ATAZANAVIR SULFATE capsule
Atazanavir Sulfate by
Drug Labeling and Warnings
Atazanavir Sulfate by is a Prescription medication manufactured, distributed, or labeled by REMEDYREPACK INC.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ATAZANAVIR SULFATE CAPSULES safely and effectively. See full prescribing information for ATAZANAVIR SULFATE CAPSULES.
ATAZANAVIR SULFATE capsules, for oral use
Initial U.S. Approval: 2003RECENT MAJOR CHANGES
INDICATIONS AND USAGE
Atazanavir sulfate capsules are a protease inhibitor indicated for use in combination with other antiretroviral agents for the treatment of HIV-1 infection for patients 6 years and older weighing at least 15 kg. ( 1)
DOSAGE AND ADMINISTRATION
- Pretreatment testing: Renal laboratory testing should be performed in all patients prior to initiation of atazanavir sulfate capsules and continued during treatment with atazanavir sulfate capsules. Hepatic testing should be performed in patients with underlying liver disease prior to initiation of atazanavir sulfate capsules and continued during treatment with atazanavir sulfate capsules. ( 2.2)
- Treatment-naive adults: Atazanavir sulfate capsules 300 mg with ritonavir 100 mg once daily with food or atazanavir 400 mg once daily with food. ( 2.3)
- Treatment-experienced adults: Atazanavir sulfate capsules 300 mg with ritonavir 100 mg once daily with food. ( 2.3)
- Pediatric patients: Atazanavir sulfate capsule dosage is based on body weight not to exceed the adult dose and must be taken with food. ( 2.4)
- Pregnancy: Atazanavir sulfate capsules 300 mg with ritonavir 100 mg once daily with food, with dosing modifications for some concomitant medications. ( 2.6)
- Dosing modifications: may be required for concomitant therapy ( 2.3, 2.4, 2.6), renal impairment ( 2.7), and hepatic impairment ( 2.8).
CONTRAINDICATIONS
- Atazanavir sulfate capsules are contraindicated in patients with previously demonstrated hypersensitivity (e.g., Stevens-Johnson syndrome, erythema multiforme, or toxic skin eruptions) to any of the components of this product. ( 4)
- Coadministration with alfuzosin, triazolam, orally administered midazolam, ergot derivatives, rifampin, irinotecan, lurasidone (if atazanavir sulfate capsules are coadministered with ritonavir), lovastatin, simvastatin, indinavir, cisapride, pimozide, St. John’s wort, nevirapine, elbasvir/grazoprevir, glecaprevir/pibrentasvir, and sildenafil when dosed as REVATIO ®. ( 4)
WARNINGS AND PRECAUTIONS
- Cardiac conduction abnormalities: PR interval prolongation may occur in some patients. ECG monitoring should be considered in patients with preexisting conduction system disease or when administered with other drugs that may prolong the PR interval. (5.1, 7.3, 12.2, 17)
- Severe Skin Reactions: Discontinue if severe rash develops. ( 5.2, 17)
- Hyperbilirubinemia: Most patients experience asymptomatic increases in indirect bilirubin, which is reversible upon discontinuation. Do not dose reduce. If a concomitant transaminase increase occurs, evaluate for alternative etiologies. ( 5.8)
- Hepatotoxicity: Patients with hepatitis B or C infection are at risk of increased transaminases or hepatic decompensation. Monitor hepatic laboratory tests prior to therapy and during treatment. ( 2.8, 5.4, 8.8)
- Chronic kidney disease has been reported during postmarketing surveillance in HIV-infected patients treated with atazanavir, with or without ritonavir. Consider alternatives in patients at high risk for renal disease or with preexisting renal disease. Monitor renal laboratory tests prior to therapy and during treatment. Consider discontinuation of atazanavir sulfate in patients with progressive renal disease. ( 5.5)
- Nephrolithiasis and cholelithiasis have been reported. Consider temporary interruption or discontinuation. ( 5.6)
- The concomitant use of atazanavir sulfate/ritonavir and certain other medications may result in known or potentially significant drug interactions. Consult the full prescribing information prior to and during treatment for potential drug interactions. ( 5.7, 7.3)
- Patients receiving atazanavir sulfate may develop new onset or exacerbations of diabetes mellitus/hyperglycemia ( 5.9), immune reconstitution syndrome ( 5.10), and redistribution/accumulation of body fat ( 5.11).
- Hemophilia: Spontaneous bleeding may occur and additional factor VIII may be required. ( 5.12)
ADVERSE REACTIONS
Most common adverse reactions (≥ 2%) are nausea, jaundice/scleral icterus, rash, headache, abdominal pain, vomiting, insomnia, peripheral neurologic symptoms, dizziness, myalgia, diarrhea, depression, and fever. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Teva Pharmaceuticals USA, Inc. at 1-888-838-2872 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
- Pregnancy: Available human and animal data suggest that atazanavir does not increase the risk of major birth defects overall compared to the background rate. ( 8.1)
- Lactation: Breastfeeding is not recommended. ( 8.2)
- Hepatitis B or C co-infection: Monitor liver enzymes. ( 5.4, 6.1)
- Renal impairment: Atazanavir sulfate is not recommended for use in treatment-experienced patients with end-stage renal disease managed with hemodialysis. ( 2.7, 8.7)
- Hepatic impairment: Atazanavir sulfate is not recommended in patients with severe hepatic impairment. Atazanavir sulfate/ritonavir is not recommended in patients with any degree of hepatic impairment. ( 2.8, 8.8)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 8/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Overview
2.2 Testing Prior to Initiation and During Treatment with Atazanavir Sulfate Capsules
2.3 Dosage of Atazanavir Sulfate Capsules in Adult Patients
2.4 Dosage of Atazanavir Sulfate Capsules in Pediatric Patients
2.6 Dosage Adjustments in Pregnant Patients
2.7 Dosage in Patients with Renal Impairment
2.8 Dosage Adjustments in Patients with Hepatic Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Cardiac Conduction Abnormalities
5.2 Severe Skin Reactions
5.4 Hepatotoxicity
5.5 Chronic Kidney Disease
5.6 Nephrolithiasis and Cholelithiasis
5.7 Risk of Serious Adverse Reactions Due to Drug Interactions
5.8 Hyperbilirubinemia
5.9 Diabetes Mellitus/Hyperglycemia
5.10 Immune Reconstitution Syndrome
5.11 Fat Redistribution
5.12 Hemophilia
5.13 Resistance/Cross-Resistance
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Potential for Atazanavir Sulfate to Affect Other Drugs
7.2 Potential for Other Drugs to Affect Atazanavir Sulfate
7.3 Established and Other Potentially Significant Drug Interactions
7.4 Drugs with No Observed Interactions with Atazanavir Sulfate
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Age/Gender
8.7 Impaired Renal Function
8.8 Impaired Hepatic Function
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Adult Patients without Prior Antiretroviral Therapy
14.2 Adult Patients with Prior Antiretroviral Therapy
14.3 Pediatric Patients
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
Atazanavir sulfate capsules are indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection for patients 6 years and older weighing at least 15 kg.
Limitations of Use:
- Atazanavir is not recommended for use in pediatric patients below the age of 3 months due to the risk of kernicterus.
- Use of atazanavir/ritonavir in treatment-experienced patients should be guided by the number of baseline primary protease inhibitor resistance substitutions [see Microbiology ( 12.4)] .
-
2 DOSAGE AND ADMINISTRATION
2.1 Overview
- Atazanavir sulfate capsules must be taken with food.
- Do not open the capsules.
- The recommended oral dosage of atazanavir sulfate capsules depends on the treatment history of the patient and the use of other coadministered drugs. When coadministered with H 2-receptor antagonists or proton-pump inhibitors, dose separation may be required [see Dosage and Administration ( 2.3, 2.4, and 2.6) and Drug Interactions ( 7)].
- Atazanavir sulfate capsules without ritonavir are not recommended for treatment-experienced adult or pediatric patients with prior virologic failure [see Clinical Studies ( 14)].
- Efficacy and safety of atazanavir sulfate capsules with ritonavir when ritonavir is administered in doses greater than 100 mg once daily have not been established. The use of higher ritonavir doses may alter the safety profile of atazanavir (cardiac effects, hyperbilirubinemia) and, therefore, is not recommended. Prescribers should consult the complete prescribing information for ritonavir when using ritonavir.
2.2 Testing Prior to Initiation and During Treatment with Atazanavir Sulfate Capsules
Renal laboratory testing should be performed in all patients prior to initiation of atazanavir sulfate capsules and continued during treatment with atazanavir sulfate capsules. Renal laboratory testing should include serum creatinine, estimated creatinine clearance, and urinalysis with microscopic examination [see Warnings and Precautions (5.5, 5.6)] .
Hepatic laboratory testing should be performed in patients with underlying liver disease prior to initiation of atazanavir sulfate capsules and continued during treatment with atazanavir sulfate capsules [see Warnings and Precautions (5.4)] .
2.3 Dosage of Atazanavir Sulfate Capsules in Adult Patients
Table 1 displays the recommended dosage of atazanavir sulfate capsules in treatment-naive and treatment-experienced adults. Table 1 also displays recommended dosage of atazanavir sulfate capsules and ritonavir when given concomitantly with other antiretroviral drugs and H 2-receptor antagonists (H2RA). Ritonavir is required with several atazanavir sulfate capsule dosage regimens (see the ritonavir complete prescribing information about the safe and effective use of ritonavir). The use of atazanavir sulfate capsules in treatment-experienced adult patients without ritonavir is not recommended.
Table 1: Recommended Atazanavir Sulfate Capsules and Ritonavir Dosage in Adults 1 Atazanavir Capsules Once Daily
Dosage
Ritonavir Once Daily
Dosage
Treatment-Naive Adult Patients
recommended regimen
300 mg
100 mg
unable to tolerate ritonavir
400 mg
N/A
in combination with efavirenz
400 mg
100 mg
Treatment-Experienced Adult Patients
recommended regimen
300 mg
100 mg
in combination with both H2RA
and tenofovir DF
400 mg
100 mg
- See Drug Interactions ( 7) for instructions concerning coadministration of acid-reducing medications (e.g., H2RA or proton pump inhibitors [PPIs]), and other antiretroviral drugs (e.g., efavirenz, tenofovir DF, and didanosine).
2.4 Dosage of Atazanavir Sulfate Capsules in Pediatric Patients
The recommended daily dosage of atazanavir capsules and ritonavir in pediatric patients (6 years of age to less than 18 years of age) is based on body weight (see Table 2).
Table 2: Recommended Dosage of Atazanavir Sulfate Capsules and Ritonavir in Pediatric Patients (6 to less than 18 years of age) 1, 2 Body Weight
Atazanavir Sulfate Capsule Daily Dosage
Ritonavir Daily Dosage
Treatment-Naive and Treatment-Experienced3
Less than 15 kg
Capsules not recommended
N/A
At least 15 kg to less than 35 kg
200 mg
100 mg
At least 35 kg
300 mg
100 mg
Treatment-Naive, at least 13 years old and cannot tolerate ritonavir3
At least 40 kg
400 mg
N/A
1. Administer atazanavir sulfate capsules and ritonavir simultaneously with food.
2. The same recommendations regarding the timing and maximum doses of concomitant PPIs and H2RAs in adults also apply to pediatric patients. See Drug Interactions ( 7) for instructions concerning coadministration of acid-reducing medications (e.g., H2RA or PPIs), and other antiretroviral drugs (e.g., efavirenz, tenofovir DF, and didanosine).
3. In treatment-experienced patients, atazanavir sulfate capsules must be administered with ritonavir.
When transitioning between formulations, a change in dose may be needed. Consult the dosing table for the specific formulation.
2.6 Dosage Adjustments in Pregnant Patients
Table 4 includes the recommended dosage of atazanavir sulfate capsules and ritonavir in treatment-naive and treatment-experienced pregnant patients. In these patients, atazanavir sulfate capsules must be administered with ritonavir. There are no dosage adjustments for postpartum patients (see Table 1 for the recommended atazanavir sulfate capsules dosage in adults) [see Use in Specific Populations ( 8.1)].
Table 4: Recommended Dosage of Atazanavir Sulfate Capsules and Ritonavir in Pregnant Patients1
Atazanavir Sulfate Capsules
Once Daily
Dosage
Ritonavir
Once Daily
Dosage
Treatment-Naive and Treatment-Experienced
Recommended Regimen
300 mg
100 mg
Treatment-Experienced During the Second or Third Trimester When Coadministered with either H2RA or Tenofovir DF2
In combination with either H2RA
or tenofovir DF
400 mg
100 mg
1. See Drug Interactions ( 7) for instructions concerning coadministration of acid-reducing medications (e.g., H2RA or PPIs), and other antiretroviral drugs (e.g., efavirenz, tenofovir DF, and didanosine).
2. Atazanavir sulfate capsules are not recommended for treatment-experienced pregnant patients during the second and third trimester taking atazanavir sulfate capsules with both tenofovir DF and H2RA.
2.7 Dosage in Patients with Renal Impairment
For patients with renal impairment, including those with severe renal impairment who are not managed with hemodialysis, no dose adjustment is required for atazanavir sulfate capsules. Treatment-naive patients with end-stage renal disease managed with hemodialysis should receive atazanavir sulfate capsules 300 mg with ritonavir 100 mg. Atazanavir sulfate capsules are not recommended in HIV-treatment‑experienced patients with end-stage renal disease managed with hemodialysis [see Use in Specific Populations ( 8.7)].
2.8 Dosage Adjustments in Patients with Hepatic Impairment
Table 5 displays the recommended atazanavir sulfate capsule dosage in treatment-naive patients with hepatic impairment. The use of atazanavir sulfate capsules in patients with severe hepatic impairment (Child-Pugh Class C) is not recommended. The coadministration of atazanavir sulfate capsules with ritonavir in patients with any degree of hepatic impairment is not recommended.
Table 5: Recommended Dosage of Atazanavir Sulfate Capsules in Treatment-Naive Adults with Hepatic Impairment
Atazanavir Capsules Once Daily Dosage
Mild hepatic impairment (Child-Pugh Class A)
400 mg
Moderate hepatic impairment (Child-Pugh Class B)
300 mg
Severe hepatic impairment (Child-Pugh Class C)
Atazanavir sulfate capsules with or without ritonavir is not recommended
-
3 DOSAGE FORMS AND STRENGTHS
- 150 mg capsule with light-turquoise-blue-opaque cap and aqua-blue-opaque body, imprinted “5526” on the body and “TEVA” on the cap.
- 200 mg capsule with light-turquoise body and light-turquoise cap, imprinted “93” over “5527” on both the cap and the body.
- 300 mg capsule with light-blue-opaque body and red-opaque cap, imprinted “93” over “5528” on both the cap and the body.
-
4 CONTRAINDICATIONS
Atazanavir sulfate capsules are contraindicated:
- in patients with previously demonstrated clinically significant hypersensitivity (e.g., Stevens-Johnson syndrome, erythema multiforme, or toxic skin eruptions) to any of the components of atazanavir sulfate capsules [see Warnings and Precautions ( 5.2)] .
- when coadministered with drugs that are highly dependent on CYP3A or UGT1A1 for clearance, and for which elevated plasma concentrations of the interacting drugs are associated with serious and/or life-threatening events (see Table 6).
- when coadministered with drugs that strongly induce CYP3A and may lead to lower exposure and loss of efficacy of atazanavir sulfate (see Table 6).
Table 6 displays drugs that are contraindicated with atazanavir sulfate.
Table 6: Drugs that are Contraindicated with Atazanavir Sulfate (Information in the table applies to Atazanavir Sulfate with or without ritonavir, unless otherwise indicated) Drug Class
Drugs within class that are contraindicated with atazanavir sulfate
Clinical Comment
Alpha 1-Adrenoreceptor Antagonist
Alfuzosin
Potential for increased alfuzosin concentrations, which can result in hypotension.
Antimycobacterials
Rifampin
Rifampin substantially decreases plasma concentrations of atazanavir, which may result in loss of therapeutic effect and development of resistance.
Antineoplastics
Irinotecan
Atazanavir inhibits UGT1A1 and may interfere with the metabolism of irinotecan, resulting in increased irinotecan toxicities.
Antipsychotics
Lurasidone
Pimozide
Potential for serious and/or life-threatening reactions if atazanavir sulfate is coadministered with ritonavir.
Potential for serious and/or life-threatening reactions such as cardiac arrhythmias.
Benzodiazepines
Triazolam, orally administered midazolam 1
Triazolam and orally administered midazolam are extensively metabolized by CYP3A4. Coadministration of triazolam or orally administered midazolam with atazanavir sulfate may cause large increases in the concentration of these benzodiazepines. Potential for serious and/or life-threatening events such as prolonged or increased sedation or respiratory depression.
Ergot Derivatives
Dihydroergotamine, ergotamine, ergonovine, methylergonovine
Potential for serious and/or life-threatening events such as acute ergot toxicity characterized by peripheral vasospasm and ischemia of the extremities and other tissues.
GI Motility Agent
Cisapride
Potential for serious and/or life-threatening reactions such as cardiac arrhythmias.
Hepatitis C Direct-Acting Antivirals
Elbasvir/grazoprevir
May increase the risk of ALT elevations due to a significant increase in grazoprevir plasma concentrations.
Glecaprevir/pibrentasvir May increase the risk of ALT elevations due to an increase in glecaprevir and pibrentasvir concentrations. Herbal Products
St. John’s wort ( Hypericum perforatum)
Coadministration of St. John’s wort and atazanavir sulfate may result in loss of therapeutic effect and development of resistance.
HMG-CoA Reductase Inhibitors
Lovastatin, simvastatin
Potential for serious reactions such as myopathy, including rhabdomyolysis.
PDE5 Inhibitor
Sildenafil 2 when dosed as REVATIO ® for the treatment of pulmonary arterial hypertension
Potential for sildenafil-associated adverse events (which include visual disturbances, hypotension, priapism, and syncope).
Protease Inhibitors
Indinavir
Both atazanavir sulfate and indinavir are associated with indirect (unconjugated) hyperbilirubinemia.
Non-nucleoside Reverse Transcriptase Inhibitors
Nevirapine
Nevirapine substantially decreases atazanavir exposure which may result in loss of therapeutic effect and development of resistance. Potential risk for nevirapine-associated adverse reactions due to increased nevirapine exposures.
1. See Drug Interactions, Table 16 ( 7) for parenterally administered midazolam.
2. See Drug Interactions, Table 16 ( 7) for sildenafil when dosed as VIAGRA ® for erectile dysfunction.
-
5 WARNINGS AND PRECAUTIONS
5.1 Cardiac Conduction Abnormalities
Atazanavir sulfate has been shown to prolong the PR interval of the electrocardiogram in some patients. In healthy volunteers and in patients, abnormalities in atrioventricular (AV) conduction were asymptomatic and generally limited to first-degree AV block. There have been reports of second-degree AV block and other conduction abnormalities [see Adverse Reactions (6.2) and Overdosage ( 10)] . In clinical trials that included electrocardiograms, asymptomatic first-degree AV block was observed in 5.9% of atazanavir-treated patients (n = 920), 5.2% of lopinavir/ritonavir-treated patients (n = 252), 10.4% of nelfinavir-treated patients (n = 48), and 3.0% of efavirenz-treated patients (n = 329). In Study AI424-045, asymptomatic first-degree AV block was observed in 5% (6/118) of atazanavir/ritonavir-treated patients and 5% (6/116) of lopinavir/ritonavir-treated patients who had on-study electrocardiogram measurements. Because of limited clinical experience in patients with preexisting conduction system disease (e.g., marked first-degree AV block or second- or third-degree AV block). ECG monitoring should be considered in these patients [see Clinical Pharmacology ( 12.2)] .
5.2 Severe Skin Reactions
In controlled clinical trials, rash (all grades, regardless of causality) occurred in approximately 20% of patients treated with atazanavir sulfate. The median time to onset of rash in clinical studies was 7.3 weeks and the median duration of rash was 1.4 weeks. Rashes were generally mild-to-moderate maculopapular skin eruptions. Treatment-emergent adverse reactions of moderate or severe rash (occurring at a rate of ≥ 2%) are presented for the individual clinical studies [see Adverse Reactions ( 6.1)]. Dosing with atazanavir sulfate was often continued without interruption in patients who developed rash. The discontinuation rate for rash in clinical trials was < 1%. Cases of Stevens-Johnson syndrome, erythema multiforme, and toxic skin eruptions, including drug rash, eosinophilia, and systemic symptoms (DRESS) syndrome, have been reported in patients receiving atazanavir sulfate [see Contraindications ( 4) and Adverse Reactions ( 6.1)]. Atazanavir sulfate should be discontinued if severe rash develops.
