XELJANZ- tofacitinib tablet, film coated XELJANZ XR- tofacitinib tablet, extended release
XELJANZ by
Drug Labeling and Warnings
XELJANZ by is a Prescription medication manufactured, distributed, or labeled by U.S. Pharmaceuticals, Pfizer Inc, Viatris Pharmaceuticals LLC, Pfizer Ireland Pharmaceuticals Unlimited Company, Pfizer Manufacturing Deutschland GmbH, Pfizer Asia Manufacturing Pte Ltd, Pharmacia & Upjohn Company LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use XELJANZ/XELJANZ XR safely and effectively. See full prescribing information for XELJANZ.
XELJANZ® (tofacitinib) tablets, for oral use
XELJANZ® XR (tofacitinib) extended-release tablets, for oral use
Initial U.S. Approval: 2012WARNING: SERIOUS INFECTIONS, MORTALITY, MALIGNANCY AND THROMBOSIS
See full prescribing information for complete boxed warning.
- Serious infections leading to hospitalization or death, including tuberculosis and bacterial, invasive fungal, viral, and other opportunistic infections, have occurred. (5.1)
- If a serious infection develops, interrupt XELJANZ/XELJANZ XR until the infection is controlled. (5.1)
- Prior to starting XELJANZ/XELJANZ XR, perform a test for latent tuberculosis; if it is positive, start treatment for tuberculosis prior to starting XELJANZ/XELJANZ XR. (5.1)
- Monitor all patients for active tuberculosis during treatment, even if the initial latent tuberculosis test is negative. (5.1)
- Thrombosis, including pulmonary embolism, deep venous thrombosis and arterial thrombosis have occurred in patients treated with XELJANZ and other Janus kinase inhibitors. Rheumatoid arthritis patients with at least one cardiovascular (CV) risk factor had a higher rate of all-cause mortality and thrombosis with XELJANZ 10 mg twice daily vs. 5 mg twice daily or TNF blockers. (5.2, 5.4)
- Lymphoma and other malignancies have been observed in patients treated with XELJANZ, including an increased rate of Epstein Barr Virus-associated post-transplant lymphoproliferative disorder. (5.3)
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
XELJANZ/XELJANZ XR is a Janus kinase (JAK) inhibitor.
-
Rheumatoid Arthritis: XELJANZ/XELJANZ XR is indicated for the treatment of adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to methotrexate. It may be used as monotherapy or in combination with methotrexate or other nonbiologic disease-modifying antirheumatic drugs (DMARDs).
- Limitations of Use: Use of XELJANZ/XELJANZ XR in combination with biologic DMARDs or potent immunosuppressants such as azathioprine and cyclosporine is not recommended. (1)
-
Psoriatic Arthritis: XELJANZ/XELJANZ XR is indicated for the treatment of adult patients with active psoriatic arthritis who have had an inadequate response or intolerance to methotrexate or other disease-modifying antirheumatic drugs (DMARDs).
- Limitations of Use: Use of XELJANZ/XELJANZ XR in combination with biologic DMARDs or potent immunosuppressants such as azathioprine and cyclosporine is not recommended. (1)
-
Ulcerative Colitis: XELJANZ/XELJANZ XR is indicated for the treatment of adult patients with moderately to severely active ulcerative colitis (UC), who have had an inadequate response or who are intolerant to TNF blockers.
- Limitations of Use: Use of XELJANZ/XELJANZ XR in combination with biological therapies for UC or with potent immunosuppressants such as azathioprine and cyclosporine is not recommended. (1)
DOSAGE AND ADMINISTRATION
Administration Instructions
- Do not initiate XELJANZ/XELJANZ XR if absolute lymphocyte count <500 cells/mm3, an absolute neutrophil count (ANC) <1000 cells/mm3 or hemoglobin <9 g/dL. (2.1)
Recommended Dosage
Rheumatoid Arthritis
- XELJANZ 5 mg twice daily or XELJANZ XR 11 mg once daily. (2.2)
- Recommended dosage in patients with moderate and severe renal impairment or moderate hepatic impairment is XELJANZ 5 mg once daily. (2, 8.7, 8.8)
Psoriatic Arthritis (in combination with nonbiologic DMARDs)
- XELJANZ 5 mg twice daily or XELJANZ XR 11 mg once daily. (2.2)
- Recommended dosage in patients with moderate and severe renal impairment or moderate hepatic impairment is XELJANZ 5 mg once daily. (2, 8.7, 8.8)
Ulcerative Colitis
- Induction: XELJANZ 10 mg twice daily or XELJANZ XR 22 mg once daily for 8 weeks; evaluate patients and transition to maintenance therapy depending on therapeutic response. If needed, continue XELJANZ 10 mg twice daily or XELJANZ XR 22 mg once daily for a maximum of 16 weeks. Discontinue XELJANZ 10 mg twice daily or XELJANZ XR 22 mg once daily after 16 weeks if adequate therapeutic response is not achieved. (2.3)
- Maintenance: XELJANZ 5 mg twice daily or XELJANZ XR 11 mg once daily. For patients with loss of response during maintenance treatment, XELJANZ 10 mg twice daily or XELJANZ XR 22 mg once daily may be considered and limited to the shortest duration, with careful consideration of the benefits and risks for the individual patient. Use the lowest effective dose needed to maintain response. (2.3)
- Dosage adjustment is needed in patients with moderate and severe renal impairment or moderate hepatic impairment: see full prescribing information. (2.3)
Dosage Adjustment
- See the full prescribing information for dosage adjustments by indication for patients receiving CYP2C19 and/or CYP3A4 inhibitors; in patients with moderate or severe renal impairment or moderate hepatic impairment; and patients with lymphopenia, neutropenia, or anemia. (2.2, 2.3, 8.7, 8.8)
- Use of XELJANZ/XELJANZ XR in patients with severe hepatic impairment is not recommended in any patient population. (2.2, 2.3, 8.8)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
- Serious Infections: Avoid use of XELJANZ/XELJANZ XR during an active serious infection, including localized infections. (5.1)
- Thrombosis, including pulmonary, deep venous and arterial, some fatal: Reported more commonly in patients treated with XELJANZ 10 mg twice daily compared to 5 mg twice daily. Avoid XELJANZ/XELJANZ XR in patients at risk. Promptly evaluate patients with symptoms of thrombosis and discontinue XELJANZ/XELJANZ XR. (5.4)
- Gastrointestinal Perforations: Use with caution in patients that may be at increased risk. (5.5)
- Laboratory Monitoring: Recommended due to potential changes in lymphocytes, neutrophils, hemoglobin, liver enzymes and lipids. (5.7)
- Immunizations: Live vaccines: Avoid use with XELJANZ/XELJANZ XR. (5.8)
ADVERSE REACTIONS
Most common adverse reactions are:
- Rheumatoid and Psoriatic Arthritis: Reported during the first 3 months in rheumatoid arthritis controlled clinical trials and occurring in ≥2% of patients treated with XELJANZ monotherapy or in combination with DMARDs: upper respiratory tract infection, nasopharyngitis, diarrhea, and headache. (6.1)
- Ulcerative Colitis: Reported in ≥5% of patients treated with either 5 mg or 10 mg twice daily of XELJANZ and ≥1% greater than reported in patients receiving placebo in either the induction or maintenance clinical trials: nasopharyngitis, elevated cholesterol levels, headache, upper respiratory tract infection, increased blood creatine phosphokinase, rash, diarrhea, and herpes zoster. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer, Inc. at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 12/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: SERIOUS INFECTIONS, MORTALITY, MALIGNANCY AND THROMBOSIS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
2.2 Recommended Dosage in Rheumatoid Arthritis and Psoriatic Arthritis
2.3 Recommended Dosage in Ulcerative Colitis
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infections
5.2 Mortality
5.3 Malignancy and Lymphoproliferative Disorders
5.4 Thrombosis
5.5 Gastrointestinal Perforations
5.6 Hypersensitivity
5.7 Laboratory Abnormalities
5.8 Vaccinations
5.9 Risk of Gastrointestinal Obstruction with a Non-Deformable Extended-Release Formulation such as XELJANZ XR
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Use in Diabetics
8.7 Renal Impairment
8.8 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Rheumatoid Arthritis
14.2 Psoriatic Arthritis
14.3 Ulcerative Colitis
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: SERIOUS INFECTIONS, MORTALITY, MALIGNANCY AND THROMBOSIS
SERIOUS INFECTIONS
Patients treated with XELJANZ/XELJANZ XR are at increased risk for developing serious infections that may lead to hospitalization or death [see Warnings and Precautions (5.1), Adverse Reactions (6.1)]. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids.
If a serious infection develops, interrupt XELJANZ/XELJANZ XR until the infection is controlled.
Reported infections include:
- Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Patients should be tested for latent tuberculosis before XELJANZ/XELJANZ XR use and during therapy. Treatment for latent infection should be initiated prior to XELJANZ/XELJANZ XR use.
- Invasive fungal infections, including cryptococcosis and pneumocystosis. Patients with invasive fungal infections may present with disseminated, rather than localized, disease.
- Bacterial, viral, including herpes zoster, and other infections due to opportunistic pathogens.
The risks and benefits of treatment with XELJANZ/XELJANZ XR should be carefully considered prior to initiating therapy in patients with chronic or recurrent infection.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with XELJANZ/XELJANZ XR, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy [see Warnings and Precautions (5.1)].
MORTALITY
Rheumatoid arthritis patients 50 years of age and older with at least one cardiovascular (CV) risk factor treated with XELJANZ 10 mg twice a day had a higher rate of all-cause mortality, including sudden CV death, compared to those treated with XELJANZ 5 mg given twice daily or TNF blockers in a large, ongoing, postmarketing safety study [see Warnings and Precautions (5.2)].
MALIGNANCIES
Lymphoma and other malignancies have been observed in patients treated with XELJANZ. Epstein Barr Virus-associated post-transplant lymphoproliferative disorder has been observed at an increased rate in renal transplant patients treated with XELJANZ and concomitant immunosuppressive medications [see Warnings and Precautions (5.3)].
THROMBOSIS
Thrombosis, including pulmonary embolism, deep venous thrombosis, and arterial thrombosis have occurred in patients treated with XELJANZ and other Janus kinase inhibitors used to treat inflammatory conditions. Rheumatoid arthritis patients who were 50 years of age and older with at least one CV risk factor treated with XELJANZ 10 mg twice daily compared to XELJANZ 5 mg twice daily or TNF blockers in a large, ongoing postmarketing safety study had an observed increase in incidence of these events. Many of these events were serious and some resulted in death. Avoid XELJANZ/XELJANZ XR in patients at risk. Discontinue XELJANZ/XELJANZ XR and promptly evaluate patients with symptoms of thrombosis [see Warnings and Precautions (5.4)].
For patients with ulcerative colitis, use XELJANZ at the lowest effective dose and for the shortest duration needed to achieve/maintain therapeutic response [see Dosage and Administration (2.3)].
-
1 INDICATIONS AND USAGE
Rheumatoid Arthritis
XELJANZ/XELJANZ XR is indicated for the treatment of adult patients with moderately to severely active rheumatoid arthritis (RA) who have had an inadequate response or intolerance to methotrexate. It may be used as monotherapy or in combination with methotrexate or other nonbiologic disease-modifying antirheumatic drugs (DMARDs).
- Limitations of Use: Use of XELJANZ/XELJANZ XR in combination with biologic DMARDs or with potent immunosuppressants such as azathioprine and cyclosporine is not recommended.
Psoriatic Arthritis
XELJANZ/XELJANZ XR is indicated for the treatment of adult patients with active psoriatic arthritis (PsA) who have had an inadequate response or intolerance to methotrexate or other disease-modifying antirheumatic drugs (DMARDs).
- Limitations of Use: Use of XELJANZ/XELJANZ XR in combination with biologic DMARDs or with potent immunosuppressants such as azathioprine and cyclosporine is not recommended.
Ulcerative Colitis
XELJANZ/XELJANZ XR is indicated for the treatment of adult patients with moderately to severely active ulcerative colitis (UC), who have an inadequate response or who are intolerant to TNF blockers.
- Limitations of Use: Use of XELJANZ/XELJANZ XR in combination with biological therapies for UC or with potent immunosuppressants such as azathioprine and cyclosporine is not recommended.
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
- Do not initiate XELJANZ/XELJANZ XR in patients with an absolute lymphocyte count less than 500 cells/mm3, an absolute neutrophil count (ANC) less than 1000 cells/mm3 or who have hemoglobin levels less than 9 g/dL.
- Dose interruption is recommended for management of lymphopenia, neutropenia, and anemia [see Warnings and Precautions (5.7), Adverse Reactions (6.1)].
- Interrupt use of XELJANZ/XELJANZ XR if a patient develops a serious infection until the infection is controlled [see Warnings and Precautions (5.1)].
- Take XELJANZ/XELJANZ XR with or without food [see Clinical Pharmacology (12.3)].
- Swallow XELJANZ XR tablets whole and intact. Do not crush, split, or chew.
