FIRVANQ- vancomycin hydrochloride kit
FIRVANQ by
Drug Labeling and Warnings
FIRVANQ by is a Prescription medication manufactured, distributed, or labeled by Azurity Pharmaceuticals, Inc. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use FIRVANQ® safely and effectively. See full prescribing information for FIRVANQ.
FIRVANQ(vancomycin hydrochloride), for oral solution
Initial U.S. Approval: 1964INDICATIONS AND USAGE
FIRVANQ is a glycopeptide antibacterial indicated in adults and pediatric patients less than 18 years of age for the treatment of: ( 1)
- Clostridiumdifficile-associated diarrhea
- Enterocolitis caused by Staphylococcus aureus (including methicillin-resistant strains)
Important Limitations of Use: ( 1) ( 5.1)
- Orally administered vancomycin hydrochloride is not effective for treatment of other types of infections.
To reduce the development of drug-resistant bacteria and maintain the effectiveness of FIRVANQ and other antibacterial drugs, FIRVANQ should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. ( 1)
DOSAGE AND ADMINISTRATION
-
C. difficile-associated diarrhea:
Adult Patients (18 years of age and older): 125 mg orally 4 times daily for 10 days. ( 2.2)
Pediatric Patients (less than 18 years of age): 40 mg/kg in 3 or 4 divided doses for 7 to 10 days. The total daily dosage should not exceed 2 g. ( 2.3)
- Staphylococcal enterocolitis:
Adult Patients (18 years of age and older): 500 mg to 2 g orally in 3 or 4 divided doses for 7 to 10 days. ( 2.2)
Pediatric Patients (less than 18 years of age): 40 mg/kg in 3 or 4 divided doses for 7 to 10 days. The total daily dosage should not exceed 2 g. ( 2.3)
- See Full Prescribing Information for preparation and important administration information. ( 2.1)
DOSAGE FORMS AND STRENGTHS
Each FIRVANQ kit contains: vancomycin hydrochloride USP, powder for oral solution, equivalent to 3.75 g, 7.5 g or 15 g vancomycin, and Grape-Flavored Diluent. ( 3)
CONTRAINDICATIONS
Hypersensitivity to vancomycin ( 4)
WARNINGS AND PRECAUTIONS
- FIRVANQ™ must be given orally for treatment of
C. difficile-associated diarrhea and staphylococcal enterocolitis. Orally administered vancomycin hydrochloride is not effective for treatment of other types of infections. (
5.1)
- Clinically significant serum concentrations have been reported in some patients who have taken multiple oral doses of vancomycin hydrochloride for
C. difficile-associated diarrhea. Monitoring of serum concentrations may be appropriate in some instances. (
5.2)
- Nephrotoxicity has occurred following oral vancomycin hydrochloride therapy and can occur either during or after completion of therapy. The risk is increased in geriatric patients. Monitor renal function. (
5.3)
- Ototoxicity has occurred in patients receiving vancomycin hydrochloride. Assessment of auditory function may be appropriate in some instances. (
5.4)
- Prescribing FIRVANQ™ in the absence of a proven or strongly suspected bacterial infection is unlikely to provide benefit to the patient and increases the risk of the development of drug resistant bacteria. ( 5.6)
ADVERSE REACTIONS
The most common adverse reactions (≥ 10%) were nausea (17%), abdominal pain (15%) and hypokalemia (13%). ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact CutisPharma, Inc. at 1 800 461 7449, EXT 103; or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 10/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1Important Administration Instructions
2.2 Adults
2.3Pediatric Patients (less than 18 years of age)
2.4Preparation and Storage of Solutions of FIRVANQ
3DOSAGE FORMS AND STRENGTHS
4CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1Oral Use Only
5.2Potential for Systemic Absorption
5.3Nephrotoxicity
5.4 Ototoxicity
5.5Potential for Microbial Overgrowth
5.6Development of Drug- Resistant Bacteria
5.7 Hemorrhagic Occlusive Retinal Vasculitis (HORV)
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1Pregnancy
8.2Lactation
8.4Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Diarrhea Associated with Clostridium difficile
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1INDICATIONS AND USAGE
FIRVANQ is indicated for the treatment of Clostridiumdifficile-associated diarrhea in adults and pediatric patients less than 18 years of age.
FIRVANQ is also indicated for the treatment of enterocolitis caused by Staphylococcus aureus (including methicillin-resistant strains) in adults and pediatric patients less than 18 years of age.
Important Limitations of Use
- Parenteral administration of vancomycin is not effective for the above infections; therefore, vancomycin must be given orally for these infections.
- Orally administered vancomycin hydrochloride is not effective for treatment of other types of infections.