5.4 Hepatotoxicity
Patients with underlying hepatitis B or C viral infections or marked elevations in transaminases before treatment may be at increased risk for developing further transaminase elevations or hepatic decompensation. In these patients, hepatic laboratory testing should be conducted prior to initiating therapy with atazanavir sulfate and during treatment [see Dosage and Administration ( 2.2), Adverse Reactions ( 6.1), and Use in Specific Populations ( 8.8)] .
5.5 Chronic Kidney Disease
Chronic kidney disease in HIV-infected patients treated with atazanavir, with or without ritonavir, has been reported during postmarketing surveillance. Reports included biopsy-proven cases of granulomatous interstitial nephritis associated with the deposition of atazanavir drug crystals in the renal parenchyma. Consider alternatives to atazanavir sulfate in patients at high risk for renal disease or with preexisting renal disease. Renal laboratory testing (including serum creatinine, estimated creatinine clearance, and urinalysis with microscopic examination) should be conducted in all patients prior to initiating therapy with atazanavir sulfate and continued during treatment with atazanavir sulfate. Expert consultation is advised for patients who have confirmed renal laboratory abnormalities while taking atazanavir sulfate. In patients with progressive kidney disease, discontinuation of atazanavir sulfate may be considered [see Dosage and Administration (2.2 and 2.7) and Adverse Reactions ( 6.2)].
5.6 Nephrolithiasis and Cholelithiasis
Cases of nephrolithiasis and/or cholelithiasis have been reported during postmarketing surveillance in HIV-infected patients receiving atazanavir sulfate therapy. Some patients required hospitalization for additional management and some had complications. Because these events were reported voluntarily during clinical practice, estimates of frequency cannot be made. If signs or symptoms of nephrolithiasis and/or cholelithiasis occur, temporary interruption or discontinuation of therapy may be considered [see Adverse Reactions (6.2)].
5.7 Risk of Serious Adverse Reactions Due to Drug Interactions
Initiation of atazanavir sulfate with ritonavir, a CYP3A inhibitor, in patients receiving medications metabolized by CYP3A or initiation of medications metabolized by CYP3A in patients already receiving atazanavir sulfate with ritonavir, may increase plasma concentrations of medications metabolized by CYP3A. Initiation of medications that inhibit or induce CYP3A may increase or decrease concentrations of atazanavir sulfate with ritonavir, respectively. These interactions may lead to:
- clinically significant adverse reactions potentially leading to severe, life-threatening, or fatal events from greater exposures of concomitant medications.
- clinically significant adverse reactions from greater exposures of atazanavir sulfate with ritonavir.
- loss of therapeutic effect of atazanavir sulfate with ritonavir and possible development of resistance.
See Table 16 for steps to prevent or manage these possible and known significant drug interactions, including dosing recommendations [see Drug Interactions ( 7)]. Consider the potential for drug interactions prior to and during atazanavir sulfate/ritonavir therapy; review concomitant medications during atazanavir sulfate/ritonavir therapy; and monitor for the adverse reactions associated with the concomitant medications [see Contraindications ( 4) and Drug Interactions ( 7)].
5.8 Hyperbilirubinemia
Most patients taking atazanavir sulfate experience asymptomatic elevations in indirect (unconjugated) bilirubin related to inhibition of UDP-glucuronosyl transferase (UGT). This hyperbilirubinemia is reversible upon discontinuation of atazanavir sulfate. Hepatic transaminase elevations that occur with hyperbilirubinemia should be evaluated for alternative etiologies. No long-term safety data are available for patients experiencing persistent elevations in total bilirubin > 5 times the upper limit of normal (ULN). Alternative antiretroviral therapy to atazanavir sulfate may be considered if jaundice or scleral icterus associated with bilirubin elevations presents cosmetic concerns for patients. Dose reduction of atazanavir is not recommended since long-term efficacy of reduced doses has not been established [see Adverse Reactions ( 6.1)] .
5.9 Diabetes Mellitus/Hyperglycemia
New-onset diabetes mellitus, exacerbation of preexisting diabetes mellitus, and hyperglycemia have been reported during postmarketing surveillance in HIV-infected patients receiving protease inhibitor therapy. Some patients required either initiation or dose adjustments of insulin or oral hypoglycemic agents for treatment of these events. In some cases, diabetic ketoacidosis has occurred. In those patients who discontinued protease inhibitor therapy, hyperglycemia persisted in some cases. Because these events have been reported voluntarily during clinical practice, estimates of frequency cannot be made and a causal relationship between protease inhibitor therapy and these events has not been established [see Adverse Reactions (6.2)].
5.10 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including atazanavir sulfate. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jiroveci pneumonia, or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves’ disease, polymyositis, and Guillain-Barré syndrome) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment.
5.11 Fat Redistribution
Redistribution/accumulation of body fat including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and “cushingoid appearance” have been observed in patients receiving antiretroviral therapy. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
5.12 Hemophilia
There have been reports of increased bleeding, including spontaneous skin hematomas and hemarthrosis, in patients with hemophilia type A and B treated with protease inhibitors. In some patients additional factor VIII was given. In more than half of the reported cases, treatment with protease inhibitors was continued or reintroduced. A causal relationship between protease inhibitor therapy and these events has not been established.
5.13 Resistance/Cross-Resistance
Various degrees of cross-resistance among protease inhibitors have been observed. Resistance to atazanavir may not preclude the subsequent use of other protease inhibitors [see Microbiology ( 12.4)] .
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- cardiac conduction abnormalities [see Warnings and Precautions ( 5.1)]
- rash [see Warnings and Precautions ( 5.2)]
- hyperbilirubinemia [see Warnings and Precautions ( 5.8)]
- chronic kidney disease [see Warnings and Precautions (5.5)]
- nephrolithiasis and cholelithiasis [see Warnings and Precautions ( 5.6)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions in Treatment-Naive Adult Patients
The safety profile of atazanavir sulfate in treatment-naive adults is based on 1625 HIV-1 infected patients in clinical trials. 536 patients received atazanavir 300 mg with ritonavir 100 mg and 1089 patients received atazanavir 400 mg or higher (without ritonavir).
The most common adverse reactions were nausea, jaundice/scleral icterus, and rash.
Selected clinical adverse reactions of moderate or severe intensity reported in ≥ 2% of treatment-naive patients receiving combination therapy including atazanavir 300 mg with ritonavir 100 mg and atazanavir 400 mg (without ritonavir) are presented in Tables 7 and 8, respectively.
Table 7: Selected Adverse Reactions 1 of Moderate or Severe Intensity Reported in ≥ 2% of Adult Treatment-Naive Patients, 2 Study AI424-138
96 weeks3
atazanavir 300 mg with ritonavir 100 mg (once daily) and tenofovir DF with emtricitabine 496 weeks3
lopinavir 400 mg with ritonavir 100 mg (twice daily) and tenofovir DF with emtricitabine 4(n = 441)
(n = 437)
Digestive System
Nausea
4%
8%
Jaundice/scleral icterus
5%
5 Diarrhea
2%
12%
Skin and Appendages
Rash
3%
2%
1. Includes events of possible, probable, certain, or unknown relationship to treatment regimen.
2. Based on the regimen containing atazanavir sulfate.
3. Median time on therapy.
4. As a fixed-dose combination: 300 mg tenofovir DF, 200 mg emtricitabine once daily.
5. None reported in this treatment arm.
Table 8: Selected Adverse Reactions 1 of Moderate or Severe Intensity Reported in ≥ 2% of Adult Treatment-Naive Patients, 2 Studies AI424-034, AI424-007, and AI424-008
Study AI424-034
Studies AI424-007, -008
64 weeks3 atazanavir 400 mg once daily + lamivudine + zidovudine4
64 weeks3
efavirenz 600 mg once daily + lamivudine + zidovudine 4120 weeks3, 5 atazanavir 400 mg once daily + stavudine + lamivudine or didanosine
73 weeks3, 5
nelfinavir 750 mg TID or 1250 mg BID + stavudine + lamivudine or didanosine(n = 404)
(n = 401)
(n = 279)
(n = 191)
Body as a Whole
Headache
6%
6%
1%
2%
Digestive System
Nausea
14%
12%
6%
4%
Jaundice/
scleral icterus7%
6 7%
6 Vomiting
4%
7%
3%
3%
Abdominal
pain4%
4%
4%
2%
Diarrhea
1%
2%
3%
16%
Nervous System
Insomnia
3%
3%
< 1%
6 Dizziness
2%
7%
< 1%
6 Peripheral
neurologicsymptoms
< 1%
1%
4%
3%
Skin and Appendages
Rash
7%
10%
5%
1%
1. Includes events of possible, probable, certain, or unknown relationship to treatment regimen.
2. Based on regimens containing atazanavir sulfate.
3. Median time on therapy.
4. As a fixed-dose combination: 150 mg lamivudine, 300 mg zidovudine twice daily.
5. Includes long-term follow-up.
6. None reported in this treatment arm.
Adverse Reactions in Treatment-Experienced Adult Patients
The safety profile of atazanavir sulfate in treatment-experienced adults is based on 119 HIV-1 infected patients in clinical trials.
The most common adverse reactions are jaundice/scleral icterus and myalgia.
Selected clinical adverse reactions of moderate or severe intensity reported in ≥ 2% of treatment-experienced patients receiving atazanavir sulfate/ritonavir are presented in Table 9.
Table 9: Selected Adverse Reactions1 of Moderate or Severe Intensity Reported in ≥ 2% of Adult Treatment-Experienced Patients,2 Study AI424-045
48 weeks3
atazanavir/ritonavir 300 mg/100 mg once daily + tenofovir DF + NRTI48 weeks3
lopinavir/ritonavir 400 mg/100 mg twice daily4 + tenofovir DF + NRTI
(n = 119)
(n = 118)
Body as a Whole
Fever
2%
5 Digestive System
Jaundice/scleral icterus
9%
5 Diarrhea
3%
11%
Nausea
3%
2%
Nervous System
Depression
2%
< 1%
Musculoskeletal System
Myalgia
4%
5 1. Includes events of possible, probable, certain, or unknown relationship to treatment regimen.
2. Based on the regimen containing atazanavir sulfate.
3. Median time on therapy.
4. As a fixed-dose combination.
5. None reported in this treatment arm.
Laboratory Abnormalities in Treatment-Naive Patients
The percentages of adult treatment-naive patients treated with combination therapy including atazanavir 300 mg with ritonavir 100 mg and atazanavir 400 mg (without ritonavir) with Grade 3 to 4 laboratory abnormalities are presented in Tables 10 and 11, respectively.
Table 10: Grade 3 to 4 Laboratory Abnormalities Reported in ≥ 2% of Adult Treatment-Naive Patients,1 Study AI424-138
96 weeks2
atazanavir 300 mg with ritonavir 100 mg (once daily) and tenofovir DF with emtricitabine4
96 weeks2
lopinavir 400 mg with ritonavir 100 mg (twice daily) and tenofovir DF with emtricitabine 4Variable
Limit3
(n = 441)
(n = 437)
Chemistry
High
SGOT/AST
≥ 5.1 x ULN
3%
1%
SGPT/ALT
≥ 5.1 x ULN
3%
2%
Total Bilirubin
≥ 2.6 x ULN
44%
< 1%
Lipase
≥ 2.1 x ULN
2%
2%
Creatine Kinase
≥ 5.1 x ULN
8%
7%
Total Cholesterol
≥ 240 mg/dL
11%
25%
Hematology
Low
Neutrophils
< 750 cells/mm 3
5%
2%
1. Based on the regimen containing atazanavir sulfate.
2. Median time on therapy.
3. ULN = upper limit of normal.
4. As a fixed-dose combination: 300 mg tenofovir DF, 200 mg emtricitabine once daily.
Table 11: Grade 3 to 4 Laboratory Abnormalities Reported in ≥ 2% of Adult Treatment-Naive Patients,1 Studies AI424-034, AI424-007, and AI424-008
Study AI424-034
Studies AI424-007, -008
64 weeks2 atazanavir 400 mg once daily + lamivudine + zidovudine5
64 weeks2 efavirenz 600 mg once daily + lamivudine + zidovudine5
120 weeks2, 3 atazanavir 400 mg once daily + stavudine + lamivudine or + stavudine + didanosine
73 weeks2, 3
nelfinavir 750 mg TID or 1250 mg BID + stavudine + lamivudine or + stavudine + didanosineVariable
Limit4
(n = 404)
(n = 401)
(n = 279)
(n = 191)
Chemistry
High
SGOT/AST
≥ 5.1 x ULN
2%
2%
7%
5%
SGPT/ALT
≥ 5.1 x ULN
4%
3%
9%
7%
Total
Bilirubin
≥ 2.6 x ULN
35%
< 1%
47%
3%
Amylase
≥ 2.1 x ULN
6 6 14%
10%
Lipase
≥ 2.1 x ULN
< 1%
1%
4%
5%
Creatine
Kinase
≥ 5.1 x ULN
6%
6%
11%
9%
Total
Cholesterol
≥ 240 mg/dL
6%
24%
19%
48%
Triglycerides≥ 751 mg/dL
< 1%
3%
4%
2%
Hematology
Low
Hemoglobin
< 8.0 g/dL
5%
3%
< 1%
4%
Neutrophils
< 750 cells/mm 3
7%
9%
3%
7%
1. Based on regimen(s) containing atazanavir sulfate.
2. Median time on therapy.
3. Includes long-term follow-up.
4. ULN = upper limit of normal.
5. As a fixed-dose combination: 150 mg lamivudine, 300 mg zidovudine twice daily.
6. None reported in this treatment arm.
Change in Lipids from Baseline in Treatment-Naive Patients
For Study AI424-138 and Study AI424-034, changes from baseline in LDL-cholesterol, HDL-cholesterol, total cholesterol, and triglycerides are shown in Tables 12 and 13, respectively.
Table 12: Lipid Values, Mean Change From Baseline, Study AI424-138
atazanavir sulfate/ritonavir1, 2
lopinavir/ritonavir2, 3
Baseline
Week 48
Week 96
Baseline
Week 48
Week 96
mg/dL
mg/dL
Change4
mg/dL
Change4
mg/dL
mg/dL
Change4
mg/dL
Change4
(n = 4285)
(n = 3725)
(n = 3725)
(n = 3425)
(n = 3425)
(n = 4245)
(n = 3355)
(n = 3355)
(n = 2915)
(n = 2915)
LDL- Cholesterol 6
92
105
+14%
105
+14%
93
111
+19%
110
+17%
HDL- Cholesterol 6
37
46
+29%
44
+21%
36
48
+37%
46
+29%
Total Cholesterol 6
149
169
+13%
169
+13%
150
187
+25%
186
+25%
Triglycerides 6
126
145
+15%
140
+13%
129
194
+52%
184
+50%
1. Atazanavir 300 mg with ritonavir 100 mg once daily with the fixed-dose combination: 300 mg tenofovir DF, 200 mg emtricitabine once daily.
2. Values obtained after initiation of serum lipid-reducing agents were not included in these analyses. At baseline, serum lipid-reducing agents were used in 1% in the lopinavir/ritonavir treatment arm and 1% in the atazanavir sulfate/ritonavir arm. Through Week 48, serum lipid-reducing agents were used in 8% in the lopinavir/ritonavir treatment arm and 2% in the atazanavir sulfate/ritonavir arm. Through Week 96, serum lipid-reducing agents were used in 10% in the lopinavir/ritonavir treatment arm and 3% in the atazanavir sulfate/ritonavir arm.
3. Lopinavir 400 mg with ritonavir 100 mg twice daily with the fixed-dose combination 300 mg tenofovir DF, 200 mg emtricitabine once daily.
4. The change from baseline is the mean of within-patient changes from baseline for patients with both baseline and Week 48 or Week 96 values and is not a simple difference of the baseline and Week 48 or Week 96 mean values, respectively.
5. Number of patients with LDL-cholesterol measured.
6. Fasting.
Table 13: Lipid Values, Mean Change From Baseline, Study AI424-034
atazanavir sulfate1, 2
efavirenz2, 3
Baseline mg/dL
Week 48 mg/dL
Week 48 Change4
Baseline mg/dL
Week 48 mg/dL
Week 48 Change4
(n = 3835)
(n = 2835)
(n = 2725)
(n = 3785)
(n = 2645)
(n = 2535)
LDL-Cholesterol 6
98
98
+1%
98
114
+18%
HDL-Cholesterol
39
43
+13%
38
46
+24%
Total Cholesterol
164
168
+2%
162
195
+21%
Triglycerides 6
138
124
-9%
129
168
+23%
1. Atazanavir 400 mg once daily with the fixed-dose combination: 150 mg lamivudine, 300 mg zidovudine twice daily.
2. Values obtained after initiation of serum lipid-reducing agents were not included in these analyses. At baseline, serum lipid-reducing agents were used in 0% in the efavirenz treatment arm and < 1% in the atazanavir sulfate arm. Through Week 48, serum lipid-reducing agents were used in 3% in the efavirenz treatment arm and 1% in the atazanavir sulfate arm.
3. Efavirenz 600 mg once daily with the fixed-dose combination: 150 mg lamivudine, 300 mg zidovudine twice daily.
4. The change from baseline is the mean of within-patient changes from baseline for patients with both baseline and Week 48 values and is not a simple difference of the baseline and Week 48 mean values.
5. Number of patients with LDL-cholesterol measured.
6. Fasting.
Laboratory Abnormalities in Treatment-Experienced Patients
The percentages of adult treatment-experienced patients treated with combination therapy including atazanavir sulfate/ritonavir with Grade 3 to 4 laboratory abnormalities are presented in Table 14.
Table 14: Grade 3 to 4 Laboratory Abnormalities Reported in ≥ 2% of Adult Treatment-Experienced Patients, Study AI424-0451
48 weeks2
atazanavir/ritonavir 300 mg/100 mg once daily + tenofovir DF + NRTI48 weeks2
lopinavir/ritonavir 400 mg/100 mg twice daily 4 + tenofovir DF + NRTIVariable
Limit3
(n = 119)
(n = 118)
Chemistry
High
SGOT/AST
≥ 5.1 x ULN
3%
3%
SGPT/ALT
≥ 5.1 x ULN
4%
3%
Total Bilirubin
≥ 2.6 x ULN
49%
< 1%
Lipase
≥ 2.1 x ULN
5%
6%
Creatine Kinase
≥ 5.1 x ULN
8%
8%
Total Cholesterol
≥ 240 mg/dL
25%
26%
Triglycerides
≥ 751 mg/dL
8%
12%
Glucose
≥ 251 mg/dL
5%
< 1%
Hematology
Low
Platelets
< 50,000 cells/mm 3
2%
3%
Neutrophils
< 750 cells/mm 3
7%
8%
1. Based on regimen(s) containing atazanavir sulfate.
2. Median time on therapy.
3. ULN = upper limit of normal.
4. As a fixed-dose combination.
Change in Lipids from Baseline in Treatment-Experienced Patients
For Study AI424-045, changes from baseline in LDL-cholesterol, HDL-cholesterol, total cholesterol, and triglycerides are shown in Table 15. The observed magnitude of dyslipidemia was less with atazanavir sulfate/ritonavir than with lopinavir/ritonavir. However, the clinical impact of such findings has not been demonstrated.
Table 15: Lipid Values, Mean Change From Baseline, Study AI424-045
atazanavir sulfate/ritonavir1, 2
lopinavir/ritonavir2, 3
Baseline mg/dL
Week 48 mg/dL
Week 48 Change4
Baseline mg/dL
Week 48 mg/dL
Week 48 Change4
(n = 1115)
(n = 755)
(n = 745)
(n = 1085)
(n = 765)
(n = 735)
LDL-Cholesterol 6
108
98
-10%
104
103
+1%
HDL-Cholesterol
40
39
-7%
39
41
+2%
Total Cholesterol
188
170
-8%
181
187
+6%
Triglycerides 6
215
161
-4%
196
224
+30%
1. Atazanavir 300 mg once daily + ritonavir + tenofovir DF + 1 NRTI.
2. Values obtained after initiation of serum lipid-reducing agents were not included in these analyses. At baseline, serum lipid-reducing agents were used in 4% in the lopinavir/ritonavir treatment arm and 4% in the atazanavir sulfate/ritonavir arm. Through Week 48, serum lipid-reducing agents were used in 19% in the lopinavir/ritonavir treatment arm and 8% in the atazanavir sulfate/ritonavir arm.