2.2 Recommended Dosage in Rheumatoid Arthritis and Psoriatic Arthritis
Table 1 displays the recommended adult daily dosage of XELJANZ and XELJANZ XR and dosage adjustments for patients receiving CYP2C19 and/or CYP3A4 inhibitors, in patients with moderate or severe renal impairment (including but not limited to those with severe insufficiency who are undergoing hemodialysis) or moderate hepatic impairment, with lymphopenia, neutropenia, or anemia.
Table 1: Recommended Dosage of XELJANZ and XELJANZ XR in Patients with Rheumatoid Arthritis* and Psoriatic Arthritis† XELJANZ XELJANZ XR - * XELJANZ/XELJANZ XR may be used as monotherapy or in combination with methotrexate or other nonbiologic disease-modifying antirheumatic drugs (DMARDs) in rheumatoid arthritis.
- † XELJANZ/XELJANZ XR is used in combination with nonbiologic disease modifying antirheumatic drugs (DMARDs) in psoriatic arthritis. The efficacy of XELJANZ/XELJANZ XR as a monotherapy has not been studied in psoriatic arthritis.
- ‡ Use of XELJANZ/XELJANZ XR in patients with severe hepatic impairment is not recommended.
Adult patients 5 mg twice daily 11 mg once daily Patients receiving: - Strong CYP3A4 inhibitors (e.g., ketoconazole), or
- a moderate CYP3A4 inhibitor(s) with a strong CYP2C19 inhibitor(s) (e.g., fluconazole)
5 mg once daily Reduce to XELJANZ 5 mg once daily Patients with: - moderate or severe renal impairment [see Use in Specific Populations (8.7)]
- moderate hepatic impairment [see Use in Specific Populations (8.8)]‡
5 mg once daily Reduce to XELJANZ 5 mg once daily For patients undergoing hemodialysis, dose should be administered after the dialysis session on dialysis days. If a dose was taken before the dialysis procedure, supplemental doses are not recommended in patients after dialysis. Patients with lymphocyte count less than 500 cells/mm3, confirmed by repeat testing Discontinue dosing. Patients with ANC 500 to 1000 cells/mm3 Interrupt dosing.
When ANC is greater than 1000, resume 5 mg twice daily.Interrupt dosing.
When ANC is greater than 1000, resume 11 mg once daily.Patients with ANC less than 500 cells/mm3 Discontinue dosing. Patients with hemoglobin less than 8 g/dL or a decrease of more than 2 g/dL Interrupt dosing until hemoglobin values have normalized. 2.3 Recommended Dosage in Ulcerative Colitis
Table 2 displays the recommended adult daily dosage of XELJANZ/XELJANZ XR and dosage adjustments for patients receiving CYP2C19 and/or CYP3A4 inhibitors, with moderate or severe renal impairment (including but not limited to those with severe insufficiency who are undergoing hemodialysis) or moderate hepatic impairment, with lymphopenia, neutropenia or anemia.
Table 2: Recommended Dosage of XELJANZ/XELJANZ XR in Patients with UC XELJANZ XELJANZ XR - * Use of XELJANZ/XELJANZ XR in patients with severe hepatic impairment is not recommended.
Adult patients Induction: 10 mg twice daily for at least 8 weeks [see Clinical Studies (14.3)]; evaluate patients and transition to maintenance therapy depending on therapeutic response. If needed continue 10 mg twice daily for a maximum of 16 weeks. Discontinue 10 mg twice daily after 16 weeks if adequate therapeutic response is not achieved.
Maintenance: 5 mg twice daily.
For patients with loss of response during maintenance treatment, a dosage of 10 mg twice daily may be considered and limited to the shortest duration, with careful consideration of the benefits and risks for the individual patient. Use the lowest effective dose needed to maintain response.Induction: 22 mg once daily for at least 8 weeks; evaluate patients and transition to maintenance therapy depending on therapeutic response. If needed continue 22 mg once daily for a maximum of 16 weeks. Discontinue 22 mg once daily after 16 weeks if adequate therapeutic response is not achieved.
Maintenance: 11 mg once daily.
For patients with loss of response during maintenance treatment, a dosage of 22 mg once daily may be considered and limited to the shortest duration, with careful consideration of the benefits and risks for the individual patient. Use the lowest effective dose needed to maintain response.Patients receiving: - Strong CYP3A4 inhibitors (e.g., ketoconazole), or
- a moderate CYP3A4 inhibitor(s) with a strong CYP2C19 inhibitor(s) (e.g., fluconazole)
If taking 10 mg twice daily, reduce to 5 mg twice daily.
If taking 5 mg twice daily, reduce to 5 mg once daily.If taking 22 mg once daily, reduce to 11 mg once daily.
If taking 11 mg once daily, reduce to XELJANZ 5 mg once dailyPatients with: - moderate or severe renal impairment [see Use in Specific Populations (8.7)]
- moderate hepatic impairment [see Use in Specific Populations (8.8)]*
If taking 10 mg twice daily, reduce to 5 mg twice daily.
If taking 5 mg twice daily, reduce to 5 mg once daily.If taking 22 mg once daily, reduce to 11 mg once daily.
If taking 11 mg once daily, reduce to XELJANZ 5 mg once daily.For patients undergoing hemodialysis, dose should be administered after the dialysis session on dialysis days. If a dose was taken before the dialysis procedure, supplemental doses are not recommended in patients after dialysis. Patients with lymphocyte count less than 500 cells/mm3, confirmed by repeat testing Discontinue dosing. Patients with ANC 500 to 1000 cells/mm3 If taking 10 mg twice daily, reduce to 5 mg twice daily. When ANC is greater than 1000, increase to 10 mg twice daily based on clinical response.
If taking 5 mg twice daily, interrupt dosing. When ANC is greater than 1000, resume 5 mg twice daily.If taking 22 mg once daily, reduce to 11 mg once daily. When ANC is greater than 1000, increase to 22 mg once daily based on clinical response.
If taking 11 mg once daily, interrupt dosing. When ANC is greater than 1000, resume 11 mg once daily.Patients with ANC less than 500 cells/mm3 Discontinue dosing. Patients with hemoglobin less than 8 g/dL or a decrease of more than 2 g/dL Interrupt dosing until hemoglobin values have normalized. Switching from XELJANZ Tablets to XELJANZ XR Tablets
Patients treated with XELJANZ 5 mg twice daily may be switched to XELJANZ XR 11 mg once daily the day following the last dose of XELJANZ 5 mg. Patients treated with XELJANZ 10 mg twice daily may be switched to XELJANZ XR 22 mg once daily the day following the last dose of XELJANZ 10 mg.
-
3 DOSAGE FORMS AND STRENGTHS
XELJANZ Tablets:
- 5 mg tofacitinib: White, round, immediate-release film-coated tablets, debossed with "Pfizer" on one side, and "JKI 5" on the other side.
- 10 mg tofacitinib: Blue, round, immediate-release film-coated tablets, debossed with "Pfizer" on one side, and "JKI 10" on the other side.
XELJANZ XR Tablets:
- 11 mg tofacitinib: Pink, oval, extended-release film-coated tablets with a drilled hole at one end of the tablet band and "JKI 11" printed on one side of the tablet.
- 22 mg tofacitinib: Beige, oval, extended-release film-coated tablets with a drilled hole at one end of the tablet band and "JKI 22" printed on one side of the tablet.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infections
Serious and sometimes fatal infections due to bacterial, mycobacterial, invasive fungal, viral, or other opportunistic pathogens have been reported in patients receiving XELJANZ. The most common serious infections reported with XELJANZ included pneumonia, cellulitis, herpes zoster, urinary tract infection, diverticulitis, and appendicitis. Among opportunistic infections, tuberculosis and other mycobacterial infections, cryptococcosis, histoplasmosis, esophageal candidiasis, pneumocystosis, multidermatomal herpes zoster, cytomegalovirus infections, BK virus infection, and listeriosis were reported with XELJANZ. Some patients have presented with disseminated rather than localized disease, and were often taking concomitant immunomodulating agents such as methotrexate or corticosteroids.
In the UC population, XELJANZ treatment with 10 mg twice daily was associated with greater risk of serious infections compared to 5 mg twice daily. Additionally, opportunistic herpes zoster infections (including meningoencephalitis, ophthalmologic, and disseminated cutaneous) were seen in patients who were treated with XELJANZ 10 mg twice daily.
Other serious infections that were not reported in clinical studies may also occur (e.g., coccidioidomycosis).
Avoid use of XELJANZ/XELJANZ XR in patients with an active, serious infection, including localized infections. The risks and benefits of treatment should be considered prior to initiating XELJANZ/XELJANZ XR in patients:
- with chronic or recurrent infection
- who have been exposed to tuberculosis
- with a history of a serious or an opportunistic infection
- who have resided or traveled in areas of endemic tuberculosis or endemic mycoses; or
- with underlying conditions that may predispose them to infection.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with XELJANZ/XELJANZ XR. XELJANZ/XELJANZ XR should be interrupted if a patient develops a serious infection, an opportunistic infection, or sepsis. A patient who develops a new infection during treatment with XELJANZ/XELJANZ XR should undergo prompt and complete diagnostic testing appropriate for an immunocompromised patient; appropriate antimicrobial therapy should be initiated, and the patient should be closely monitored.
Caution is also recommended in patients with a history of chronic lung disease, or in those who develop interstitial lung disease, as they may be more prone to infections.
Risk of infection may be higher with increasing degrees of lymphopenia and consideration should be given to lymphocyte counts when assessing individual patient risk of infection. Discontinuation and monitoring criteria for lymphopenia are recommended [see Dosage and Administration (2.2, 2.3)].
Tuberculosis
Patients should be evaluated and tested for latent or active infection prior to and per applicable guidelines during administration of XELJANZ/XELJANZ XR.
Anti-tuberculosis therapy should also be considered prior to administration of XELJANZ/XELJANZ XR in patients with a past history of latent or active tuberculosis in whom an adequate course of treatment cannot be confirmed, and for patients with a negative test for latent tuberculosis but who have risk factors for tuberculosis infection. Consultation with a physician with expertise in the treatment of tuberculosis is recommended to aid in the decision about whether initiating anti-tuberculosis therapy is appropriate for an individual patient.
Patients should be closely monitored for the development of signs and symptoms of tuberculosis, including patients who tested negative for latent tuberculosis infection prior to initiating therapy.
Patients with latent tuberculosis should be treated with standard antimycobacterial therapy before administering XELJANZ/XELJANZ XR.
Viral Reactivation
Viral reactivation, including cases of herpes virus reactivation (e.g., herpes zoster), were observed in clinical studies with XELJANZ. Postmarketing cases of hepatitis B reactivation have been reported in patients treated with XELJANZ. The impact of XELJANZ/XELJANZ XR on chronic viral hepatitis reactivation is unknown. Patients who screened positive for hepatitis B or C were excluded from clinical trials. Screening for viral hepatitis should be performed in accordance with clinical guidelines before starting therapy with XELJANZ/XELJANZ XR. The risk of herpes zoster is increased in patients treated with XELJANZ/XELJANZ XR and appears to be higher in patients treated with XELJANZ in Japan and Korea.
5.2 Mortality
Rheumatoid arthritis patients 50 years of age and older with at least one cardiovascular (CV) risk factor treated with XELJANZ 10 mg twice a day had a higher rate of all-cause mortality, including sudden CV death, compared to those treated with XELJANZ 5 mg given twice daily or TNF blockers in a large, ongoing, postmarketing safety study.
A dosage of XELJANZ 10 mg twice daily or XELJANZ XR 22 mg once daily is not recommended for the treatment of RA or PsA [see Dosage and Administration (2.2)].
For the treatment of UC, use XELJANZ at the lowest effective dose and for the shortest duration needed to achieve/maintain therapeutic response [see Dosage and Administration (2.3)].
5.3 Malignancy and Lymphoproliferative Disorders
Consider the risks and benefits of XELJANZ/XELJANZ XR treatment prior to initiating therapy in patients with a known malignancy other than a successfully treated non-melanoma skin cancer (NMSC) or when considering continuing XELJANZ/XELJANZ XR in patients who develop a malignancy. Malignancies were observed in clinical studies of XELJANZ [see Adverse Reactions (6.1)].
In the seven controlled rheumatoid arthritis clinical studies, 11 solid cancers and one lymphoma were diagnosed in 3328 patients receiving XELJANZ with or without DMARD, compared to 0 solid cancers and 0 lymphomas in 809 patients in the placebo with or without DMARD group during the first 12 months of exposure. Lymphomas and solid cancers have also been observed in the long-term extension studies in rheumatoid arthritis patients treated with XELJANZ.
During the 2 PsA controlled clinical studies there were 3 malignancies (excluding NMSC) in 474 patients receiving XELJANZ plus nonbiologic DMARD (6 to 12 months exposure) compared with 0 malignancies in 236 patients in the placebo plus nonbiologic DMARD group (3 months exposure) and 0 malignancies in 106 patients in the adalimumab plus nonbiologic DMARD group (12 months exposure). No lymphomas were reported. Malignancies have also been observed in the long-term extension study in psoriatic arthritis patients treated with XELJANZ.
