TAGRISSO- osimertinib tablet, film coated
TAGRISSO by
Drug Labeling and Warnings
TAGRISSO by is a Prescription medication manufactured, distributed, or labeled by AstraZeneca Pharmaceuticals LP, AstraZeneca PLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TAGRISSO safely and effectively. See full prescribing information for TAGRISSO.
TAGRISSO® (osimertinib) tablets, for oral use
Initial U.S. Approval: 2015INDICATIONS AND USAGE
TAGRISSO is a kinase inhibitor indicated for
- the first-line treatment of patients with metastatic NSCLC whose tumors have epidermal growth factor receptor (EGFR) exon 19 deletions or exon 21 L858R mutations, as detected by an FDA-approved test. (1.1, 2.1)
- the treatment of patients with metastatic EGFR T790M mutation-positive NSCLC, as detected by an FDA-approved test, whose disease has progressed on or after EGFR TKI therapy. (1.2, 2.1)
DOSAGE AND ADMINISTRATION
Recommended dosage: 80 mg orally once daily, with or without food. (2.2)
DOSAGE FORMS AND STRENGTHS
Tablets: 80 mg and 40 mg. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Interstitial Lung Disease (ILD)/Pneumonitis: Occurred in 3.9% of patients. Permanently discontinue TAGRISSO in patients diagnosed with ILD/Pneumonitis. (5.1)
- QTc Interval Prolongation: Monitor electrocardiograms and electrolytes in patients who have a history or predisposition for QTc prolongation, or those who are taking medications that are known to prolong the QTc interval. Withhold then restart at a reduced dose or permanently discontinue TAGRISSO. (2.4, 5.2)
- Cardiomyopathy: Occurred in 2.6% of patients. Conduct cardiac monitoring, including left ventricular ejection fraction (LVEF) assessment in patients with cardiac risk factors. (2.4, 5.3)
- Keratitis: Promptly refer patients with signs and symptoms of keratitis to an ophthalmologist for evaluation. (5.4)
- Erythema Multiforme and Stevens-Johnson Syndrome: Withhold TAGRISSO if erythema multiforme major (EMM) or Stevens-Johnson syndrome (SJS) is suspected and permanently discontinue if confirmed. (2.4, 5.5)
- Embryo-Fetal Toxicity: TAGRISSO can cause fetal harm. Advise females of potential risk to the fetus and to use effective contraception during treatment with TAGRISSO and for 6 weeks after final dose. Advise males to use effective contraception for 4 months, after the last dose of TAGRISSO. (5.6,8.1, 8.3)
ADVERSE REACTIONS
Most common adverse reactions (≥20%) were diarrhea, rash, dry skin, nail toxicity, stomatitis, fatigue and decreased appetite. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AstraZeneca at 1-800-236-9933 or www.TAGRISSO.com or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
Lactation: Do not breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 12/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 First-line Treatment of EGFR Mutation-Positive Metastatic Non-Small Cell Lung Cancer (NSCLC)
1.2 Previously Treated EGFR T790M Mutation-Positive Metastatic NSCLC
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
2.2 Recommended Dosage Regimen
2.3 Administration to Patients Who Have Difficulty Swallowing Solids
2.4 Dosage Modifications
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Interstitial Lung Disease/Pneumonitis
5.2 QTc Interval Prolongation
5.3 Cardiomyopathy
5.4 Keratitis
5.5 Erythema Multiforme and Stevens-Johnson Syndrome
5.6 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on Osimertinib
7.2 Effect of Osimertinib on Other Drugs
7.3 Drugs That Prolong the QTc Interval
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Previously Untreated EGFR Mutation-Positive Metastatic Non-Small Cell Lung Cancer
14.2 Previously Treated EGFR T790M Mutation-Positive Metastatic Non-Small Cell Lung Cancer
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 First-line Treatment of EGFR Mutation-Positive Metastatic Non-Small Cell Lung Cancer (NSCLC)
TAGRISSO is indicated for the first-line treatment of patients with metastatic non-small cell lung cancer (NSCLC) whose tumors have epidermal growth factor receptor (EGFR) exon 19 deletions or exon 21 L858R mutations, as detected by an FDA-approved test [see Dosage and Administration (2.1)].
1.2 Previously Treated EGFR T790M Mutation-Positive Metastatic NSCLC
TAGRISSO is indicated for the treatment of patients with metastatic EGFR T790M mutation-positive NSCLC, as detected by an FDA-approved test, whose disease has progressed on or after EGFR tyrosine kinase inhibitor (TKI) therapy [see Dosage and Administration (2.1)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients for the first-line treatment of metastatic EGFR-positive NSCLC with TAGRISSO based on the presence of EGFR exon 19 deletions or exon 21 L858R mutations in tumor or plasma specimens [see Clinical Studies (14)]. If these mutations are not detected in a plasma specimen, test tumor tissue if feasible.
Select patients for the treatment of metastatic EGFR T790M mutation-positive NSCLC with TAGRISSO following progression on or after EGFR TKI therapy based on the presence of an EGFR T790M mutation in tumor or plasma specimens [see Clinical Studies (14)]. Testing for the presence of the T790M mutation in plasma specimens is recommended only in patients for whom a tumor biopsy cannot be obtained. If this mutation is not detected in a plasma specimen, re-evaluate the feasibility of biopsy for tumor tissue testing.
Information on FDA-approved tests for the detection of EGFR mutations is available at http://www.fda.gov/companiondiagnostics.
2.2 Recommended Dosage Regimen
The recommended dosage of TAGRISSO is 80 mg tablet once a day until disease progression or unacceptable toxicity. TAGRISSO can be taken with or without food.
If a dose of TAGRISSO is missed, do not make up the missed dose and take the next dose as scheduled.
2.3 Administration to Patients Who Have Difficulty Swallowing Solids
Disperse tablet in 60 mL (2 ounces) of non-carbonated water only. Stir until tablet is dispersed into small pieces (the tablet will not completely dissolve) and swallow immediately. Do not crush, heat, or ultrasonicate during preparation. Rinse the container with 120 mL to 240 mL (4 to 8 ounces) of water and immediately drink.
If administration via nasogastric tube is required, disperse the tablet as above in 15 mL of non-carbonated water, and then use an additional 15 mL of water to transfer any residues to the syringe. The resulting 30 mL liquid should be administered as per the nasogastric tube instructions with appropriate water flushes (approximately 30 mL).
2.4 Dosage Modifications
Adverse Reactions
Table 1. Recommended Dosage Modifications for TAGRISSO - * Adverse reactions graded by the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0 (NCI CTCAE v4.0).
- † QTc = QT interval corrected for heart rate
- ‡ ECGs = Electrocardiograms
Target Organ
Adverse Reaction*
Dosage Modification
Pulmonary [see Warnings and Precautions (5.1)]
Interstitial lung disease (ILD)/Pneumonitis
Permanently discontinue TAGRISSO.
QTc† interval greater than 500 msec on at least 2 separate ECGs‡
Withhold TAGRISSO until QTc interval is less than 481 msec or recovery to baseline if baseline QTc is greater than or equal to 481 msec, then resume at 40 mg dose.
QTc interval prolongation with signs/symptoms of life-threatening arrhythmia
Permanently discontinue TAGRISSO.
Symptomatic congestive heart failure
Permanently discontinue TAGRISSO.
Cutaneous [see Warnings and Precautions (5.5)]
Stevens-Johnson syndrome (SJS), Erythema Multiforme Major (EMM)
Withhold TAGRISSO if suspected and permanently discontinue if confirmed.
Other [see Adverse Reactions (6.1)]
Adverse reaction of Grade 3 or greater severity
Withhold TAGRISSO for up to 3 weeks.
If improvement to Grade 0-2 within 3 weeks
Resume at 80 mg or 40 mg daily.