To reduce the development of drug-resistant bacteria and maintain the effectiveness of FIRVANQ and other antibacterial drugs, FIRVANQ should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
-
2
DOSAGE AND ADMINISTRATION
2.1Important Administration Instructions
Prior to oral administration, the supplied FIRVANQ powder must be reconstituted by the healthcare provider (i.e., a pharmacist) to produce the oral solution [ see Dosage and Administration (2.4)].
2.2 Adults
-
C. difficile-associated diarrhea: The recommended dose is 125 mg administered orally 4 times daily for 10 days.
- Staphylococcal enterocolitis: Total daily dosage is 500 mg to 2 g administered orally in 3 or 4 divided doses for 7 to 10 days.
2.3Pediatric Patients (less than 18 years of age)
For both C. difficile-associated diarrhea and staphylococcal enterocolitis, the usual daily dosage of FIRVANQ is 40 mg/kg in 3 or 4 divided doses for 7 to 10 days. The total daily dosage should not exceed 2 g.
2.4Preparation and Storage of Solutions of FIRVANQ
Each FIRVANQ kit contains 1 bottle of vancomycin hydrochloride USP powder and 1 bottle of pre-measured Grape-Flavored Diluent to be added to the vancomycin bottle. A healthcare provider (i.e., a pharmacist) must reconstitute vancomycin hydrochloride USP powder with the Grape-Flavored Diluent provided in the kit. FIRVANQ is available in various strengths and volumes in the kit as shown in Table 1.
Table 1: Vancomycin Concentration and Volume after Reconstitution Vancomycin Concentration after Reconstitution Final Volume of FIRVANQ after Reconstitution Vancomycin Strength per Bottle Diluent for FIRVANQ 25 mg/mL 150 mL 3.75 g 147 mL 300 mL 7.5 g 295 mL 50 mg/mL 150 mL 7.5 g 145 mL 300 mL 15 g 289 mL Steps for the Preparation of Solutions of FIRVANQ
- Hold the neck of the bottle containing the vancomycin hydrochloride USP powder for oral solution (see Table 1), and tap the bottom edges on a hard surface to loosen the powder.
- Remove the cap from the bottle.
- Tap the top of the induction seal liner to loosen any powder which may have adhered to the liner.
- Carefully and slowly peel back the inner foil seal liner from the bottle.
- Shake the Grape-Flavored Diluent (see Table 1) for a few seconds.
- Open the diluent bottle and carefully and slowly peel back the inner foil seal from the diluent bottle.
- Transfer about half the contents of the Grape-Flavored Diluent into the vancomycin hydrochloride, USP powder bottle (“Powder Bottle”).
- Replace the Powder Bottle cap, tighten onto the Powder Bottle, and shake the Powder Bottle vertically for approximately 45 seconds. NOTE: Do NOT use the diluent cap on the Powder Bottle.
- Re-open the Powder Bottle and add the remaining Grape‑Flavored Diluent into the Powder Bottle.
- Replace the Powder Bottle cap, tighten onto the Powder Bottle, and shake the Powder Bottle for approximately 30 seconds. NOTE: Do NOT use the diluent cap on the Powder Bottle as it may cause the solution to leak from the bottle.
- Apply appropriate bar coded dispensing labels onto Powder Bottle and dispense FIRVANQ oral solution to the patient.
- Instruct the patient to shake the reconstituted solution of FIRVANQ well before each use and to use an oral dosing device that measures the appropriate volume of the oral solution in milliliters.
- Instruct the patient to discard the reconstituted solution of FIRVANQ solution after 14 days, or if it appears hazy or contains particulates.
-
C. difficile-associated diarrhea: The recommended dose is 125 mg administered orally 4 times daily for 10 days.
- 3DOSAGE FORMS AND STRENGTHS
- 4CONTRAINDICATIONS
-
5
WARNINGS AND PRECAUTIONS
5.1Oral Use Only
FIRVANQ must be given orally for treatment of C. difficile-associated diarrhea and staphylococcal enterocolitis. Orally administered vancomycin is not effective for treatment of other types of infections.
Parenteral administration of vancomycin is not effective for treatment of C. difficile-associated diarrhea and staphylococcal enterocolitis. If parenteral vancomycin therapy is desired, use an intravenous preparation of vancomycin and consult the package insert accompanying that preparation.
5.2Potential for Systemic Absorption
Significant systemic absorption has been reported in some patients (e.g., patients with renal insufficiency and/or colitis) who have taken multiple oral doses of vancomycin hydrochloride for C. difficile-associated diarrhea. In these patients, serum vancomycin concentrations reached therapeutic levels for the treatment of systemic infections. Some patients with inflammatory disorders of the intestinal mucosa also may have significant systemic absorption of vancomycin. These patients may be at risk for the development of adverse reactions associated with higher doses of FIRVANQ; therefore, monitoring of serum concentrations of vancomycin may be appropriate in some instances, e.g., in patients with renal insufficiency and/or colitis or in those receiving concomitant therapy with an aminoglycoside antibacterial drug.
