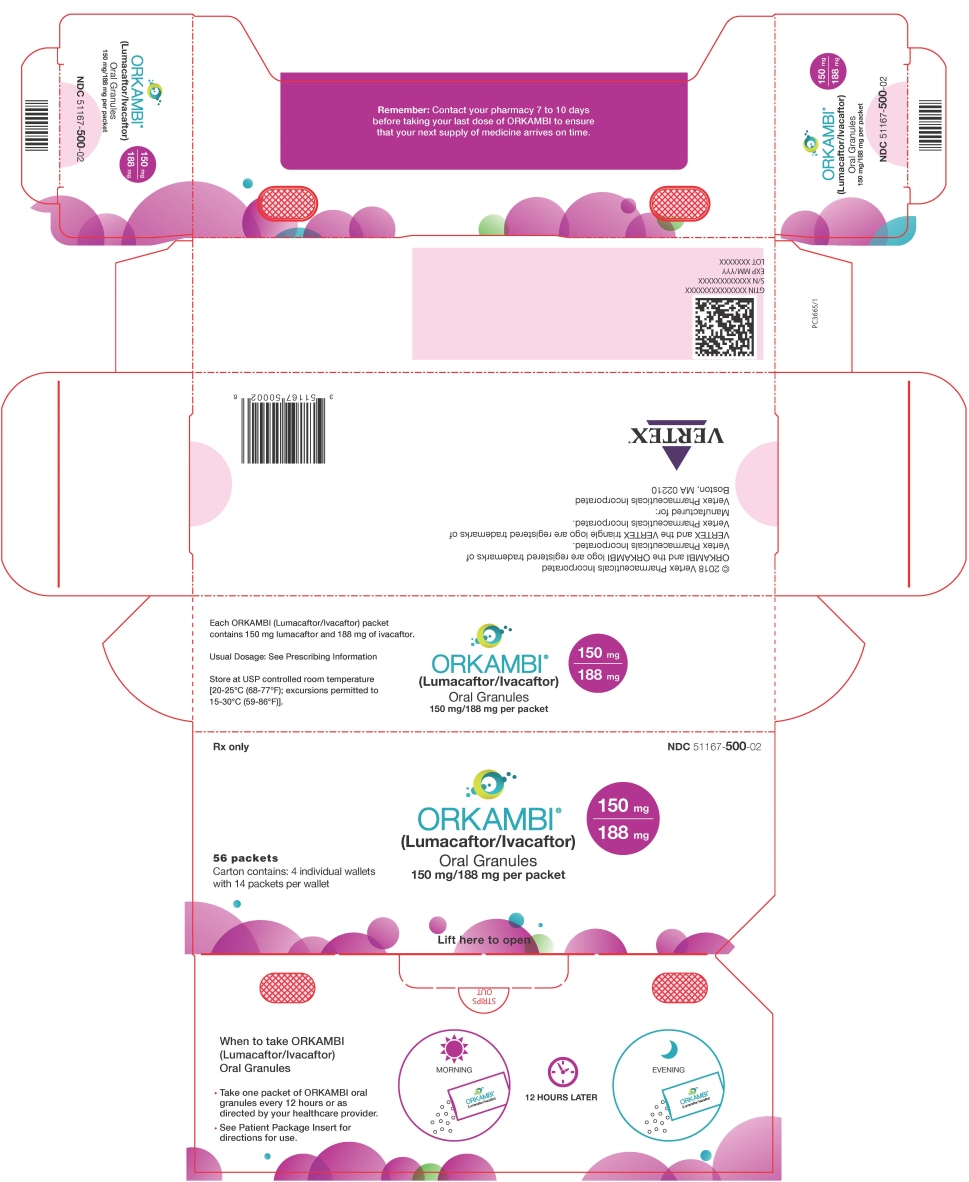
-
INGREDIENTS AND APPEARANCE
ORKAMBI
lumacaftor and ivacaftor tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 51167-809 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength lumacaftor (UNII: EGP8L81APK) (lumacaftor - UNII:EGP8L81APK) lumacaftor 200 mg ivacaftor (UNII: 1Y740ILL1Z) (ivacaftor - UNII:1Y740ILL1Z) ivacaftor 125 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) HYPROMELLOSE ACETATE SUCCINATE 06081224 (3 MM2/S) (UNII: 6N003M473W) MAGNESIUM STEARATE (UNII: 70097M6I30) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) SODIUM LAURYL SULFATE (UNII: 368GB5141J) CARMINIC ACID (UNII: CID8Z8N95N) FD&C Blue No. 1 (UNII: H3R47K3TBD) FD&C Blue No. 2 (UNII: L06K8R7DQK) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) AMMONIA (UNII: 5138Q19F1X) FERROSOFERRIC OXIDE (UNII: XM0M87F357) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SHELLAC (UNII: 46N107B71O) Product Characteristics Color PINK Score no score Shape OVAL Size 14mm Flavor Imprint Code 2V125 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 51167-809-01 4 in 1 BOX 07/06/2015 1 7 in 1 CARTON 1 4 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA206038 07/06/2015 ORKAMBI
lumacaftor and ivacaftor tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 51167-700 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength lumacaftor (UNII: EGP8L81APK) (lumacaftor - UNII:EGP8L81APK) lumacaftor 100 mg ivacaftor (UNII: 1Y740ILL1Z) (ivacaftor - UNII:1Y740ILL1Z) ivacaftor 125 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) HYPROMELLOSE ACETATE SUCCINATE 06081224 (3 MM2/S) (UNII: 6N003M473W) MAGNESIUM STEARATE (UNII: 70097M6I30) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) SODIUM LAURYL SULFATE (UNII: 368GB5141J) CARMINIC ACID (UNII: CID8Z8N95N) FD&C Blue No. 1 (UNII: H3R47K3TBD) FD&C Blue No. 2 (UNII: L06K8R7DQK) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) AMMONIA (UNII: 5138Q19F1X) FERROSOFERRIC OXIDE (UNII: XM0M87F357) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SHELLAC (UNII: 46N107B71O) Product Characteristics Color PINK Score no score Shape OVAL Size 14mm Flavor Imprint Code 1V125 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 51167-700-02 4 in 1 BOX 10/03/2016 1 7 in 1 CARTON 1 4 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA206038 10/03/2016 ORKAMBI