If no improvement within 3 weeks
Permanently discontinue TAGRISSO.
Drug Interactions
Strong CYP3A4 Inducers
If concurrent use is unavoidable, increase TAGRISSO dosage to 160 mg daily when co-administering with a strong CYP3A inducer. Resume TAGRISSO at 80 mg 3 weeks after discontinuation of the strong CYP3A4 inducer [see Drug Interactions (7) and Clinical Pharmacology (12.3)]. - 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Interstitial Lung Disease/Pneumonitis
Interstitial lung disease (ILD)/pneumonitis occurred in 3.9% of the 1142 TAGRISSO-treated patients; 0.4% of cases were fatal.
Withhold TAGRISSO and promptly investigate for ILD in patients who present with worsening of respiratory symptoms which may be indicative of ILD (e.g., dyspnea, cough and fever). Permanently discontinue TAGRISSO if ILD is confirmed [see Dosage and Administration (2.4) and Adverse Reactions (6)].
5.2 QTc Interval Prolongation
Heart rate-corrected QT (QTc) interval prolongation occurs in patients treated with TAGRISSO. Of the 1142 patients treated with TAGRISSO in clinical trials, 0.9% were found to have a QTc > 500 msec, and 3.6% of patients had an increase from baseline QTc > 60 msec [see Clinical Pharmacology (12.2)]. No QTc-related arrhythmias were reported.
Clinical trials of TAGRISSO did not enroll patients with baseline QTc of > 470 msec. Conduct periodic monitoring with ECGs and electrolytes in patients with congenital long QTc syndrome, congestive heart failure, electrolyte abnormalities, or those who are taking medications known to prolong the QTc interval. Permanently discontinue TAGRISSO in patients who develop QTc interval prolongation with signs/symptoms of life-threatening arrhythmia [see Dosage and Administration (2.4)].
5.3 Cardiomyopathy
Across clinical trials, cardiomyopathy (defined as cardiac failure, chronic cardiac failure, congestive heart failure, pulmonary edema or decreased ejection fraction) occurred in 2.6% of the 1142 TAGRISSO-treated patients; 0.1% of cardiomyopathy cases were fatal.
A decline in left ventricular ejection fraction (LVEF) ≥ 10% from baseline and to less than 50% LVEF occurred in 3.9% of 908 patients who had baseline and at least one follow-up LVEF assessment.
Conduct cardiac monitoring, including assessment of LVEF at baseline and during treatment, in patients with cardiac risk factors. Assess LVEF in patients who develop relevant cardiac signs or symptoms during treatment. For symptomatic congestive heart failure, permanently discontinue TAGRISSO [see Dosage and Administration (2.4)].
5.4 Keratitis
Keratitis was reported in 0.7% of 1142 patients treated with TAGRISSO in clinical trials. Promptly refer patients with signs and symptoms suggestive of keratitis (such as eye inflammation, lacrimation, light sensitivity, blurred vision, eye pain and/or red eye) to an ophthalmologist.
5.5 Erythema Multiforme and Stevens-Johnson Syndrome
Postmarketing cases consistent with Stevens-Johnson syndrome (SJS) and erythema multiforme major (EMM) have been reported in patients receiving TAGRISSO. Withhold TAGRISSO if SJS or EMM is suspected and permanently discontinue if confirmed.
5.6 Embryo-Fetal Toxicity
Based on data from animal studies and its mechanism of action, TAGRISSO can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, osimertinib caused post-implantation fetal loss when administered during early development at a dose exposure 1.5 times the exposure at the recommended clinical dose. When males were treated prior to mating with untreated females, there was an increase in preimplantation embryonic loss at plasma exposures of approximately 0.5 times those observed at the recommended dose of 80 mg once daily. Verify pregnancy status of females of reproductive potential prior to initiating TAGRISSO. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TAGRISSO and for 6 weeks after the final dose. Advise males with female partners of reproductive potential to use effective contraception for 4 months after the final dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
Interstitial Lung Disease/Pneumonitis [see Warnings and Precautions (5.1)]
QTc Interval Prolongation [see Warnings and Precautions (5.2)]
Cardiomyopathy [see Warnings and Precautions (5.3)]
Keratitis [see Warnings and Precautions (5.4)]
Erythema multiforme and Stevens-Johnson syndrome [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data in the Warnings and Precautions section reflect exposure to TAGRISSO in 1142 patients with EGFR mutation-positive NSCLC who received TAGRISSO at the recommended dose of 80 mg once daily in two randomized, active-controlled trials [FLAURA (n=279) and AURA3 (n=279)], two single arm trials [AURA Extension (n=201) and AURA2 (n=210)], and one dose-finding study, AURA1 (n=173) [see Warnings and Precautions (5)].
The data described below reflect exposure to TAGRISSO (80 mg daily) in 558 patients with EGFR mutation-positive, metastatic NSCLC in two randomized, active-controlled trials [FLAURA (n=279) and AURA3 (n=279)]. Patients with a history of interstitial lung disease, drug induced interstitial disease or radiation pneumonitis that required steroid treatment, serious arrhythmia or baseline QTc interval greater than 470 msec on electrocardiogram were excluded from enrollment in these studies.
Previously Untreated EGFR Mutation-Positive Metastatic Non-Small Cell Lung Cancer
The safety of TAGRISSO was evaluated in FLAURA, a multicenter international double-blind randomized (1:1) active controlled trial conducted in 556 patients with EGFR exon 19 deletion or exon 21 L858R mutation-positive, unresectable or metastatic NSCLC who had not received previous systemic treatment for advanced disease. The median duration of exposure to TAGRISSO was 16.2 months.
The most common adverse reactions (≥20%) in patients treated with TAGRISSO were diarrhea (58%), rash (58%), dry skin (36%), nail toxicity (35%), stomatitis (29%), and decreased appetite (20%). Serious adverse reactions were reported in 4% of patients treated with TAGRISSO; the most common serious adverse reactions (≥1%) were pneumonia (2.9%), ILD/pneumonitis (2.1%), and pulmonary embolism (1.8%). Dose reductions occurred in 2.9% of patients treated with TAGRISSO. The most frequent adverse reactions leading to dose reductions or interruptions were prolongation of the QT interval as assessed by ECG (4.3%), diarrhea (2.5%), and lymphopenia (1.1%). Adverse reactions leading to permanent discontinuation occurred in 13% of patients treated with TAGRISSO. The most frequent adverse reaction leading to discontinuation of TAGRISSO was ILD/pneumonitis (3.9%).
Tables 2 and 3 summarize common adverse reactions and laboratory abnormalities which occurred in FLAURA. FLAURA was not designed to demonstrate a statistically significant reduction in adverse reaction rates for TAGRISSO, or for the control arm, for any adverse reaction listed in Tables 2 and 3.
Table 2. Adverse Reactions Occurring in ≥10% of Patients Receiving TAGRISSO in FLAURA* - * NCI CTCAE v4.0
- † One grade 5 (fatal) event was reported (diarrhea) for EGFR TKI comparator
- ‡ Includes rash, rash generalized, rash erythematous, rash macular, rash maculo-papular, rash papular, rash pustular, rash pruritic, rash vesicular, rash follicular, erythema, folliculitis, acne, dermatitis, dermatitis acneiform, drug eruption, skin erosion.
- § Includes dry skin, skin fissures, xerosis, eczema, xeroderma.
- ¶ Includes nail bed disorder, nail bed inflammation, nail bed infection, nail discoloration, nail pigmentation, nail disorder, nail toxicity, nail dystrophy, nail infection, nail ridging, onychalgia, onychoclasis, onycholysis, onychomadesis, onychomalacia, paronychia.
- # Includes pruritus, pruritus generalized, eyelid pruritus.
- Þ The frequency of "Prolonged QT Interval" represents reported adverse events in the FLAURA study. Frequencies of QTc intervals of >500 ms or >60 ms are presented in Section 5.2.