5.3Nephrotoxicity
Nephrotoxicity (e.g., reports of renal failure, renal impairment, blood creatinine increased) has occurred following oral vancomycin hydrochloride therapy in randomized controlled clinical trials, and can occur either during or after completion of therapy. The risk of nephrotoxicity is increased in patients over 65 years of age [ seeAdverse Reactions(6.1) and Use in Specific Populations(8.5)].
In patients over 65 years of age, including those with normal renal function prior to treatment, renal function should be monitored during and following treatment with FIRVANQ to detect potential vancomycin induced nephrotoxicity.
5.4 Ototoxicity
Ototoxicity has occurred in patients receiving vancomycin. It may be transient or permanent. It has been reported mostly in patients who have been given high intravenous doses, who have an underlying hearing loss, or who are receiving concomitant therapy with another ototoxic agent, such as an aminoglycoside. Serial tests of auditory function may be helpful in order to minimize the risk of ototoxicity [ see Adverse Reactions (6.2)].
5.5Potential for Microbial Overgrowth
Use of FIRVANQ may result in the overgrowth of non-susceptible bacteria. If superinfection occurs during therapy, appropriate measures should be taken.
5.6Development of Drug- Resistant Bacteria
Prescribing FIRVANQ in the absence of a proven or strongly suspected bacterial infection is unlikely to provide benefit to the patient and increases the risk of the development of drug-resistant bacteria.
5.7 Hemorrhagic Occlusive Retinal Vasculitis (HORV)
Hemorrhagic occlusive retinal vasculitis, including permanent loss of vision, occurred in patients receiving intracameral or intravitreal administration of vancomycin during or after cataract surgery. The safety and efficacy of vancomycin administered by the intracameral or intravitreal route have not been established by adequate and well-controlled studies. Vancomycin is not indicated for prophylaxis of endophthalmitis.
-
6
ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
The data described below reflect exposure to vancomycin hydrochloride in 260 adult subjects in two Phase 3 clinical trials for the treatment of C. difficile-associated diarrhea. In both trials, subjects received vancomycin hydrochloride 125 mg orally four times daily. The mean duration of treatment was 9.4 days. The median age of patients was 67, ranging between 19 and 96 years of age. Patients were predominantly Caucasian (93%) and 52% were male.
Adverse reactions occurring in ≥ 5% of vancomycin hydrochloride-treated subjects are shown in Table 2. The most common adverse reactions associated with vancomycin hydrochloride (≥ 10%) were nausea, abdominal pain, and hypokalemia.
Table 2: Common (≥ 5%) Adverse Reactions* for Vancomycin Hydrochloride Reported in Clinical Trials for Treatment of C. difficile Associated Diarrhea System/Organ Class Adverse Reaction Vancomycin Hydrochloride (%) (N=260) Gastrointestinal disorders Nausea 17 Abdominal pain 15 Vomiting 9 Diarrhea 9 Flatulence 8 General disorders and administration site conditions Pyrexia 9 Edema peripheral 6 Fatigue 5 Infections and infestations Urinary tract infection 8 Metabolism and nutrition disorders Hypokalemia 13 Musculoskeletal and connective tissue disorders Back pain 6 Nervous system disorders Headache 7 * Adverse reaction rates were derived from the incidence of treatment-emergent adverse events. Nephrotoxicity (e.g., reports of renal failure, renal impairment, blood creatinine increased) occurred in 5% of subjects treated with vancomycin hydrochloride. Nephrotoxicity following vancomycin hydrochloride typically first occurred within one week after completion of treatment (median day of onset was Day 16). Nephrotoxicity following vancomycin hydrochloride occurred in 6% of subjects over 65 years of age and 3% of subjects 65 years of age and younger [ see Warnings and Precautions (5.3)]. Nephrotoxicity can also occur during oral vancomycin administration.
The incidences of hypokalemia, urinary tract infection, peripheral edema, insomnia, constipation, anemia, depression, vomiting, and hypotension were higher among subjects over 65 years of age than in subjects 65 years of age and younger [ see Use in Specific Populations (8.5)].
Discontinuation of study drug due to adverse events occurred in 7% of subjects treated with vancomycin hydrochloride. The most common adverse events leading to discontinuation of vancomycin hydrochloride were C. difficile colitis (< 1%), nausea (< 1%), and vomiting (< 1%).
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of vancomycin hydrochloride. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Ototoxicity: Cases of hearing loss associated with intravenously administered vancomycin have been reported. Most of these patients had kidney dysfunction or a preexisting hearing loss or were receiving concomitant treatment with an ototoxic drug [ see Warnings and Precautions (5.4)]. Vertigo, dizziness, and tinnitus have been reported.