3. Lopinavir/ritonavir (400 mg/100 mg) BID + tenofovir DF + 1 NRTI.
4. The change from baseline is the mean of within-patient changes from baseline for patients with both baseline and Week 48 values and is not a simple difference of the baseline and Week 48 mean values.
5. Number of patients with LDL-cholesterol measured.
6. Fasting.
Adverse Reactions in Pediatric Patients: Atazanavir Sulfate Capsules
The safety and tolerability of atazanavir sulfate capsules with and without ritonavir have been established in pediatric patients at least 6 years of age from the open-label, multicenter clinical trial PACTG 1020A.
The safety profile of atazanavir sulfate in pediatric patients (6 to less than 18 years of age) taking the capsule formulation was generally similar to that observed in clinical studies of atazanavir sulfate in adults. The most common Grade 2 to 4 adverse events (≥ 5%, regardless of causality) reported in pediatric patients were cough (21%), fever (18%), jaundice/scleral icterus (15%), rash (14%), vomiting (12%), diarrhea (9%), headache (8%), peripheral edema (7%), extremity pain (6%), nasal congestion (6%), oropharyngeal pain (6%), wheezing (6%), and rhinorrhea (6%).
Asymptomatic second-degree atrioventricular block was reported in < 2% of patients. The most common Grade 3 to 4 laboratory abnormalities occurring in pediatric patients taking the capsule formulation were elevation of total bilirubin (≥ 3.2 mg/dL, 58%), neutropenia (9%), and hypoglycemia (4%). All other Grade 3 to 4 laboratory abnormalities occurred with a frequency of less than 3%.
Adverse Reactions in Patients Co-Infected with Hepatitis B and/or Hepatitis C Virus
In Study AI424-138, 60 patients treated with atazanavir/ritonavir 300 mg/100 mg once daily, and 51 patients treated with lopinavir/ritonavir 400 mg/100 mg twice daily, each with fixed dose tenofovir DF-emtricitabine, were seropositive for hepatitis B and/or C at study entry. ALT levels > 5 times ULN developed in 10% (6/60) of the atazanavir sulfate/ritonavir-treated patients and 8% (4/50) of the lopinavir/ritonavir-treated patients. AST levels > 5 times ULN developed in 10% (6/60) of the atazanavir sulfate/ritonavir-treated patients and none (0/50) of the lopinavir/ritonavir-treated patients.
In Study AI424-045, 20 patients treated with atazanavir/ritonavir 300 mg/100 mg once daily, and 18 patients treated with lopinavir/ritonavir 400 mg/100 mg twice daily, were seropositive for hepatitis B and/or C at study entry. ALT levels > 5 times ULN developed in 25% (5/20) of the atazanavir sulfate/ritonavir-treated patients and 6% (1/18) of the lopinavir/ritonavir-treated patients. AST levels > 5 times ULN developed in 10% (2/20) of the atazanavir sulfate/ritonavir-treated patients and 6% (1/18) of the lopinavir/ritonavir-treated patients.
In Studies AI424-008 and AI424-034, 74 patients treated with 400 mg of atazanavir once daily, 58 who received efavirenz, and 12 who received nelfinavir were seropositive for hepatitis B and/or C at study entry. ALT levels > 5 times ULN developed in 15% of the atazanavir sulfate-treated patients, 14% of the efavirenz-treated patients, and 17% of the nelfinavir-treated patients. AST levels > 5 times ULN developed in 9% of the atazanavir sulfate-treated patients, 5% of the efavirenz-treated patients, and 17% of the nelfinavir‑treated patients. Within atazanavir sulfate and control regimens, no difference in frequency of bilirubin elevations was noted between seropositive and seronegative patients [see Warnings and Precautions ( 5.8)] .
6.2 Postmarketing Experience
The following events have been identified during postmarketing use of atazanavir sulfate. Because these reactions are reported voluntarily from a population of unknown size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Body as a Whole: edema
Cardiovascular System: second-degree AV block, third-degree AV block, left bundle branch block, QT c prolongation [see Warnings and Precautions ( 5.1)]
Gastrointestinal System: pancreatitis
Hepatic System: hepatic function abnormalities
Hepatobiliary Disorders: cholelithiasis [see Warnings and Precautions ( 5.6)] , cholecystitis, cholestasis
Metabolic System and Nutrition Disorders: diabetes mellitus, hyperglycemia [see Warnings and Precautions ( 5.9)]
Musculoskeletal System: arthralgia
Renal System: nephrolithiasis [see Warnings and Precautions ( 5.6)], interstitial nephritis, granulomatous interstitial nephritis, chronic kidney disease [see Warnings and Precautions (5.5)]
Skin and Appendages: alopecia, maculopapular rash [see Contraindications ( 4) and Warnings and Precautions ( 5.2)], pruritus, angioedema
-
7 DRUG INTERACTIONS
7.1 Potential for Atazanavir Sulfate to Affect Other Drugs
Atazanavir is an inhibitor of CYP3A and UGT1A1. Coadministration of atazanavir sulfate and drugs primarily metabolized by CYP3A or UGT1A1 may result in increased plasma concentrations of the other drug that could increase or prolong its therapeutic and adverse effects.
Atazanavir is a weak inhibitor of CYP2C8. Use of atazanavir sulfate without ritonavir is not recommended when coadministered with drugs highly dependent on CYP2C8 with narrow therapeutic indices (e.g., paclitaxel, repaglinide). When atazanavir sulfate with ritonavir is coadministered with substrates of CYP2C8, clinically significant interactions are not expected [see Clinical Pharmacology, Table 22 ( 12.3)].
The magnitude of CYP3A-mediated drug interactions on coadministered drug may change when atazanavir sulfate is coadministered with ritonavir. See the complete prescribing information for ritonavir for information on drug interactions with ritonavir.
7.2 Potential for Other Drugs to Affect Atazanavir Sulfate
Atazanavir is a CYP3A4 substrate; therefore, drugs that induce CYP3A4 may decrease atazanavir plasma concentrations and reduce atazanavir sulfate’s therapeutic effect.
Atazanavir solubility decreases as pH increases. Reduced plasma concentrations of atazanavir are expected if proton-pump inhibitors, antacids, buffered medications, or H 2-receptor antagonists are administered with atazanavir sulfate [see Dosage and Administration ( 2.3, 2.4, and 2.6)].
7.3 Established and Other Potentially Significant Drug Interactions
Table 16 provides dosing recommendations in adults as a result of drug interactions with atazanavir sulfate. These recommendations are based on either drug interaction studies or predicted interactions due to the expected magnitude of interaction and potential for serious events or loss of efficacy.
Table 16: Established and Other Potentially Significant Drug Interactions: Alteration in Dose or Regimen May Be Recommended Based on Drug Interaction Studies 1 or Predicted Interactions (Information in the table applies to Atazanavir Sulfate with or without ritonavir, unless otherwise indicated)
Concomitant Drug Class: Specific Drugs
Effect on Concentration of Atazanavir or Concomitant Drug
Clinical Comment
HIV Antiviral Agents
Nucleoside Reverse Transcriptase Inhibitors (NRTIs):
didanosine buffered formulationsenteric-coated (EC) capsules
↓ atazanavir
↓ didanosine
Coadministration of atazanavir sulfate with didanosine buffered tablets resulted in a marked decrease in atazanavir exposure. It is recommended that atazanavir sulfate be given (with food) 2 h before or 1 h after didanosine buffered formulations. Simultaneous administration of didanosine EC and atazanavir sulfate with food results in a decrease in didanosine exposure. Thus, atazanavir sulfate and didanosine EC should be administered at different times.
Nucleotide Reverse Transcriptase Inhibitors: tenofovir disoproxil fumarate (DF)
↓ atazanavir
↑ tenofovir
Tenofovir DF may decrease the AUC and C min of atazanavir. When coadministered with tenofovir DF in adults, it is recommended that atazanavir 300 mg be given with ritonavir 100 mg and tenofovir DF 300 mg (all as a single daily dose with food). Atazanavir sulfate increases tenofovir concentrations. The mechanism of this interaction is unknown. Higher tenofovir concentrations could potentiate tenofovir-associated adverse reactions, including renal disorders. Patients receiving atazanavir sulfate and tenofovir DF should be monitored for tenofovir-associated adverse reactions. For pregnant women taking atazanavir sulfate with ritonavir and tenofovir DF, see Dosage and Administration ( 2.6) .
Non-nucleoside Reverse Transcriptase Inhibitors (NNRTIs): efavirenz
↓ atazanavir
Efavirenz decreases atazanavir exposure.
In treatment-naive adult patients:
If atazanavir sulfate is combined with efavirenz, atazanavir 400 mg (two 200 mg capsules) should be administered with ritonavir 100 mg simultaneously once daily with food, and efavirenz 600 mg should be administered once daily on an empty stomach, preferably at bedtime.
In treatment-experienced adult patients:
Coadministration of atazanavir sulfate with efavirenz in treatment-experienced patients is not recommended due to decreased atazanavir exposure.
Protease Inhibitors: saquinavir (soft gelatin capsules)
↑ saquinavir
Appropriate dosing recommendations for this combination, with or without ritonavir, with respect to efficacy and safety have not been established. In a clinical study, saquinavir 1200 mg coadministered with atazanavir 400 mg and tenofovir DF 300 mg (all given once daily) plus nucleoside analogue reverse transcriptase inhibitors did not provide adequate efficacy [see Clinical Studies ( 14.2)] .
Ritonavir
↑ atazanavir
If atazanavir sulfate is coadministered with ritonavir, it is recommended that atazanavir 300 mg once daily be given with ritonavir 100 mg once daily with food in adults. See the complete prescribing information for ritonavir for information on drug interactions with ritonavir.
Others
↑ other protease inhibitor
Although not studied, the coadministration of atazanavir sulfate/ritonavir and an additional protease inhibitor would be expected to increase exposure to the other protease inhibitor. Such coadministration is not recommended.
HCV Antiviral Agents
Protease Inhibitors:
boceprevir
↓ atazanavir
↓ ritonavir
Concomitant administration of boceprevir and atazanavir/ritonavir resulted in reduced steady-state exposures to atazanavir and ritonavir. Coadministration of atazanavir sulfate/ritonavir and boceprevir is not recommended.
sofosbuvir, velpatasvir, voxilaprevir ↑ voxilaprevir Coadministration with atazanavir sulfate is not recommended. Other Agents
Antacids and buffered medications
↓ atazanavir
Reduced plasma concentrations of atazanavir are expected if antacids, including buffered medications, are administered with atazanavir sulfate. Atazanavir sulfate should be administered 2 hours before or 1 hour after these medications.
Antiarrhythmics: amiodarone, bepridil, lidocaine (systemic), quinidine
↑ amiodarone, bepridil, lidocaine (systemic), quinidine
Coadministration with atazanavir sulfate has the potential to produce serious and/or life-threatening adverse events and has not been studied. Caution is warranted and therapeutic concentration monitoring of these drugs is recommended if they are used concomitantly with atazanavir sulfate.
Anticoagulants: warfarin
↑ warfarin
Coadministration with atazanavir sulfate has the potential to produce serious and/or life-threatening bleeding and has not been studied. It is recommended that International Normalized Ratio (INR) be monitored.
Antidepressants: tricyclic antidepressants
↑ tricyclic antidepressants
Coadministration with atazanavir sulfate has the potential to produce serious and/or life-threatening adverse events and has not been studied. Concentration monitoring of these drugs is recommended if they are used concomitantly with atazanavir sulfate.
Trazodone
↑ trazodone
Concomitant use of trazodone and atazanavir sulfate with or without ritonavir may increase plasma concentrations of trazodone. Nausea, dizziness, hypotension, and syncope have been observed following coadministration of trazodone and ritonavir. If trazodone is used with a CYP3A4 inhibitor such as atazanavir sulfate, the combination should be used with caution and a lower dose of trazodone should be considered.
Antiepileptics:
carbamazepine
↓ atazanavir
↑ carbamazepine
Plasma concentrations of atazanavir may be decreased when carbamazepine is administered with atazanavir sulfate without ritonavir. Coadministration of carbamazepine and atazanavir sulfate without ritonavir is not recommended. Ritonavir may increase plasma levels of carbamazepine. If patients beginning treatment with atazanavir sulfate/ritonavir have been titrated to a stable dose of carbamazepine, a dose reduction for carbamazepine may be necessary.
phenytoin, phenobarbital
↓ atazanavir
↓ phenytoin
↓ phenobarbital
Plasma concentrations of atazanavir may be decreased when phenytoin or phenobarbital is administered with atazanavir sulfate without ritonavir. Coadministration of phenytoin or phenobarbital and atazanavir sulfate without ritonavir is not recommended. Ritonavir may decrease plasma levels of phenytoin and phenobarbital. When atazanavir sulfate with ritonavir is coadministered with either phenytoin or phenobarbital, a dose adjustment of phenytoin or phenobarbital may be required.
Lamotrigine
↓ lamotrigine
Coadministration of lamotrigine and atazanavir sulfate with ritonavir may decrease lamotrigine plasma concentrations. Dose adjustment of lamotrigine may be required when coadministered with atazanavir sulfate and ritonavir. Coadministration of lamotrigine and atazanavir sulfate without ritonavir is not expected to decrease lamotrigine plasma concentrations. No dose adjustment of lamotrigine is required when coadministered with atazanavir sulfate without ritonavir.
Antifungals:
ketoconazole, itraconazoleAtazanavir sulfate/ ritonavir:
↑ ketoconazole
↑ itraconazole
Coadministration of ketoconazole has only been studied with atazanavir sulfate without ritonavir (negligible increase in atazanavir AUC and C max). Due to the effect of ritonavir on ketoconazole, high doses of ketoconazole and itraconazole (> 200 mg/day) should be used cautiously with atazanavir sulfate/ritonavir.
Voriconazole
Atazanavir sulfate/ ritonavir in subjects with a functional CYP2C19 allele:
↓ voriconazole
↓ atazanavir
Atazanavir sulfate/ ritonavir in subjects without a functional CYP2C19 allele:
↑ voriconazole
↓ atazanavir
The use of voriconazole in patients receiving atazanavir sulfate/ritonavir is not recommended unless an assessment of the benefit/risk to the patient justifies the use of voriconazole. Patients should be carefully monitored for voriconazole-associated adverse reactions and loss of either voriconazole or atazanavir efficacy during the coadministration of voriconazole and atazanavir sulfate/ritonavir. Coadministration of voriconazole with atazanavir sulfate (without ritonavir) may affect atazanavir concentrations; however, no data are available.
Antigout: colchicine
↑ colchicine
The coadministration of atazanavir sulfate with colchicine in patients with renal or hepatic impairment is not recommended.
Recommended adult dosage of colchicine when administered with atazanavir sulfate:
Treatment of gout flares:
0.6 mg (1 tablet) for 1 dose, followed by 0.3 mg (half tablet) 1 hour later. Not to be repeated before 3 days.
Prophylaxis of gout flares:
If the original regimen was 0.6 mg twice a day, the regimen should be adjusted to 0.3 mg once a day.
If the original regimen was 0.6 mg once a day, the regimen should be adjusted to 0.3 mg once every other day.
Treatment of familial Mediterranean fever (FMF):
Maximum daily dose of 0.6 mg (may be given as 0.3 mg twice a day).
Antimycobacterials: rifabutin
↑ rifabutin
A rifabutin dose reduction of up to 75% (e.g., 150 mg every other day or 3 times per week) is recommended. Increased monitoring for rifabutin-associated adverse reactions including neutropenia is warranted.
Antipsychotics: quetiapine and lurasidone
↑ quetiapine
atazanavir sulfate
↑ lurasidone
atazanavir sulfate/
ritonavir
↑ lurasidone
Initiation of atazanavir sulfate with ritonavir in patients taking quetiapine:
Consider alternative antiretroviral therapy to avoid increases in quetiapine exposures. If coadministration is necessary, reduce the quetiapine dose to 1/6 of the current dose and monitor for quetiapine-associated adverse reactions. Refer to the quetiapine prescribing information for recommendations on adverse reaction monitoring.
Initiation of quetiapine in patients taking atazanavir sulfate with ritonavir:
Refer to the quetiapine prescribing information for initial dosing and titration of quetiapine.
Atazanavir sulfate without ritonavir
If coadministration is necessary, reduce the lurasidone dose. Refer to the lurasidone prescribing information for concomitant use with moderate CYP3A4 inhibitors.
Atazanavir sulfate/ritonavir
Use of lurasidone is contraindicated.
Benzodiazepines: parenterally administered midazolam 2
↑ midazolam
Concomitant use of parenteral midazolam with atazanavir sulfate may increase plasma concentrations of midazolam. Coadministration should be done in a setting which ensures close clinical monitoring and appropriate medical management in case of respiratory depression and/or prolonged sedation. Dosage reduction for midazolam should be considered, especially if more than a single dose of midazolam is administered. Coadministration of oral midazolam with atazanavir sulfate is CONTRAINDICATED.
Calcium channel blockers: diltiazem
↑ diltiazem and desacetyl-diltiazem
Caution is warranted. A dose reduction of diltiazem by 50% should be considered. ECG monitoring is recommended. Coadministration of atazanavir sulfate/ritonavir with diltiazem has not been studied.
felodipine, nifedipine, nicardipine, and verapamil
↑ calcium channel blocker
Caution is warranted. Dose titration of the calcium channel blocker should be considered. ECG monitoring is recommended.
Endothelin receptor antagonists:
bosentan↓ atazanavir
↑ bosentan
Plasma concentrations of atazanavir may be decreased when bosentan is administered with atazanavir sulfate without ritonavir. Coadministration of bosentan and atazanavir sulfate without ritonavir is not recommended.
Coadministration of bosentan in adult patients on atazanavir sulfate/ritonavir:
For patients who have been receiving atazanavir sulfate/ritonavir for at least 10 days, start bosentan at 62.5 mg once daily or every other day based on individual tolerability.
Coadministration of atazanavir sulfate/ritonavir in adult patients on bosentan:
Discontinue bosentan at least 36 hours before starting atazanavir sulfate/ritonavir. At least 10 days after starting atazanavir sulfate/ritonavir, resume bosentan at 62.5 mg once daily or every other day based on individual tolerability.
HMG-CoA reductase inhibitors: atorvastatin, rosuvastatin
↑ atorvastatin
↑ rosuvastatin
Titrate atorvastatin dose carefully and use the lowest necessary dose. Rosuvastatin dose should not exceed 10 mg/day. The risk of myopathy, including rhabdomyolysis, may be increased when HIV protease inhibitors, including atazanavir sulfate, are used in combination with these drugs.
H 2-Receptor antagonists
↓ atazanavir
Plasma concentrations of atazanavir were substantially decreased when atazanavir 400 mg once daily was administered simultaneously with famotidine 40 mg twice daily in adults, which may result in loss of therapeutic effect and development of resistance.
In treatment-naive adult patients:
Atazanavir 300 mg with ritonavir 100 mg once daily with food should be administered simultaneously with, and/or at least 10 hours after, a dose of the H 2-receptor antagonist (H2RA). An H2RA dose comparable to famotidine 20 mg once daily up to a dose comparable to famotidine 40 mg twice daily can be used with atazanavir 300 mg with ritonavir 100 mg in treatment-naive patients.
OR
For patients unable to tolerate ritonavir, atazanavir 400 mg once daily with food should be administered at least 2 hours before and at least 10 hours after a dose of the H2RA. No single dose of the H2RA should exceed a dose comparable to famotidine 20 mg, and the total daily dose should not exceed a dose comparable to famotidine 40 mg. The use of atazanavir sulfate without ritonavir in pregnant women is not recommended.
In treatment-experienced adult patients:
Whenever an H2RA is given to a patient receiving atazanavir sulfate with ritonavir, the H2RA dose should not exceed a dose comparable to famotidine 20 mg twice daily, and the atazanavir sulfate and ritonavir doses should be administered simultaneously with, and/or at least 10 hours after, the dose of the H2RA.
- Atazanavir 300 mg with ritonavir 100 mg once daily (all as a single dose with food) if taken with an H2RA.
- Atazanavir 400 mg with ritonavir 100 mg once daily (all as a single dose with food) if taken with both tenofovir DF and an H2RA.
- Atazanavir 400 mg with ritonavir 100 mg once daily (all as a single dose with food) if taken with either tenofovir DF or an H2RA for pregnant women during the second and third trimester. Atazanavir sulfate is not recommended for pregnant women during the second and third trimester taking atazanavir sulfate with both tenofovir DF and an H2RA.