- ß Includes fatigue, asthenia.
Adverse Reaction
TAGRISSO
(N=279)
EGFR TKI comparator
(gefitinib or erlotinib)
(N=277)
Any Grade
(%)
Grade 3 or higher (%)
Any Grade (%)
Grade 3 or higher (%)
Gastrointestinal Disorders
Diarrhea†
58
2.2
57
2.5
Stomatitis
29
0.7
20
0.4
Nausea
14
0
19
0
Constipation
15
0
13
0
Vomiting
11
0
11
1.4
Skin Disorders
Rash‡
58
1.1
78
6.9
Dry skin§
36
0.4
36
1.1
Nail toxicity¶
35
0.4
33
0.7
Pruritus#
17
0.4
17
0
Metabolism and Nutrition Disorders
Decreased appetite
20
2.5
19
1.8
Respiratory, Thoracic and Mediastinal Disorders
Cough
17
0
15
0.4
Dyspnea
13
0.4
7
1.4
Neurologic Disorders
Headache
12
0.4
7
0
Cardiac Disorders
Prolonged QT IntervalÞ
10
2.2
4
0.7
General Disorders and Administration Site Conditions
Fatigueß
21
1.4
15
1.4
Pyrexia
10
0
4
0.4
Infection and Infestation Disorders
Upper Respiratory Tract Infection
10
0
7
0
Table 3. Laboratory Abnormalities Worsening from Baseline in ≥ 20% of Patients in FLAURA - * NCI CTCAE v4.0
- † Each test incidence, except for hyperglycemia, is based on the number of patients who had both baseline and at least one on-study laboratory measurement available (TAGRISSO range: 267 - 273 and EGFR TKI comparator range: 256 - 268)
- ‡ Hyperglycemia is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: TAGRISSO (179) and EGFR comparator (191)
TAGRISSO
(N=279)
EGFR TKI comparator
(gefitinib or erlotinib)
(N=277)
Change from Baseline
All Grades (%)
Change from Baseline to
Grade 3 or Grade 4 (%)
Change from Baseline
All Grades
(%)
Change from Baseline to Grade 3
or Grade 4
(%)
Hematology
Lymphopenia
63
5.6
36
4.2
Anemia
59
0.7
47
0.4
Thrombocytopenia
51
0.7
12
0.4
Neutropenia
41
3.0
10
0
Chemistry
Hyperglycemia‡
37
0
31
0.5
Hypermagnesemia
30
0.7
11
0.4
Hyponatremia
26
1.1
27
1.5
Increased AST
22
1.1
43
4.1
Increased ALT
21
0.7
52
8
Hypokalemia
16
0.4
22
1.1
Hyperbilirubinemia
14
0
29
1.1
Previously Treated EGFR T790M Mutation-Positive Metastatic Non-Small Cell Lung Cancer
The safety of TAGRISSO was evaluated in AURA3, a multicenter international open label randomized (2:1) controlled trial conducted in 419 patients with unresectable or metastatic EGFR T790M mutation-positive NSCLC who had progressive disease following first line EGFR TKI treatment. A total of 279 patients received TAGRISSO 80 mg orally once daily until intolerance to therapy, disease progression, or investigator determination that the patient was no longer benefiting from treatment. A total of 136 patients received pemetrexed plus either carboplatin or cisplatin every three weeks for up to 6 cycles; patients without disease progression after 4 cycles of chemotherapy could continue maintenance pemetrexed until disease progression, unacceptable toxicity, or investigator determination that the patient was no longer benefiting from treatment. Left Ventricular Ejection Fraction (LVEF) was evaluated at screening and every 12 weeks. The median duration of treatment was 8.1 months for patients treated with TAGRISSO and 4.2 months for chemotherapy-treated patients. The trial population characteristics were: median age 62 years, age less than 65 (58%), female (64%), Asian (65%), never smokers (68%), and ECOG PS 0 or 1 (100%).
The most common adverse reactions (≥20%) in patients treated with TAGRISSO were diarrhea (41%), rash (34%), dry skin (23%), nail toxicity (22%), and fatigue (22%). Serious adverse reactions were reported in 18% of patients treated with TAGRISSO and 26% in the chemotherapy group. No single serious adverse reaction was reported in 2% or more patients treated with TAGRISSO. One patient (0.4%) treated with TAGRISSO experienced a fatal adverse reaction (ILD/pneumonitis).
Dose reductions occurred in 2.9% of patients treated with TAGRISSO. The most frequent adverse reactions leading to dose reductions or interruptions were prolongation of the QT interval as assessed by ECG (1.8%), neutropenia (1.1%), and diarrhea (1.1%). Adverse reactions resulting in permanent discontinuation of TAGRISSO occurred in 7% of patients treated with TAGRISSO. The most frequent adverse reaction leading to discontinuation of TAGRISSO was ILD/pneumonitis (3%).
Tables 4 and 5 summarize common adverse reactions and laboratory abnormalities which occurred in TAGRISSO-treated patients in AURA3. AURA3 was not designed to demonstrate a statistically significant reduction in adverse reaction rates for TAGRISSO, or for the control arm, for any adverse reaction listed in Tables 4 and 5.
Table 4. Adverse Reactions Occurring in ≥10% of Patients Receiving TAGRISSO in AURA3* - * NCI CTCAE v4.0.
- † No grade 4 events were reported.
- ‡ Includes rash, rash generalized, rash erythematous, rash macular, rash maculo-papular, rash papular, rash pustular, erythema, folliculitis, acne, dermatitis and acneform dermatitis.
- § Includes dry skin, eczema, skin fissures, xerosis.
- ¶ Includes nail disorders, nail bed disorders, nail bed inflammation, nail bed tenderness, nail discoloration, nail disorder, nail dystrophy, nail infection, nail ridging, nail toxicity, onychoclasis, onycholysis, onychomadesis, paronychia.
- # Includes pruritus, pruritus generalized, eyelid pruritus.
- Þ Includes fatigue, asthenia.
Adverse Reaction
TAGRISSO
(N=279)
Chemotherapy
(Pemetrexed/Cisplatin or
Pemetrexed/Carboplatin)
(N=136)
All Grades†
(%)
Grade 3/4†
(%)
All Grades†
(%)
Grade 3/4†
(%)
Gastrointestinal Disorders
Diarrhea
41
1.1
11
1.5
Nausea
16
0.7
49
3.7
Stomatitis
15
0
15
1.5
Constipation
14
0
35
0
Vomiting
11
0.4
20
2.2
Skin Disorders
Rash‡
34
0.7
5.9
0
Dry skin§
23
0
4.4
0
Nail toxicity¶
22
0
1.5
0
Pruritus#
13
0
5.1
0
Metabolism and Nutrition Disorders
Decreased appetite
18
1.1
36
2.9
Respiratory, Thoracic and Mediastinal Disorders
Cough
17
0
14
0
Musculoskeletal and Connective Tissue Disorders
Back pain
10
0.4
9
0.7
General Disorders and Administration Site Conditions
FatigueÞ
22
1.8
40
5.1
Table 5. Laboratory Abnormalities Worsening from Baseline in ≥20% of Patients in AURA3 - * NCI CTCAE v4.0
- † Each test incidence, except for hyperglycemia, is based on the number of patients who had both baseline and at least one on-study laboratory measurement available (TAGRISSO 279, chemotherapy 131)
- ‡ Hyperglycemia is based on the number of patients who had both baseline and at least one on-study laboratory measurement available (TAGRISSO 270, Chemotherapy 5; fasting glucose was not a protocol requirement for patients in the chemotherapy arm)
TAGRISSO
(N=279)
Chemotherapy (Pemetrexed/Cisplatin or Pemetrexed/Carboplatin)
(N=131)
Change from Baseline All Grades
(%)
Change from Baseline to Grade 3 or Grade 4
(%)
Change from Baseline
All Grades
(%)
Change from Baseline to Grade 3 or Grade 4
(%)
Hematology
Anemia
43
0
79
3.1
Lymphopenia
63
8.2
61
9.9
Thrombocytopenia
46
0.7
48
7.4
Neutropenia
27
2.2
49
12
Chemistry
Hypermagnesemia†
27
1.8
9.2
1.5
Hyponatremia†
26
2.2
36
1.5
Hyperglycemia‡
20
0
NA
NA
Hypokalemia†
9.0
1.4
18
1.5
NA=not applicable
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of TAGRISSO. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cutaneous:Stevens-Johnson syndrome, erythema multiforme
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on Osimertinib
Strong CYP3A Inducers
Co-administering TAGRISSO with a strong CYP3A4 inducer decreased the exposure of osimertinib compared to administering TAGRISSO alone [see Clinical Pharmacology (12.3)]. Decreased osimertinib exposure may lead to reduced efficacy.