Hematopoietic: Reversible neutropenia, usually starting 1 week or more after onset of intravenous therapy with vancomycin or after a total dose of more than 25 g, has been reported. Neutropenia appears to be promptly reversible when vancomycin is discontinued. Thrombocytopenia has been reported.
Miscellaneous: Anaphylaxis, drug fever, chills, nausea, eosinophilia, rashes (including exfoliative dermatitis), Stevens-Johnson syndrome, toxic epidermal necrolysis, and vasculitis have been reported with the administration of vancomycin.
A condition has been reported with oral vancomycin that is similar to the IV–induced syndrome with symptoms consistent with anaphylactoid reactions, including hypotension, wheezing, dyspnea, urticaria, pruritus, flushing of the upper body (“Red Man Syndrome”), pain and muscle spasm of the chest and back. These reactions usually resolve within 20 minutes but may persist for several hours.
- 7 DRUG INTERACTIONS
-
8
USE IN SPECIFIC POPULATIONS
8.1Pregnancy
Risk Summary
There are no available data on FIRVANQ use in pregnant women to inform a drug associated risk of major birth defects or miscarriage. Available published data on vancomycin use in pregnancy during the second and third trimesters have not shown an association with adverse pregnancy related outcomes (see Data). Vancomycin did not show adverse developmental effects when administered intravenously to pregnant rats and rabbits during organogenesis at doses less than or equal to the recommended maximum human dose based on body surface area (see Data).
All pregnancies have a background risk of birth defect, loss or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Human Data
A published study evaluated hearing loss and nephrotoxicity in infants of pregnant intravenous drug users treated with vancomycin for suspected or documented methicillin-resistant S. aureus in the second or third trimester. The comparison groups were 10 non-intravenous drug-dependent patients who received no treatment, and 10 untreated intravenous drug-dependent patients served as substance abuse controls. No infant in the vancomycin exposed group had abnormal sensorineural hearing at 3 months of age or nephrotoxicity.
A published prospective study assessed outcomes in 55 pregnant women with a positive Group B Streptococcus culture and a high-risk penicillin allergy with resistance to clindamycin or unknown sensitivity who were administered vancomycin at the time of delivery. Vancomycin dosing ranged from the standard 1 g intravenously every 12 hours to 20 mg/kg intravenous every 8 hours (maximum individual dose 2 g). No major adverse reactions were recorded either in the mothers or their newborns. None of the newborns had sensorineural hearing loss. Neonatal renal function was not examined, but all of the newborns were discharged in good condition.
Animal Data
Vancomycin did not cause fetal malformations when administered during organogenesis to pregnant rats (gestation days 6 to 15) and rabbits (gestation days 6 to 18) at the equivalent recommended maximum human dose (based on body surface area comparisons) of 200 mg/kg/day IV to rats or 120 mg/kg/day IV to rabbits. No effects on fetal weight or development were seen in rats at the highest dose tested or in rabbits given 80 mg/kg/day (approximately 1 and 0.8 times the recommended maximum human dose based on body surface area, respectively). Maternal toxicity was observed in rats (at doses 120 mg/kg and above) and rabbits (at 80 mg/kg and above).
8.2Lactation
Risk Summary
There are insufficient data to inform the levels of vancomycin in human milk. However, systemic absorption of vancomycin following oral administration is expected to be minimal [see Clinical Pharmacology (12.3)]. There are no data on the effects of FIRVANQ on the breastfed infant or milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for FIRVANQ and any potential adverse effects on the breastfed infant from Firvanq ™ or from the underlying maternal condition.
8.4Pediatric Use
FIRVANQ is indicated in pediatric patients less than 18 years of age for the treatment of C. difficile-associated diarrhea and enterocolitis caused by S. aureus (including methicillin-resistant strains) [ see Indications and Usage (1) and Dosage and Administration (2.3)].
8.5 Geriatric Use
In clinical trials, 54% of vancomycin hydrochloride-treated subjects were > 65 years of age. Of these, 40% were between the ages of > 65 and 75, and 60% were > 75 years of age.
Clinical studies with vancomycin hydrochloride in C. difficile-associated diarrhea have demonstrated that geriatric subjects are at increased risk of developing nephrotoxicity following treatment with oral vancomycin hydrochloride, which may occur during or after completion of therapy. In patients over 65 years of age, including those with normal renal function prior to treatment, renal function should be monitored during and following treatment with vancomycin hydrochloride to detect potential vancomycin induced nephrotoxicity [ see Warnings and Precautions(5.3), Adverse Reactions (6.1) and Clinical Studies (14.1)].