During the UC controlled clinical studies (8-week induction and 52-week maintenance studies), which included 1220 patients, 0 cases of solid cancer or lymphoma were observed in XELJANZ-treated patients. In the long-term extension study, malignancies (including solid cancers and lymphomas) were observed more often in patients treated with XELJANZ 10 mg twice daily.
In Phase 2B, controlled dose-ranging trials in de-novo renal transplant patients, all of whom received induction therapy with basiliximab, high-dose corticosteroids, and mycophenolic acid products, Epstein Barr Virus-associated post-transplant lymphoproliferative disorder was observed in 5 out of 218 patients treated with XELJANZ (2.3%) compared to 0 out of 111 patients treated with cyclosporine.
Other malignancies were observed in clinical studies and the postmarketing setting, including, but not limited to, lung cancer, breast cancer, melanoma, prostate cancer, and pancreatic cancer.
Non-Melanoma Skin Cancer
Non-melanoma skin cancers (NMSCs) have been reported in patients treated with XELJANZ. Periodic skin examination is recommended for patients who are at increased risk for skin cancer. In the UC population, treatment with XELJANZ 10 mg twice daily was associated with greater risk of NMSC.
5.4 Thrombosis
Thrombosis, including pulmonary embolism, deep venous thrombosis, and arterial thrombosis, have occurred in patients treated with XELJANZ and other Janus kinase (JAK) inhibitors used to treat inflammatory conditions. Patients with rheumatoid arthritis 50 years of age and older with at least one CV risk factor treated with XELJANZ 10 mg twice daily compared to XELJANZ 5 mg twice daily or TNF blockers in a large, ongoing postmarketing study had an observed increase in incidence of these events. Many of these events were serious and some resulted in death [see Warnings and Precautions (5.2)].
A dosage of XELJANZ 10 mg twice daily or XELJANZ XR 22 mg once daily is not recommended for the treatment of RA or PsA [see Dosage and Administration (2.2)].
In a long-term extension study in patients with UC, four cases of pulmonary embolism were reported in patients taking XELJANZ 10 mg twice daily, including one death in a patient with advanced cancer.
Promptly evaluate patients with symptoms of thrombosis and discontinue XELJANZ/XELJANZ XR in patients with symptoms of thrombosis.
Avoid XELJANZ/XELJANZ XR in patients that may be at increased risk of thrombosis. For the treatment of UC, use XELJANZ at the lowest effective dose and for the shortest duration needed to achieve/maintain therapeutic response [see Dosage and Administration (2.3)].
5.5 Gastrointestinal Perforations
Events of gastrointestinal perforation have been reported in clinical studies with XELJANZ, although the role of JAK inhibition in these events is not known. In these studies, many patients with rheumatoid arthritis were receiving background therapy with Nonsteroidal Anti-Inflammatory Drugs (NSAIDs).
There was no discernable difference in frequency of gastrointestinal perforation between the placebo and the XELJANZ arms in clinical trials of patients with UC, and many of them were receiving background corticosteroids.
XELJANZ/XELJANZ XR should be used with caution in patients who may be at increased risk for gastrointestinal perforation (e.g., patients with a history of diverticulitis or taking NSAIDs). Patients presenting with new onset abdominal symptoms should be evaluated promptly for early identification of gastrointestinal perforation [see Adverse Reactions (6.1)].
5.6 Hypersensitivity
Reactions such as angioedema and urticaria that may reflect drug hypersensitivity have been observed in patients receiving XELJANZ/XELJANZ XR. Some events were serious. If a serious hypersensitivity reaction occurs, promptly discontinue tofacitinib while evaluating the potential cause or causes of the reaction [see Adverse Reactions (6.2)].
5.7 Laboratory Abnormalities
Lymphocyte Abnormalities
Treatment with XELJANZ was associated with initial lymphocytosis at one month of exposure followed by a gradual decrease in mean absolute lymphocyte counts below the baseline of approximately 10% during 12 months of therapy. Lymphocyte counts less than 500 cells/mm3 were associated with an increased incidence of treated and serious infections.
Avoid initiation of XELJANZ/XELJANZ XR treatment in patients with a low lymphocyte count (i.e., less than 500 cells/mm3). In patients who develop a confirmed absolute lymphocyte count less than 500 cells/mm3, treatment with XELJANZ/XELJANZ XR is not recommended.
Monitor lymphocyte counts at baseline and every 3 months thereafter. For recommended modifications based on lymphocyte counts [see Dosage and Administration (2.2, 2.3)].
Neutropenia
Treatment with XELJANZ was associated with an increased incidence of neutropenia (less than 2000 cells/mm3) compared to placebo.
Avoid initiation of XELJANZ/XELJANZ XR treatment in patients with a low neutrophil count (i.e., ANC less than 1000 cells/mm3). For patients who develop a persistent ANC of 500 to 1000 cells/mm3, interrupt XELJANZ/XELJANZ XR dosing until ANC is greater than or equal to 1000 cells/mm3. In patients who develop an ANC less than 500 cells/mm3, treatment with XELJANZ/XELJANZ XR is not recommended.
Monitor neutrophil counts at baseline and after 4–8 weeks of treatment and every 3 months thereafter. For recommended modifications based on ANC results [see Dosage and Administration (2.2, 2.3)].
Anemia
Avoid initiation of XELJANZ/XELJANZ XR treatment in patients with a low hemoglobin level (i.e., less than 9 g/dL). Treatment with XELJANZ/XELJANZ XR should be interrupted in patients who develop hemoglobin levels less than 8 g/dL or whose hemoglobin level drops greater than 2 g/dL on treatment.
Monitor hemoglobin at baseline and after 4–8 weeks of treatment and every 3 months thereafter. For recommended modifications based on hemoglobin results [see Dosage and Administration (2)].
Liver Enzyme Elevations
Treatment with XELJANZ was associated with an increased incidence of liver enzyme elevation compared to placebo. Most of these abnormalities occurred in studies with background DMARD (primarily methotrexate) therapy.
Routine monitoring of liver tests and prompt investigation of the causes of liver enzyme elevations is recommended to identify potential cases of drug-induced liver injury. If drug-induced liver injury is suspected, the administration of XELJANZ/XELJANZ XR should be interrupted until this diagnosis has been excluded.
Lipid Elevations
Treatment with XELJANZ was associated with dose-dependent increases in lipid parameters including total cholesterol, low-density lipoprotein (LDL) cholesterol, and high-density lipoprotein (HDL) cholesterol. Maximum effects were generally observed within 6 weeks. There were no clinically relevant changes in LDL/HDL cholesterol ratios. The effect of these lipid parameter elevations on cardiovascular morbidity and mortality has not been determined.
Assessment of lipid parameters should be performed approximately 4–8 weeks following initiation of XELJANZ/XELJANZ XR therapy.
Manage patients according to clinical guidelines [e.g., National Cholesterol Educational Program (NCEP)] for the management of hyperlipidemia.
5.8 Vaccinations
Avoid use of live vaccines concurrently with XELJANZ/XELJANZ XR. The interval between live vaccinations and initiation of tofacitinib therapy should be in accordance with current vaccination guidelines regarding immunosuppressive agents.
A patient experienced dissemination of the vaccine strain of varicella zoster virus, 16 days after vaccination with live attenuated (Zostavax) virus vaccine and 2 days after treatment start with tofacitinib 5 mg twice daily. The patient was varicella virus naïve, as evidenced by no previous history of varicella infection and no anti-varicella antibodies at baseline. Tofacitinib was discontinued and the patient recovered after treatment with standard doses of antiviral medication.
Update immunizations in agreement with current immunization guidelines prior to initiating XELJANZ/XELJANZ XR therapy.
5.9 Risk of Gastrointestinal Obstruction with a Non-Deformable Extended-Release Formulation such as XELJANZ XR
As with any other non-deformable material, caution should be used when administering XELJANZ XR to patients with pre-existing severe gastrointestinal narrowing (pathologic or iatrogenic). There have been rare reports of obstructive symptoms in patients with known strictures in association with the ingestion of other drugs utilizing a non-deformable extended-release formulation.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Serious Infections [see Warnings and Precautions (5.1)]
- Mortality [see Warnings and Precautions (5.2)]
- Malignancy and Lymphoproliferative Disorders [see Warnings and Precautions (5.3)]
- Thrombosis [see Warnings and Precautions (5.4)]
- Gastrointestinal Perforations [see Warnings and Precautions (5.5)]
- Hypersensitivity [see Warnings and Precautions (5.6)]
- Laboratory Abnormalities [see Warnings and Precautions (5.7)]
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not predict the rates observed in a broader patient population in clinical practice.
Rheumatoid Arthritis
The clinical studies described in the following sections were conducted using XELJANZ. Although other doses of XELJANZ have been studied, the recommended dose of XELJANZ is 5 mg twice daily. The recommended dose for XELJANZ XR is 11 mg once daily. A dosage of XELJANZ 10 mg twice daily or XELJANZ XR 22 mg once daily is not a recommended regimen for the treatment of rheumatoid arthritis [see Dosage and Administration (2.2)].
The following data includes two Phase 2 and five Phase 3 double-blind, controlled, multicenter trials. In these trials, patients were randomized to doses of XELJANZ 5 mg twice daily (292 patients) and 10 mg twice daily (306 patients) monotherapy, XELJANZ 5 mg twice daily (1044 patients) and 10 mg twice daily (1043 patients) in combination with DMARDs (including methotrexate) and placebo (809 patients). All seven protocols included provisions for patients taking placebo to receive treatment with XELJANZ at Month 3 or Month 6 either by patient response (based on uncontrolled disease activity) or by design, so that adverse events cannot always be unambiguously attributed to a given treatment. Therefore, some analyses that follow include patients who changed treatment by design or by patient response from placebo to XELJANZ in both the placebo and XELJANZ group of a given interval. Comparisons between placebo and XELJANZ were based on the first 3 months of exposure, and comparisons between XELJANZ 5 mg twice daily and XELJANZ 10 mg twice daily were based on the first 12 months of exposure.
The long-term safety population includes all patients who participated in a double-blind, controlled trial (including earlier development phase studies) and then participated in one of two long-term safety studies. The design of the long-term safety studies allowed for modification of XELJANZ doses according to clinical judgment. This limits the interpretation of the long-term safety data with respect to dose.
The most common serious adverse reactions were serious infections [see Warnings and Precautions (5.1)].
The proportion of patients who discontinued treatment due to any adverse reaction during the 0 to 3 months exposure in the double-blind, placebo-controlled trials was 4% for patients taking XELJANZ and 3% for placebo-treated patients.
Overall Infections
In the seven controlled trials, during the 0 to 3 months exposure, the overall frequency of infections was 20% and 22% in the 5 mg twice daily and 10 mg twice daily groups, respectively, and 18% in the placebo group.
The most commonly reported infections with XELJANZ were upper respiratory tract infections, nasopharyngitis, and urinary tract infections (4%, 3%, and 2% of patients, respectively).
Serious Infections
In the seven controlled trials, during the 0 to 3 months exposure, serious infections were reported in 1 patient (0.5 events per 100 patient-years) who received placebo and 11 patients (1.7 events per 100 patient-years) who received XELJANZ 5 mg or 10 mg twice daily. The rate difference between treatment groups (and the corresponding 95% confidence interval) was 1.1 (-0.4, 2.5) events per 100 patient-years for the combined 5 mg twice daily and 10 mg twice daily XELJANZ group minus placebo.
In the seven controlled trials, during the 0 to 12 months exposure, serious infections were reported in 34 patients (2.7 events per 100 patient-years) who received 5 mg twice daily of XELJANZ and 33 patients (2.7 events per 100 patient-years) who received 10 mg twice daily of XELJANZ. The rate difference between XELJANZ doses (and the corresponding 95% confidence interval) was -0.1 (-1.3, 1.2) events per 100 patient-years for 10 mg twice daily XELJANZ minus 5 mg twice daily XELJANZ.
The most common serious infections included pneumonia, cellulitis, herpes zoster, and urinary tract infection [see Warnings and Precautions (5.1)].
Tuberculosis
In the seven controlled trials, during the 0 to 3 months exposure, tuberculosis was not reported in patients who received placebo, 5 mg twice daily of XELJANZ, or 10 mg twice daily of XELJANZ.
In the seven controlled trials, during the 0 to 12 months exposure, tuberculosis was reported in 0 patients who received 5 mg twice daily of XELJANZ and 6 patients (0.5 events per 100 patient-years) who received 10 mg twice daily of XELJANZ. The rate difference between XELJANZ doses (and the corresponding 95% confidence interval) was 0.5 (0.1, 0.9) events per 100 patient-years for 10 mg twice daily XELJANZ minus 5 mg twice daily XELJANZ.