Avoid co-administering TAGRISSO with strong CYP3A inducers. Increase the TAGRISSO dosage when co-administering with a strong CYP3A4 inducer if concurrent use is unavoidable [see Dosage and Administration (2.4)]. No dose adjustments are required when TAGRISSO is used with moderate and/or weak CYP3A inducers.
7.2 Effect of Osimertinib on Other Drugs
Co-administering TAGRISSO with a breast cancer resistant protein (BCRP) or P-glycoprotein (P-gp) substrate increased the exposure of the substrate compared to administering it alone [see Clinical Pharmacology (12.3)]. Increased BCRP or P-gp substrate exposure may increase the risk of exposure-related toxicity.
Monitor for adverse reactions of the BCRP or P-gp substrate, unless otherwise instructed in its approved labeling, when co-administered with TAGRISSO.
7.3 Drugs That Prolong the QTc Interval
The effect of co-administering medicinal products known to prolong the QTc interval with TAGRISSO is unknown. When feasible, avoid concomitant administration of drugs known to prolong the QTc interval with known risk of Torsades de pointes. If not feasible to avoid concomitant administration of such drugs, conduct periodic ECG monitoring [see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on data from animal studies and its mechanism of action [see Clinical Pharmacology (12.1)], TAGRISSO can cause fetal harm when administered to a pregnant woman. There are no available data on TAGRISSO use in pregnant women. Administration of osimertinib to pregnant rats was associated with embryolethality and reduced fetal growth at plasma exposures 1.5 times the exposure at the recommended clinical dose (see Data). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically-recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
When administered to pregnant rats prior to embryonic implantation through the end of organogenesis (gestation days 2-20) at a dose of 20 mg/kg/day, which produced plasma exposures of approximately 1.5 times the clinical exposure, osimertinib caused post-implantation loss and early embryonic death. When administered to pregnant rats from implantation through the closure of the hard palate (gestation days 6 to 16) at doses of 1 mg/kg/day and above (0.1 times the AUC observed at the recommended clinical dose of 80 mg once daily), an equivocal increase in the rate of fetal malformations and variations was observed in treated litters relative to those of concurrent controls. When administered to pregnant dams at doses of 30 mg/kg/day during organogenesis through lactation Day 6, osimertinib caused an increase in total litter loss and postnatal death. At a dose of 20 mg/kg/day, osimertinib administration during the same period resulted in increased postnatal death as well as a slight reduction in mean pup weight at birth that increased in magnitude between lactation days 4 and 6.
8.2 Lactation
Risk Summary
There are no data on the presence of osimertinib or its active metabolites in human milk, the effects of osimertinib on the breastfed infant or on milk production. Administration to rats during gestation and early lactation was associated with adverse effects, including reduced growth rates and neonatal death [see Use in Specific Populations (8.1)]. Because of the potential for serious adverse reactions in breastfed infants from osimertinib, advise women not to breastfeed during treatment with TAGRISSO and for 2 weeks after the final dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating TAGRISSO.
Contraception
TAGRISSO can cause fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
Females
Advise females of reproductive potential to use effective contraception during treatment with TAGRISSO and for 6 weeks after the final dose [see Use in Specific Populations (8.1)].
Males
Advise male patients with female partners of reproductive potential to use effective contraception during and for 4 months following the final dose of TAGRISSO [see Nonclinical Toxicology (13.1)].
Infertility
Based on animal studies, TAGRISSO may impair fertility in females and males of reproductive potential. The effects on female fertility showed a trend toward reversibility. It is not known whether the effects on male fertility are reversible [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of TAGRISSO in pediatric patients have not been established.
8.5 Geriatric Use
Forty-three percent (43%) of the 1142 patients in FLAURA (n=279), AURA3 (n=279), AURA Extension (n=201), AURA2 (n=210), and AURA1, (n=173) were 65 years of age and older. No overall differences in effectiveness were observed based on age. Exploratory analysis suggests a higher incidence of Grade 3 and 4 adverse reactions (13.4% versus 9.3%) and more frequent dose modifications for adverse reactions (13.4% versus 7.6%) in patients 65 years or older as compared to those younger than 65 years.
8.6 Renal Impairment
No dose adjustment is recommended in patients with creatinine clearance (CLcr) 15 - 89 mL/min, as estimated by Cockcroft-Gault. There is no recommended dose of TAGRISSO for patients with end-stage renal disease (CLcr < 15 mL/min) [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dose adjustment is recommended in patients with mild to moderate hepatic impairment (Child-Pugh A and B or total bilirubin ≤ ULN and AST > ULN or total bilirubin 1 to 3 times ULN and any AST).There is no recommended dose for TAGRISSO for patients with severe hepatic impairment (total bilirubin between 3 to 10 times ULN and any AST) [see Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
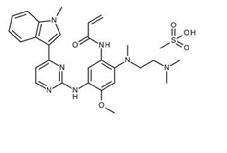
Osimertinib is a kinase inhibitor for oral use. The molecular formula for osimertinib mesylate is C28H33N7O2CH4O3S, and the molecular weight is 596 g/mol. The chemical name is N-(2-{2-dimethylaminoethyl-methylamino}-4-methoxy-5-{[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino}phenyl)prop-2-enamide mesylate salt. Osimertinib has the following structural formula (as osimertinib mesylate):
TAGRISSO tablets contain 40 or 80 mg of osimertinib, equivalent to 47.7 and 95.4 mg of osimertinib mesylate, respectively. Inactive ingredients in the tablet core are mannitol, microcrystalline cellulose, low-substituted hydroxpropyl cellulose and sodium stearyl fumarate. The tablet coating consists of polyvinyl alcohol, titanium dioxide, macrogol 3350, talc, ferric oxide yellow, ferric oxide red and ferric oxide black.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Osimertinib is a kinase inhibitor of the epidermal growth factor receptor (EGFR), which binds irreversibly to certain mutant forms of EGFR (T790M, L858R, and exon 19 deletion) at approximately 9-fold lower concentrations than wild-type. Two pharmacologically-active metabolites (AZ7550 and AZ5104 circulating at approximately 10% of the parent) with similar inhibitory profiles to osimertinib have been identified in the plasma after oral administration of osimertinib. AZ7550 showed a similar potency to osimertinib, while AZ5104 showed greater potency against exon 19 deletion and T790M mutants (approximately 8-fold) and wild-type (approximately 15-fold) EGFR. In vitro, osimertinib also inhibited the activity of HER2, HER3, HER4, ACK1, and BLK at clinically relevant concentrations.