Patients over 65 years of age may take longer to respond to therapy compared to patients 65 years of age and younger [ see Clinical Studies (14.1)]. Clinicians should be aware of the importance of appropriate duration of vancomycin hydrochloride treatment in patients over 65 years of age and not discontinue or switch to alternative treatment prematurely.
-
10
OVERDOSAGE
Supportive care is advised, with maintenance of glomerular filtration. Vancomycin is poorly removed by dialysis. Hemofiltration and hemoperfusion with polysulfone resin have been reported to result in increased vancomycin clearance.
For current information on the management of overdosage, contact the National Poison Control Center at 1-800-222-1222 or www.poison.org.
-
11DESCRIPTION
FIRVANQ for oral administration contains the hydrochloride salt of vancomycin, a tricyclic glycopeptide antibiotic derived from Amycolatopsis orientalis (formerly Nocardia orientalis), which has the chemical formula C 66H 75Cl 2N 9O 24HCl. The molecular weight of vancomycin hydrochloride is 1485.71 g/mol.
Vancomycin hydrochloride has the structural formula:
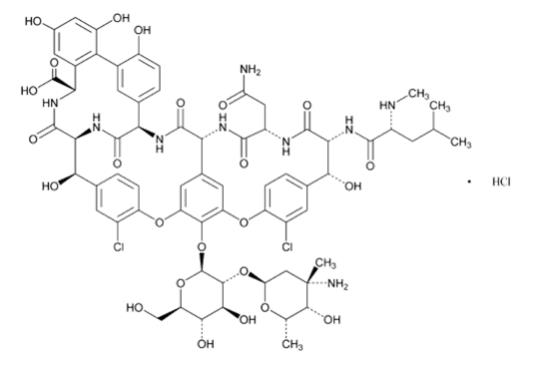
Each FIRVANQ kit contains a bottle of vancomycin hydrochloride USP, as white to almost white or tan to brown powder for oral solution, and a bottle of pre-measured Grape-Flavored Diluent, in the strengths and volumes listed in Table 3.
Table 3: Vancomycin Strength, Diluent Volume and Vancomycin Concentration after Reconstitution Vancomycin Strength per Bottle Equivalent Amount of Vancomycin Hydrochloride per Bottle Diluent for FIRVANQ Vancomycin Concentration after Reconstitution 3.75 g 3.8 g 147 mL 25 mg/mL 7.5 g 7.7 g 295 mL 7.5 g 7.7 g 145 mL 50 mg/mL 15 g 15.4 g 289 mL The Grape-Flavored Diluent used to reconstitute the oral solution contains: artificial grape flavor, citric acid (anhydrous), D&C Yellow No. 10, FD&C Red No. 40, purified water, sodium benzoate and sucralose.
-
12
CLINICAL PHARMACOLOGY
12.3 Pharmacokinetics
Vancomycin is poorly absorbed after oral administration. During multiple dosing of vancomycin hydrochloride capsules at 250 mg every 8 hours for 7 doses, fecal concentrations of vancomycin in volunteers exceeded 100 mcg/g in the majority of samples. No blood concentrations were detected and urinary recovery did not exceed 0.76%. In anephric subjects with no inflammatory bowel disease who received vancomycin oral solution 2 g for 16 days, blood concentrations of vancomycin were ≤ 0.66 mcg/mL in 2 of 5 subjects. No measurable blood concentrations were attained in the other 3 subjects. Following doses of 2 g daily, concentrations of drug were > 3100 mcg/g in the feces and < 1 mcg/mL in the serum of subjects with normal renal function who had C. difficile-associated diarrhea. After multiple-dose oral administration of vancomycin, measurable serum concentrations may occur in patients with active C. difficile-associated diarrhea, and, in the presence of renal impairment, the possibility of accumulation exists. It should be noted that the total systemic and renal clearances of vancomycin are reduced in the elderly [s ee Use in Specific Populations (8.5)].
12.4 Microbiology
Mechanism of Action
The bactericidal action of vancomycin against the vegetative cells of C. difficile and S. aureus results primarily from inhibition of cell-wall biosynthesis. In addition, vancomycin alters bacterial-cell-membrane permeability and RNA synthesis.
Mechanism of Resistance
C. difficile
Isolates of C. difficile generally have vancomycin minimal inhibitory concentrations (MICs) of < 1 mcg/mL; however, vancomycin MICs ranging from 4 mcg/mL to 16 mcg/mL have been reported. The mechanism which mediates C. difficile's decreased susceptibility to vancomycin has not been fully elucidated.
S. aureus
S. aureus isolates with vancomycin MICs as high as 1024 mcg/mL have been reported. The exact mechanism of this resistance is not clear but is believed to be due to cell wall thickening and potentially the transfer of genetic material.