Cases of disseminated tuberculosis were also reported. The median XELJANZ exposure prior to diagnosis of tuberculosis was 10 months (range from 152 to 960 days) [see Warnings and Precautions (5.1)].
Opportunistic Infections (excluding tuberculosis)
In the seven controlled trials, during the 0 to 3 months exposure, opportunistic infections were not reported in patients who received placebo, 5 mg twice daily of XELJANZ, or 10 mg twice daily of XELJANZ.
In the seven controlled trials, during the 0 to 12 months exposure, opportunistic infections were reported in 4 patients (0.3 events per 100 patient-years) who received 5 mg twice daily of XELJANZ and 4 patients (0.3 events per 100 patient-years) who received 10 mg twice daily of XELJANZ. The rate difference between XELJANZ doses (and the corresponding 95% confidence interval) was 0 (-0.5, 0.5) events per 100 patient-years for 10 mg twice daily XELJANZ minus 5 mg twice daily XELJANZ.
The median XELJANZ exposure prior to diagnosis of an opportunistic infection was 8 months (range from 41 to 698 days) [see Warnings and Precautions (5.1)].
Malignancy
In the seven controlled trials, during the 0 to 3 months exposure, malignancies excluding NMSC were reported in 0 patients who received placebo and 2 patients (0.3 events per 100 patient-years) who received either XELJANZ 5 mg or 10 mg twice daily. The rate difference between treatment groups (and the corresponding 95% confidence interval) was 0.3 (-0.1, 0.7) events per 100 patient-years for the combined 5 mg and 10 mg twice daily XELJANZ group minus placebo.
In the seven controlled trials, during the 0 to 12 months exposure, malignancies excluding NMSC were reported in 5 patients (0.4 events per 100 patient-years) who received 5 mg twice daily of XELJANZ and 7 patients (0.6 events per 100 patient-years) who received 10 mg twice daily of XELJANZ. The rate difference between XELJANZ doses (and the corresponding 95% confidence interval) was 0.2 (-0.4, 0.7) events per 100 patient-years for 10 mg twice daily XELJANZ minus 5 mg twice daily XELJANZ. One of these malignancies was a case of lymphoma that occurred during the 0 to 12 month period in a patient treated with XELJANZ 10 mg twice daily.
The most common types of malignancy, including malignancies observed during the long-term extension, were lung and breast cancer, followed by gastric, colorectal, renal cell, prostate cancer, lymphoma, and malignant melanoma [see Warnings and Precautions (5.3)].
Laboratory Abnormalities
Lymphopenia
In the controlled clinical trials, confirmed decreases in absolute lymphocyte counts below 500 cells/mm3 occurred in 0.04% of patients for the 5 mg twice daily and 10 mg twice daily XELJANZ groups combined during the first 3 months of exposure.
Confirmed lymphocyte counts less than 500 cells/mm3 were associated with an increased incidence of treated and serious infections [see Warnings and Precautions (5.7)].
Neutropenia
In the controlled clinical trials, confirmed decreases in ANC below 1000 cells/mm3 occurred in 0.07% of patients for the 5 mg twice daily and 10 mg twice daily XELJANZ groups combined during the first 3 months of exposure.
There were no confirmed decreases in ANC below 500 cells/mm3 observed in any treatment group.
There was no clear relationship between neutropenia and the occurrence of serious infections.
In the long-term safety population, the pattern and incidence of confirmed decreases in ANC remained consistent with what was seen in the controlled clinical trials [see Warnings and Precautions (5.7)].
Liver Enzyme Elevations
Confirmed increases in liver enzymes greater than 3 times the upper limit of normal (3× ULN) were observed in patients treated with XELJANZ. In patients experiencing liver enzyme elevation, modification of treatment regimen, such as reduction in the dose of concomitant DMARD, interruption of XELJANZ, or reduction in XELJANZ dose, resulted in decrease or normalization of liver enzymes.
In the controlled monotherapy trials (0–3 months), no differences in the incidence of ALT or AST elevations were observed between the placebo, and XELJANZ 5 mg, and 10 mg twice daily groups.
In the controlled background DMARD trials (0–3 months), ALT elevations greater than 3× ULN were observed in 1.0%, 1.3% and 1.2% of patients receiving placebo, 5 mg, and 10 mg twice daily, respectively. In these trials, AST elevations greater than 3× ULN were observed in 0.6%, 0.5% and 0.4% of patients receiving placebo, 5 mg, and 10 mg twice daily, respectively.
One case of drug-induced liver injury was reported in a patient treated with XELJANZ 10 mg twice daily for approximately 2.5 months. The patient developed symptomatic elevations of AST and ALT greater than 3× ULN and bilirubin elevations greater than 2× ULN, which required hospitalizations and a liver biopsy.
Lipid Elevations
In the controlled clinical trials, dose-related elevations in lipid parameters (total cholesterol, LDL cholesterol, HDL cholesterol, triglycerides) were observed at one month of exposure and remained stable thereafter. Changes in lipid parameters during the first 3 months of exposure in the controlled clinical trials are summarized below:
- Mean LDL cholesterol increased by 15% in the XELJANZ 5 mg twice daily arm and 19% in the XELJANZ 10 mg twice daily arm.
- Mean HDL cholesterol increased by 10% in the XELJANZ 5 mg twice daily arm and 12% in the XELJANZ 10 mg twice daily arm.
- Mean LDL/HDL ratios were essentially unchanged in XELJANZ-treated patients.
In a controlled clinical trial, elevations in LDL cholesterol and ApoB decreased to pretreatment levels in response to statin therapy.
In the long-term safety population, elevations in lipid parameters remained consistent with what was seen in the controlled clinical trials.
Serum Creatinine Elevations
In the controlled clinical trials, dose-related elevations in serum creatinine were observed with XELJANZ treatment. The mean increase in serum creatinine was <0.1 mg/dL in the 12-month pooled safety analysis; however with increasing duration of exposure in the long-term extensions, up to 2% of patients were discontinued from XELJANZ treatment due to the protocol-specified discontinuation criterion of an increase in creatinine by more than 50% of baseline. The clinical significance of the observed serum creatinine elevations is unknown.
Other Adverse Reactions
Adverse reactions occurring in 2% or more of patients on 5 mg twice daily or 10 mg twice daily XELJANZ and at least 1% greater than that observed in patients on placebo with or without DMARD are summarized in Table 3.
Table 3: Common Adverse Reactions* in Clinical Trials of XELJANZ for the Treatment of Rheumatoid Arthritis With or Without Concomitant DMARDs (0–3 Months) Preferred Term XELJANZ
5 mg Twice DailyXELJANZ
10 mg Twice Daily†Placebo N = 1336
(%)N = 1349
(%)N = 809
(%)N reflects randomized and treated patients from the seven clinical trials. - * reported in ≥2% of patients treated with either dose of XELJANZ and ≥1% greater than that reported for placebo.
- † the recommended dose of XELJANZ for the treatment of rheumatoid arthritis is 5 mg twice daily [see Dosage and Administration (2)].
Upper respiratory tract infection 4 4 3 Nasopharyngitis 4 3 3 Diarrhea 4 3 2 Headache 4 3 2 Hypertension 2 2 1 Other adverse reactions occurring in controlled and open-label extension studies included:
Blood and lymphatic system disorders: Anemia
Infections and infestations: Diverticulitis
Metabolism and nutrition disorders: Dehydration
Psychiatric disorders: Insomnia
Nervous system disorders: Paresthesia
Respiratory, thoracic and mediastinal disorders: Dyspnea, cough, sinus congestion, interstitial lung disease (cases were limited to patients with rheumatoid arthritis and some were fatal)
Gastrointestinal disorders: Abdominal pain, dyspepsia, vomiting, gastritis, nausea
Hepatobiliary disorders: Hepatic steatosis
Skin and subcutaneous tissue disorders: Rash, erythema, pruritus
Musculoskeletal, connective tissue and bone disorders: Musculoskeletal pain, arthralgia, tendonitis, joint swelling
Neoplasms benign, malignant and unspecified (including cysts and polyps): Non-melanoma skin cancers
General disorders and administration site conditions: Pyrexia, fatigue, peripheral edema
Clinical Experience in Methotrexate-Naïve Patients
Study RA-VI was an active-controlled clinical trial in methotrexate-naïve patients [see Clinical Studies (14)]. The safety experience in these patients was consistent with Studies RA-I through V.
Psoriatic Arthritis
XELJANZ 5 mg twice daily and 10 mg twice daily were studied in 2 double-blind Phase 3 clinical trials in patients with active psoriatic arthritis (PsA). Although other doses of XELJANZ have been studied, the recommended dose of XELJANZ is 5 mg twice daily. The recommended dose for XELJANZ XR is 11 mg once daily. A dosage of XELJANZ 10 mg twice daily or XELJANZ XR 22 mg once daily is not recommended for the treatment of PsA [see Dosage and Administration (2.2)].
Study PsA-I (NCT01877668) had a duration of 12 months and enrolled patients who had an inadequate response to a nonbiologic DMARD and who were naïve to treatment with a TNF blocker. Study PsA-I included a 3-month placebo-controlled period and also included adalimumab 40 mg subcutaneously once every 2 weeks for 12 months.
Study PsA-II (NCT01882439) had a duration of 6 months and enrolled patients who had an inadequate response to at least one approved TNF blocker. This clinical trial included a 3-month placebo controlled period.
In these combined Phase 3 clinical trials, 238 patients were randomized and treated with XELJANZ 5 mg twice daily and 236 patients were randomized and treated with XELJANZ 10 mg twice daily. All patients in the clinical trials were required to receive treatment with a stable dose of a nonbiologic DMARD [the majority (79%) received methotrexate]. The study population randomized and treated with XELJANZ (474 patients) included 45 (9.5%) patients aged 65 years or older and 66 (13.9%) patients with diabetes at baseline.
The safety profile observed in patients with active psoriatic arthritis treated with XELJANZ was consistent with the safety profile observed in rheumatoid arthritis patients.
Ulcerative Colitis
XELJANZ has been studied in patients with moderately to severely active UC in 4 randomized, double-blind, placebo-controlled trials (UC-I, UC-II, UC-III, and dose-ranging UC-V) and an open-label long-term extension study (UC-IV) [see Clinical Studies (14.3)].
Adverse reactions reported in ≥5% of patients treated with either 5 mg or 10 mg twice daily of XELJANZ and ≥1% greater than reported in patients receiving placebo in either the induction or maintenance clinical trials were: nasopharyngitis, elevated cholesterol levels, headache, upper respiratory tract infection, increased blood creatine phosphokinase, rash, diarrhea, and herpes zoster.
Induction Trials (Study UC-I, UC-II, and UC-V):
Common adverse reactions reported in ≥2% of patients treated with XELJANZ 10 mg twice daily and ≥1% greater than that reported in patients receiving placebo in the 3 induction trials were: headache, nasopharyngitis, elevated cholesterol levels, acne, increased blood creatine phosphokinase, and pyrexia.
Maintenance Trial (Study UC-III)
Common adverse reactions reported in ≥4% of patients treated with either dose of XELJANZ and ≥1% greater than reported in patients receiving placebo are shown in Table 4.
Table 4: Common Adverse Reactions* in -UC Patients during the Maintenance Trial (Study UC-III) Preferred Term XELJANZ
5 mg Twice DailyXELJANZ
10 mg Twice DailyPlacebo N = 198
(%)N = 196
(%)N = 198
(%)- * reported in ≥4% of patients treated with either dose of XELJANZ and ≥1% greater than reported for placebo.
- † includes hypercholesterolemia, hyperlipidemia, blood cholesterol increased, dyslipidemia, blood triglycerides increased, low density lipoprotein increased, low density lipoprotein abnormal, or lipids increased.
Nasopharyngitis 10 14 6 Elevated cholesterol levels† 5 9 1 Headache 9 3 6 Upper respiratory tract infection 7 6 4 Increased blood creatine phosphokinase 3 7 2 Rash 3 6 4 Diarrhea 2 5 3 Herpes zoster 1 5 1 Gastroenteritis 3 4 3 Anemia 4 2 2 Nausea 1 4 3 In the long-term extension study, malignancies (including solid cancers, lymphomas and NMSC) were observed more often in patients treated with XELJANZ 10 mg twice daily [see Warnings and Precautions (5.3)]. Four cases of pulmonary embolism were reported in patients taking XELJANZ 10 mg twice daily, including one fatality in a patient with advanced cancer [see Warnings and Precautions (5.4)].
Dose-dependent adverse reactions seen in patients treated with XELJANZ 10 mg twice daily, in comparison to 5 mg twice daily, include the following: herpes zoster infections, serious infections, and NMSC [see Warnings and Precautions (5.1, 5.3)].
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of XELJANZ/XELJANZ XR. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune system disorders: Drug hypersensitivity (events such as angioedema and urticaria have been observed).
-
7 DRUG INTERACTIONS
Table 5 includes drugs with clinically important drug interactions when administered concomitantly with XELJANZ/XELJANZ XR and instructions for preventing or managing them.