In cultured cells and animal tumor implantation models, osimertinib exhibited anti-tumor activity against NSCLC lines harboring EGFR-mutations (T790M/L858R, L858R, T790M/exon 19 deletion, and exon 19 deletion) and, to a lesser extent, wild-type EGFR amplifications. Osimertinib distributed to the brain in multiple animal species (monkey, rat, and mouse) with brain to plasma AUC ratios of approximately 2 following oral dosing. These data are consistent with observations of tumor regression and increased survival in osimertinib- versus control-treated animals in a pre-clinical mutant-EGFR intracranial mouse metastasis xenograft model (PC9; exon 19 deletion).
12.2 Pharmacodynamics
Based on an analysis of dose-exposure response relationships over the dose range of 20 mg (0.25 times the recommended dose) to 240 mg (3 times the recommended dose), no apparent relationship between osimertinib exposure and overall response rate, duration of response and progression-free survival was identified; however, there were limited data available at the 20 mg dose. Over the same dose range, increased exposure led to increased probability of adverse reactions, specifically rash, diarrhea and ILD.
Cardiac Electrophysiology
The QTc interval prolongation potential of osimertinib was assessed in 210 patients who received TAGRISSO 80 mg daily in AURA2. A central tendency analysis of the QTcF data at steady-state demonstrated that the maximum mean change from baseline was 16.2 msec (upper bound of two-sided 90% confidence interval (CI) 17.6 msec). A pharmacokinetic/pharmacodynamic analysis in AURA2 suggested a concentration-dependent QTc interval prolongation of 14 msec (upper bound of two-sided 90% CI: 16 msec) at a dose of TAGRISSO 80 mg.
12.3 Pharmacokinetics
The area under the plasma concentration-time curve (AUC) and maximal plasma concentration (Cmax) of osimertinib increased dose proportionally over 20 to 240 mg dose range (i.e., 0.25 to 3 times the recommended dosage) after oral administration and exhibited linear pharmacokinetics (PK). Administration of TAGRISSO orally once daily resulted in approximately 3-fold accumulation with steady-state exposures achieved after 15 days of dosing. At steady state, the Cmax to Cmin (minimal concentration) ratio was 1.6-fold.
Absorption
The median time to Cmax of osimertinib was 6 hours (range 3-24 hours).
Following administration of a 20 mg TAGRISSO tablet with a high-fat, high-calorie meal (containing approximately 58 grams of fat and 1000 calories), the Cmax and AUC of osimertinib were comparable to that under fasting conditions.
Distribution
The mean volume of distribution at steady-state (Vss/F) of osimertinib was 918 L. Plasma protein binding of osimertinib was 95%.
Elimination
Osimertinib plasma concentrations decreased with time and a population estimated mean half-life of osimertinib was 48 hours, and oral clearance (CL/F) was 14.3 (L/h).
Metabolism
The main metabolic pathways of osimertinib were oxidation (predominantly CYP3A) and dealkylation in vitro. Two pharmacologically active metabolites (AZ7550 and AZ5104) have been identified in the plasma after TAGRISSO oral administration. The geometric mean exposure (AUC) of each metabolite (AZ5104 and AZ7550) was approximately 10% of the exposure of osimertinib at steady-state.
Excretion
Osimertinib is primarily eliminated in the feces (68%) and to a lesser extent in the urine (14%). Unchanged osimertinib accounted for approximately 2% of the elimination.
Specific Populations
No clinically significant differences in the pharmacokinetics of osimertinib were observed based on age, sex, ethnicity, body weight, baseline albumin, line of therapy, smoking status, renal function (creatinine clearance (CLcr) ≥15 mL/min by Cockcroft-Gault), or hepatic impairment (Child-Pugh A and B, or total bilirubin ≤ ULN and AST > ULN or total bilirubin between 1 to 3 times ULN and any AST). The pharmacokinetics of osimertinib in patients with end-stage renal disease (CLcr < 15 mL/min) or severe hepatic impairment (total bilirubin 3 to 10 times ULN and any AST) are unknown [see Use in Specific Populations (8.6) and (8.7)].
Drug Interaction Studies
Effect of Other Drugs on TAGRISSO in Clinical Pharmacokinetic Studies
Strong CYP3A Inducers: The steady-state AUC of osimertinib was reduced by 78% in patients when co-administered with rifampin (600 mg daily for 21 days) [see Drug Interactions (7.1)].
Strong CYP3A Inhibitors: Co-administering TAGRISSO with 200 mg itraconazole twice daily (a strong CYP3A4 inhibitor) had no clinically significant effect on the exposure of osimertinib (AUC increased by 24% and Cmax decreased by 20%).
Gastric Acid Reducing Agents: The exposure of osimertinib was not affected by concurrent administration of a single 80 mg TAGRISSO tablet following 40 mg omeprazole administration for 5 days.
Effect of Osimertinib on Other Drugs in Clinical Pharmacokinetic Studies
BCRP substrates: Co-administering TAGRISSO with rosuvastatin (a BCRP substrate) increased rosuvastatin AUC by 35% and Cmax by 72% [see Drug Interactions (7.2)].
P-gp substrates: Co-administering TAGRISSO with fexofenadine (a P-gp substrate) increased fexofenadine AUC and Cmax by 56% and 76% after a single dose and 27% and 25% at steady state, respectively.
CYP3A4 substrates: Co-administering TAGRISSO with simvastatin (a CYP3A4 substrate) had no clinically significant effect on the exposure of simvastatin.
In Vitro Studies
CYP450 Metabolic Pathways: Osimertinib does not inhibit CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6 and 2E1. Osimertinib induced CYP1A2 enzymes.
Transporter Systems: Osimertinib is a substrate of P-glycoprotein and BCRP and is not a substrate of OATP1B1 and OATP1B3. Osimertinib is an inhibitor of BCRP and does not inhibit OAT1, OAT3, OATP1B1, OATP1B3, MATE1, MATE2K and OCT2.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been performed with osimertinib. Osimertinib did not cause genetic damage in in vitro and in vivo assays.
Based on studies in animals, male fertility may be impaired by treatment with TAGRISSO. Degenerative changes were present in the testes in rats and dogs exposed to osimertinib for 1 month or more with evidence of reversibility in the rat. Following administration of osimertinib to rats for approximately 10 weeks at a dose of 40 mg/kg, at exposures 0.5 times the AUC observed at the recommended clinical dose of 80 mg once daily, there was a reduction in male fertility, demonstrated by increased pre-implantation loss in untreated females mated to treated males.
Based on studies in animals, female fertility may be impaired by treatment with TAGRISSO. In repeat dose toxicity studies, histological evidence of anestrus, corpora lutea degeneration in the ovaries and epithelial thinning in the uterus and vagina were seen in rats exposed to osimertinib for 1 month or more at exposures 0.3 times the AUC observed at the recommended clinical dose of 80 mg once daily. Findings in the ovaries seen following 1 month of dosing exhibited evidence of reversibility. In a female fertility study in rats, administration of osimertinib from 2 weeks prior to mating through Day 8 of gestation at a dose of 20 mg/kg/day (approximately 1.5 times the Cmax at the recommended dose of 80 mg once daily) had no effects on oestrus cycling or the number of females becoming pregnant, but caused early embryonic deaths. These findings showed evidence of reversibility when females were mated 1 month after treatment discontinuation.
-
14 CLINICAL STUDIES
14.1 Previously Untreated EGFR Mutation-Positive Metastatic Non-Small Cell Lung Cancer
The efficacy of TAGRISSO was demonstrated in a randomized, multicenter, double-blind, active-controlled trial (FLAURA [NCT02296125]) in patients with EGFR exon 19 deletion or exon 21 L858R mutation-positive, metastatic NSCLC, who had not received previous systemic treatment for metastatic disease. Patients were required to have measurable disease per RECIST v1.1, a WHO performance status of 0-1, and EGFR exon 19 deletion or exon 21 L858R mutation in tumor prospectively identified by the cobas® EGFR Mutation Test in a central laboratory or by an investigational assay at a CLIA-certified or accredited laboratory. Patients with CNS metastases not requiring steroids and with stable neurologic status for at least two weeks after completion of definitive surgery or radiotherapy were eligible. Patients were assessed at the investigator’s discretion for CNS metastases if they had a history of, or suspected, CNS metastases at study entry.