Vancomycin has been shown to be active against susceptible isolates of the following bacteria in clinical infections [ see Indications and Usage (1)].
Anaerobic gram-positive bacteria
C. difficile isolates associated with C. difficile-associated diarrhea.Gram-positive bacteria
S. aureus (including methicillin-resistant isolates) associated with enterocolitis. -
13
NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No long-term carcinogenesis studies in animals have been conducted.
At concentrations up to 1000 mcg/mL, vancomycin had no mutagenic effect in vitro in the mouse lymphoma forward mutation assay or the primary rat hepatocyte unscheduled DNA synthesis assay. The concentrations tested in vitro were above the peak plasma vancomycin concentrations of 20 to 40 mcg/mL usually achieved in humans after slow infusion of the maximum recommended dose of 1 g. Vancomycin had no mutagenic effect in vivo in the Chinese hamster sister chromatid exchange assay (400 mg/kg IP) or the mouse micronucleus assay (800 mg/kg IP).
No definitive fertility studies have been conducted.
-
14
CLINICAL STUDIES
14.1 Diarrhea Associated with Clostridium difficile
In two trials, vancomycin hydrochloride 125 mg orally four times daily for 10 days was evaluated in 266 adult subjects with C. difficile-associated diarrhea (CDAD). Enrolled subjects were 18 years of age or older and received no more than 48 hours of treatment with oral vancomycin hydrochloride or oral/intravenous metronidazole in the 5 days preceding enrollment. CDAD was defined as ≥ 3 loose or watery bowel movements within the 24 hours preceding enrollment, and the presence of either C. difficile toxin A or B, or pseudomembranes on endoscopy within the 72 hours preceding enrollment. Subjects with fulminant C. difficile disease, sepsis with hypotension, ileus, peritoneal signs or severe hepatic disease were excluded.
Efficacy analyses were performed on the Full Analysis Set (FAS), which included randomized subjects who received at least one dose of vancomycin hydrochloride and had any post-dosing investigator evaluation data (N = 259; 134 in Trial 1 and 125 in Trial 2).
The demographic profile and baseline CDAD characteristics of enrolled subjects were similar in the two trials. Vancomycin hydrochloride-treated subjects had a median age of 67 years, were mainly white (93%), and male (52%). CDAD was classified as severe (defined as 10 or more unformed bowel movements per day or white blood cell count (WBC) ≥ 15000/mm 3) in 25% of subjects, and 47% were previously treated for CDAD.
Efficacy was assessed by using clinical success, defined as diarrhea resolution and the absence of severe abdominal discomfort due to CDAD, on Day 10. An additional efficacy endpoint was the time to resolution of diarrhea, defined as the beginning of diarrhea resolution that was sustained through the end of the prescribed active treatment period.
The results for clinical success for vancomycin hydrochloride-treated subjects in both trials are shown in Table 4.
Table 4: Clinical Success Rates (Full Analysis Set) Clinical Success Rate
Vancomycin Hydrochloride % (N)95% Confidence Interval Trial 1 81.3 (134) (74.4, 88.3) Trial 2 80.8 (125) (73.5, 88.1) The median time to resolution of diarrhea was 5 days and 4 days in Trial 1 and Trial 2, respectively. For subjects older than 65 years of age, the median time to resolution was 6 days and 4 days in Trial 1 and Trial 2, respectively. In subjects with diarrhea resolution at end-of-treatment with vancomycin hydrochloride, recurrence of CDAD during the following four weeks occurred in 25 of 107 (23%) and 18 of 102 (18%) in Trial 1 and Trial 2, respectively.
Restriction Endonuclease Analysis (REA) was used to identify C. difficile baseline isolates in the BI group. In Trial 1, the vancomycin hydrochloride-treated subjects were classified at baseline as follows 31 (23%) with BI strain, 69 (52%) with non-BI strain, and 34 (25%) with unknown strain. Clinical success rates were 87% for BI strain, 81% for non-BI strain, and 76% for unknown strain. In subjects with diarrhea resolution at end-of-treatment with vancomycin hydrochloride, recurrence of CDAD during the following four weeks occurred in 7 of 26 subjects with BI strain, 12 of 56 subjects with non-BI strain, and 6 of 25 subjects with unknown strain.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Each FIRVANQ kit contains a bottle of vancomycin hydrochloride USP, as white to almost white or tan to brown powder for oral solution, and a bottle of pre-measured Grape-Flavored Diluent, in the strengths and volumes listed in Table 5.
Table 5: Vancomycin Strength, Diluent Volume and National Drug Code (NDC) Numbers Vancomycin Strength per Bottle Diluent for FIRVANQ™ NDC Numbers 3.75 g 147 mL 65628-204-05 7.5 g 295 mL 65628-205-10 7.5 g 145 mL 65628-206-05 15 g 289 mL 65628-208-10 Storage and Handling
Store FIRVANQ Kit prior and reconstituted solution at refrigerated conditions, 2°C to 8°C (36°F to 46°F).