Hormonal contraceptives: ethinyl estradiol and norgestimate or norethindrone
↓ ethinyl estradiol
↑ norgestimate 3
Use with caution if coadministration of atazanavir sulfate or atazanavir sulfate/ritonavir with oral contraceptives is considered. If an oral contraceptive is administered with atazanavir sulfate plus ritonavir, it is recommended that the oral contraceptive contain at least 35 mcg of ethinyl estradiol. If atazanavir sulfate is administered without ritonavir, the oral contraceptive should contain no more than 30 mcg of ethinyl estradiol.
↑ ethinyl estradiol
↑ norethindrone 4
Potential safety risks include substantial increases in progesterone exposure. The long-term effects of increases in concentration of the progestational agent are unknown and could increase the risk of insulin resistance, dyslipidemia, and acne.
Coadministration of atazanavir sulfate or atazanavir sulfate/ritonavir with other hormonal contraceptives (e.g., contraceptive patch, contraceptive vaginal ring, or injectable contraceptives) or oral contraceptives containing progestogens other than norethindrone or norgestimate, or less than 25 mcg of ethinyl estradiol, has not been studied; therefore, alternative methods of contraception are recommended.
Immunosuppressants: cyclosporine, sirolimus, tacrolimus
↑ immunosuppressants
Therapeutic concentration monitoring is recommended for these immunosuppressants when coadministered with atazanavir sulfate.
Inhaled beta agonist: salmeterol
↑ salmeterol
Coadministration of salmeterol with atazanavir sulfate is not recommended. Concomitant use of salmeterol and atazanavir sulfate may result in increased risk of cardiovascular adverse reactions associated with salmeterol, including QT prolongation, palpitations, and sinus tachycardia.
Inhaled/nasal steroid: fluticasone
Atazanavir sulfate
↑ fluticasone
Concomitant use of fluticasone propionate and atazanavir sulfate (without ritonavir) may increase plasma concentrations of fluticasone propionate. Use with caution. Consider alternatives to fluticasone propionate, particularly for long-term use.
Atazanavir sulfate/ritonavir
↑ fluticasone
Concomitant use of fluticasone propionate and atazanavir sulfate/ritonavir may increase plasma concentrations of fluticasone propionate, resulting in significantly reduced serum cortisol concentrations. Systemic corticosteroid effects, including Cushing’s syndrome and adrenal suppression, have been reported during postmarketing use in patients receiving ritonavir and inhaled or intranasally administered fluticasone propionate. Coadministration of fluticasone propionate and atazanavir sulfate/ritonavir is not recommended unless the potential benefit to the patient outweighs the risk of systemic corticosteroid side effects [see Warnings and Precautions (5.1)].
Macrolide antibiotics: clarithromycin
↑ clarithromycin
↓ 14-OH clarithromycin
↑ atazanavir
Increased concentrations of clarithromycin may cause QT c prolongations; therefore, a dose reduction of clarithromycin by 50% should be considered when it is coadministered with atazanavir sulfate. In addition, concentrations of the active metabolite 14-OH clarithromycin are significantly reduced; consider alternative therapy for indications other than infections due to Mycobacterium avium complex. Coadministration of atazanavir sulfate/ritonavir with clarithromycin has not been studied.
Opioids: Buprenorphine
↑ buprenorphine
↑ norbuprenorphine
Coadministration of buprenorphine and atazanavir sulfate with or without ritonavir increases the plasma concentration of buprenorphine and norbuprenorphine. Coadministration of atazanavir sulfate plus ritonavir with buprenorphine warrants clinical monitoring for sedation and cognitive effects. A dose reduction of buprenorphine may be considered. Coadministration of buprenorphine and atazanavir sulfate with ritonavir is not expected to decrease atazanavir plasma concentrations. Coadministration of buprenorphine and atazanavir sulfate without ritonavir may decrease atazanavir plasma concentrations. The coadministration of atazanavir sulfate and buprenorphine without ritonavir is not recommended.
PDE5 inhibitors: sildenafil, tadalafil, vardenafil
↑ sildenafil
↑ tadalafil
↑ vardenafil
Coadministration with atazanavir sulfate has not been studied but may result in an increase in PDE5 inhibitor-associated adverse reactions, including hypotension, syncope, visual disturbances, and priapism.
Use of PDE5 inhibitors for pulmonary arterial hypertension (PAH):
Use of REVATIO ® (sildenafil) for the treatment of pulmonary hypertension (PAH) is contraindicated with atazanavir sulfate [see Contraindications (4)].
The following dose adjustments are recommended for the use of ADCIRCA ® (tadalafil) with atazanavir sulfate:
Coadministration of ADCIRCA ® in patients on atazanavir sulfate (with or without ritonavir):
- For patients receiving atazanavir sulfate (with or without ritonavir) for at least one week, start ADCIRCA
® at 20 mg once daily. Increase to 40 mg once daily based on individual tolerability.
Coadministration of atazanavir sulfate (with or without ritonavir) in patients on ADCIRCA ®:
- Avoid the use of ADCIRCA ® when starting atazanavir sulfate (with or without ritonavir). Stop ADCIRCA ® at least 24 hours before starting atazanavir sulfate (with or without ritonavir). At least one week after starting atazanavir sulfate (with or without ritonavir), resume ADCIRCA ® at 20 mg once daily. Increase to 40 mg once daily based on individual tolerability.
Use of PDE5 inhibitors for erectile dysfunction:
Use VIAGRA ® (sildenafil) with caution at reduced doses of 25 mg every 48 hours with increased monitoring for adverse events.
Use CIALIS ® (tadalafil) with caution at reduced doses of 10 mg every 72 hours with increased monitoring for adverse events.
Atazanavir sulfate/ritonavir: Use vardenafil with caution at reduced doses of no more than 2.5 mg every 72 hours with increased monitoring for adverse reactions.
Atazanavir sulfate: Use vardenafil with caution at reduced doses of no more than 2.5 mg every 24 hours with increased monitoring for adverse reactions.
Proton-pump inhibitors: omeprazole
↓ atazanavir
Plasma concentrations of atazanavir were substantially decreased when atazanavir 400 mg or atazanavir 300 mg/ritonavir 100 mg once daily was administered with omeprazole 40 mg once daily in adults, which may result in loss of therapeutic effect and development of resistance.
In treatment-naive adult patients:
The proton-pump inhibitor (PPI) dose should not exceed a dose comparable to omeprazole 20 mg and must be taken approximately 12 hours prior to the atazanavir 300 mg with ritonavir 100 mg dose.
In treatment-experienced adult patients:
The use of PPIs in treatment-experienced patients receiving atazanavir sulfate is not recommended.
1. For magnitude of interactions see Clinical Pharmacology, Tables 21 and 22 (12.3).
2. See Contraindications (4), Table 6 for orally administered midazolam.
3. In combination with atazanavir 300 mg and ritonavir 100 mg once daily.
4. In combination with atazanavir 400 mg once daily.
7.4 Drugs with No Observed Interactions with Atazanavir Sulfate
No clinically significant drug interactions were observed when atazanavir sulfate was coadministered with methadone, fluconazole, acetaminophen, atenolol, or the nucleoside reverse transcriptase inhibitors lamivudine or zidovudine [see Clinical Pharmacology, Tables 21 and 22 ( 12.3)] .
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to atazanavir sulfate during pregnancy. Healthcare providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry (APR) at 1-800-258-4263.
Risk Summary
Atazanavir has been evaluated in a limited number of women during pregnancy. Available human and animal data suggest that atazanavir does not increase the risk of major birth defects overall compared to the background rate [see Data]. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively. No treatment-related malformations were observed in rats and rabbits, for which the atazanavir exposures were 0.7 to 1.2 times of those at the human clinical dose (300 mg/day atazanavir boosted with 100 mg/day ritonavir). When atazanavir was administered to rats during pregnancy and throughout lactation, reversible neonatal growth retardation was observed [see Data].
Clinical ConsiderationsDose Adjustments during Pregnancy and the Postpartum Period
- Atazanavir sulfate must be administered with ritonavir in pregnant women.
- For pregnant patients, no dosage adjustment is required for atazanavir sulfate with the following exceptions:
- For treatment-experienced pregnant women during the second or third trimester, when atazanavir sulfate is coadministered with either an H 2-receptor antagonist or tenofovir DF, atazanavir 400 mg with ritonavir 100 mg once daily is recommended. There are insufficient data to recommend an atazanavir sulfate dose for use with both an H 2-receptor antagonist and tenofovir DF in treatment-experienced pregnant women.
- No dosage adjustment is required for postpartum patients. However, patients should be closely monitored for adverse events because atazanavir exposures could be higher during the first 2 months after delivery
[see Dosage and Administration (
2.6) and Clinical Pharmacology (
12.3)]
.
Maternal Adverse Reactions
Cases of lactic acidosis syndrome, sometimes fatal, and symptomatic hyperlactatemia have occurred in pregnant women using atazanavir sulfate in combination with nucleoside analogues, which are associated with an increased risk of lactic acidosis syndrome.
Hyperbilirubinemia occurs frequently in patients who take atazanavir sulfate [see Warnings and Precautions ( 5.8)], including pregnant women [see Data].
Advise pregnant women of the potential risks of lactic acidosis syndrome and hyperbilirubinemia.
Fetal/Neonatal Adverse Reactions
All infants, including neonates exposed to atazanavir sulfate in utero, should be monitored for the development of severe hyperbilirubinemia during the first few days of life [see Data].
Data
Human Data
In clinical trial AI424-182, atazanavir/ritonavir (300 mg/100 mg or 400 mg/100 mg) in combination with zidovudine/lamivudine was administered to 41 HIV-infected pregnant women during the second or third trimester. Among the 39 women who completed the study, 38 women achieved an HIV RNA less than 50 copies/mL at time of delivery. Six of 20 (30%) women on atazanavir/ritonavir 300 mg/100 mg and 13 of 21 (62%) women on atazanavir/ritonavir 400 mg/100 mg experienced hyperbilirubinemia (total bilirubin greater than or equal to 2.6 times ULN). There were no cases of lactic acidosis observed in clinical trial AI424-182.
Atazanavir drug concentrations in fetal umbilical cord blood were approximately 12% to 19% of maternal concentrations. Among the 40 infants born to 40 HIV-infected pregnant women, all had test results that were negative for HIV-1 DNA at the time of delivery and/or during the first 6 months postpartum. All 40 infants received antiretroviral prophylactic treatment containing zidovudine. No evidence of severe hyperbilirubinemia (total bilirubin levels greater than 20 mg/dL) or acute or chronic bilirubin encephalopathy was observed among neonates in this study. However, 10/36 (28%) infants (6 greater than or equal to 38 weeks gestation and 4 less than 38 weeks gestation) had bilirubin levels of 4 mg/dL or greater within the first day of life.
Lack of ethnic diversity was a study limitation. In the study population, 33/40 (83%) infants were Black/African American, who have a lower incidence of neonatal hyperbilirubinemia than Caucasians and Asians. In addition, women with Rh incompatibility were excluded, as well as women who had a previous infant who developed hemolytic disease and/or had neonatal pathologic jaundice (requiring phototherapy).
Additionally, of the 38 infants who had glucose samples collected in the first day of life, 3 had adequately collected serum glucose samples with values of less than 40 mg/dL that could not be attributed to maternal glucose intolerance, difficult delivery, or sepsis.
Based on prospective reports from the APR of approximately 1600 live births following exposure to atazanavir-containing regimens (including 1037 live births in infants exposed in the first trimester and 569 exposed in second/third trimesters), there was no difference between atazanavir and overall birth defects compared with the background birth defect rate. In the U.S. general population, the estimated background risk of major birth defects in clinically recognized pregnancies is 2 to 4%.
Animal Data
In animal reproduction studies, there was no evidence of mortality or teratogenicity in offspring born to animals at systemic drug exposure levels (AUC) 0.7 (in rabbits) to 1.2 (in rats) times those observed at the human clinical dose (300 mg/day atazanavir boosted with 100 mg/day ritonavir). In pre- and postnatal development studies in the rat, atazanavir caused neonatal growth retardation during lactation that reversed after weaning. Maternal drug exposure at this dose was 1.3 times the human exposure at the recommended clinical exposure. Minimal maternal toxicity occurred at this exposure level.
8.2 Lactation
Risk Summary
The Centers for Disease Control and Prevention recommend that HIV-1 infected mothers not breastfeed their infants to avoid risking postnatal transmission of HIV-1. Atazanavir has been detected in human milk. No data are available regarding atazanavir effects on milk production. Atazanavir was present in the milk of lactating rats and was associated with neonatal growth retardation that reversed after weaning.
Because of both the potential for HIV-1 transmission and the potential for serious adverse reactions in breastfed infants, advise women not to breastfeed.
8.4 Pediatric Use
Atazanavir sulfate is indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection in pediatric patients 6 years of age and older and weighing at least 15 kg. Atazanavir sulfate is not recommended for use in pediatric patients below the age of 3 months due to the risk of kernicterus [see Indications and Usage ( 1)]. All atazanavir sulfate contraindications, warnings, and precautions apply to pediatric patients [see Contraindications ( 4) and Warnings and Precautions ( 5)].
The safety, pharmacokinetic profile, and virologic response of atazanavir sulfate in pediatric patients at least 6 years of age and older and weighing at least 15 kg were established in an open-label, multicenter clinical trial: PACTG 1020A [see Clinical Pharmacology ( 12.3) and Clinical Studies ( 14.3)]. The safety profile in pediatric patients was generally similar to that observed in adults [see Adverse Reactions ( 6.1)]. See Dosage and Administration ( 2.4) for dosing recommendations for the use of atazanavir sulfate capsules.
8.5 Geriatric Use
Clinical studies of atazanavir sulfate did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients. Based on a comparison of mean single-dose pharmacokinetic values for C max and AUC, a dose adjustment based upon age is not recommended. In general, appropriate caution should be exercised in the administration and monitoring of atazanavir sulfate in elderly patients reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Age/Gender
A study of the pharmacokinetics of atazanavir was performed in young (n = 29; 18 to 40 years) and elderly (n = 30; ≥ 65 years) healthy subjects. There were no clinically significant pharmacokinetic differences observed due to age or gender.
-
10 OVERDOSAGE
Human experience of acute overdose with atazanavir sulfate is limited. Single doses up to 1200 mg (three times the 400 mg maximum recommended dose) have been taken by healthy volunteers without symptomatic untoward effects. A single self-administered overdose of 29.2 g of atazanavir sulfate in an HIV-infected patient (73 times the 400-mg recommended dose) was associated with asymptomatic bifascicular block and PR interval prolongation. These events resolved spontaneously. At atazanavir sulfate doses resulting in high atazanavir exposures, jaundice due to indirect (unconjugated) hyperbilirubinemia (without associated liver function test changes) or PR interval prolongation may be observed [see Warnings and Precautions ( 5.1, 5.8) and Clinical Pharmacology ( 12.2)].
Treatment of overdosage with atazanavir sulfate should consist of general supportive measures, including monitoring of vital signs and ECG, and observations of the patient’s clinical status. If indicated, elimination of unabsorbed atazanavir should be achieved by emesis or gastric lavage. Administration of activated charcoal may also be used to aid removal of unabsorbed drug. There is no specific antidote for overdose with atazanavir sulfate. Since atazanavir is extensively metabolized by the liver and is highly protein bound, dialysis is unlikely to be beneficial in significant removal of this medicine.
-
11 DESCRIPTION
The active ingredient in atazanavir sulfate capsules is atazanavir sulfate, which is an HIV-1 protease inhibitor.
The chemical name for atazanavir sulfate is (3 S,8 S,9 S,12 S)-3,12-bis(1,1-dimethylethyl)-8-hydroxy-4,11-dioxo-9-(phenylmethyl)-6-[[4-(2-pyridinyl)phenyl]methyl]-2,5,6,10,13-pentaazatetradecanedioic acid dimethyl ester, sulfate (1:1). Atazanavir sulfate has the following structural formula:
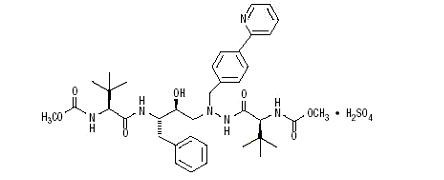
C 38H 52N 6O 7H 2SO 4 M.W. 704.9 (free base) 802.9 (sulfuric acid salt)
Atazanavir sulfate is a white to pale yellow crystalline powder. It is slightly soluble in water (4 to 5 mg/mL, free base equivalent) with the pH of a saturated solution in water being about 1.9 at 24 ± 3° C.
Atazanavir sulfate capsules are available for oral administration in strengths containing the equivalent of 150 mg, 200 mg or 300 mg of atazanavir as atazanavir sulfate and the following inactive ingredients: crospovidone, D&C yellow #10 aluminum lake, FD&C blue #1, FD&C blue #1 aluminum lake, FD&C blue #2 aluminum lake, FD&C red #40 aluminum lake, gelatin, iron oxide black, lactose monohydrate, magnesium stearate, propylene glycol, shellac, sodium lauryl sulfate, sorbitan monolaurate, and titanium dioxide. Additionally, the 300 mg capsules also contain FD&C red #40.
-
12 CLINICAL PHARMACOLOGY
12.2 Pharmacodynamics
Cardiac Electrophysiology
Concentration- and dose-dependent prolongation of the PR interval in the electrocardiogram has been observed in healthy volunteers receiving atazanavir. In a placebo-controlled study (AI424-076), the mean (± SD) maximum change in PR interval from the predose value was 24 (± 15) msec following oral dosing with 400 mg of atazanavir (n = 65) compared to 13 (± 11) msec following dosing with placebo (n = 67). The PR interval prolongations in this study were asymptomatic. There is limited information on the potential for a pharmacodynamic interaction in humans between atazanavir and other drugs that prolong the PR interval of the electrocardiogram [see Warnings and Precautions ( 5.1)].
Electrocardiographic effects of atazanavir were determined in a clinical pharmacology study of 72 healthy subjects. Oral doses of 400 mg (maximum recommended dosage) and 800 mg (twice the maximum recommended dosage) were compared with placebo; there was no concentration-dependent effect of atazanavir on the QT c interval (using Fridericia’s correction). In 1793 HIV-infected patients receiving antiretroviral regimens, QT c prolongation was comparable in the atazanavir and comparator regimens. No atazanavir-treated healthy subject or HIV-infected patient in clinical trials had a QT c interval > 500 msec [see Warnings and Precautions ( 5.1)].
12.3 Pharmacokinetics
The pharmacokinetics of atazanavir were evaluated in healthy adult volunteers and in HIV-infected patients after administration of atazanavir 400 mg once daily and after administration of atazanavir 300 mg with ritonavir 100 mg once daily (see Table 17).
Table 17: Steady-State Pharmacokinetics of Atazanavir in Healthy Subjects or HIV-Infected Patients in the Fed State
Parameter400 mg once daily
300 mg with ritonavir 100 mg once daily
Healthy Subjects
HIV-Infected Patients
Healthy Subjects
HIV-Infected Patients
(n = 14)
(n = 13)
(n = 28)
(n = 10)
C max (ng/mL)
Geometric mean (CV%)
5199 (26)
2298 (71)
6129 (31)
4422 (58)
Mean (SD)
5358 (1371)
3152 (2231)
6450 (2031)
5233 (3033)
T max (h)
Median
2.5
2.0
2.7
3.0
AUC (ngh/mL)
Geometric mean (CV%)
28132 (28)
14874 (91)
57039 (37)
46073 (66)
Mean (SD)
29303 (8263)
22262 (20159)
61435 (22911)
53761 (35294)
T-half (h)
Mean (SD)
7.9 (2.9)
6.5 (2.6)
18.1 (6.2) 1
8.6 (2.3)
C min (ng/mL)
Geometric mean (CV%)
159 (88)
120 (109)
1227 (53)
636 (97)
Mean (SD)
218 (191)
273 (298) 2
1441 (757)
862 (838)
1. n = 26.
2. n = 12.
Figure 1 displays the mean plasma concentrations of atazanavir at steady state after atazanavir 400 mg once daily (as two 200 mg capsules) with a light meal and after atazanavir 300 mg (as two 150 mg capsules) with ritonavir 100 mg once daily with a light meal in HIV-infected adult patients.