Patients were randomized (1:1) to receive TAGRISSO 80 mg orally once daily or to receive gefitinib 250 mg orally once daily or erlotinib 150 mg orally once daily until disease progression or unacceptable toxicity. Randomization was stratified by EGFR mutation type (exon 19 deletion or exon 21 L858R mutation) and ethnicity (Asian or non-Asian). Patients randomized to the control arm were offered TAGRISSO at the time of disease progression if tumor samples tested positive for the EGFR T790M mutation. The major efficacy outcome measure was progression-free survival (PFS), as assessed by investigator. Additional efficacy outcome measures included overall survival (OS) and overall response rate (ORR).
A total of 556 patients were randomized to TAGRISSO (n=279) or to control (gefitinib n=183; erlotinib n=94). The median age was 64 years (range 26-93 years); 54% were < 65 years of age; 63% were female; 62% were Asian and 64% were never smokers. Baseline WHO performance status was 0 (41%) or 1 (59%); 5% had Stage IIIb and 95% had Stage IV; and 7% received prior systemic cytotoxic chemotherapy as neoadjuvant or adjuvant therapy. With regard to EGFR tumor testing, 63% were exon 19 deletion and 37% were exon 21 L858R; 5 patients (<1%) also had a concomitant de novo T790M mutation. EGFR mutation status was confirmed centrally using the cobas EGFR Mutation Test in 90% of patients. Of those randomized to investigator’s choice of erlotinib or gefitinib, 55 patients (20%) received TAGRISSO as the next line of antineoplastic therapy.
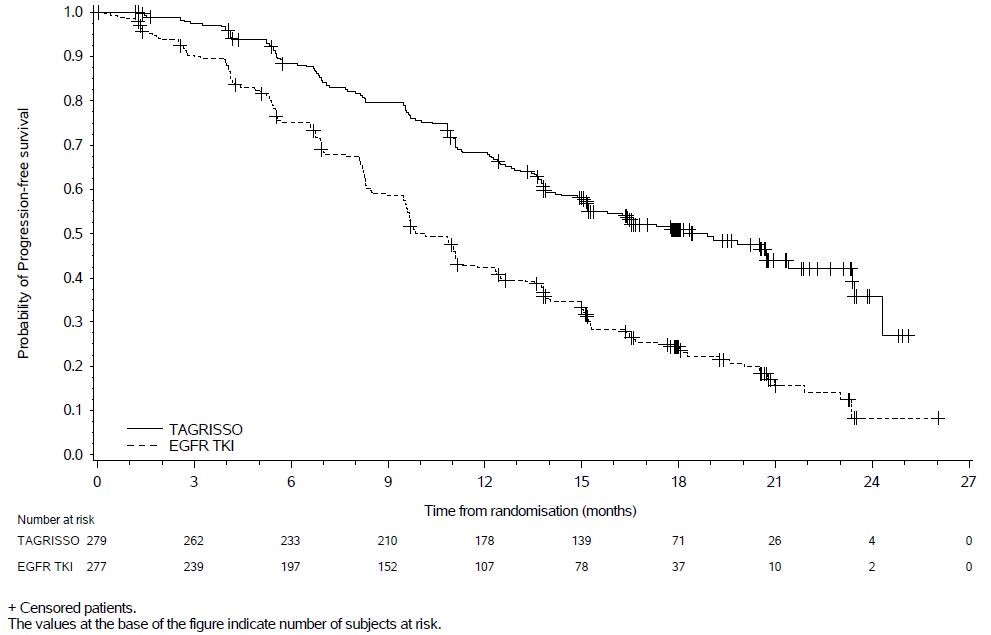
FLAURA demonstrated a statistically significant improvement in PFS for patients randomized to TAGRISSO as compared to erlotinib or gefitinib (see Table 6 and Figure 1). Overall survival data were not mature at the time of the final PFS analysis.
Table 6. Efficacy Results in FLAURA according to Investigator Assessment Efficacy Parameter TAGRISSO
(N=279)EGFR TKI
(gefitinib or erlotinib)
(N=277)- * Without documented radiological disease progression
- † Stratified by ethnicity (Asian vs. non-Asian) and mutation status (Ex19del vs. L858R)
- ‡ Pike estimator
- § Stratified log-rank test
- ¶ Confirmed responses
Progression-Free Survival (PFS)
PFS events (%)
136 (49)
206 (74)
Progressive disease (%)
125 (45)
192 (69)
Death* (%)
11 (4)
14 (5)
Median PFS in months (95% CI)
18.9 (15.2, 21.4)
10.2 (9.6, 11.1)
0.46 (0.37, 0.57)
P < 0.0001
Overall Response Rate (ORR)¶
ORR, % (95% CI)†
77 (71, 82)
69 (63, 74)
Complete response, %
2%
1%
Partial response, %
75%
68%
Duration of Response (DoR)¶
Median in months (95% CI)
17.6 (13.8, 22.0)
9.6 (8.3, 11.1)
Figure 1. Kaplan-Meier Curves of PFS by Investigator Assessment in FLAURA
In a supportive analysis of PFS according to blinded independent central review, median PFS was 17.7 months in the TAGRISSO arm compared to 9.7 months in the EGFR TKI comparator arm (HR=0.45; 95% CI: 0.36, 0.57).
Of 556 patients, 200 patients (36%) had baseline brain scans reviewed by BICR; this included 106 patients in the TAGRISSO arm and 94 patients in the investigator choice of EGFR TKI arm. Of these 200 patients, 41 had measurable CNS lesions per RECIST v1.1. Results of pre-specified exploratory analyses of CNS ORR and DoR by BICR in the subset of patients with measurable CNS lesions at baseline are summarized in Table 7.
Table 7. CNS ORR and DOR by BICR in Patients with Measurable CNS Lesions at Baseline in FLAURA - * According to RECIST v1.1.
- † Based on confirmed response.
- ‡ Based on patients with response only; DoR defined as the time from the date of first documented response (complete response or partial response) until progression or death event.
TAGRISSO
N=22
EGFR TKI
(gefitinib or erlotinib)
N=19
CNS ORR, % (95% CI)
77% (55, 92)
63% (38, 84)
Complete response
18%
0%
Duration of CNS Response‡
Number of responders
17
12
Response Duration ≥6 months
88%
50%
Response Duration ≥12 months
47%
33%
14.2 Previously Treated EGFR T790M Mutation-Positive Metastatic Non-Small Cell Lung Cancer
The efficacy of TAGRISSO was demonstrated in a randomized, multicenter open-label, active-controlled trial in patients with metastatic EGFR T790M mutation-positive NSCLC who had progressed on prior systemic therapy, including an EGFR TKI (AURA3). All patients were required to have EGFR T790M mutation-positive NSCLC identified by the cobas® EGFR Mutation Test performed in a central laboratory prior to randomization.
A total of 419 patients were randomized 2:1 to receive TAGRISSO (n=279) or platinum-based doublet chemotherapy (n=140). Randomization was stratified by ethnicity (Asian vs. non-Asian). Patients in the TAGRISSO arm received TAGRISSO 80 mg orally once daily until intolerance to therapy, disease progression, or investigator determination that the patient was no longer benefiting from treatment. Patients in the chemotherapy arm received pemetrexed 500 mg/m2 with carboplatin AUC5 or pemetrexed 500mg/m2 with cisplatin 75 mg/m2 on Day 1 of every 21-day cycle for up to 6 cycles. Patients whose disease had not progressed after four cycles of platinum-based chemotherapy could have received pemetrexed maintenance therapy (pemetrexed 500 mg/m2 on Day 1 of every 21-day cycle).