Table 5: Clinical Relevant Interactions Affecting XELJANZ and XELJANZ XR When Coadministered with Other Drugs Strong CP3A4 Inhibitors (e.g., ketoconazole) Clinical Impact Increased exposure to tofacitinib Intervention Dosage adjustment of XELJANZ/XELJANZ XR is recommended [see Dosage and Administration (2), Clinical Pharmacology, Figure 3 (12.3)] Moderate CYP3A4 Inhibitors Coadministered with Strong CYP2C19 Inhibitors (e.g., fluconazole) Clinical Impact Increased exposure to tofacitinib Intervention Dosage adjustment of XELJANZ/XELJANZ XR is recommended [see Dosage and Administration (2), Clinical Pharmacology, Figure 3 (12.3)] Strong CYP3A4 Inducers (e.g., rifampin) Clinical Impact Decreased exposure to tofacitinib and may result in loss of or reduced clinical response Intervention Coadministration with XELJANZ/XELJANZ XR is not recommended [see Clinical Pharmacology, Figure 3 (12.3)] Immunosuppressive Drugs (e.g., azathioprine, tacrolimus, cyclosporine) Clinical Impact Risk of added immunosuppression; coadministration with biologic DMARDs or potent immunosuppressants has not been studied in patients with rheumatoid arthritis, psoriatic arthritis, or UC. Intervention Coadministration with XELJANZ/XELJANZ XR is not recommended [see Indications and Usage (1), Clinical Pharmacology, Figure 3 (12.3)] -
8 USE IN SPECIFIC POPULATIONS
All information provided in this section is applicable to XELJANZ and XELJANZ XR as they contain the same active ingredient (tofacitinib).
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to XELJANZ/XELJANZ XR during pregnancy. Patients should be encouraged to enroll in the XELJANZ/XELJANZ XR pregnancy registry if they become pregnant. To enroll or obtain information from the registry, patients can call the toll free number 1-877-311-8972.
Risk Summary
Available data with XELJANZ/XELJANZ XR use in pregnant women are insufficient to establish a drug associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. There are risks to the mother and the fetus associated with rheumatoid arthritis and UC in pregnancy (see Clinical Considerations). In animal reproduction studies, fetocidal and teratogenic effects were noted when pregnant rats and rabbits received tofacitinib during the period of organogenesis at exposures multiples of 73-times and 6.3-times the maximum recommended dose of 10 mg twice daily, respectively. Further, in a peri- and post-natal study in rats, tofacitinib resulted in reductions in live litter size, postnatal survival, and pup body weights at exposure multiples of approximately 73-times the recommended dose of 5 mg twice daily and approximately 36 times the maximum recommended dose of 10 mg twice daily, respectively (see Data).
The estimated background risks of major birth defects and miscarriage for the indicated populations are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The background risks in the U.S. general population of major birth defects and miscarriages are 2 to 4% and 15 to 20% of clinically recognized pregnancies, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Published data suggest that increased disease activity is associated with the risk of developing adverse pregnancy outcomes in women with rheumatoid arthritis or ulcerative colitis. Adverse pregnancy outcomes include preterm delivery (before 37 weeks of gestation), low birth weight (less than 2500 g) infants, and small for gestational age at birth.
Data
Animal Data
In a rat embryofetal developmental study, in which pregnant rats received tofacitinib during organogenesis, tofacitinib was teratogenic at exposure levels approximately 146 times the recommended dose of 5 mg twice daily, and approximately 73 times the maximum recommended dose of 10 mg twice daily (on an AUC basis at oral doses of 100 mg/kg/day in rats). Teratogenic effects consisted of external and soft tissue malformations of anasarca and membranous ventricular septal defects, respectively; and skeletal malformations or variations (absent cervical arch; bent femur, fibula, humerus, radius, scapula, tibia, and ulna; sternoschisis; absent rib; misshapen femur; branched rib; fused rib; fused sternebra; and hemicentric thoracic centrum). In addition, there was an increase in post-implantation loss, consisting of early and late resorptions, resulting in a reduced number of viable fetuses. Mean fetal body weight was reduced. No developmental toxicity was observed in rats at exposure levels approximately 58 times the recommended dose of 5 mg twice daily, and approximately 29 times the maximum recommended dose of 10 mg twice daily (on an AUC basis at oral doses of 30 mg/kg/day in pregnant rats).
In a rabbit embryofetal developmental study in which pregnant rabbits received tofacitinib during the period of organogenesis, tofacitinib was teratogenic at exposure levels approximately 13 times the recommended dose of 5 mg twice daily, and approximately 6.3 times the maximum recommended dose of 10 mg twice daily (on an AUC basis at oral doses of 30 mg/kg/day in rabbits) in the absence of signs of maternal toxicity. Teratogenic effects included thoracogastroschisis, omphalocele, membranous ventricular septal defects, and cranial/skeletal malformations (microstomia, microphthalmia), mid-line and tail defects. In addition, there was an increase in post-implantation loss associated with late resorptions. No developmental toxicity was observed in rabbits at exposure levels approximately 3 times the recommended dose of 5 mg twice daily, and approximately 1.5 times the maximum recommended dose of 10 mg twice daily (on an AUC basis at oral doses of 10 mg/kg/day in pregnant rabbits).
In a peri- and postnatal development study in pregnant rats that received tofacitinib from gestation day 6 through day 20 of lactation, there were reductions in live litter size, postnatal survival, and pup body weights at exposure levels approximately 73 times the recommended dose of 5 mg twice daily, and approximately 36 times the maximum recommended dose of 10 mg twice daily (on an AUC basis at oral doses of 50 mg/kg/day in rats). There was no effect on behavioral and learning assessments, sexual maturation or the ability of the F1 generation rats to mate and produce viable F2 generation fetuses in rats at exposure levels approximately 17 times the recommended dose of 5 mg twice daily, and approximately 8.3 times the maximum recommended dose of 10 mg twice daily (on an AUC basis at oral doses of 10 mg/kg/day in rats).
8.2 Lactation
Risk Summary
There are no data on the presence of tofacitinib in human milk, the effects on a breastfed infant, or the effects on milk production. Tofacitinib is present in the milk of lactating rats (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk. Given the serious adverse reactions seen in adults treated with XELJANZ/XELJANZ XR, such as increased risk of serious infections, advise patients that breastfeeding is not recommended during treatment and for at least 18 hours after the last dose of XELJANZ or 36 hours after the last dose of XELJANZ XR (approximately 6 elimination half-lives).
8.3 Females and Males of Reproductive Potential
Contraception
Females
In an animal reproduction study, tofacitinib at AUC multiples of 13 times the recommended dose of 5 mg twice daily and 6.3 times the maximum recommended dose of 10 mg twice daily demonstrated adverse embryo-fetal findings [see Use in Specific Populations (8.1)]. However, there is uncertainty as to how these animal findings relate to females of reproductive potential treated with the recommended clinical dose. Consider pregnancy planning and prevention for females of reproductive potential.
Infertility
Females
Based on findings in rats, treatment with XELJANZ/XELJANZ XR may result in reduced fertility in females of reproductive potential. It is not known if this effect is reversible [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of XELJANZ/XELJANZ XR in pediatric patients have not been established.
8.5 Geriatric Use
Of the 3315 patients who enrolled in rheumatoid arthritis Studies I to V, a total of 505 rheumatoid arthritis patients were 65 years of age and older, including 71 patients 75 years and older. The frequency of serious infection among XELJANZ-treated subjects 65 years of age and older was higher than among those under the age of 65.
Of the 1156 XELJANZ-treated patients in the UC program, a total of 77 patients (7%) were 65 years of age or older. The number of patients aged 65 years and older was not sufficient to determine whether they responded differently from younger patients.
As there is a higher incidence of infections in the elderly population in general, caution should be used when treating the elderly [see Warnings and Precautions (5.1)].
8.6 Use in Diabetics
As there is a higher incidence of infection in diabetic population in general, caution should be used when treating patients with diabetes.
8.7 Renal Impairment
Moderate and Severe Impairment
XELJANZ-treated patients with moderate or severe renal impairment had greater tofacitinib blood concentrations than XELJANZ-treated patients with normal renal function. Therefore, dosage adjustment of XELJANZ/XELJANZ XR is recommended in patients with moderate or severe renal impairment (including but not limited to those with severe insufficiency who are undergoing hemodialysis) [see Dosage and Administration (2.2, 2.3)].
8.8 Hepatic Impairment
Severe Impairment
XELJANZ/XELJANZ XR has not been studied in patients with severe hepatic impairment; therefore, use of XELJANZ/XELJANZ XR in patients with severe hepatic impairment is not recommended.
Moderate Impairment
XELJANZ-treated patients with moderate hepatic impairment had greater tofacitinib blood concentration than XELJANZ-treated patients with normal hepatic function [see Clinical Pharmacology (12.3)]. Higher blood concentrations may increase the risk of some adverse reactions. Therefore, dosage adjustment of XELJANZ/XELJANZ XR is recommended in patients with moderate hepatic impairment [see Dosage and Administration (2.2, 2.3)].
-
10 OVERDOSAGE
There is no specific antidote for overdose with XELJANZ/XELJANZ XR. In case of an overdose, it is recommended that the patient be monitored for signs and symptoms of adverse reactions.
In a study in subjects with end stage renal disease (ESRD) undergoing hemodialysis, plasma tofacitinib concentrations declined more rapidly during the period of hemodialysis and dialyzer efficiency, calculated as dialyzer clearance/blood flow entering the dialyzer, was high [mean (SD) = 0.73 (0.15)]. However, due to the significant non-renal clearance of tofacitinib, the fraction of total elimination occurring by hemodialysis was small, and thus limits the value of hemodialysis for treatment of overdose with XELJANZ/XELJANZ XR.
-
11 DESCRIPTION
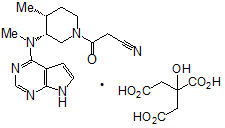
XELJANZ/XELJANZ XR (tofacitinib) tablets are formulated with the citrate salt of tofacitinib, a JAK inhibitor.
Tofacitinib citrate is a white to off-white powder with the following chemical name: (3R,4R)-4-methyl-3-(methyl-7H-pyrrolo [2,3-d]pyrimidin-4-ylamino)-ß-oxo-1-piperidinepropanenitrile, 2-hydroxy-1,2,3-propanetricarboxylate (1:1).
The solubility of tofacitinib citrate in water is 2.9 mg/mL.
Tofacitinib citrate has a molecular weight of 504.5 Daltons (or 312.4 Daltons as the tofacitinib free base) and a molecular formula of C16H20N6O∙C6H8O7. The chemical structure of tofacitinib citrate is:
XELJANZ is supplied for oral administration as a 5 mg white round, immediate-release film-coated tablet. Each tablet of XELJANZ contains 5 mg tofacitinib (equivalent to 8.08 mg tofacitinib citrate) and the following inactive ingredients: croscarmellose sodium, HPMC 2910/Hypromellose 6cP, lactose monohydrate, macrogol/PEG3350, magnesium stearate, microcrystalline cellulose, titanium dioxide, and triacetin.
XELJANZ is supplied for oral administration as a 10 mg blue round, immediate-release film-coated tablet. Each 10 mg tablet of XELJANZ contains 10 mg tofacitinib (equivalent to 16.16 mg of tofacitinib citrate) and the following inactive ingredients: croscarmellose sodium, FD&C Blue #1/Brilliant Blue FCF Aluminum Lake, FD&C Blue #2/Indigo Carmine Aluminum Lake, HPMC 2910/Hypromellose 6cP, lactose monohydrate, macrogol/PEG3350, magnesium stearate, microcrystalline cellulose, titanium dioxide, and triacetin.
XELJANZ XR is supplied for oral administration as a 11 mg pink, oval, extended-release film-coated tablet with a drilled hole at one end of the tablet band. Each 11 mg tablet of XELJANZ XR contains 11 mg tofacitinib (equivalent to 17.77 mg tofacitinib citrate) and the following inactive ingredients: cellulose acetate, copovidone, hydroxyethyl cellulose, hydroxypropyl cellulose, HPMC 2910/Hypromellose, magnesium stearate, red iron oxide, sorbitol, titanium dioxide and triacetin. Printing ink contains, ammonium hydroxide, ferrosoferric oxide/black iron oxide, propylene glycol, and shellac glaze.