Figure 1: Mean (SD) Steady-State Plasma Concentrations of Atazanavir 400 mg (n = 13) and 300 mg With Ritonavir (n = 10) for HIV-Infected Adult Patients
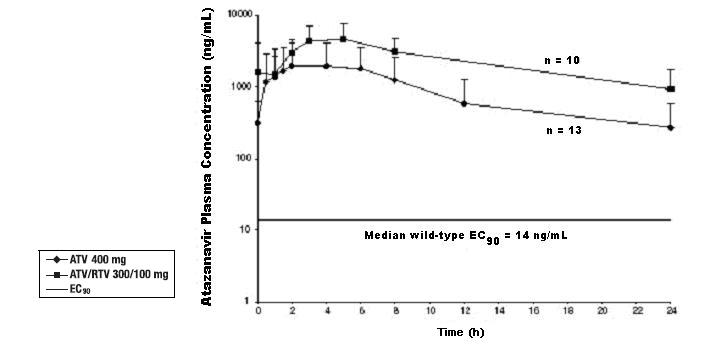
Absorption
Atazanavir is rapidly absorbed with a T max of approximately 2.5 hours. Atazanavir demonstrates nonlinear pharmacokinetics with greater than dose-proportional increases in AUC and C max values over the dose range of 200 to 800 mg once daily. Steady state is achieved between Days 4 and 8, with an accumulation of approximately 2.3 fold.
Food Effect
Administration of atazanavir sulfate with food enhances bioavailability and reduces pharmacokinetic variability. Administration of a single 400 mg dose of atazanavir with a light meal (357 kcal, 8.2 g fat, 10.6 g protein) resulted in a 70% increase in AUC and 57% increase in C max relative to the fasting state. Administration of a single 400 mg dose of atazanavir with a high-fat meal (721 kcal, 37.3 g fat, 29.4 g protein) resulted in a mean increase in AUC of 35% with no change in C max relative to the fasting state. Administration of atazanavir sulfate with either a light meal or high-fat meal decreased the coefficient of variation of AUC and C max by approximately one-half compared to the fasting state.
Coadministration of a single 300 mg dose of atazanavir and a 100 mg dose of ritonavir with a light meal (336 kcal, 5.1 g fat, 9.3 g protein) resulted in a 33% increase in the AUC and a 40% increase in both the C max and the 24 hour concentration of atazanavir relative to the fasting state. Coadministration with a high-fat meal (951 kcal, 54.7 g fat, 35.9 g protein) did not affect the AUC of atazanavir relative to fasting conditions and the C max was within 11% of fasting values. The 24 hour concentration following a high-fat meal was increased by approximately 33% due to delayed absorption; the median T max increased from 2.0 to 5.0 hours. Coadministration of atazanavir sulfate with ritonavir with either a light or a high-fat meal decreased the coefficient of variation of AUC and C max by approximately 25% compared to the fasting state.
Distribution
Atazanavir is 86% bound to human serum proteins and protein binding is independent of concentration. Atazanavir binds to both alpha-1-acid glycoprotein (AAG) and albumin to a similar extent (89% and 86%, respectively). In a multiple-dose study in HIV-infected patients dosed with atazanavir 400 mg once daily with a light meal for 12 weeks, atazanavir was detected in the cerebrospinal fluid and semen. The cerebrospinal fluid/plasma ratio for atazanavir (n = 4) ranged between 0.0021 and 0.0226 and seminal fluid/plasma ratio (n = 5) ranged between 0.11 and 4.42.
Metabolism
Atazanavir is extensively metabolized in humans. The major biotransformation pathways of atazanavir in humans consisted of monooxygenation and dioxygenation. Other minor biotransformation pathways for atazanavir or its metabolites consisted of glucuronidation, N-dealkylation, hydrolysis, and oxygenation with dehydrogenation. Two minor metabolites of atazanavir in plasma have been characterized. Neither metabolite demonstrated in vitro antiviral activity. In vitro studies using human liver microsomes suggested that atazanavir is metabolized by CYP3A.
Elimination
Following a single 400 mg dose of 14C-atazanavir, 79% and 13% of the total radioactivity was recovered in the feces and urine, respectively. Unchanged drug accounted for approximately 20% and 7% of the administered dose in the feces and urine, respectively. The mean elimination half-life of atazanavir in healthy volunteers (n = 214) and HIV-infected adult patients (n = 13) was approximately 7 hours at steady state following a dose of 400 mg daily with a light meal.
Specific Populations
Renal Impairment
In healthy subjects, the renal elimination of unchanged atazanavir was approximately 7% of the administered dose. Atazanavir sulfate has been studied in adult subjects with severe renal impairment (n = 20), including those on hemodialysis, at multiple doses of 400 mg once daily. The mean atazanavir C max was 9% lower, AUC was 19% higher, and C min was 96% higher in subjects with severe renal impairment not undergoing hemodialysis (n = 10), than in age-, weight-, and gender-matched subjects with normal renal function. In a 4 hour dialysis session, 2.1% of the administered dose was removed. When atazanavir was administered either prior to, or following hemodialysis (n = 10), the geometric means for C max, AUC, and C min were approximately 25% to 43% lower compared to subjects with normal renal function. The mechanism of this decrease is unknown. Atazanavir sulfate is not recommended for use in HIV-treatment-experienced patients with end-stage renal disease managed with hemodialysis [see Dosage and Administration ( 2.7)].
Hepatic Impairment
Atazanavir sulfate has been studied in adult subjects with moderate-to-severe hepatic impairment (14 Child-Pugh B and 2 Child-Pugh C subjects) after a single 400 mg dose. The mean AUC (0-∞) was 42% greater in subjects with impaired hepatic function than in healthy volunteers. The mean half-life of atazanavir in hepatically impaired subjects was 12.1 hours compared to 6.4 hours in healthy volunteers. A dose reduction to 300 mg is recommended for patients with moderate hepatic impairment (Child-Pugh Class B) who have not experienced prior virologic failure as increased concentrations of atazanavir are expected. Atazanavir sulfate is not recommended for use in patients with severe hepatic impairment. The pharmacokinetics of atazanavir sulfate in combination with ritonavir has not been studied in subjects with hepatic impairment; thus, coadministration of atazanavir sulfate with ritonavir is not recommended for use in patients with any degree of hepatic impairment [see Dosage and Administration ( 2.8)].
Pediatrics
The pharmacokinetic parameters for atazanavir at steady state in pediatric patients taking the capsule formulation were predicted by a population pharmacokinetic model and are summarized in Table 19 by weight ranges that correspond to the recommended doses [see Dosage and Administration ( 2.4)].
Table 19: Predicted Steady-State Pharmacokinetics of Atazanavir (Capsule Formulation) With Ritonavir in HIV-Infected Pediatric Patients
Body Weightatazanavir/ritonavir
C max ng/mL
Geometric MeanAUC ngh/mL
Geometric MeanC min ng/mL
Geometric Mean(range in kg)
Dose (mg)
(CV%)
(CV%)
(CV%)
15 to < 35
200/100
3303 (86%)
37235 (84%)
538 (99%)
≥ 35
300/100
2980 (82%)
37643 (83%)
653 (89%)
PregnancyThe pharmacokinetic data from HIV-infected pregnant women receiving atazanavir sulfate capsules with ritonavir are presented in Table 20.
Table 20: Steady-State Pharmacokinetics of Atazanavir With Ritonavir in HIV-Infected Pregnant Women in the Fed State
Pharmacokinetic ParameterAtazanavir 300 mg with ritonavir 100 mg
2nd Trimester
(n = 51)
3rd Trimester
(n = 20)
Postpartum2
(n = 34)
C max ng/mL
3078.85
3291.46
5721.21
Geometric mean (CV%)
(50)
(48)
(31)
AUC ngh/mL
27657.1
34251.5
61990.4
Geometric mean (CV%)
(43)
(43)
(32)
C min ng/mL 3
538.70
668.48
1462.59
Geometric mean (CV%)
(46)
(50)
(45)
1. Available data during the 2nd trimester are limited.
2. Atazanavir peak concentrations and AUCs were found to be approximately 28% to 43% higher during the postpartum period (4 to 12 weeks) than those observed historically in HIV-infected, non-pregnant patients. Atazanavir plasma trough concentrations were approximately 2.2 fold higher during the postpartum period when compared to those observed historically in HIV-infected, non-pregnant patients.
3. C min is concentration 24 hours post-dose.
Drug Interaction Data
Atazanavir is a metabolism-dependent CYP3A inhibitor, with a K inact value of 0.05 to 0.06 min -1 and K i value of 0.84 to 1.0 µM. Atazanavir is also a direct inhibitor for UGT1A1 (K i = 1.9 µM) and CYP2C8 (K i = 2.1 µM).
Atazanavir has been shown in vivo not to induce its own metabolism, nor to increase the biotransformation of some drugs metabolized by CYP3A. In a multiple-dose study, atazanavir sulfate decreased the urinary ratio of endogenous 6β-OH cortisol to cortisol versus baseline, indicating that CYP3A production was not induced.
Clinically significant interactions are not expected between atazanavir and substrates of CYP2C19, CYP2C9, CYP2D6, CYP2B6, CYP2A6, CYP1A2, or CYP2E1. Clinically significant interactions are not expected between atazanavir when administered with ritonavir and substrates of CYP2C8. See the complete prescribing information for ritonavir for information on other potential drug interactions with ritonavir.
Based on known metabolic profiles, clinically significant drug interactions are not expected between atazanavir and dapsone, trimethoprim/sulfamethoxazole, azithromycin, or erythromycin. Atazanavir does not interact with substrates of CYP2D6 (e.g., nortriptyline, desipramine, metoprolol).
Drug interaction studies were performed with atazanavir sulfate and other drugs likely to be coadministered and some drugs commonly used as probes for pharmacokinetic interactions. The effects of coadministration of atazanavir sulfate on the AUC, C max, and C min are summarized in Tables 21 and 22. Neither didanosine EC nor diltiazem had a significant effect on atazanavir exposures (see Table 22 for effect of atazanavir on didanosine EC or diltiazem exposures). Atazanavir sulfate did not have a significant effect on the exposures of didanosine (when administered as the buffered tablet), stavudine, or fluconazole. For information regarding clinical recommendations, see Drug Interactions ( 7) .
Table 21: Drug Interactions: Pharmacokinetic Parameters for Atazanavir in the Presence of Coadministered Drugs1
Coadministered Drug
Coadministered Drug Dose/Schedule
Atazanavir Dose/Schedule
Ratio (90% Confidence Interval) of Atazanavir Pharmacokinetic Parameters With/Without Coadministered Drug;
No Effect = 1.00C max
AUC
C min
atenolol
50 mg QD, d 7 to 11 (n = 19) and d 19 to 23
400 mg QD, d 1 to 11 (n = 19)
1.00
(0.89, 1.12)0.93
(0.85, 1.01)0.74
(0.65, 0.86)boceprevir
800 mg TID,
d 1 to 6, 25 to 31
300 mg QD/ritonavir
100 mg QD,
d 10 to 31atazanavir: 0.75
(0.64 to 0.88)ritonavir: 0.73
(0.64 to 0.83)
atazanavir: 0.65
(0.55 to 0.78)ritonavir: 0.64
(0.58 to 0.72)
atazanavir: 0.51
(0.44 to 0.61)ritonavir: 0.55
(0.45 to 0.67)
clarithromycin
500 mg BID, d 7 to 10 (n = 29) and d 18 to 21
400 mg QD,
d 1 to 10
(n = 29)1.06
(0.93, 1.20)1.28
(1.16, 1.43)1.91
(1.66, 2.21)didanosine (ddI) (buffered tablets) plus stavudine (d4T) 2
ddI: 200 mg x 1 dose, d4T: 40 mg x 1 dose (n = 31)
400 mg x 1 dose simultaneously with ddI and d4T (n = 31)
0.11
(0.06, 0.18)0.13
(0.08, 0.21)0.16
(0.10, 0.27)ddI: 200 mg x 1 dose, d4T: 40 mg x 1 dose (n = 32)
400 mg x 1 dose 1 h after ddI + d4T
(n = 32)1.12
(0.67, 1.18)1.03
(0.64, 1.67)1.03
(0.61, 1.73)efavirenz
600 mg QD, d 7 to 20 (n = 27)
400 mg QD,
d 1 to 20
(n = 27)0.41
(0.33, 0.51)0.26
(0.22, 0.32)0.07
(0.05, 0.10)600 mg QD, d 7 to 20 (n = 13)
400 mg QD,
d 1 to 6 (n = 23) then 300 mg/ritonavir 100 mg QD, 2 h before efavirenz,
d 7 to 20
(n = 13)1.14
(0.83, 1.58)1.39
(1.02, 1.88)1.48
(1.24, 1.76)600 mg QD,
d 11 to 24 (pm)
(n = 14)300 mg QD/ritonavir 100 mg QD,
d 1 to 10 (pm)
(n = 22), then 400 mg QD/ritonavir 100 mg QD,
d 11 to 24 (pm), (simultaneously with efavirenz) (n = 14)1.17
(1.08, 1.27)1.00
(0.91, 1.10)0.58
(0.49, 0.69)famotidine
40 mg BID, d 7 to 12 (n = 15)
400 mg QD,
d 1 to 6 (n = 45), d 7 to 12 (simultaneous administration) (n = 15)0.53
(0.34, 0.82)0.59
(0.40, 0.87)0.58
(0.37, 0.89)40 mg BID, d 7 to 12 (n = 14)
400 mg QD (pm), d 1 to 6
(n = 14),
d 7 to 12 (10 h after, 2 h before famotidine)
(n = 14)1.08
(0.82, 1.41)0.95
(0.74, 1.21)0.79
(0.60, 1.04)40 mg BID, d 11 to 20 (n = 14) 3
300 mg QD/ritonavir 100 mg QD,
d 1 to 10
(n = 46),
d 11 to 20 4 (simultaneous administration) (n = 14)0.86
(0.79, 0.94)0.82
(0.75, 0.89)0.72
(0.64, 0.81)20 mg BID, d 11 to 17 (n = 18)
300 mg QD/ritonavir 100 mg QD/tenofovir DF 300 mg QD,
d 1 to 10 (am)
(n = 39),
d 11 to 17 (am) (simultaneous administration with am famotidine)
(n = 18) 4, 50.91
(0.84, 0.99)0.90
(0.82, 0.98)0.81
(0.69, 0.94)40 mg QD (pm),
d 18 to 24
(n = 20)300 mg QD/ritonavir 100 mg QD/tenofovir DF 300 mg QD,
d 1 to 10 (am)
(n = 39),
d 18 to 24 (am) (12 h after pm famotidine)
(n = 20) 50.89
(0.81, 0.97)0.88
(0.80, 0.96)0.77
(0.63, 0.93)40 mg BID,
d 18 to 24
(n = 18)300 mg QD/ritonavir 100 mg QD/tenofovir DF 300 mg QD,
d 1 to 10 (am)
(n = 39),
d 18 to 24 (am) (10 h after pm famotidine and 2 h before am famotidine)
(n = 18) 50.74
(0.66, 0.84)0.79
(0.70, 0.88)0.72
(0.63, 0.83)40 mg BID, d 11 to 20 (n = 15)
300 mg QD/ritonavir 100 mg QD,
d 1 to 10 (am) (n = 46), then 400 mg QD/ritonavir 100 mg QD,
d 11 to 20 (am)
(n = 15)1.02
(0.87, 1.18)1.03
(0.86, 1.22)0.86
(0.68, 1.08)grazoprevir/ elbasvir
grazoprevir 200 mg QD d 1 to 35
(n = 11)300 mg QD/ritonavir 100 mg QD,
d 1 to 35
(n = 11)1.12
(1.01, 1.24)1.43
(1.30, 1.57)1.23
(1.13, 1.34)elbasvir 50 mg QD
d 1 to 35
(n = 8)300 mg QD/ritonavir 100 mg QD,
d 1 to 35 (n = 8)1.02
(0.96, 1.08)1.07 (0.98,1.17)
1.15
(1.02, 1.29)ketoconazole
200 mg QD, d 7 to 13 (n = 14)
400 mg QD,
d 1 to 13
(n = 14)0.99
(0.77, 1.28)1.10
(0.89, 1.37)1.03
(0.53, 2.01)nevirapine 6, 7
200 mg BID,
d 1 to 23 (n = 23)300 mg QD/ritonavir 100 mg QD,
d 4 to 13, then0.72
(0.60, 0.86)0.58
(0.48, 0.71)0.28
(0.20, 0.40)400 mg QD/ritonavir 100 mg QD,
d 14 to 23
(n = 23) 81.02
(0.85, 1.24)0.81
(0.65, 1.02)0.41
(0.27, 0.60)omeprazole
40 mg QD, d 7 to 12 (n = 16) 9
400 mg QD,
d 1 to 6
(n = 48),
d 7 to 12
(n = 16)0.04
(0.04, 0.05)0.06
(0.05, 0.07)0.05
(0.03, 0.07)40 mg QD, d 11 to 20 (n = 15) 9
300 mg QD/ritonavir 100 mg QD, d 1 to 20 (n = 15)
0.28
(0.24, 0.32)0.24
(0.21, 0.27)0.22
(0.19, 0.26)20 mg QD, d 17 to 23 (am) (n = 13)
300 mg QD/ritonavir 100 mg QD,
d 7 to 16 (pm) (n = 27),
d 17 to 23 (pm) (n = 13) 10, 110.61
(0.46, 0.81)0.58
(0.44, 0.75)0.54
(0.41, 0.71)20 mg QD, d 17 to 23 (am) (n = 14)
300 mg QD/ritonavir 100 mg QD,
d 7 to 16 (am) (n = 27), then 400 mg QD/ritonavir 100 mg QD,
d 17 to 23 (am) (n = 14) 12, 130.69
(0.58, 0.83)0.70
(0.57, 0.86)0.69
(0.54, 0.88)pitavastatin
4 mg QD for 5 days
300 mg QD for 5 days
1.13
(0.96, 1.32)1.06
(0.90, 1.26)NA
rifabutin
150 mg QD, d 15 to 28 (n = 7)
400 mg QD,
d 1 to 28 (n = 7)1.34
(1.14, 1.59)1.15
(0.98, 1.34)1.13
(0.68, 1.87)rifampin
600 mg QD, d 17 to 26 (n = 16)
300 mg QD/ritonavir 100 mg QD,
d 7 to 16
(n = 48),
d 17 to 26
(n = 16)0.47
(0.41, 0.53)0.28
(0.25, 0.32)0.02
(0.02, 0.03)ritonavir 14
100 mg QD, d 11 to 20 (n = 28)
300 mg QD,
d 1 to 20
(n = 28)1.86
(1.69, 2.05)3.38
(3.13, 3.63)11.89 (10.23, 13.82)
tenofovir DF 15
300 mg QD, d 9 to 16 (n = 34)
400 mg QD,
d 2 to 16
(n = 34)0.79
(0.73, 0.86)0.75
(0.70, 0.81)0.60
(0.52, 0.68)300 mg QD, d 15 to 42 (n = 10)
300 mg/ritonavir 100 mg QD,
d 1 to 42
(n = 10)0.72 16
(0.50, 1.05)0.75 16
(0.58, 0.97)0.77 16
(0.54, 1.10)voriconazole
(Subjects with at least one functional
CYP2C19 allele)
200 mg BID, d 2 to 3, 22 to 30; 400 mg BID, d 1, 21 (n = 20)
300 mg/ritonavir 100 mg QD,
d 11 to 30
(n = 20)0.87
(0.80, 0.96)
0.88
(0.82, 0.95)
0.80
(0.72, 0.90)
voriconazole
(Subjects without a functional
CYP2C19 allele)
50 mg BID, d 2 to 3, 22 to 30; 100 mg BID, d 1, 21 (n = 8)
300 mg/ritonavir 100 mg QD,
d 11 to 30
(n = 8)0.81
(0.66, 1.00)
0.80
(0.65, 0.97)
0.69
(0.54, 0.87)
1. Data provided are under fed conditions unless otherwise noted.
2. All drugs were given under fasted conditions.
3. Atazanavir 300 mg plus ritonavir 100 mg once daily coadministered with famotidine 40 mg twice daily resulted in atazanavir geometric mean C max that was similar and AUC and C min values that were 1.79 and 4.46 fold higher relative to atazanavir 400 mg once daily alone.
4. Similar results were noted when famotidine 20 mg BID was administered 2 hours after and 10 hours before atazanavir 300 mg and ritonavir 100 mg plus tenofovir DF 300 mg.
5. Atazanavir/ritonavir/tenofovir DF was administered after a light meal.
6. Study was conducted in HIV-infected individuals.
7. Compared with atazanavir 400 mg historical data without nevirapine (n = 13), the ratio of geometric means (90% confidence intervals) for C max, AUC, and C min were 1.42 (0.98, 2.05), 1.64 (1.11, 2.42), and 1.25 (0.66, 2.36), respectively, for atazanavir/ritonavir 300 mg/100 mg; and 2.02 (1.42, 2.87), 2.28 (1.54, 3.38), and 1.80 (0.94, 3.45), respectively, for atazanavir/ritonavir 400 mg/100 mg.