The major efficacy outcome measure was progression-free survival (PFS) according to Response Evaluation Criteria in Solid Tumors (RECIST v1.1) by investigator assessment. Additional efficacy outcome measures included overall response rate (ORR), duration of response (DoR), and overall survival (OS). Patients randomized to the chemotherapy arm who had radiological progression according to both investigator and blinded independent central review (BICR) were permitted to cross over to receive treatment with TAGRISSO.
The baseline demographic and disease characteristics of the overall trial population were: median age 62 years (range: 20-90 years), ≥75 years old (15%), female (64%), White (32%), Asian (65%), never smoker (68%), WHO performance status 0 or 1 (100%). Fifty-four percent (54%) of patients had extra-thoracic visceral metastases, including 34% with central nervous system (CNS) metastases (including 11% with measurable CNS metastases) and 23% with liver metastases. Forty-two percent (42%) of patients had metastatic bone disease.
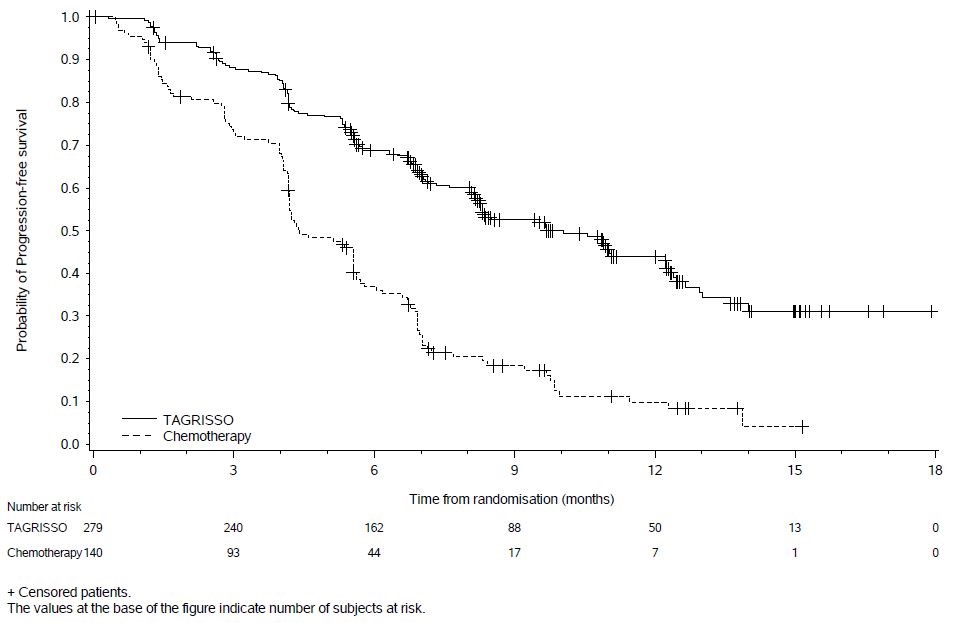
In AURA3, there was a statistically significant improvement in PFS in the patients randomized to TAGRISSO compared to chemotherapy (see Table 8 and Figure 2). Overall survival data were not mature at the time of the PFS analysis.
Table 8. Efficacy Results According to Investigator Assessment in AURA3 Efficacy Parameter TAGRISSO
(N=279)Chemotherapy
(N=140)- * Without documented radiological disease progression
- † Stratified by ethnicity (Asian vs. non-Asian)
- ‡ Pike estimator
- § Stratified log-rank test
- ¶ Logistic regression analysis
Progression-Free Survival
Number of events (%)
140 (50)
110 (79)
Progressive disease
129 (46)
104 (74)
Death*
11 (4)
6 (4)
Median PFS in months (95% CI)
10.1 (8.3, 12.3)
4.4 (4.2, 5.6)
0.30 (0.23,0.41)
<0.001
Overall Response Rate
Overall Response Rate
65%
29%
(95% CI)
(59%, 70%)
(21%, 37%)
Complete response
1%
1%
Partial response
63%
27%
<0.001
Duration of Response (DoR)
Median Duration of Response in months (95% CI)
11.0 (8.6, 12.6)
4.2 (3.0, 5.9)
Figure 2. Kaplan-Meier Curves of PFS by Investigator Assessment in AURA3
In a supportive analysis of PFS according to blinded independent central review, median PFS was 11 months in the TAGRISSO arm compared to 4.2 months in the chemotherapy arm (HR 0.28; 95% CI: 0.20, 0.38).
Of 419 patients, 205 (49%) had baseline brain scans reviewed by BICR; this included 134 (48%) patients in the TAGRISSO arm and 71 (51%) patients in the chemotherapy arm. Assessment of CNS efficacy by RECIST v1.1 was performed in the subgroup of 46/419 (11%) patients identified by BICR to have measurable CNS lesions on a baseline brain scan. Results are summarized in Table 9.
Table 9. CNS ORR and DoR by BICR in Patients with Measurable CNS Lesions at Baseline in AURA3 - * According to RECIST v1.1.
- † Based on confirmed response.
- ‡ Based on patients with response only; DoR defined as the time from the date of first documented response (complete response or partial response) until progression or death event.
TAGRISSO
N=30
Chemotherapy
N=16
CNS ORR, % (95% CI)
57% (37, 75)
25% (7, 52)
Complete response
7%
0
Number of responders
17
4
Response Duration ≥ 6 months
47%
0
Response Duration ≥ 9 months
12%
0
-
16 HOW SUPPLIED/STORAGE AND HANDLING
80 mg tablets: beige, oval and biconvex tablet marked with “AZ 80” on one side and plain on the reverse and are available in bottles of 30 (NDC: 0310-1350-30).
40 mg tablets: beige, round and biconvex tablet marked with “AZ 40” on one side and plain on the reverse and are available in bottles of 30 (NDC: 0310-1349-30).
Store TAGRISSO bottles at 25°C (77°F). Excursions permitted to 15-30°C (59-86°F) [see USP Controlled Room Temperature].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Interstitial Lung Disease/Pneumonitis
- Inform patients of the risks of severe or fatal ILD, including pneumonitis. Advise patients to contact their healthcare provider immediately to report new or worsening respiratory symptoms [see Warnings and Precautions (5.1)].
QTc Interval Prolongation
- Inform patients of symptoms that may be indicative of significant QTc prolongation including dizziness, lightheadedness, and syncope. Advise patients to report these symptoms and to inform their physician about the use of any heart or blood pressure medications [see Warnings and Precautions (5.2)].
Cardiomyopathy
- Inform patients that TAGRISSO can cause cardiomyopathy. Advise patients to immediately report any signs or symptoms of heart failure to their healthcare provider [see Warnings and Precautions (5.3)].
Keratitis
- Advise patients to contact their healthcare provider immediately if they develop eye symptoms (eye inflammation, lacrimation, light sensitivity, eye pain, red eye or changes in vision) [see Warnings and Precautions (5.4)].
- Erythema multiforme and Stevens-Johnson syndrome
- Inform patients of signs and symptoms that may be indicative of EM or SJS. Advise patients to contact their healthcare provider immediately if they develop target lesions or severe blistering or peeling of skin. [see Warnings and Precautions (5.5)].
Embryo-Fetal Toxicity
- Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females to inform their healthcare provider if they become pregnant or if pregnancy is suspected, while taking TAGRISSO [see Warnings and Precautions (5.6) and Use in Specific Populations (8.1)].
Females and Males of Reproductive Potential
- Advise females of reproductive potential to use effective contraception during treatment with TAGRISSO and for 6 weeks after the final dose [see Use in Specific Populations (8.3)].
- Advise males to use effective contraception during treatment and for 4 months after the final dose of TAGRISSO [see Use in Specific Populations (8.3)].