XELJANZ XR is supplied for oral administration as a 22 mg beige, oval, extended-release film-coated tablet with a drilled hole at one end of the tablet band. Each 22 mg tablet of XELJANZ XR contains 22 mg tofacitinib (equivalent to 35.54 mg tofacitinib citrate) and the following inactive ingredients: cellulose acetate, copovidone, FD&C Blue #2 Aluminum Lake, hydroxyethyl cellulose, hydroxypropyl cellulose, HPMC 2910/Hypromellose, magnesium stearate, red iron oxide, sorbitol, titanium dioxide, triacetin, and yellow iron oxide. Printing ink contains ammonium hydroxide, ferrosoferric oxide/black iron oxide, propylene glycol, and shellac glaze.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tofacitinib is a Janus kinase (JAK) inhibitor. JAKs are intracellular enzymes which transmit signals arising from cytokine or growth factor-receptor interactions on the cellular membrane to influence cellular processes of hematopoiesis and immune cell function. Within the signaling pathway, JAKs phosphorylate and activate Signal Transducers and Activators of Transcription (STATs) which modulate intracellular activity including gene expression. Tofacitinib modulates the signaling pathway at the point of JAKs, preventing the phosphorylation and activation of STATs. JAK enzymes transmit cytokine signaling through pairing of JAKs (e.g., JAK1/JAK3, JAK1/JAK2, JAK1/TyK2, JAK2/JAK2). Tofacitinib inhibited the in vitro activities of JAK1/JAK2, JAK1/JAK3, and JAK2/JAK2 combinations with IC50 of 406, 56, and 1377 nM, respectively. However, the relevance of specific JAK combinations to therapeutic effectiveness is not known.
12.2 Pharmacodynamics
Treatment with XELJANZ was associated with dose-dependent reductions of circulating CD16/56+ natural killer cells, with estimated maximum reductions occurring at approximately 8–10 weeks after initiation of therapy. These changes generally resolved within 2–6 weeks after discontinuation of treatment. Treatment with XELJANZ was associated with dose-dependent increases in B cell counts. Changes in circulating T-lymphocyte counts and T-lymphocyte subsets (CD3+, CD4+ and CD8+) were small and inconsistent. The clinical significance of these changes is unknown.
Total serum IgG, IgM, and IgA levels after 6-month dosing in patients with rheumatoid arthritis were lower than placebo; however, changes were small and not dose-dependent.
After treatment with XELJANZ in patients with rheumatoid arthritis, rapid decreases in serum C-reactive protein (CRP) were observed and maintained throughout dosing. Changes in CRP observed with XELJANZ treatment do not reverse fully within 2 weeks after discontinuation, indicating a longer duration of pharmacodynamic activity compared to the pharmacokinetic half-life.
Similar changes in T cells, B cells, and serum CRP have been observed in patients with active psoriatic arthritis although reversibility was not assessed. Total serum immunoglobulins were not assessed in patients with active psoriatic arthritis.
12.3 Pharmacokinetics
XELJANZ
Following oral administration of XELJANZ, peak plasma concentrations are reached within 0.5–1 hour, elimination half-life is about 3 hours and a dose-proportional increase in systemic exposure was observed in the therapeutic dose range. Steady state concentrations are achieved in 24–48 hours with negligible accumulation after twice daily administration.
XELJANZ XR
Following oral administration of XELJANZ XR, peak plasma concentrations are reached at 4 hours and half-life is about 6 to 8 hours. Steady state concentrations are achieved within 48 hours with negligible accumulation after once daily administration.
Table 6: Pharmacokinetic Parameters of XELJANZ/ XELJANZ XR Following Multiple Oral Dosing PK Parameters* (CV%) XELJANZ XELJANZ XR Dosing Regimen 5 mg
Twice Daily10 mg
Twice Daily11 mg
Once Daily22 mg
Once DailyAbbreviations: AUC24 = area under the concentration-time profile from time 0 to 24 hours; Cmax = maximum plasma concentration; Cmin = minimum plasma concentration; Tmax = time to Cmax; CV = Coefficient of variation. - * Values represent the geometric mean, except T max, for which is the median (range) is shown.
- † Values beyond 12 hours were after the evening dose which was administered 12 hours after the morning dose of twice-daily XELJANZ
AUC24 (ng.hr/mL) 263.4 (15) 539.6 (22) 269.0 (18) 596.6 (19) Cmax (ng/mL) 42.7 (26) 84.7 (18) 38.2 (15) 83.8 (25) Cmin (ng/mL) 1.41 (40) 3.10 (54) 1.07 (69) 3.11 (43) Tmax (hours) 1.0
(0.5 to14.0†)0.8
(0.5 to 14.0†)4.0
(3.0 to 4.0)4.0
(2.0 to 4.0)Absorption
XELJANZ
The absolute oral bioavailability of XELJANZ is 74%. Coadministration of XELJANZ with a high-fat meal resulted in no changes in AUC while Cmax was reduced by 32%. In clinical trials, XELJANZ was administered without regard to meals [see Dosage and Administration (2.1)].
Distribution
After intravenous administration, the volume of distribution is 87 L. The protein binding of tofacitinib is approximately 40%. Tofacitinib binds predominantly to albumin and does not appear to bind to α1-acid glycoprotein. Tofacitinib distributes equally between red blood cells and plasma.
Metabolism and Excretion
Clearance mechanisms for tofacitinib are approximately 70% hepatic metabolism and 30% renal excretion of the parent drug. The metabolism of tofacitinib is primarily mediated by CYP3A4 with minor contribution from CYP2C19. In a human radiolabeled study, more than 65% of the total circulating radioactivity was accounted for by unchanged tofacitinib, with the remaining 35% attributed to 8 metabolites, each accounting for less than 8% of total radioactivity. The pharmacologic activity of tofacitinib is attributed to the parent molecule.
Pharmacokinetics in Patient Populations
Population pharmacokinetic analyses indicated that pharmacokinetic characteristics were similar between patients with rheumatoid arthritis, psoriatic arthritis, and UC. The coefficient of variation (%) in AUC of tofacitinib were generally similar across different disease patients, ranging from 22% to 34% (Table 7).
Table 7. XELJANZ Exposure in Patient Populations at 5 mg Twice Daily and 10 mg Twice Daily Doses Pharmacokinetic Parameters*
Geometric Mean (CV%)XELJANZ 5 mg
Twice DailyXELJANZ 10 mg Twice Daily Rheumatoid Arthritis Psoriatic Arthritis Ulcerative Colitis Ulcerative Colitis Abbreviations: AUC0–24,ss=area under the plasma concentration-time curve over 24 hours at steady state; CV=coefficient of variation. - * Pharmacokinetic parameters estimated based on population pharmacokinetic analysis.
AUC0–24,ss (ng∙h/mL) 504
(22.0%)419
(34.1%)423
(22.6%)807
(24.6%)Specific Populations
Covariate evaluation as part of population PK analyses in patient populations indicated no clinically relevant change in tofacitinib exposure, after accounting for differences in renal function (i.e., creatinine clearance) between patients, based on age, weight, gender and race (Figure 1). An approximately linear relationship between body weight and volume of distribution was observed, resulting in higher peak (Cmax) and lower trough (Cmin) concentrations in lighter patients. However, this difference is not considered to be clinically relevant.
The effect of renal and hepatic impairment and other intrinsic factors on the pharmacokinetics of tofacitinib is shown in Figure 1.
Figure 1: Impact of Intrinsic Factors on Tofacitinib Pharmacokinetics
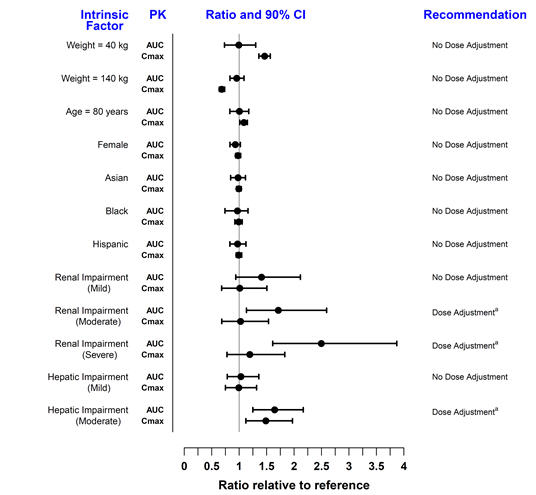
Note: Reference values for weight, age, gender, and race comparisons are 70 kg, 55 years, male, and white, respectively; reference groups for renal and hepatic impairment data are subjects with normal renal and hepatic function.
a [see Dosage and Administration (2.2, 2.3)] for dosage adjustment in RA, PsA, and UC patients.
In subjects with ESRD maintained on hemodialysis, mean AUC was approximately 40% higher compared with historical healthy subject data, consistent with approximately 30% contribution of renal clearance to the total clearance of tofacitinib. Dose adjustment is recommended in ESRD patients maintained on hemodialysis ([see Dosage and Administration (2.2, 2.3)] for dosage adjustment in RA, PsA, and UC patients).
Drug Interaction Studies
Potential for XELJANZ/XELJANZ XR to Influence the PK of Other Drugs
In vitro studies indicate that tofacitinib does not significantly inhibit or induce the activity of the major human drug-metabolizing CYPs (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) at concentrations corresponding to the steady state Cmax of a 10 mg twice daily dose. These in vitro results were confirmed by a human drug interaction study showing no changes in the pharmacokinetics of midazolam, a highly sensitive CYP3A4 substrate, when coadministered with XELJANZ.
In vitro studies indicate that tofacitinib does not significantly inhibit the activity of the major human drug-metabolizing uridine 5'-diphospho-glucuronosyltransferases (UGTs) [UGT1A1, UGT1A4, UGT1A6, UGT1A9, and UGT2B7] at concentrations exceeding 250 times the steady state Cmax of a 10 mg twice daily dose.
In rheumatoid arthritis patients, the oral clearance of tofacitinib does not vary with time, indicating that tofacitinib does not normalize CYP enzyme activity in rheumatoid arthritis patients. Therefore, coadministration with XELJANZ/XELJANZ XR is not expected to result in clinically relevant increases in the metabolism of CYP substrates in rheumatoid arthritis patients.
In vitro data indicate that the potential for tofacitinib to inhibit transporters such as P-glycoprotein, organic anionic or cationic transporters at therapeutic concentrations is low.
Dosing recommendations for coadministered drugs following administration with XELJANZ/XELJANZ XR are shown in Figure 2.
Figure 2: Impact of Tofacitinib on the Pharmacokinetics of Other Drugs
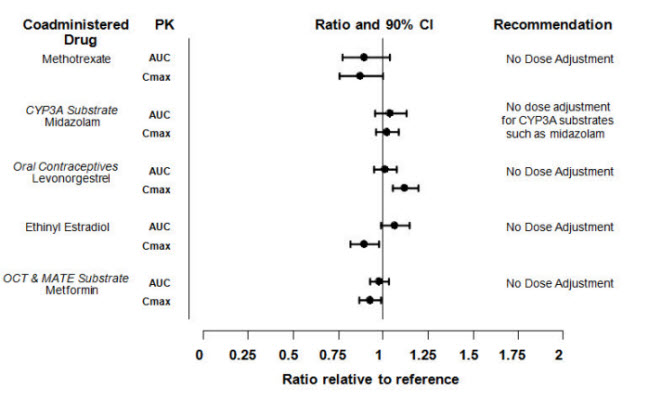
Note: Reference group is administration of concomitant medication alone; OCT = Organic Cationic Transporter; MATE = Multidrug and Toxic Compound Extrusion
Potential for Other Drugs to Influence the Pharmacokinetics of Tofacitinib
Since tofacitinib is metabolized by CYP3A4, interaction with drugs that inhibit or induce CYP3A4 is likely. Inhibitors of CYP2C19 alone or P-glycoprotein are unlikely to substantially alter the pharmacokinetics of tofacitinib (see Figure 3).
Figure 3: Impact of Other Drugs on the Pharmacokinetics of Tofacitinib
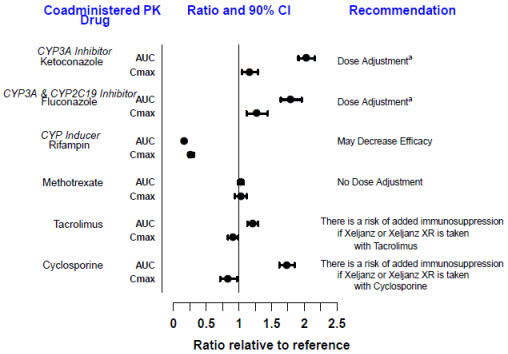
Note: Reference group is administration of tofacitinib alone.
a [see Dosage and Administration (2.2, 2.3), Drug Interactions (7)]. -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 39-week toxicology study in monkeys, tofacitinib at exposure levels approximately 6 times the recommended dose of 5 mg twice daily, and approximately 3 times the 10 mg twice daily dose (on an AUC basis at oral doses of 5 mg/kg twice daily) produced lymphomas. No lymphomas were observed in this study at exposure levels 1 times the recommended dose of 5 mg twice daily, and approximately 0.5 times the 10 mg twice daily dose (on an AUC basis at oral doses of 1 mg/kg twice daily).
The carcinogenic potential of tofacitinib was assessed in 6-month rasH2 transgenic mouse carcinogenicity and 2-year rat carcinogenicity studies. Tofacitinib, at exposure levels approximately 34 times the recommended dose of 5 mg twice daily, and approximately 17 times the 10 mg twice daily dose (on an AUC basis at oral doses of 200 mg/kg/day) was not carcinogenic in mice.