8. Parallel group design; n = 23 for atazanavir/ritonavir plus nevirapine, n = 22 for atazanavir 300 mg/ritonavir 100 mg without nevirapine. Subjects were treated with nevirapine prior to study entry.
9. Omeprazole 40 mg was administered on an empty stomach 2 hours before atazanavir sulfate.
10. Omeprazole 20 mg was administered 30 minutes prior to a light meal in the morning and atazanavir 300 mg plus ritonavir 100 mg in the evening after a light meal, separated by 12 hours from omeprazole.
11. Atazanavir 300 mg plus ritonavir 100 mg once daily separated by 12 hours from omeprazole 20 mg daily resulted in increases in atazanavir geometric mean AUC (10%) and C min (2.4 fold), with a decrease in C max (29%) relative to atazanavir 400 mg once daily in the absence of omeprazole (study days 1 to 6).
12. Omeprazole 20 mg was given 30 minutes prior to a light meal in the morning and atazanavir 400 mg plus ritonavir 100 mg once daily after a light meal, 1 hour after omeprazole. Effects on atazanavir concentrations were similar when atazanavir 400 mg plus ritonavir 100 mg was separated from omeprazole 20 mg by 12 hours.
13. Atazanavir 400 mg plus ritonavir 100 mg once daily administered with omeprazole 20 mg once daily resulted in increases in atazanavir geometric mean AUC (32%) and C min (3.3 fold), with a decrease in C max (26%) relative to atazanavir 400 mg once daily in the absence of omeprazole (study days 1 to 6).
14. Compared with atazanavir 400 mg QD historical data, administration of atazanavir/ritonavir 300 mg/100 mg QD increased the atazanavir geometric mean values of C max, AUC, and C min by 18%, 103%, and 671%, respectively.
15. Note that similar results were observed in studies where administration of tenofovir DF and atazanavir sulfate was separated by 12 hours.
16. Ratio of atazanavir plus ritonavir plus tenofovir DF to atazanavir plus ritonavir. Atazanavir 300 mg plus ritonavir 100 mg results in higher atazanavir exposure than atazanavir 400 mg (see footnote 15). The geometric mean values of atazanavir pharmacokinetic parameters when coadministered with ritonavir and tenofovir DF were: C max = 3190 ng/mL, AUC = 34459 ngh/mL, and C min = 491 ng/mL. Study was conducted in HIV-infected individuals.
NA = not available.
Table 22: Drug Interactions: Pharmacokinetic Parameters for Coadministered Drugs in the Presence of Atazanavir Sulfate 1 Coadministered Drug
Coadministered Drug Dose/Schedule
Atazanavir Dose/Schedule
Ratio (90% Confidence Interval) of Coadministered Drug Pharmacokinetic Parameters With/Without Atazanavir Sulfate; No Effect = 1.00
C max
AUC
C min
acetaminophen
1 gm BID,
d 1 to 20 (n = 10)300 mg QD/ritonavir 100 mg QD,
d 11 to 20
(n = 10)0.87
(0.77, 0.99)0.97
(0.91, 1.03)1.26
(1.08, 1.46)atenolol
50 mg QD,
d 7 to 11 (n = 19) and
d 19 to 23400 mg QD,
d 1 to 11
(n = 19)1.34
(1.26, 1.42)1.25
(1.16, 1.34)1.02
(0.88, 1.19)boceprevir
800 mg TID,
d 1 to 6, 25 to 31
300 mg QD/ritonavir
100 mg QD,
d 10 to 31
0.93
(0.80, 1.08)
0.95
(0.87, 1.05)
0.82
(0.68, 0.98)
clarithromycin
500 mg BID,
d 7 to 10 (n = 21) and d 18 to 21400 mg QD,
d 1 to 10
(n = 21)1.50
(1.32, 1.71)OH-clarithromycin: 0.28
(0.24, 0.33)1.94
(1.75, 2.16)OH-clarithromycin:
0.30 (0.26, 0.34)2.60
(2.35, 2.88)OH-clarithromycin: 0.38
(0.34, 0.42)ddI (enteric-coated [EC] capsules) 2
400 mg
d 1 (fasted),
d 8 (fed) (n = 34)400 mg QD,
d 2 to 8 (n = 34)0.64
(0.55, 0.74)0.66
(0.60, 0.74)1.13
(0.91, 1.41)400 mg d 1 (fasted), d 19 (fed) (n = 31)
300 mg QD/ritonavir 100 mg QD,
d 9 to 19
(n = 31)
0.62
(0.52, 0.74)0.66
(0.59, 0.73)1.25
(0.92, 1.69)diltiazem
180 mg QD, d 7 to 11 (n = 28) and d 19 to 23
400 mg QD,
d 1 to 11
(n = 28)
1.98
(1.78, 2.19)2.25
(2.09, 2.16)2.42
(2.14, 2.73)desacetyl-diltiazem: 2.72
(2.44, 3.03)
desacetyl-diltiazem: 2.65
(2.45, 2.87)
desacetyl-diltiazem:
2.21 (2.02, 2.42)ethinyl estradiol & norethindrone 3
Ortho-Novum ® 7/7/7 QD, d 1 to 29
(n = 19)400 mg QD,
d 16 to 29
(n = 19)ethinyl estradiol:
1.15
(0.99, 1.32)ethinyl estradiol:
1.48
(1.31, 1.68)ethinyl estradiol:
1.91
(1.57, 2.33)norethindrone: 1.67
(1.42, 1.96)norethindrone: 2.10
(1.68, 2.62)norethindrone: 3.62
(2.57, 5.09)ethinyl estradiol & norgestimate 4
Ortho Tri-Cyclen ® QD,
d 1 to 28 (n = 18), then Ortho Tri-Cyclen ® LO QD,
d 29 to 42 5
(n = 14)300 mg QD/ritonavir 100 mg QD,
d 29 to 42
(n = 14)
ethinyl estradiol:
0.84
(0.74, 0.95)ethinyl estradiol:
0.81
(0.75, 0.87)ethinyl estradiol:
0.63
(0.55, 0.71)17-deacetyl norgestimate: 6 1.68
(1.51, 1.88)
17-deacetyl norgestimate: 6 1.85
(1.67, 2.05)
17-deacetyl norgestimate: 6 2.02
(1.77, 2.31)
glecaprevir/ pibrentasvir
300 mg glecaprevir
(n = 12)
300 mg QD/ritonavir 100 mg QD
(n = 12)
≥ 4.06 7 (3.15, 5.23)
≥ 6.53 7 (5.24, 8.14)
≥ 14.3 7 (9.85, 20.7)
120 mg pibrentasvir
(n = 12)
300 mg QD/ritonavir 100 mg QD
(n = 12)
≥ 1.29 7 (1.15, 1.45)
≥ 1.64 7 (1.48, 1.82)
≥ 2.29 7 (1.95, 2.68)
grazoprevir/ elbasvir
grazoprevir 200 mg QD d 1 to 35
(n = 12)300 mg QD/ritonavir 100 mg QD
d 1 to 35
(n = 12)
6.24
(4.42, 8.81)10.58
(7.78, 14.39)11.64
(7.96, 17.02)elbasvir 50 mg QD d 1 to 35
(n = 10)300 mg QD/ritonavir 100 mg QD
d 1 to 35
(n = 10)
4.15
(3.46, 4.97)4.76
(4.07, 5.56)6.45
(5.51 7.54)methadone
Stable maintenance dose, d 1 to 15
(n = 16)400 mg QD,
d 2 to 15
(n = 16)
(R)-methadone 8
0.91
(0.84, 1.0)
(R)-methadone 8 1.03
(0.95, 1.10)
(R)-methadone 8 1.11
(1.02, 1.20)
total: 0.85 (0.78, 0.93)
total: 0.94 (0.87, 1.02)
total: 1.02 (0.93, 1.12)
nevirapine 9,10
200 mg BID,
d 1 to 23
(n = 23)300 mg QD/ ritonavir
100 mg QD,
d 4 to 13, then
1.17
(1.09, 1.25)
1.25
(1.17, 1.34)
1.32
(1.22, 1.43)
400 mg QD/ ritonavir
100 mg QD,
d 14 to 23
(n = 23)
1.21
(1.11, 1.32)
1.26
(1.17, 1.36)
1.35
(1.25, 1.47)
omeprazole 11
40 mg single dose, d 7 and d 20 (n = 16)
400 mg QD,
d 1 to 12
(n = 16)
1.24
(1.04, 1.47)
1.45
(1.20, 1.76)
NA
rifabutin
300 mg QD,
d 1 to 10 then
150 mg QD,
d 11 to 20
(n = 3)
600 mg QD, 12
d 11 to 20
(n = 3)
1.18
(0.94, 1.48)
2.10
(1.57, 2.79)
3.43
(1.98, 5.96)
25-O-desacetyl
rifabutin: 8.20 (5.90, 11.40)
25-O-desacetyl
rifabutin: 22.01 (15.97, 30.34)
25-O-desacetyl
rifabutin: 75.6
(30.1, 190.0)
150 mg twice weekly, d 1 to 15 (n = 7)
300 mg QD/ritonavir 100 mg QD,
d 1 to 17
(n = 7)2.49 13
(2.03, 3.06)
25-O-desacetyl
rifabutin: 7.77 (6.13, 9.83)1.48 13
(1.19, 1.84)
25-O-desacetyl
rifabutin: 10.90 (8.14, 14.61)1.40 13
(1.05, 1.87)
25-O-desacetyl
rifabutin: 11.45(8.15, 16.10)
pitavastatin
4 mg QD for
5 days
300 mg QD for 5 days
1.60
(1.39, 1.85)
1.31
(1.23, 1.39)
NA
rosiglitazone 14
4 mg single dose, d 1, 7, 17
(n = 14)400 mg QD,
d 2 to 7, then
1.08
(1.03, 1.13)
1.35
(1.26, 1.44)
NA
300 mg QD/ ritonavir
100 mg QD,
d 8 to 17
(n = 14)
0.97
(0.91, 1.04)
0.83
(0.77, 0.89)
NA
rosuvastatin
10 mg single dose
300 mg QD/ ritonavir
100 mg QD for 7 days
↑7 fold 15
↑3 fold 15
NA
saquinavir 16 (soft gelatin capsules)
1200 mg QD,
d 1 to 13
(n = 7)400 mg QD,
d 7 to 13
(n = 7)4.39
(3.24, 5.95)
5.49
(4.04, 7.47)
6.86
(5.29, 8.91)
sofosbuvir/ velpatasvir/ voxilaprevir
400 mg sofosbuvir single dose
(n = 15)
300 mg/100 mg ritonavir single dose
(n = 15)
1.29
(1.09, 1.52) sofosbuvir metabolite
GS-331007 1.05
(0.99, 1.12)
1.40
(1.25, 1.57) sofosbuvir metabolite
GS-331007 1.25
(1.16, 1.36)
NA
100 mg velpatasvir single dose
(n = 15)
300 mg/100 mg ritonavir single dose
(n = 15)
1.29
(1.07, 1.56)
1.93
(1.58, 2.36)
NA
100 mg voxilaprevir single dose
(n = 15)
300 mg/100 mg ritonavir single dose
(n = 15)
4.42
(3.65, 5.35)
4.31
(3.76, 4.93)
NA
tenofovir DF 17
300 mg QD,
d 9 to 16 (n = 33) and d 24 to 30
(n = 33)
400 mg QD,
d 2 to 16
(n = 33)
1.14
(1.08, 1.20)
1.24
(1.21, 1.28)
1.22
(1.15, 1.30)
300 mg QD,
d 1 to 7 (pm)
(n = 14)
d 25 to 34 (pm)
(n = 12)
300 mg QD/ritonavir 100 mg QD,
d 25 to 34 (am) (n = 12) 18
1.34
(1.20, 1.51)
1.37
(1.30, 1.45)
1.29
(1.21, 1.36)
voriconazole (Subjects with at least one functional CYP2C19 allele)
200 mg BID,
d 2 to 3, 22 to 30; 400 mg BID,
d 1, 21
(n = 20)
300 mg/ritonavir 100 mg QD,
d 11 to 30
(n = 20)
0.90
(0.78, 1.04)
0.67
(0.58, 0.78)
0.61
(0.51, 0.72)
voriconazole (Subjects without a functional CYP2C19 allele)
50 mg BID,
d 2 to 3, 22 to 30; 100 mg BID,
d 1, 21
(n = 8)
300 mg/ritonavir 100 mg QD,
d 11 to 30
(n = 8)
4.38
(3.55, 5.39)
5.61
(4.51, 6.99)
7.65
(5.71, 10.2)
lamivudine + zidovudine
150 mg lamivudine + 300 mg zidovudine BID, d 1 to 12
(n = 19)
400 mg QD,
d 7 to 12
(n = 19)
lamivudine: 1.04
(0.92, 1.16)
lamivudine: 1.03
(0.98, 1.08)
lamivudine: 1.12
(1.04, 1.21)
zidovudine: 1.05
(0.88, 1.24)
zidovudine: 1.05
(0.96, 1.14)
zidovudine: 0.69
(0.57, 0.84)
zidovudine glucuronide: 0.95
(0.88, 1.02)
zidovudine glucuronide: 1.00
(0.97, 1.03)
zidovudine glucuronide: 0.82
(0.62, 1.08)
1. Data provided are under fed conditions unless otherwise noted.
2. 400 mg ddI EC and atazanavir were administered together with food on Days 8 and 19.
3. Upon further dose normalization of ethinyl estradiol 25 mcg with atazanavir relative to ethinyl estradiol 35 mcg without atazanavir, the ratio of geometric means (90% confidence intervals) for C max, AUC, and C min were 0.82 (0.73, 0.92), 1.06 (0.95, 1.17), and 1.35 (1.11, 1.63), respectively.
4. Upon further dose normalization of ethinyl estradiol 35 mcg with atazanavir/ritonavir relative to ethinyl estradiol 25 mcg without atazanavir/ritonavir, the ratio of geometric means (90% confidence intervals) for C max, AUC, and C min were 1.17 (1.03, 1.34), 1.13 (1.05, 1.22), and 0.88 (0.77, 1.00), respectively.
5. All subjects were on a 28 day lead-in period; one full cycle of Ortho Tri-Cyclen ®. Ortho Tri-Cyclen ® contains 35 mcg of ethinyl estradiol. Ortho Tri-Cyclen ® LO contains 25 mcg of ethinyl estradiol. Results were dose normalized to an ethinyl estradiol dose of 35 mcg.
6. 17-deacetyl norgestimate is the active component of norgestimate.
7. Effect of atazanavir and ritonavir on the first dose of glecaprevir and pibrentasvir is reported.
8. (R)-methadone is the active isomer of methadone.
9. Study was conducted in HIV-infected individuals.
10. Subjects were treated with nevirapine prior to study entry.
11. Omeprazole was used as a metabolic probe for CYP2C19. Omeprazole was given 2 hours after atazanavir sulfate on Day 7; and was given alone 2 hours after a light meal on Day 20.
12. Not the recommended therapeutic dose of atazanavir.
13. When compared to rifabutin 150 mg QD alone d 1 to 10 (n = 14). Total of rifabutin + 25-O-desacetyl-rifabutin: AUC 2.19 (1.78, 2.69).
14. Rosiglitazone used as a probe substrate for CYP2C8.
15. Mean ratio (with/without coadministered drug). ↑ indicates an increase in rosuvastatin exposure.
16. The combination of atazanavir and saquinavir 1200 mg QD produced daily saquinavir exposures similar to the values produced by the standard therapeutic dosing of saquinavir at 1200 mg TID. However, the C max is about 79% higher than that for the standard dosing of saquinavir (soft gelatin capsules) alone at 1200 mg TID.
17. Note that similar results were observed in a study where administration of tenofovir DF and atazanavir sulfate was separated by 12 hours.
18. Administration of tenofovir DF and atazanavir sulfate was temporally separated by 12 hours.
NA = not available.
12.4 Microbiology
Mechanism of Action
Atazanavir (ATV) is an azapeptide HIV-1 protease inhibitor (PI). The compound selectively inhibits the virus-specific processing of viral Gag and Gag-Pol polyproteins in HIV-1 infected cells, thus preventing formation of mature virions.
Antiviral Activity in Cell Culture
Atazanavir exhibits anti-HIV-1 activity with a mean 50% effective concentration (EC 50) in the absence of human serum of 2 to 5 nM against a variety of laboratory and clinical HIV-1 isolates grown in peripheral blood mononuclear cells, macrophages, CEM-SS cells, and MT-2 cells. ATV has activity against HIV-1 Group M subtype viruses A, B, C, D, AE, AG, F, G, and J isolates in cell culture. ATV has variable activity against HIV-2 isolates (1.9 to 32 nM), with EC 50 values above the EC 50 values of failure isolates. Two-drug combination antiviral activity studies with ATV showed no antagonism in cell culture with PIs (amprenavir, indinavir, lopinavir, nelfinavir, ritonavir, and saquinavir) , NNRTIs (delavirdine, efavirenz, and nevirapine), NRTIs (abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir DF, and zidovudine), the HIV-1 fusion inhibitor enfuvirtide, and two compounds used in the treatment of viral hepatitis, adefovir and ribavirin, without enhanced cytotoxicity.
Resistance
In Cell Culture: HIV-1 isolates with a decreased susceptibility to ATV have been selected in cell culture and obtained from patients treated with ATV or atazanavir/ritonavir (ATV/RTV). HIV-1 isolates with 93 to 183 fold reduced susceptibility to ATV from three different viral strains were selected in cell culture by 5 months. The substitutions in these HIV-1 viruses that contributed to ATV resistance include I50L, N88S, I84V, A71V, and M46I. Changes were also observed at the protease cleavage sites following drug selection. Recombinant viruses containing the I50L substitution without other major PI substitutions were growth impaired and displayed increased susceptibility in cell culture to other PIs (amprenavir, indinavir, lopinavir, nelfinavir, ritonavir, and saquinavir). The I50L and I50V substitutions yielded selective resistance to ATV and amprenavir, respectively, and did not appear to be cross-resistant.
Clinical Studies of Treatment-Naive Patients: Comparison of Ritonavir-Boosted Atazanavir Sulfate vs. Unboosted Atazanavir Sulfate: Study AI424-089 compared atazanavir 300 mg once daily with ritonavir 100 mg vs. atazanavir 400 mg once daily when administered with lamivudine and extended-release stavudine in HIV-infected treatment-naive patients. A summary of the number of virologic failures and virologic failure isolates with ATV resistance in each arm is shown in Table 23.
Table 23: Summary of Virologic Failures 1 at Week 96 in Study AI424-089: Comparison of Ritonavir Boosted Atazanavir Sulfate vs. Unboosted Atazanavir Sulfate: Randomized Patients Atazanavir 300 mg + ritonavir
100 mg
Atazanavir
400 mg
(n = 95)
(n = 105)
Virologic Failure (≥ 50 copies/mL) at Week 96
15 (16%)
34 (32%)
Virologic Failure with Genotypes and Phenotypes Data
5
17
Virologic Failure Isolates with ATV-resistance at Week 96
0/5 (0%) 2
4/17 (24%) 2
Virologic Failure Isolates with I50L Emergence at Week 96 3
0/5 (0%) 2
2/17 (12%) 2
Virologic Failure Isolates with Lamivudine Resistance at Week 96
2/5 (40%) 2
11/17 (65%) 2
1. Virologic failure includes patients who were never suppressed through Week 96 and on study at Week 96, had virologic rebound or discontinued due to insufficient viral load response.
2. Percentage of Virologic Failure Isolates with genotypic and phenotypic data.
3. Mixture of I50I/L emerged in 2 other ATV 400 mg-treated patients. Neither isolate was phenotypically resistant to ATV.