Discard the reconstituted solution after 14 days, or if it appears hazy or contains particulates.
Do not freeze. Keep container tightly closed. Protect from light.
[ see Dosage and Administration (2.4)].
-
17 PATIENT COUNSELING INFORMATION
Antibacterial Resistance
Patients should be counseled that antibacterial drugs including FIRVANQ should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When FIRVANQ is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by FIRVANQ or other antibacterial drugs in the future.
Important Administration Instructions
Instruct the patient or caregiver to:
- Shake the reconstituted solutions of FIRVANQ well before each use and to use an oral dosing device that measures the appropriate volume of the oral solution in milliliters.
- Store the reconstituted solutions of FIRVANQ in the refrigerator (with temperatures set to between 2°C to 8°C (36°F to 46°F)) when not in use.
- Discard reconstituted solutions of FIRVANQ after 14 days, or if it appears hazy or contains particulates.
Manufactured for:
841 Woburn St.
Wilmington, MA 01887 USA -
PRINCIPAL DISPLAY PANEL
NDC: 65628-204-05
FIRVANQ
(vancomycin hydrochloride) for oral solution
For Oral Use Only
Vancomycin Hydrochloride Powder for Oral Solution Kit
Vancomycin 25 mg/mL in Grape Flavored Diluent
EACH KIT INCLUDES:
1 bottle containing Vancomycin Hydrochloride USP,
powder for oral solution, equivalent to 3.75 g Vancomycin
1 bottle containing 147 mL Grape Flavored Diluent
for reconstitution
When reconstituted, each mL contains 25 mg Vancomycin
Must be Refrigerated
150 mL
final volume after reconstitution
Rx Only
-
PRINCIPAL DISPLAY PANEL
NDC: 65628-205-10
FIRVANQ
(vancomycin hydrochloride) for oral solution
For Oral Use Only
Vancomycin Hydrochloride Powder for Oral Solution Kit
Vancomycin 25 mg/mL in Grape Flavored Diluent
EACH KIT INCLUDES:
1 bottle containing Vancomycin Hydrochloride USP,
powder for oral solution, equivalent to 7.5 g Vancomycin
1 bottle containing 295 mL Grape Flavored Diluent
for reconstitution
When reconstituted, each mL contains 25 mg Vancomycin
Must be Refrigerated
300 mL
final volume after reconstitution
Rx Only
-
PRINCIPAL DISPLAY PANEL
NDC: 65628-206-05
FIRVANQ
(vancomycin hydrochloride) for oral solution
For Oral Use Only
Vancomycin Hydrochloride Powder for Oral Solution Kit
Vancomycin 50 mg/mL in Grape Flavored Diluent
EACH KIT INCLUDES:
1 bottle containing Vancomycin Hydrochloride USP,
powder for oral solution, equivalent to 7.5 g Vancomycin
1 bottle containing 145 mL Grape Flavored Diluent
for reconstitution
When reconstituted, each mL contains 50 mg Vancomycin
Must be Refrigerated
150 mL
final volume after reconstitution
Rx Only
-
PRINCIPAL DISPLAY PANEL
NDC: 65628-208-10
FIRVANQ
(vancomycin hydrochloride) for oral solution
For Oral Use Only
Vancomycin Hydrochloride Powder for Oral Solution Kit
Vancomycin 50 mg/mL in Grape Flavored Diluent
EACH KIT INCLUDES:
1 bottle containing Vancomycin Hydrochloride USP,
powder for oral solution, equivalent to 15 g Vancomycin
1 bottle containing 289 mL Grape Flavored Diluent
for reconstitution
When reconstituted, each mL contains 50 mg Vancomycin
Must be Refrigerated
300 mL
final volume after reconstitution
Rx Only
-
INGREDIENTS AND APPEARANCE
FIRVANQ
vancomycin hydrochloride kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 65628-204 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 65628-204-05 1 in 1 CARTON; Type 0: Not a Combination Product 01/26/2018 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 BOTTLE 3.75 g Part 2 1 BOTTLE 147 mL Part 1 of 2 VANCOMYCIN HYDROCHLORIDE
vancomycin hydrochloride powder, for solutionProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength VANCOMYCIN HYDROCHLORIDE (UNII: 71WO621TJD) (VANCOMYCIN - UNII:6Q205EH1VU) VANCOMYCIN 1 g in 1 g Product Characteristics Color white (white to almost white or tan to brown) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 3.75 g in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208910 01/26/2018 Part 2 of 2 GRAPE FLAVORED DILUENT
grape flavored diluent solutionProduct Information Route of Administration ORAL Inactive Ingredients Ingredient Name Strength ANHYDROUS CITRIC ACID (UNII: XF417D3PSL) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) FD&C RED NO. 40 (UNII: WZB9127XOA) WATER (UNII: 059QF0KO0R) SODIUM BENZOATE (UNII: OJ245FE5EU) SUCRALOSE (UNII: 96K6UQ3ZD4) Product Characteristics Color Score Shape Size Flavor GRAPE Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 147 mL in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208910 01/26/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208910 01/26/2018 FIRVANQ