In the 24-month oral carcinogenicity study in Sprague-Dawley rats, tofacitinib caused benign Leydig cell tumors, hibernomas (malignancy of brown adipose tissue), and benign thymomas at doses greater than or equal to 30 mg/kg/day (approximately 42 times the exposure levels at the recommended dose of 5 mg twice daily, and approximately 21 times the 10 mg twice daily dose on an AUC basis). The relevance of benign Leydig cell tumors to human risk is not known.
Tofacitinib was not mutagenic in the bacterial reverse mutation assay. It was positive for clastogenicity in the in vitro chromosome aberration assay with human lymphocytes in the presence of metabolic enzymes, but negative in the absence of metabolic enzymes. Tofacitinib was negative in the in vivo rat micronucleus assay and in the in vitro CHO-HGPRT assay and the in vivo rat hepatocyte unscheduled DNA synthesis assay.
In rats, tofacitinib at exposure levels approximately 17 times the recommended dose of 5 mg twice daily, and approximately 8.3 times the 10 mg twice daily dose (on an AUC basis at oral doses of 10 mg/kg/day) reduced female fertility due to increased post-implantation loss. There was no impairment of female rat fertility at exposure levels of tofacitinib equal to the recommended dose of 5 mg twice daily, and approximately 0.5 times the 10 mg twice daily dose (on an AUC basis at oral doses of 1 mg/kg/day). Tofacitinib exposure levels at approximately 133 times the recommended dose of 5 mg twice daily, and approximately 67 times the 10 mg twice daily dose (on an AUC basis at oral doses of 100 mg/kg/day) had no effect on male fertility, sperm motility, or sperm concentration.
-
14 CLINICAL STUDIES
14.1 Rheumatoid Arthritis
The XELJANZ clinical development program included two dose-ranging trials and five confirmatory trials. Although other doses have been studied, the recommended dose of XELJANZ is 5 mg twice daily. XELJANZ 10 mg twice daily is not recommended for the treatment of rheumatoid arthritis [see Dosage and Administration (2.2)].
Dose-Ranging Trials
Dose selection for XELJANZ was based on two pivotal dose-ranging trials.
Dose-Ranging Study 1 was a 6-month monotherapy trial in 384 patients with active rheumatoid arthritis who had an inadequate response to a DMARD. Patients who previously received adalimumab therapy were excluded. Patients were randomized to 1 of 7 monotherapy treatments: XELJANZ 1, 3, 5, 10 or 15 mg twice daily, adalimumab 40 mg subcutaneously every other week for 10 weeks followed by XELJANZ 5 mg twice daily for 3 months, or placebo.
Dose-Ranging Study 2 was a 6-month trial in which 507 patients with active rheumatoid arthritis who had an inadequate response to MTX alone received one of 6 dose regimens of XELJANZ (20 mg once daily; 1, 3, 5, 10 or 15 mg twice daily), or placebo added to background MTX.
The results of XELJANZ-treated patients achieving ACR20 responses in Studies 1 and 2 are shown in Figure 4. Although a dose-response relationship was observed in Study 1, the proportion of patients with an ACR20 response did not clearly differ between the 10 mg and 15 mg doses. In Study 2, a smaller proportion of patients achieved an ACR20 response in the placebo and XELJANZ 1 mg groups compared to patients treated with the other XELJANZ doses. However, there was no difference in the proportion of responders among patients treated with XELJANZ 3, 5, 10, 15 mg twice daily or 20 mg once daily doses.
Figure 4: Proportion of Patients with ACR20 Response at Month 3 in Dose-Ranging Studies 1 and 2
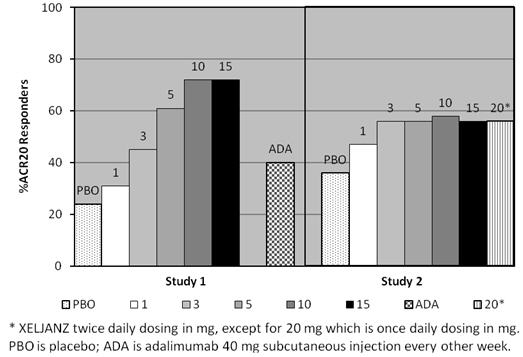
Study 1 was a dose-ranging monotherapy trial not designed to provide comparative effectiveness data and should not be interpreted as evidence of superiority to adalimumab.
Confirmatory Trials
Study RA-I (NCT00814307) was a 6-month monotherapy trial in which 610 patients with moderate to severe active rheumatoid arthritis who had an inadequate response to a DMARD (nonbiologic or biologic) received XELJANZ 5 or 10 mg twice daily or placebo. At the Month 3 visit, all patients randomized to placebo treatment were advanced in a blinded fashion to a second predetermined treatment of XELJANZ 5 or 10 mg twice daily. The primary endpoints at Month 3 were the proportion of patients who achieved an ACR20 response, changes in Health Assessment Questionnaire – Disability Index (HAQ-DI), and rates of Disease Activity Score DAS28-4(ESR) less than 2.6.
Study RA-II (NCT00856544) was a 12-month trial in which 792 patients with moderate to severe active rheumatoid arthritis who had an inadequate response to a nonbiologic DMARD received XELJANZ 5 or 10 mg twice daily or placebo added to background DMARD treatment (excluding potent immunosuppressive treatments such as azathioprine or cyclosporine). At the Month 3 visit, nonresponding patients were advanced in a blinded fashion to a second predetermined treatment of XELJANZ 5 or 10 mg twice daily. At the end of Month 6, all placebo patients were advanced to their second predetermined treatment in a blinded fashion. The primary endpoints were the proportion of patients who achieved an ACR20 response at Month 6, changes in HAQ-DI at Month 3, and rates of DAS28-4(ESR) less than 2.6 at Month 6.
Study RA-III (NCT00853385) was a 12-month trial in 717 patients with moderate to severe active rheumatoid arthritis who had an inadequate response to MTX. Patients received XELJANZ 5 or 10 mg twice daily, adalimumab 40 mg subcutaneously every other week, or placebo added to background MTX. Placebo patients were advanced as in Study II. The primary endpoints were the proportion of patients who achieved an ACR20 response at Month 6, HAQ-DI at Month 3, and DAS28-4(ESR) less than 2.6 at Month 6.
Study RA-IV (NCT00847613) was a 2-year trial with a planned analysis at 1 year in which 797 patients with moderate to severe active rheumatoid arthritis who had an inadequate response to MTX received XELJANZ 5 or 10 mg twice daily or placebo added to background MTX. Placebo patients were advanced as in Study II. The primary endpoints were the proportion of patients who achieved an ACR20 response at Month 6, mean change from baseline in van der Heijde-modified total Sharp Score (mTSS) at Month 6, HAQ-DI at Month 3, and DAS28-4(ESR) less than 2.6 at Month 6.
Study RA-V (NCT00960440) was a 6-month trial in which 399 patients with moderate to severe active rheumatoid arthritis who had an inadequate response to at least one approved TNF blocking biologic agent received XELJANZ 5 or 10 mg twice daily or placebo added to background MTX. At the Month 3 visit, all patients randomized to placebo treatment were advanced in a blinded fashion to a second predetermined treatment of XELJANZ 5 or 10 mg twice daily. The primary endpoints at Month 3 were the proportion of patients who achieved an ACR20 response, HAQ-DI, and DAS28-4(ESR) less than 2.6.
Study RA-VI (NCT01039688) was a 2-year monotherapy trial with a planned analysis at 1 year in which 952 MTX-naïve patients with moderate to severe active rheumatoid arthritis received XELJANZ 5 or 10 mg twice daily or MTX dose-titrated over 8 weeks to 20 mg weekly. The primary endpoints were mean change from baseline in van der Heijde-modified Total Sharp Score (mTSS) at Month 6 and the proportion of patients who achieved an ACR70 response at Month 6.
Clinical Response
The percentages of XELJANZ-treated patients achieving ACR20, ACR50, and ACR70 responses in Studies RA-I, IV, and V are shown in Table 8. Similar results were observed with Studies RA-II and III. In trials RA-I through V, patients treated with 5 mg twice daily XELJANZ had higher ACR20, ACR50, and ACR70 response rates versus placebo, with or without background DMARD treatment, at Month 3 and Month 6. Higher ACR20 response rates were observed within 2 weeks compared to placebo. In the 12-month trials, ACR response rates in XELJANZ-treated patients were consistent at 6 and 12 months.
Table 8: Proportion of Patients with an ACR Response Percent of Patients Monotherapy in Nonbiologic or Biologic DMARD Inadequate Responders* MTX Inadequate Responders† TNF Blocker Inadequate Responders‡ Study I Study IV Study V N§ PBO XELJANZ
5 mg Twice DailyPBO + MTX XELJANZ 5 mg Twice Daily + MTX PBO + MTX XELJANZ 5 mg Twice Daily + MTX 122 243 160 321 132 133 - * Inadequate response to at least one DMARD (biologic or nonbiologic) due to lack of efficacy or toxicity.
- † Inadequate response to MTX defined as the presence of sufficient residual disease activity to meet the entry criteria.
- ‡ Inadequate response to a least one TNF blocker due to lack of efficacy and/or intolerance.
- § N is number of randomized and treated patients.
- ¶ NA Not applicable, as data for placebo treatment is not available beyond 3 months in Studies I and V due to placebo advancement.
ACR20 Month 3 26% 59% 27% 55% 24% 41% Month 6 NA¶ 69% 25% 50% NA 51% ACR50 Month 3 12% 31% 8% 29% 8% 26% Month 6 NA 42% 9% 32% NA 37% ACR70 Month 3 6% 15% 3% 11% 2% 14% Month 6 NA 22% 1% 14% NA 16% In Study RA-IV, a greater proportion of patients treated with XELJANZ 5 mg twice daily plus MTX achieved a low level of disease activity as measured by a DAS28-4(ESR) less than 2.6 at 6 months compared to those treated with MTX alone (Table 9).
Table 9: Proportion of Patients with DAS28-4(ESR) Less Than 2.6 with Number of Residual Active Joints Study IV DAS28-4(ESR) Less Than 2.6 Placebo + MTX XELJANZ 5 mg Twice Daily + MTX 160 321 Proportion of responders at Month 6 (n) 1% (2) 6% (19) Of responders, proportion with 0 active joints (n) 50% (1) 42% (8) Of responders, proportion with 1 active joint (n) 0 5% (1) Of responders, proportion with 2 active joints (n) 0 32% (6) Of responders, proportion with 3 or more active joints (n) 50% (1) 21% (4) The results of the components of the ACR response criteria for Study RA-IV are shown in Table 10. Similar results were observed for XELJANZ in Studies RA-I, II, III, V, and VI.
Table 10: Components of ACR Response at Month 3 Study IV XELJANZ 5 mg Twice Daily + MTX Placebo + MTX N=321 N=160 Component (mean) * Baseline Month 3* Baseline Month 3* - * Data shown is mean (Standard Deviation) at Month 3.
- † Visual analog scale: 0 = best, 100 = worst.
- ‡ Health Assessment Questionnaire Disability Index: 0 = best, 3 = worst; 20 questions; categories: dressing and grooming, arising, eating, walking, hygiene, reach, grip, and activities.
Number of tender joints
(0–68)24
(14)13
(14)23
(13)18
(14)Number of swollen joints
(0–66)14
(8)6
(8)14
(9)10
(9)Pain† 58
(23)34
(23)55
(24)47
(24)Patient global assessment† 58
(24)35
(23)54
(23)47
(24)Disability index
(HAQ-DI)‡1.41
(0.68)0.99
(0.65)1.32
(0.67)1.19
(0.68)Physician global assessment† 59
(16)30
(19)56
(18)43
(22)CRP (mg/L) 15.3
(19.0)7.1
(19.1)13.7
(14.9)14.6
(18.7)The percent of ACR20 responders by visit for Study RA-IV is shown in Figure 5. Similar responses were observed for XELJANZ in Studies RA-I, II, III, V, and VI.
Figure 5: Percentage of ACR20 Responders by Visit for Study RA-IV
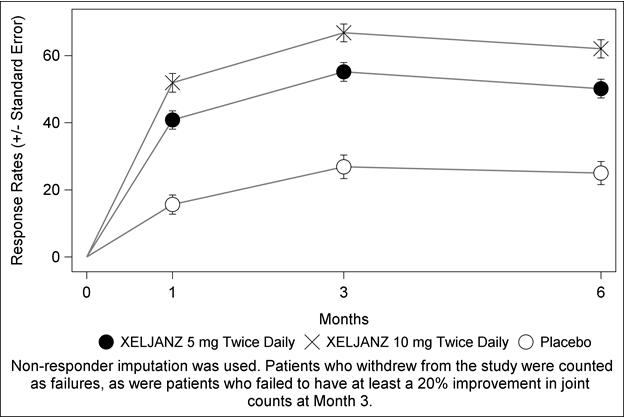
Radiographic Response
Two studies were conducted to evaluate the effect of XELJANZ on structural joint damage. In Study RA-IV and Study RA-VI, progression of structural joint damage was assessed radiographically and expressed as change from baseline in mTSS and its components, the erosion score and joint space narrowing score, at Months 6 and 12. The proportion of patients with no radiographic progression (mTSS change less than or equal to 0) was also assessed.