Clinical Studies of Treatment-Naive Patients Receiving Atazanavir 300 mg With Ritonavir 100 mg: In Phase III Study AI424-138, an as-treated genotypic and phenotypic analysis was conducted on samples from patients who experienced virologic failure (HIV-1 RNA ≥ 400 copies/mL) or discontinued before achieving suppression on ATV/RTV (n = 39; 9%) and LPV/RTV (n = 39; 9%) through 96 weeks of treatment. In the ATV/RTV arm, one of the virologic failure isolates had a 56 fold decrease in ATV susceptibility emerge on therapy with the development of PI resistance-associated substitutions L10F, V32I, K43T, M46I, A71I, G73S, I85I/V, and L90M. The NRTI resistance-associated substitution M184V also emerged on treatment in this isolate conferring emtricitabine resistance. Two ATV/RTV-virologic failure isolates had baseline phenotypic ATV resistance and IAS-defined major PI resistance-associated substitutions at baseline. The I50L substitution emerged on study in one of these failure isolates and was associated with a 17 fold decrease in ATV susceptibility from baseline and the other failure isolate with baseline ATV resistance and PI substitutions (M46M/I and I84I/V) had additional IAS-defined major PI substitutions (V32I, M46I, and I84V) emerge on ATV treatment associated with a 3 fold decrease in ATV susceptibility from baseline. Five of the treatment failure isolates in the ATV/RTV arm developed phenotypic emtricitabine resistance with the emergence of either the M184I (n = 1) or the M184V (n = 4) substitution on therapy and none developed phenotypic tenofovir disoproxil resistance. In the LPV/RTV arm, one of the virologic failure patient isolates had a 69 fold decrease in LPV susceptibility emerge on therapy with the development of PI substitutions L10V, V11I, I54V, G73S, and V82A in addition to baseline PI substitutions L10L/I, V32I, I54I/V, A71I, G73G/S, V82V/A, L89V, and L90M. Six LPV/RTV virologic failure isolates developed the M184V substitution and phenotypic emtricitabine resistance and two developed phenotypic tenofovir disoproxil resistance.
Clinical Studies of Treatment-Naive Patients Receiving Atazanavir 400 mg Without Ritonavir: ATV-resistant clinical isolates from treatment-naive patients who experienced virologic failure on atazanavir 400 mg treatment without ritonavir often developed an I50L substitution (after an average of 50 weeks of ATV therapy), often in combination with an A71V substitution, but also developed one or more other PI substitutions (e.g., V32I, L33F, G73S, V82A, I85V, or N88S) with or without the I50L substitution. In treatment-naive patients, viral isolates that developed the I50L substitution, without other major PI substitutions, showed phenotypic resistance to ATV but retained in cell culture susceptibility to other PIs (amprenavir, indinavir, lopinavir, nelfinavir, ritonavir, and saquinavir); however, there are no clinical data available to demonstrate the effect of the I50L substitution on the efficacy of subsequently administered PIs.
Clinical Studies of Treatment-Experienced Patients: In studies of treatment-experienced patients treated with ATV or ATV/RTV, most ATV-resistant isolates from patients who experienced virologic failure developed substitutions that were associated with resistance to multiple PIs and displayed decreased susceptibility to multiple PIs. The most common protease substitutions to develop in the viral isolates of patients who failed treatment with ATV 300 mg once daily and RTV 100 mg once daily (together with tenofovir DF and an NRTI) included V32I, L33F/V/I, E35D/G, M46I/L, I50L, F53L/V, I54V, A71V/T/I, G73S/T/C, V82A/T/L, I85V, and L89V/Q/M/T. Other substitutions that developed on ATV/RTV treatment including E34K/A/Q, G48V, I84V, N88S/D/T, and L90M occurred in less than 10% of patient isolates. Generally, if multiple PI resistance substitutions were present in the HIV-1 virus of the patient at baseline, ATV resistance developed through substitutions associated with resistance to other PIs and could include the development of the I50L substitution. The I50L substitution has been detected in treatment-experienced patients experiencing virologic failure after long-term treatment. Protease cleavage site changes also emerged on ATV treatment but their presence did not correlate with the level of ATV resistance.
Cross-Resistance
Cross-resistance among PIs has been observed. Baseline phenotypic and genotypic analyses of clinical isolates from ATV clinical trials of PI-experienced patients showed that isolates cross-resistant to multiple PIs were cross-resistant to ATV. Greater than 90% of the isolates with substitutions that included I84V or G48V were resistant to ATV. Greater than 60% of isolates containing L90M, G73S/T/C, A71V/T, I54V, M46I/L, or a change at V82 were resistant to ATV, and 38% of isolates containing a D30N substitution in addition to other changes were resistant to ATV. Isolates resistant to ATV were also cross-resistant to other PIs with > 90% of the isolates resistant to indinavir, lopinavir, nelfinavir, ritonavir, and saquinavir, and 80% resistant to amprenavir. In treatment-experienced patients, PI-resistant viral isolates that developed the I50L substitution in addition to other PI resistance-associated substitution were also cross-resistant to other PIs.
Baseline Genotype/Phenotype and Virologic Outcome Analyses
Genotypic and/or phenotypic analysis of baseline virus may aid in determining ATV susceptibility before initiation of ATV/RTV therapy. An association between virologic response at 48 weeks and the number and type of primary PI resistance-associated substitutions detected in baseline HIV-1 isolates from antiretroviral-experienced patients receiving ATV/RTV once daily or lopinavir (LPV)/RTV twice daily in Study AI424-045 is shown in Table 24.
Overall, both the number and type of baseline PI substitutions affected response rates in treatment-experienced patients. In the ATV/RTV group, patients had lower response rates when 3 or more baseline PI substitutions, including a substitution at position 36, 71, 77, 82, or 90, were present compared to patients with 1 to 2 PI substitutions, including one of these substitutions.
Table 24: HIV RNA Response by Number and Type of Baseline PI Substitution, Antiretroviral-Experienced Patients in Study AI424-045, As-Treated Analysis Number and Type of Baseline PI Substitutions1
Virologic Response = HIV RNA < 400 copies/mL2
ATV/RTV
LPV/RTV
(n = 110)
(n = 113)
3 or more primary PI substitutions including3:
D30N
75% (6/8)
50% (3/6)
M36I/V
19% (3/16)
33% (6/18)
M46I/L/T
24% (4/17)
23% (5/22)
I54V/L/T/M/A
31% (5/16)
31% (5/16)
A71V/T/I/G
34% (10/29)
39% (12/31)
G73S/A/C/T
14% (1/7)
38% (3/8)
V77I
47% (7/15)
44% (7/16)
V82A/F/T/S/I
29% (6/21)
27% (7/26)
I84V/A
11% (1/9)
33% (2/6)
N88D
63% (5/8)
67% (4/6)
L90M
10% (2/21)
44% (11/25)
Number of baseline primary PI substitutions1
All patients, as-treated
58% (64/110)
59% (67/113)
0 to 2 PI substitutions
75% (50/67)
75% (50/67)
3 to 4 PI substitutions
41% (14/34)
43% (12/28)
5 or more PI substitutions
0% (0/9)
28% (5/18)
1. Primary substitutions include any change at D30, V32, M36, M46, I47, G48, I50, I54, A71, G73, V77, V82, I84, N88, and L90.
2. Results should be interpreted with caution because the subgroups were small.
3. There were insufficient data (n < 3) for PI substitutions V32I, I47V, G48V, I50V, and F53L.
The response rates of antiretroviral-experienced patients in Study AI424-045 were analyzed by baseline phenotype (shift in susceptibility in cell culture relative to reference, Table 25). The analyses are based on a select patient population with 62% of patients receiving an NNRTI-based regimen before study entry compared to 35% receiving a PI-based regimen. Additional data are needed to determine clinically relevant break points for atazanavir sulfate.
Table 25: Baseline Phenotype by Outcome, Antiretroviral-Experienced Patients in Study AI424-045, As-Treated Analysis Baseline Phenotype1
Virologic Response = HIV RNA < 400 copies/mL2
ATV/RTV
LPV/RTV
(n = 111)
(n = 111)
0 to 2
71% (55/78)
70% (56/80)
> 2 to 5
53% (8/15)
44% (4/9)
> 5 to 10
13% (1/8)
33% (3/9)
> 10
10% (1/10)
23% (3/13)
1. Fold change susceptibility in cell culture relative to the wild-type reference.
2. Results should be interpreted with caution because the subgroups were small.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Long-term carcinogenicity studies in mice and rats were carried out with atazanavir for two years. In the mouse study, drug-related increases in hepatocellular adenomas were found in females at 360 mg/kg/day. The systemic drug exposure (AUC) at the NOAEL (no observable adverse effect level) in females, (120 mg/kg/day) was 2.8 times and in males (80 mg/kg/day) was 2.9 times higher than those in humans at the clinical dose (300 mg/day atazanavir boosted with 100 mg/day ritonavir, non-pregnant patients). In the rat study, no drug-related increases in tumor incidence were observed at doses up to 1200 mg/kg/day, for which AUCs were 1.1 (males) or 3.9 (females) times those measured in humans at the clinical dose.
Mutagenesis
Atazanavir tested positive in an in vitro clastogenicity test using primary human lymphocytes, in the absence and presence of metabolic activation. Atazanavir tested negative in the in vitro Ames reverse-mutation assay, in vivo micronucleus and DNA repair tests in rats, and in vivo DNA damage test in rat duodenum (comet assay).
Impairment of Fertility
At the systemic drug exposure levels (AUC) 0.9 (in male rats) or 2.3 (in female rats) times that of the human clinical dose, (300 mg/day atazanavir boosted with 100 mg/day ritonavir) significant effects on mating, fertility, or early embryonic development were not observed.
-
14 CLINICAL STUDIES
14.1 Adult Patients without Prior Antiretroviral Therapy
Study AI424-138: a 96 week study comparing the antiviral efficacy and safety of atazanavir sulfate/ritonavir with lopinavir/ritonavir, each in combination with fixed-dose tenofovir DF‑emtricitabine in HIV-1 infected treatment-naive subjects. Study AI424-138 was a 96 week, open-label, randomized, multicenter study, comparing atazanavir (300 mg once daily) with ritonavir (100 mg once daily) to lopinavir with ritonavir (400 mg/100 mg twice daily), each in combination with fixed-dose tenofovir DF with emtricitabine (300 mg/200 mg once daily), in 878 antiretroviral treatment-naive treated patients. Patients had a mean age of 36 years (range: 19 to 72), 49% were Caucasian, 18% Black, 9% Asian, 23% Hispanic/Mestizo/mixed race, and 68% were male. The median baseline plasma CD4+ cell count was 204 cells/mm 3 (range: 2 to 810 cells/mm 3) and the mean baseline plasma HIV-1 RNA level was 4.94 log 10 copies/mL (range: 2.60 to 5.88 log 10 copies/mL). Treatment response and outcomes through Week 96 are presented in Table 26.
Table 26: Outcomes of Treatment Through Week 96 in Treatment-Naive Adults (Study AI424-138) Outcome
atazanavir 300 mg +
ritonavir 100 mg(once daily) with tenofovir DF/emtricitabine
(once daily)1
lopinavir 400 mg + ritonavir 100 mg
(twice daily) with tenofovir DF/emtricitabine
(once daily)1
(n = 441)
(n = 437)
96 Weeks
96 Weeks
Responder 2, 3, 4
75%
68%
Virologic failure 5
17%
19%
Rebound
8%
10%
Never suppressed
through Week 96
9%
9%
Death
1%
1%
Discontinued due to adverse event
3%
5%
Discontinued for other reasons 6
4%
7%
1. As a fixed-dose combination: 300 mg tenofovir DF, 200 mg emtricitabine once daily.
2. Patients achieved HIV RNA < 50 copies/mL at Week 96. Roche Amplicor ®, v1.5 ultra-sensitive assay.
3. Pre-specified ITT analysis at Week 48 using as-randomized cohort: ATV/RTV 78% and LPV/RTV 76% (difference estimate: 1.7% [95% confidence interval: -3.8%, 7.1%]).
4. Pre-specified ITT analysis at Week 96 using as-randomized cohort: ATV/RTV 74% and LPV/RTV 68% (difference estimate: 6.1% [95% confidence interval: 0.3%, 12.0%]).
5. Includes viral rebound and failure to achieve confirmed HIV RNA < 50 copies/mL through Week 96.
6. Includes lost to follow-up, patient's withdrawal, noncompliance, protocol violation, and other reasons.
Through 96 weeks of therapy, the proportion of responders among patients with high viral loads (i.e., baseline HIV RNA ≥ 100,000 copies/mL) was comparable for the atazanavir sulfate/ritonavir (165 of 223 patients, 74%) and lopinavir/ritonavir (148 of 222 patients, 67%) arms. At 96 weeks, the median increase from baseline in CD4+ cell count was 261 cells/mm 3 for the atazanavir sulfate/ritonavir arm and 273 cells/mm 3 for the lopinavir/ritonavir arm.
Study AI424-034: Atazanavir sulfate once daily compared to efavirenz once daily, each in combination with fixed-dose lamivudine + zidovudine twice daily. Study AI424-034 was a randomized, double-blind, multicenter trial comparing atazanavir (400 mg once daily) to efavirenz (600 mg once daily), each in combination with a fixed-dose combination of lamivudine (3TC) (150 mg) and zidovudine (ZDV) (300 mg) given twice daily, in 810 antiretroviral treatment-naive patients. Patients had a mean age of 34 years (range: 18 to 73), 36% were Hispanic, 33% were Caucasian, and 65% were male. The mean baseline CD4+ cell count was 321 cells/mm 3 (range: 64 to 1424 cells/mm 3) and the mean baseline plasma HIV-1 RNA level was 4.8 log 10 copies/mL (range: 2.2 to 5.9 log 10 copies/mL). Treatment response and outcomes through Week 48 are presented in Table 27.
Table 27: Outcomes of Randomized Treatment Through Week 48 in Treatment-Naive Adults (Study AI424-034) Outcome
atazanavir 400 mg once daily + lamivudine + zidovudine1
efavirenz 600 mg once daily + lamivudine + zidovudine1
(n = 405)
(n = 405)
Responder 2
67% (32%)
62% (37%)
Virologic failure 3
20%
21%
Rebound
17%
16%
Never suppressed through Week 48
3%
5%
Death
–
< 1%
Discontinued due to adverse event
5%
7%
Discontinued for other reasons 4
8%
10%
1. As a fixed-dose combination: 150 mg lamivudine, 300 mg zidovudine twice daily.
2. Patients achieved and maintained confirmed HIV RNA < 400 copies/mL (< 50 copies/mL) through Week 48. Roche Amplicor ® HIV-1 Monitor™ Assay, test version 1.0 or 1.5 as geographically appropriate.
3. Includes viral rebound and failure to achieve confirmed HIV RNA < 400 copies/mL through Week 48.
4. Includes lost to follow-up, patient’s withdrawal, noncompliance, protocol violation, and other reasons.
Through 48 weeks of therapy, the proportion of responders among patients with high viral loads (i.e., baseline HIV RNA ≥ 100,000 copies/mL) was comparable for the atazanavir sulfate and efavirenz arms. The mean increase from baseline in CD4+ cell count was 176 cells/mm 3 for the atazanavir sulfate arm and 160 cells/mm 3 for the efavirenz arm.
Study AI424-008: Atazanavir 400 mg once daily compared to atazanavir 600 mg once daily, and compared to nelfinavir 1250 mg twice daily, each in combination with stavudine and lamivudine twice daily. Study AI424-008 was a 48 week, randomized, multicenter trial, blinded to dose of atazanavir sulfate, comparing atazanavir at two dose levels (400 mg and 600 mg once daily) to nelfinavir (1250 mg twice daily), each in combination with stavudine (40 mg) and lamivudine (150 mg) given twice daily, in 467 antiretroviral treatment-naive patients. Patients had a mean age of 35 years (range: 18 to 69), 55% were Caucasian, and 63% were male. The mean baseline CD4+ cell count was 295 cells/mm 3 (range: 4 to 1003 cells/mm 3) and the mean baseline plasma HIV-1 RNA level was 4.7 log 10 copies/mL (range: 1.8 to 5.9 log 10 copies/mL). Treatment response and outcomes through Week 48 are presented in Table 28.
Table 28: Outcomes of Randomized Treatment Through Week 48 in Treatment-Naive Adults (Study AI424-008) Outcome
atazanavir 400 mg once daily + lamivudine + stavudine
nelfinavir 1250 mg twice daily + lamivudine + stavudine
(n = 181)
(n = 91)
Responder 1
67% (33%)
59% (38%)
Virologic failure 2
24%
27%
Rebound
14%
14%
Never suppressed through Week 48
10%
13%
Death
< 1%
–
Discontinued due to adverse event
1%
3%
Discontinued for other reasons 3
7%
10%
1. Patients achieved and maintained confirmed HIV RNA < 400 copies/mL (< 50 copies/mL) through Week 48. Roche Amplicor ® HIV-1 Monitor™ Assay, test version 1.0 or 1.5 as geographically appropriate.
2. Includes viral rebound and failure to achieve confirmed HIV RNA < 400 copies/mL through Week 48.
3. Includes lost to follow-up, patient's withdrawal, noncompliance, protocol violation, and other reasons.
Through 48 weeks of therapy, the mean increase from baseline in CD4+ cell count was 234 cells/mm 3 for the atazanavir 400 mg arm and 211 cells/mm 3 for the nelfinavir arm.
14.2 Adult Patients with Prior Antiretroviral Therapy
Study AI424-045: Atazanavir sulfate once daily + ritonavir once daily compared to atazanavir sulfate once daily + saquinavir (soft gelatin capsules) once daily, and compared to lopinavir + ritonavir twice daily, each in combination with tenofovir DF + one NRTI. Study AI424-045 was a randomized, multicenter trial comparing atazanavir (300 mg once daily) with ritonavir (100 mg once daily) to atazanavir (400 mg once daily) with saquinavir soft gelatin capsules (1200 mg once daily), and to lopinavir + ritonavir (400/100 mg twice daily), each in combination with tenofovir DF and one NRTI, in 347 (of 358 randomized) patients who experienced virologic failure on HAART regimens containing PIs, NNRTIs, and NRTIs. The mean time of prior exposure to antiretrovirals was 139 weeks for PIs, 85 weeks for NNRTIs, and 283 weeks for NRTIs. The mean age was 41 years (range: 24 to 74); 60% were Caucasian, and 78% were male. The mean baseline CD4+ cell count was 338 cells/mm 3 (range: 14 to 1543 cells/mm 3) and the mean baseline plasma HIV-1 RNA level was 4.4 log 10 copies/mL (range: 2.6 to 5.88 log 10 copies/mL).
Treatment outcomes through Week 48 for the atazanavir sulfate/ritonavir and lopinavir/ritonavir treatment arms are presented in Table 29. Atazanavir sulfate/ritonavir and lopinavir/ritonavir were similar for the primary efficacy outcome measure of time-averaged difference in change from baseline in HIV RNA level. Study AI424-045 was not large enough to reach a definitive conclusion that atazanavir sulfate/ritonavir and lopinavir/ritonavir are equivalent on the secondary efficacy outcome measure of proportions below the HIV RNA lower limit of quantification [see Microbiology, Tables 24 and 25 ( 12.4)] .
Table 29: Outcomes of Treatment Through Week 48 in Study AI424-045 (Patients with Prior Antiretroviral Experience) Outcome
atazanavir 300 mg + ritonavir 100 mg once daily + tenofovir DF + 1 NRTI
lopinavir/ritonavir (400/100 mg) twice daily + tenofovir DF + 1 NRTI
Difference1 (atazanavir sulfate ‑ lopinavir/ritonavir)
(n = 119)
(n = 118)
(CI)
HIV RNA Change from Baseline
(log 10 copies/mL) 2
-1.58
-1.70
+0.12 3 (-0.17, 0.41)
CD4+ Change from Baseline (cells/mm 3) 4
116
123
-7 (-67, 52)
Percent of Patients Responding 5
HIV RNA < 400
copies/mL 2
55%
57%
-2.2% (-14.8%, 10.5%)
HIV RNA < 50
copies/mL 2
38%
45%
-7.1% (-19.6%, 5.4%)
1. Time-averaged difference through Week 48 for HIV RNA; Week 48 difference in HIV RNA percentages and CD4+ mean changes, atazanavir sulfate/ritonavir vs lopinavir/ritonavir; CI = 97.5% confidence interval for change in HIV RNA; 95% confidence interval otherwise.
2. Roche Amplicor ® HIV-1 Monitor™ Assay, test version 1.5.
3. Protocol-defined primary efficacy outcome measure.
4. Based on patients with baseline and Week 48 CD4+ cell count measurements (atazanavir sulfate/ritonavir, n = 85; lopinavir/ritonavir, n = 93).
5. Patients achieved and maintained confirmed HIV-1 RNA < 400 copies/mL (< 50 copies/mL) through Week 48.
No patients in the atazanavir sulfate/ritonavir treatment arm and three patients in the lopinavir/ritonavir treatment arm experienced a new-onset CDC Category C event during the study.
In Study AI424-045, the mean change from baseline in plasma HIV-1 RNA for atazanavir 400 mg with saquinavir (n = 115) was -1.55 log 10 copies/mL, and the time-averaged difference in change in HIV-1 RNA levels versus lopinavir/ritonavir was 0.33. The corresponding mean increase in CD4+ cell count was 72 cells/mm 3. Through 48 weeks of treatment, the proportion of patients in this treatment arm with plasma HIV-1 RNA < 400 (< 50) copies/mL was 38% (26%). In this study, coadministration of atazanavir sulfate and saquinavir did not provide adequate efficacy [see Drug Interactions ( 7)] .