vancomycin hydrochloride kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 65628-205 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 65628-205-10 1 in 1 CARTON; Type 0: Not a Combination Product 01/26/2018 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 BOTTLE 7.5 g Part 2 1 BOTTLE 295 mL Part 1 of 2 VANCOMYCIN HYDROCHLORIDE
vancomycin hydrochloride powder, for solutionProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength VANCOMYCIN HYDROCHLORIDE (UNII: 71WO621TJD) (VANCOMYCIN - UNII:6Q205EH1VU) VANCOMYCIN 1 g in 1 g Product Characteristics Color white Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 7.5 g in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208910 01/26/2018 Part 2 of 2 GRAPE FLAVORED DILUENT
grape flavored diluent solutionProduct Information Route of Administration ORAL Inactive Ingredients Ingredient Name Strength ANHYDROUS CITRIC ACID (UNII: XF417D3PSL) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) FD&C RED NO. 40 (UNII: WZB9127XOA) WATER (UNII: 059QF0KO0R) SODIUM BENZOATE (UNII: OJ245FE5EU) SUCRALOSE (UNII: 96K6UQ3ZD4) Product Characteristics Color Score Shape Size Flavor GRAPE Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 295 mL in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208910 01/26/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208910 01/26/2018 FIRVANQ
vancomycin hydrochloride kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 65628-206 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 65628-206-05 1 in 1 CARTON; Type 0: Not a Combination Product 01/26/2018 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 BOTTLE 7.5 g Part 2 1 BOTTLE 145 mL Part 1 of 2 VANCOMYCIN HYDROCHLORIDE
vancomycin hydrochloride powder, for solutionProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength VANCOMYCIN HYDROCHLORIDE (UNII: 71WO621TJD) (VANCOMYCIN - UNII:6Q205EH1VU) VANCOMYCIN 1 g in 1 g Product Characteristics Color white Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 7.5 g in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208910 01/26/2018 Part 2 of 2 GRAPE FLAVORED DILUENT
grape flavored diluent solutionProduct Information Route of Administration ORAL Inactive Ingredients Ingredient Name Strength ANHYDROUS CITRIC ACID (UNII: XF417D3PSL) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) FD&C RED NO. 40 (UNII: WZB9127XOA) WATER (UNII: 059QF0KO0R) SODIUM BENZOATE (UNII: OJ245FE5EU) SUCRALOSE (UNII: 96K6UQ3ZD4) Product Characteristics Color Score Shape Size Flavor GRAPE Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 145 mL in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208910 01/26/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208910 01/26/2018 FIRVANQ
vancomycin hydrochloride kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 65628-208 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 65628-208-10 1 in 1 CARTON; Type 0: Not a Combination Product 01/26/2018 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 BOTTLE 15 g Part 2 1 BOTTLE 289 mL Part 1 of 2 VANCOMYCIN HYDROCHLORIDE
vancomycin hydrochloride powder, for solutionProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength VANCOMYCIN HYDROCHLORIDE (UNII: 71WO621TJD) (VANCOMYCIN - UNII:6Q205EH1VU) VANCOMYCIN 1 g in 1 g Product Characteristics Color white Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 15 g in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208910 01/26/2018 Part 2 of 2 GRAPE FLAVORED DILUENT
grape flavored diluent solutionProduct Information Route of Administration ORAL Inactive Ingredients Ingredient Name Strength ANHYDROUS CITRIC ACID (UNII: XF417D3PSL) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) FD&C RED NO. 40 (UNII: WZB9127XOA) WATER (UNII: 059QF0KO0R) SODIUM BENZOATE (UNII: OJ245FE5EU) SUCRALOSE (UNII: 96K6UQ3ZD4) Product Characteristics Color Score Shape Size Flavor GRAPE Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 289 mL in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208910 01/26/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208910 01/26/2018 Labeler - CutisPharma, Inc. (090598256)
Trademark Results [FIRVANQ]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 FIRVANQ 87715534 5651067 Live/Registered |
CutisPharma, Inc. 2017-12-11 |
 FIRVANQ 87451656 5525903 Live/Registered |
CutisPharma, Inc. 2017-05-16 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.