lumacaftor and ivacaftor granuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 51167-900 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength lumacaftor (UNII: EGP8L81APK) (lumacaftor - UNII:EGP8L81APK) lumacaftor 100 mg ivacaftor (UNII: 1Y740ILL1Z) (ivacaftor - UNII:1Y740ILL1Z) ivacaftor 125 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) HYPROMELLOSE ACETATE SUCCINATE 06081224 (3 MM2/S) (UNII: 6N003M473W) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) SODIUM LAURYL SULFATE (UNII: 368GB5141J) Product Characteristics Color WHITE Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 51167-900-01 56 in 1 CARTON 08/14/2018 1 1 in 1 PACKET; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA211358 08/14/2018 ORKAMBI
lumacaftor and ivacaftor granuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 51167-500 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength lumacaftor (UNII: EGP8L81APK) (lumacaftor - UNII:EGP8L81APK) lumacaftor 150 mg ivacaftor (UNII: 1Y740ILL1Z) (ivacaftor - UNII:1Y740ILL1Z) ivacaftor 188 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) HYPROMELLOSE ACETATE SUCCINATE 06081224 (3 MM2/S) (UNII: 6N003M473W) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) SODIUM LAURYL SULFATE (UNII: 368GB5141J) Product Characteristics Color WHITE Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 51167-500-02 56 in 1 CARTON 08/14/2018 1 1 in 1 PACKET; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA211358 08/14/2018 Labeler - Vertex Pharmaceuticals Incorporated (602478257) Establishment Name Address ID/FEI Business Operations Vertex Pharmaceuticals Incorporated 602478257 MANUFACTURE(51167-809) , ANALYSIS(51167-809, 51167-700, 51167-900, 51167-500)
Trademark Results [ORKAMBI]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ORKAMBI 86399751 4828608 Live/Registered |
Vertex Pharmaceuticals Incorporated 2014-09-19 |
 ORKAMBI 86179790 4822946 Live/Registered |
Vertex Pharmaceuticals Incorporated 2014-01-30 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.