Study AI424-045 also compared changes from baseline in lipid values [see Adverse Reactions ( 6.1)] .
Study AI424-043: Study AI424-043 was a randomized, open-label, multicenter trial comparing atazanavir (400 mg once daily) to lopinavir/ritonavir (400/100 mg twice daily), each in combination with two NRTIs, in 300 patients who experienced virologic failure to only one prior PI-containing regimen. Through 48 weeks, the proportion of patients with plasma HIV-1 RNA < 400 (< 50) copies/mL was 49% (35%) for patients randomized to atazanavir sulfate (n = 144) and 69% (53%) for patients randomized to lopinavir/ritonavir (n = 146). The mean change from baseline was -1.59 log 10 copies/mL in the atazanavir sulfate treatment arm and -2.02 log 10 copies/mL in the lopinavir/ritonavir arm. Based on the results of this study, atazanavir sulfate without ritonavir was inferior to lopinavir/ritonavir in PI-experienced patients with prior virologic failure and is not recommended for such patients.
14.3 Pediatric Patients
Pediatric Trials with Atazanavir Sulfate Capsules
Assessment of the pharmacokinetics, safety, tolerability, and virologic response of atazanavir sulfate capsules was based on data from the open-label, multicenter clinical trial PACTG 1020A which included patients from 6 years to 21 years of age. In this study, 105 patients (43 antiretroviral-naive and 62 antiretroviral-experienced) received once daily atazanavir sulfate capsule formulation, with or without ritonavir, in combination with two NRTIs.
One-hundred five (105) patients (6 to less than 18 years of age) treated with the atazanavir sulfate capsule formulation, with or without ritonavir, were evaluated. Using an ITT analysis, the overall proportions of antiretroviral-naive and -experienced patients with HIV RNA < 400 copies/mL at Week 96 were 51% (22/43) and 34% (21/62), respectively. The overall proportions of antiretroviral-naive and -experienced patients with HIV RNA < 50 copies/mL at Week 96 were 47% (20/43) and 24% (15/62), respectively. The median increase from baseline in absolute CD4 count at 96 weeks of therapy was 335 cells/mm 3 in antiretroviral-naive patients and 220 cells/mm 3 in antiretroviral-experienced patients.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Atazanavir sulfate capsules are available as:
150 mg – capsules with light-turquoise-blue-opaque cap and aqua-blue-opaque body, imprinted “5526” on the body and “TEVA” on the cap, in bottles of 60 (NDC: 0093-5526-06).
200 mg – capsules with light-turquoise body and light-turquoise cap, imprinted “93” over “5527” on both the cap and the body, in bottles of 60 (NDC: 0093-5527-06).
300 mg – capsules with light-blue-opaque body and red-opaque cap, imprinted “93” over “5528” on both the cap and the body, in bottles of 30 (NDC: 0093-5528-56).
Dispense in a tight, light-resistant container as defined in the USP, with a child-resistant closure (as required).
Store at 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Atazanavir sulfate capsules are not a cure for HIV infection. Advise patients to remain under the care of a healthcare provider while using atazanavir sulfate capsules.
Cardiac Conduction Abnormalities
Inform patients that atazanavir may produce changes in the electrocardiogram (e.g., PR prolongation). Tell patients to consult their healthcare provider if they are experiencing symptoms such as dizziness or lightheadedness [see Warnings and Precautions ( 5.1)].Severe Skin Reaction
Inform patients that there have been reports of severe skin reactions (e.g., Stevens-Johnson syndrome, erythema multiforme, and toxic skin eruptions) with atazanavir sulfate capsules use. Advise patients that if signs or symptoms of severe skin reactions or hypersensitivity reactions develop, they must discontinue atazanavir sulfate capsules and seek medical evaluation immediately [see Warnings and Precautions ( 5.2) and Adverse Reactions ( 6.1)] .Hyperbilirubinemia
Inform patients that asymptomatic elevations in indirect bilirubin have occurred in patients receiving atazanavir sulfate capsules. This may be accompanied by yellowing of the skin or whites of the eyes and alternative antiretroviral therapy may be considered if the patient has cosmetic concerns [see Warnings and Precautions ( 5.8)] .
Chronic Kidney Disease
Inform patients that treatment with atazanavir sulfate capsules may lead to the development of chronic kidney disease, and to maintain adequate hydration while taking atazanavir sulfate capsules [see Warnings and Precautions ( 5.5)].
Nephrolithiasis and Cholelithiasis
Inform patients that kidney stones and/or gallstones have been reported with atazanavir sulfate use. Some patients with kidney stones and/or gallstones required hospitalization for additional management and some had complications. Discontinuation of atazanavir sulfate capsules may be necessary as part of the medical management of these adverse events [see Warnings and Precautions ( 5.6)].Drug Interactions
Atazanavir sulfate capsules may lead to significant interaction with some drugs; therefore, advise patients to report the use of any other prescription, nonprescription medication, or herbal products, particularly St. John’s wort, to their healthcare provider prior to use [see Contraindications ( 4), Warnings and Precautions ( 5.7)].Immune Reconstitution Syndrome
Advise patients to inform their healthcare provider immediately of any symptoms of infection, as in some patients with advanced HIV infection (AIDS), signs and symptoms of inflammation from previous infections may occur soon after anti-HIV treatment is started [see Warnings and Precautions (5.10)] .
Fat Redistribution
Inform patients that redistribution or accumulation of body fat may occur in patients receiving antiretroviral therapy including protease inhibitors and that the cause and long-term health effects of these conditions are not known at this time [see Warnings and Precautions ( 5.11)].
Dosing Instructions
Advise patients to take atazanavir sulfate capsules with food every day and take other concomitant antiretroviral therapy as prescribed. Atazanavir sulfate capsules must always be used in combination with other antiretroviral drugs. Advise patients that they should not alter the dose or discontinue therapy without consulting with their healthcare provider. Tell patients if a dose of atazanavir sulfate capsules is missed, they should take the dose as soon as possible and then return to their normal schedule; however, if a dose is skipped the patient should not double the next dose.
Pregnancy
Inform pregnant patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to atazanavir sulfate capsules during pregnancy. Healthcare providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry [see Use in Specific Populations ( 8.1)] .Lactation
Instruct women with HIV-1 infection not to breastfeed because HIV-1 can be passed to the baby in the breast milk. Atazanavir can also be passed to the baby in breast milk and it is not known whether it could harm the baby [see Use in Specific Populations ( 8.2)] .All brand names listed are the registered trademarks of their respective owners and are not trademarks of Teva Pharmaceuticals USA, Inc.
Manufactured In Croatia By:
Pliva Hrvatska d.o.o.
Zagreb, Croatia
Manufactured For:
Teva Pharmaceuticals USA, Inc.
North Wales, PA 19454
Rev. D 5/2018
-
PATIENT INFORMATION
ATAZANAVIR (a ta ZAN a vir) SULFATE CAPSULES
Important: Ask your healthcare provider or pharmacist about medicines that should not be taken with atazanavir sulfate capsules. For more information, see “Who should not take atazanavir sulfate capsules?” and “What should I tell my healthcare provider before taking atazanavir sulfate capsules?”
What are atazanavir sulfate capsules?
Atazanavir sulfate capsules are a prescription HIV-1 (Human Immunodeficiency Virus-type 1) medicine that is used with other antiretroviral medicines to treat HIV-1 infection in adults and children at least 6 years of age and older and who weigh at least 15 kg. HIV-1 is the virus that causes AIDS (Acquired Immunodeficiency Syndrome).
Atazanavir sulfate capsules should not be used in children younger than 3 months of age.
When used with other antiretroviral medicines to treat HIV-1 infection, atazanavir sulfate capsules may help:
- reduce the amount of HIV-1 in your blood. This is called “viral load”.
- increase the number of CD4+ (T) cells in your blood that help fight off other infections.
Reducing the amount of HIV-1 and increasing the CD4+ (T) cells in your blood may help improve your immune system. This may reduce your risk of death or getting infections that can happen when your immune system is weak (opportunistic infections).
Atazanavir sulfate capsules do not cure HIV-1 infection or AIDS. You must keep taking HIV-1 medicines to control HIV-1 infection and decrease HIV-related illnesses.
Who should not take atazanavir sulfate capsules?
Do not take atazanavir sulfate capsules if you:
- are allergic to atazanavir or any of the ingredients in atazanavir sulfate capsules. See the end of this leaflet for a complete list of ingredients in atazanavir sulfate capsules.
- are taking any of the following medicines. Taking atazanavir sulfate capsules with these medicines may affect how atazanavir sulfate capsules work. Atazanavir sulfate capsules may cause serious life-threatening side effects or death when used with these medicines:
- alfuzosin (UROXATRAL ®)
- cisapride (PROPULSID ®)
- elbasvir/grazoprevir (ZEPATIER ®)
- ergot medicines including:
- ergotamine tartrate (CAFERGOT ®, MIGERGOT ®, ERGOMAR ®, ERGOSTAT ®, MEDIHALER ®, Ergotamine, WIGRAINE ®, WIGRETTES ®)
- dihydroergotamine mesylate (D.H.E. 45 ®, MIGRANAL ®)
- methylergonovine (METHERGINE ®)
- glecaprevir/pibrentasvir (MAVYRET ®)
- indinavir (CRIXIVAN ®)
- irinotecan (CAMPTOSAR ®)
- lurasidone (LATUDA ®) if atazanavir sulfate capsules are used with ritonavir (NORVIR ®)
- lovastatin (ADVICOR ®, ALTOPREV ®, MEVACOR ®)
- midazolam (VERSED ®), when taken by mouth for sedation
- nevirapine (VIRAMUNE ®, VIRAMUNE XR ®)
- pimozide (ORAP ®)
- rifampin (RIFADIN ®, RIFAMATE ®, RIFATER ®, RIMACTANE ®)
- sildenafil (REVATIO ®), when used for the treatment of pulmonary arterial hypertension
- simvastatin (SIMCOR ®, VYTORIN ®, ZOCOR ®)
- St. John’s wort ( Hypericum perforatum)
- triazolam (HALCION ®)
Serious problems can happen if you or your child takes any of the medicines listed above with atazanavir sulfate capsules.
What should I tell my healthcare provider before taking atazanavir sulfate capsules?
Before taking atazanavir sulfate capsules, tell your healthcare provider if you:
- have heart problems
- have liver problems, including hepatitis B or C virus infection
- are receiving dialysis treatment
- have diabetes
- have hemophilia
- have any other medical conditions
- are pregnant or plan to become pregnant. Talk to your healthcare provider about taking atazanavir sulfate capsules during your pregnancy or if you are planning to become pregnant while you are taking atazanavir sulfate capsules.
- Hormonal forms of birth control, such as injections, vaginal rings or implants, contraceptive patch, and some birth control pills may not work during treatment with atazanavir sulfate capsules. Talk to your healthcare provider about forms of birth control that may be used during treatment with atazanavir sulfate capsules.
- Pregnancy Registry. There is a pregnancy registry for women who take antiretroviral medicines during pregnancy. The purpose of this registry is to collect information about the health of you and your baby. Talk to your healthcare provider about how you can take part in this registry.
- After your baby is born, tell your healthcare provider if your baby’s skin or the white part of the eyes turns yellow.
- are breastfeeding or plan to breastfeed. Do not breastfeed if you are taking atazanavir sulfate capsules. You should not breastfeed if you have HIV-1 because of the risk of passing HIV-1 to your baby. Atazanavir can pass into your breast milk. Talk to your healthcare provider about the best way to feed your baby.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Some medicines interact with atazanavir sulfate capsules. Keep a list of your medicines to show your healthcare provider and pharmacist. You can ask your healthcare provider or pharmacist for a list of medicines that interact with atazanavir sulfate capsules. Do not start taking a new medicine without telling your healthcare provider. Your healthcare provider can tell you if it is safe to take atazanavir sulfate capsules with other medicines.
How should I take atazanavir sulfate capsules?
- Take atazanavir sulfate capsules exactly as your healthcare provider tells you to.
- Do not change your dose or stop taking atazanavir sulfate capsules unless your healthcare provider tells you to.
- Stay under the care of your healthcare provider during treatment with atazanavir sulfate capsules.
- Atazanavir sulfate capsules must be used with other antiretroviral medicines.
- Take atazanavir sulfate capsules 1 time each day.
- Atazanavir sulfate capsules come as capsules.
- Take atazanavir sulfate capsules with food.
- Swallow the capsules whole. Do not open the capsules.
- If you miss a dose of atazanavir sulfate capsules, take it as soon as you remember. Then take the next dose at your regular time. Do not take 2 doses at the same time.
- If you take too many atazanavir sulfate capsules, call your healthcare provider or go to the nearest hospital emergency room right away.
When your supply of atazanavir sulfate capsules starts to run low, get more from your healthcare provider or pharmacy. It is important not to run out of atazanavir sulfate capsules. The amount of HIV-1 in your blood may increase if the medicine is stopped for even a short time. The virus may become resistant to atazanavir sulfate capsules and harder to treat.
What are the possible side effects of atazanavir sulfate capsules?
Atazanavir sulfate capsules can cause serious side effects, including:
- A change in the way your heart beats (heart rhythm change). Tell your healthcare provider right away if you get dizzy or lightheaded. These could be symptoms of a heart problem.
-
Skin rash. Skin rash is common with atazanavir sulfate capsules but can sometimes be severe. Skin rash usually goes away within 2 weeks without any change in treatment. Severe rash may develop in association with other symptoms which could be serious. If you develop a severe rash or a rash with any of the following symptoms, stop taking atazanavir sulfate capsules and call your healthcare provider right away:
- general feeling of discomfort or “flu-like” symptoms
- fever
- muscle or joint aches
- red or inflamed eyes, like “pink eye” (conjunctivitis)
- blisters
- mouth sores
- swelling of your face
- painful, warm, or red lump under your skin
- Yellowing of your skin or the white part of your eyes is common with atazanavir sulfate capsules, and is usually not harmful in adults and infants older than 3 months of age; but it could also be a symptom of a serious problem. These effects may be due to increases in bilirubin levels in your blood (bilirubin is made by the liver). Although these effects may not be damaging to your liver, skin, or eyes, tell your healthcare provider right away if your skin or the white part of your eyes turns yellow.
-
Liver problems. If you have liver problems, including hepatitis B or C infection, your liver problems may get worse when you take atazanavir sulfate capsules. Your healthcare provider will do blood tests to check your liver before you start atazanavir sulfate capsules and during treatment. Tell your healthcare provider right away if you get any of the following symptoms:
- dark “tea-colored” urine
- your skin or the white part of your eyes turns yellow
- light colored stools
- nausea
- itching
- stomach-area pain
- Chronic kidney disease. Atazanavir sulfate capsules may affect how well your kidneys work. Your healthcare provider will do blood and urine tests to check your kidneys before you start atazanavir sulfate capsules and during treatment.
- Kidney stones have happened in some people who take atazanavir sulfate capsules. Tell your healthcare provider right away if you get symptoms of kidney stones which may include, pain in your low back or low stomach-area, blood in your urine, or pain when you urinate.
-
Gallbladder problems have happened in some people who take atazanavir sulfate capsules. Tell your healthcare provider right away if you get symptoms of gallbladder problems which may include:
- pain in the right or middle upper stomach area
- fever
- nausea and vomiting
- your skin or the white part of your eyes turns yellow
- Diabetes and high blood sugar (hyperglycemia) have happened or have worsened in some people who take protease inhibitor medicines like atazanavir sulfate capsules. Some people have had to start taking medicine to treat diabetes or have had to change their diabetes medicine.
- Changes in your immune system (Immune Reconstitution Syndrome) can happen when you start taking HIV-1 medicines. Your immune system may get stronger and begin to fight infections that have been hidden in your body for a long time. Tell your healthcare provider if you start having new symptoms after starting atazanavir sulfate capsules.
- Changes in body fat can happen in people taking HIV-1 medicines. These changes may include increased amount of fat in the upper back and neck (“buffalo hump”), breast, and around the main part of your body (trunk). Loss of fat from the legs, arms, and face may also happen. The exact cause and long-term health effects of these conditions are not known.
- Increased bleeding problems in people with hemophilia have happened when taking protease inhibitors like atazanavir sulfate capsules.
The most common side effects of atazanavir sulfate capsules include:
- nausea
- dizziness
- headache
- muscle pain
- stomach-area pain
- diarrhea
- vomiting
- depression
- trouble sleeping
- fever
- numbness, tingling, or burning of hands or feet
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of atazanavir sulfate capsules. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store atazanavir sulfate capsules?
- Store atazanavir sulfate capsules at room temperature, 68° to 77°F (20° to 25°C).
- Keep capsules in a tightly closed container.
Keep atazanavir sulfate capsules and all medicines out of the reach of children.
General information about the safe and effective use of atazanavir sulfate capsules
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use atazanavir sulfate capsules for a condition for which they were not prescribed. Do not give atazanavir sulfate capsules to other people, even if they have the same symptoms that you have. They may harm them. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about atazanavir sulfate capsules that is written for health professionals.
For more information, call 1-888-838-2872.What are the ingredients in atazanavir sulfate capsules?
Active ingredient: atazanavir sulfate
Inactive ingredients: crospovidone, D&C yellow #10 aluminum lake, FD&C blue #1, FD&C blue #1 aluminum lake, FD&C blue #2 aluminum lake, FD&C red #40 aluminum lake, gelatin, iron oxide black, lactose monohydrate, magnesium stearate, propylene glycol, shellac, sodium lauryl sulfate, sorbitan monolaurate, and titanium dioxide. Additionally, the 300 mg capsules also contain FD&C red #40.
Manufactured In Croatia By:
Pliva Hrvatska d.o.o.
Zagreb, Croatia
Manufactured For:
Teva Pharmaceuticals USA, Inc.
North Wales, PA 19454
Orap ® is a registered trademark of Teva Pharmaceuticals USA, Inc.
All brand names listed are the registered trademarks of their respective owners and are not trademarks of Teva Pharmaceuticals USA, Inc.
This Patient Information has been approved by the U.S. Food and Drug Administration.
Rev. D 5/2018
-
PRINCIPAL DISPLAY PANEL
DRUG: Atazanavir Sulfate
GENERIC: Atazanavir Sulfate
DOSAGE: CAPSULE
ADMINSTRATION: ORAL
NDC: 70518-0946-0
COLOR: blue
SHAPE: CAPSULE
SCORE: No score
SIZE: 23 mm
IMPRINT: 93;5528;93;5528
PACKAGING: 30 in 1 BLISTER PACK
ACTIVE INGREDIENT(S):
- ATAZANAVIR SULFATE 300mg in 1
INACTIVE INGREDIENT(S):
- CROSPOVIDONE (15 MPA.S AT 5%)
- MAGNESIUM STEARATE
- PROPYLENE GLYCOL
- LACTOSE MONOHYDRATE
- SHELLAC
- SODIUM LAURYL SULFATE
- SORBITAN MONOLAURATE
- D&C YELLOW NO. 10
- FD&C BLUE NO. 1
- FD&C BLUE NO. 2
- FERROSOFERRIC OXIDE
- FD&C RED NO. 40
- GELATIN, UNSPECIFIED
- TITANIUM DIOXIDE

-
INGREDIENTS AND APPEARANCE
ATAZANAVIR SULFATE
atazanavir sulfate capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 70518-0946(NDC:0093-5528) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ATAZANAVIR SULFATE (UNII: 4MT4VIE29P) (ATAZANAVIR - UNII:QZU4H47A3S) ATAZANAVIR 300 mg Inactive Ingredients Ingredient Name Strength CROSPOVIDONE (15 MPA.S AT 5%) (UNII: 68401960MK) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) FD&C RED NO. 40 (UNII: WZB9127XOA) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) FERROSOFERRIC OXIDE (UNII: XM0M87F357) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SHELLAC (UNII: 46N107B71O) SODIUM LAURYL SULFATE (UNII: 368GB5141J) SORBITAN MONOLAURATE (UNII: 6W9PS8B71J) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color blue (light-blue) , red Score no score Shape CAPSULE Size 23mm Flavor Imprint Code 93;5528;93;5528 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 70518-0946-0 30 in 1 BLISTER PACK; Type 0: Not a Combination Product 01/10/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA091673 01/10/2018 Labeler - REMEDYREPACK INC. (829572556)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.