Lactation
- Advise women not to breastfeed during treatment with TAGRISSO and for 2 weeks after the final dose [see Use in Specific Populations (8.2)].
Distributed by:
AstraZeneca Pharmaceuticals LP
Wilmington, DE 19850TAGRISSO is a registered trademark of the AstraZeneca group of companies.
©AstraZeneca 2019
-
PATIENT PACKAGE INSERT
Patient Information
TAGRISSO® (tuh-GRISS-oh)
(osimertinib)
tabletsWhat is the most important information I should know about TAGRISSO?
TAGRISSO may cause serious side effects, including:
- lung problems. TAGRISSO may cause lung problems that may lead to death. Symptoms may be similar to those symptoms from lung cancer. Tell your doctor right away if you have any new or worsening lung symptoms, including trouble breathing, shortness of breath, cough, or fever.
- heart problems, including heart failure. TAGRISSO may cause heart problems that may lead to death. Your doctor should check your heart function before you start taking TAGRISSO and during treatment as needed. Tell your doctor right away if you have any of the following signs and symptoms of a heart problem: feeling like your heart is pounding or racing, shortness of breath, swelling of your ankles and feet, feeling lightheaded.
- eye problems. TAGRISSO may cause eye problems. Tell your doctor right away if you have symptoms of eye problems which may include watery eyes, sensitivity to light, eye pain, eye redness, or vision changes. Your doctor may send you to see an eye specialist (ophthalmologist) if you get eye problems with TAGRISSO.
- skin problems. TAGRISSO may cause skin problems. Tell your doctor right away if you develop target lesions (skin reactions that look like rings), severe blistering or peeling of the skin.
See “What are the possible side effects of TAGRISSO?” for more information about side effects.
What is TAGRISSO?
TAGRISSO is a prescription medicine used to treat non-small cell lung cancer (NSCLC) that has spread to other parts of the body (metastatic):
- as your first treatment if your tumor has a certain abnormal epidermal growth factor receptor (EGFR) gene(s)
- or
- if you have a certain type of EGFR gene and had previous treatment with an EGFR tyrosine kinase inhibitor (TKI) medicine that did not work or is no longer working.
Your doctor will perform a test to make sure that TAGRISSO is right for you.
It is not known if TAGRISSO is safe and effective in children.
Before taking TAGRISSO, tell your doctor about all of your medical conditions, including if you:
- have lung or breathing problems.
- have heart problems, including a condition called long QTc syndrome.
- have problems with your electrolytes, such as sodium, potassium, calcium or magnesium.
- have a history of eye problems.
-
are pregnant or plan to become pregnant. TAGRISSO can harm your unborn baby. Tell your doctor right away if you become pregnant during treatment with TAGRISSO or think you may be pregnant.
- Females who are able to become pregnant should use effective birth control during treatment with TAGRISSO and for 6 weeks after the final dose of TAGRISSO.
- Males who have female partners that are able to become pregnant should use effective birth control during treatment with TAGRISSO and for 4 months after the final dose of TAGRISSO.
- are breastfeeding or plan to breastfeed. It is not known if TAGRISSO passes into your breast milk. Do not breastfeed during treatment with TAGRISSO and for 2 weeks after your final dose of TAGRISSO. Talk to your doctor about the best way to feed your baby during this time.
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins, or herbal supplements. Especially tell your doctor if you take a heart or blood pressure medicine.
How should I take TAGRISSO?
- Take TAGRISSO exactly as your doctor tells you to take it.
- Your doctor may change your dose, temporarily stop, or permanently stop treatment with TAGRISSO if you have side effects.
- Take TAGRISSO 1 time each day.
- You can take TAGRISSO with or without food.
- If you miss a dose of TAGRISSO, do not make up for the missed dose. Take your next dose at your regular time.
-
If you cannot swallow TAGRISSO tablets whole:
- o place your dose of TAGRISSO in a container that contains 60 mL (2 ounces) of water. Do not use carbonated water or any other liquids.
- o stir the TAGRISSO tablet and water until the TAGRISSO tablet is in small pieces (the tablet will not completely dissolve). Do not crush, heat, or use ultrasound to prepare the mixture.
- o drink the TAGRISSO and water mixture right away.
- o add 120 mL to 240 mL (4 to 8 ounces) of water into the container and drink to make sure that you take your full dose of TAGRISSO.
What are the possible side effects of TAGRISSO?
TAGRISSO may cause serious side effects, including:
-
- See “What is the most important information I should know about TAGRISSO?”
- Severe blistering or peeling of skin – seek medical attention right away if you develop these symptoms.
- Target lesions, which are skin reactions that look like rings – seek medical attention right away if you develop these symptoms.
The most common side effects of TAGRISSO are:
-
- diarrhea
- rash
- dry skin
- changes in your nails, including: redness, tenderness, pain, inflammation, brittleness, separation from nailbed, and shedding of nails
- mouth sores
- tiredness
- decreased appetite
Tell your doctor if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of TAGRISSO. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store TAGRISSO?
- Store TAGRISSO at room temperature between 68°F to 77°F (20°C to 25°C).
- Safely throw away medicine that is out of date or that you no longer need.
- Keep TAGRISSO and all medicines out of the reach of children.
General information about the safe and effective use of TAGRISSO.
- Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use TAGRISSO for a condition for which it was not prescribed. Do not give TAGRISSO to other people, even if they have the same symptoms you have. It may harm them. You can ask your doctor or pharmacist for information about TAGRISSO that is written for a healthcare professional.
What are the ingredients in TAGRISSO?
Active ingredient: osimertinib
Inactive ingredients: mannitol, microcrystalline cellulose, low-substituted hydroxypropyl cellulose, and sodium stearyl fumarate. Tablet coating contains: polyvinyl alcohol, titanium dioxide, macrogol 3350, talc, ferric oxide yellow, ferric oxide red and ferric oxide black.
For more information, go to www.Tagrisso.com or call 1-800-236-9933.
Distributed by: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
©AstraZeneca 2018
- This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 12/2019
- PACKAGE/LABEL PRINCIPAL DISPLAY PANEL – 40 mg
- PACKAGE/LABEL PRINCIPAL DISPLAY PANEL – 80 mg
-
INGREDIENTS AND APPEARANCE
TAGRISSO
osimertinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0310-1349 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OSIMERTINIB (UNII: 3C06JJ0Z2O) (OSIMERTINIB - UNII:3C06JJ0Z2O) OSIMERTINIB 40 Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) TALC (UNII: 7SEV7J4R1U) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) FERROSOFERRIC OXIDE (UNII: XM0M87F357) LOW-SUBSTITUTED HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 2165RE0K14) Product Characteristics Color BROWN (beige) Score no score Shape ROUND (biconvex) Size 9mm Flavor Imprint Code AZ;40 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0310-1349-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 11/13/2015 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208065 11/13/2015 TAGRISSO
osimertinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0310-1350 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OSIMERTINIB (UNII: 3C06JJ0Z2O) (OSIMERTINIB - UNII:3C06JJ0Z2O) OSIMERTINIB 80 Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) TALC (UNII: 7SEV7J4R1U) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) FERROSOFERRIC OXIDE (UNII: XM0M87F357) LOW-SUBSTITUTED HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 2165RE0K14) Product Characteristics Color BROWN (beige) Score no score Shape OVAL (biconvex) Size 15mm Flavor Imprint Code AZ;80 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0310-1350-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 11/13/2015 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208065 11/13/2015 Labeler - AstraZeneca Pharmaceuticals LP (054743190) Registrant - AstraZeneca PLC (230790719)


Trademark Results [TAGRISSO]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 TAGRISSO 86827065 5117285 Live/Registered |
AstraZeneca AB 2015-11-20 |
 TAGRISSO 86435845 4930229 Live/Registered |
AstraZeneca AB 2014-10-27 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.