In Study RA-IV, XELJANZ 5 mg twice daily reduced the mean progression of structural damage (not statistically significant) as shown in Table 10. Analyses of erosion and joint space narrowing scores were consistent with the overall results.
In the placebo plus MTX group, 74% of patients experienced no radiographic progression at Month 6 compared to 84% of patients treated with XELJANZ plus MTX 5 mg twice daily.
In Study RA-VI, XELJANZ monotherapy inhibited the progression of structural damage compared to MTX at Months 6 and 12 as shown in Table 10. Analyses of erosion and joint space narrowing scores were consistent with the overall results.
In the MTX group, 55% of patients experienced no radiographic progression at Month 6 compared to 73% of patients treated with XELJANZ 5 mg twice daily.
Table 11: Radiographic Changes at Months 6 and 12 - * SD = Standard Deviation
- † Difference between least squares means XELJANZ minus placebo or MTX (95% CI = 95% confidence interval)
- ‡ Month 6 and Month 12 data are mean change from baseline.
Study IV Placebo
N=139
Mean (SD)*XELJANZ 5 mg Twice Daily
N=277
Mean (SD) *XELJANZ 5 mg Twice Daily
Mean Difference from Placebo† (CI)mTSS‡ Baseline 33 (42) 31 (48) - Month 6 0.5 (2.0) 0.1 (1.7) -0.3 (-0.7, 0.0) Study VI MTX
N=166
Mean (SD)*XELJANZ 5 mg Twice Daily
N=346
Mean (SD) *XELJANZ 5 mg Twice Daily
Mean Difference from MTX† (CI)mTSS‡ Baseline 17 (29) 20 (40) - Month 6 0.8 (2.7) 0.2 (2.3) -0.7 (-1.0, -0.3) Month 12 1.3 (3.7) 0.4 (3.0) -0.9 (-1.4, -0.4) Physical Function Response
Improvement in physical functioning was measured by the HAQ-DI. Patients receiving XELJANZ 5 mg twice daily demonstrated greater improvement from baseline in physical functioning compared to placebo at Month 3.
The mean (95% CI) difference from placebo in HAQ-DI improvement from baseline at Month 3 in Study RA-III was -0.22 (-0.35, -0.10) in patients receiving 5 mg XELJANZ twice daily. Similar results were obtained in Studies RA-I, II, IV and V. In the 12-month trials, HAQ-DI results in XELJANZ-treated patients were consistent at 6 and 12 months.
Other Health-Related Outcomes
General health status was assessed by the Short Form health survey (SF-36). In Studies RA-I, IV, and V, patients receiving XELJANZ 5 mg twice daily demonstrated greater improvement from baseline compared to placebo in physical component summary (PCS), mental component summary (MCS) scores and in all 8 domains of the SF-36 at Month 3.
14.2 Psoriatic Arthritis
The XELJANZ clinical development program to assess efficacy and safety included 2 multicenter, randomized, double-blind, placebo-controlled confirmatory trials in 816 patients 18 years of age and older (PsA-I and PsA-II). Although other doses have been studied, the recommended dose of XELJANZ is 5 mg twice daily. XELJANZ 10 mg twice daily is not recommended for the treatment of psoriatic arthritis [see Dosage and Administration (2.2)]. All patients had active psoriatic arthritis for at least 6 months based upon the Classification Criteria for Psoriatic Arthritis (CASPAR), at least 3 tender/painful joints and at least 3 swollen joints, and active plaque psoriasis. Patients randomized and treated across the 2 clinical trials represented different psoriatic arthritis subtypes at screening, including <5 joints or asymmetric involvement (21%), ≥5 joints involved (90%), distal interphalangeal (DIP) joint involvement (61%), arthritis mutilans (8%), and spondylitis (19%). Patients in these clinical trials had a diagnosis of psoriatic arthritis for a mean (SD) of 7.7 (7.2) years. At baseline, 80% and 53% of patients had enthesitis and dactylitis, respectively. At baseline, all patients were required to receive treatment with a stable dose of a nonbiologic DMARD (79% received methotrexate, 13% received sulfasalazine, 7% received leflunomide, 1% received other nonbiologic DMARDs). In both clinical trials, the primary endpoints were the ACR20 response and the change from baseline in HAQ-DI at Month 3.
Study PsA-I was a 12-month clinical trial in 422 patients who had an inadequate response to a nonbiologic DMARD (67% and 33% were inadequate responders to 1 nonbiologic DMARD and ≥2 nonbiologic DMARDs, respectively) and who were naïve to treatment with a TNF blocker. Patients were randomized in a 2:2:2:1:1 ratio to receive XELJANZ 5 mg twice daily, XELJANZ 10 mg twice daily, adalimumab 40 mg subcutaneously once every 2 weeks, placebo to XELJANZ 5 mg twice daily treatment sequence, or placebo to XELJANZ 10 mg twice daily treatment sequence, respectively; study drug was added to background nonbiologic DMARD treatment. At the Month 3 visit, all patients randomized to placebo treatment were advanced in a blinded fashion to a predetermined XELJANZ dose of 5 mg or 10 mg twice daily. Study PsA-I was not designed to demonstrate noninferiority or superiority to adalimumab.
Study PsA-II was a 6-month clinical trial in 394 patients who had an inadequate response to at least 1 approved TNF blocker (66%, 19%, and 15% were inadequate responders to 1 TNF blocker, 2 TNF blockers and ≥3 TNF blockers, respectively). Patients were randomized in a 2:2:1:1 ratio to receive XELJANZ 5 mg twice daily, XELJANZ 10 mg twice daily, placebo to XELJANZ 5 mg twice daily treatment sequence, or placebo to XELJANZ 10 mg twice daily treatment sequence, respectively; study drug was added to background nonbiologic DMARD treatment. At the Month 3 visit, placebo patients were advanced in a blinded fashion to a predetermined XELJANZ dose of 5 mg or 10 mg twice daily as in Study PsA-I.
Clinical Response
At Month 3, patients treated with XELJANZ 5 mg twice daily had higher (p≤0.05) response rates versus placebo for ACR20, ACR50, and ACR70 in Study PsA-I and for ACR20 and ACR50 in Study PsA-II; ACR70 response rates were also higher for XELJANZ 5 mg twice daily versus placebo in Study PsA-II, although the differences versus placebo were not statistically significant (p>0.05) (Tables 12 and 13).
Table 12: Proportion of Patients with an ACR Response in Study PsA-I* [Nonbiologic DMARD Inadequate Responders (TNF Blocker-Naïve)] Treatment Group Placebo XELJANZ
5 mg
Twice DailyN† 105 107 Response Rate Response Rate Difference (%)
95% CI from PlaceboSubjects with missing data were treated as non-responders. - * Subjects received one concomitant nonbiologic DMARD.
- † N is number of randomized and treated patients.
Month 3 ACR20 33% 50% 17.1 (4.1, 30.2) ACR50 10% 28% 18.5 (8.3, 28.7) ACR70 5% 17% 12.1 (3.9, 20.2) Table 13: Proportion of Patients with an ACR Response in Study PsA-II* (TNF Blocker Inadequate Responders) Treatment Group Placebo XELJANZ
5 mg
Twice DailyN† 131 131 Response Rate Response Rate Difference (%)
95% CI from PlaceboSubjects with missing data were treated as non-responders. - * Subjects received one concomitant nonbiologic DMARD.
- † N is number of randomized and treated patients.
Month 3 ACR20 24% 50% 26.0 (14.7, 37.2) ACR50 15% 30% 15.3 (5.4, 25.2) ACR70 10% 17% 6.9 (-1.3, 15.1) Improvements from baseline in the ACR response criteria components for both studies are shown in Table 14.
Table 14: Components of ACR Response at Baseline and Month 3 in Studies PsA-I and PsA-II Nonbiologic DMARD Inadequate Responders (TNF Blocker-Naïve) TNF Blocker Inadequate Responders Study PsA-I* Study PsA-II* Treatment Group Placebo XELJANZ
5 mg
Twice DailyPlacebo XELJANZ
5 mg
Twice DailyN at Baseline 105 107 131 131 - * Subjects received one concomitant nonbiologic DMARD.
- † Data shown are mean value at baseline and at Month 3.
- ‡ Visual analog scale (VAS): 0 = best, 100 = worst.
- § HAQ-DI = Health Assessment Questionnaire – Disability Index: 0 = best, 3 = worst; 20 questions; categories: dressing and grooming, arising, eating, walking, hygiene, reach, grip, and activities.
ACR Component† Number of tender/painful joints (0–68) Baseline 20.6 20.5 19.8 20.5 Month 3 14.6 12.2 15.1 11.5 Number of swollen joints (0–66) Baseline 11.5 12.9 10.5 12.1 Month 3 7.1 6.3 7.7 4.8 Patient assessment of arthritis pain‡ Baseline 53.2 55.7 54.9 56.4 Month 3 44.7 34.7 48.0 36.1 Patient global assessment of arthritis‡ Baseline 53.9 54.7 55.8 57.4 Month 3 44.4 35.5 49.2 36.9 HAQ-DI§ Baseline 1.11 1.16 1.25 1.26 Month 3 0.95 0.81 1.09 0.88 Physician's Global Assessment of Arthritis‡ Baseline 53.8 54.6 53.7 53.5 Month 3 35.4 29.5 36.4 27.0 CRP (mg/L) Baseline 10.4 10.5 12.1 13.8 Month 3 8.6 4.0 11.4 7.7 The percentage of ACR20 responders by visit for Study PsA-I is shown in Figure 6. Similar responses were observed in Study PsA-II. In both studies, improvement in ACR20 response on XELJANZ was observed at the first visit after baseline (Week 2).
BID=twice daily; SE=standard error.
Subjects with missing data were treated as non-responders.- * Subjects received one concomitant nonbiologic DMARD.
Figure 6: Percentage of ACR20 Responders by Visit Through Month 3 in Study PsA-I* 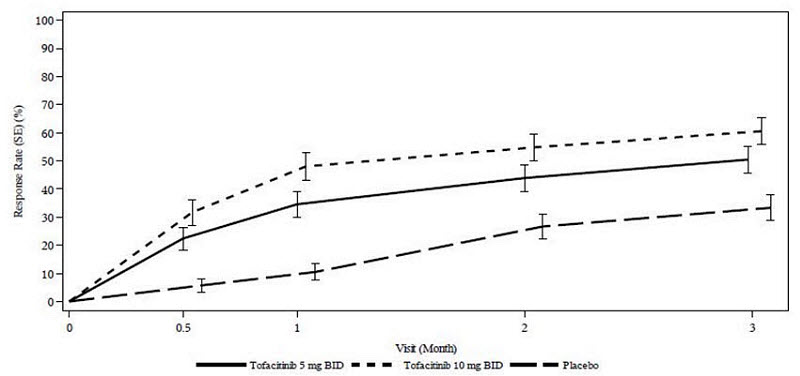
In patients with active psoriatic arthritis evidence of benefit in enthesitis and dactylitis was observed with XELJANZ treatment.
Physical Function
Improvement in physical functioning was measured by the HAQ-DI. Patients receiving XELJANZ 5 mg twice daily demonstrated significantly greater improvement (p ≤0.05) from baseline in physical functioning compared to placebo at Month 3 (Table 15).
Table 15: Change from Baseline in HAQ-DI in Studies PsA-I and PsA-II Least Squares Mean Change from Baseline In HAQ-DI at Month 3 Nonbiologic DMARD Inadequate Responders* (TNF Blocker-Naïve) TNF Blocker Inadequate Responders† Study PsA-I‡ Study PsA-II‡ Treatment Group Placebo XELJANZ 5 mg
Twice DailyPlacebo XELJANZ 5 mg
Twice DailyN§ 104 107 131 129 - * Inadequate response to at least one nonbiologic DMARD due to lack of efficacy and/or intolerability.
- † Inadequate response to at least one TNF blocker due to lack of efficacy and/or intolerability.
- ‡ Subjects received one concomitant nonbiologic DMARD.
- § N is the total number of subjects in the statistical analysis.
LSM Change from Baseline -0.18 -0.35 -0.14 -0.39 Difference from Placebo (95% CI) - -0.17
(-0.29, -0.05)- -0.25
(-0.38, -0.13)In Study PsA-I, the HAQ-DI responder rate (response defined as having improvement from baseline of ≥0.35) at Month 3 was 53% in patients receiving XELJANZ 5 mg twice daily and 31% in patients receiving placebo. Similar responses were observed in Study PsA-II.
Other Health-Related Outcomes
General health status was assessed by the Short Form health survey (SF-36). In Studies PsA-I and PsA-II, patients receiving XELJANZ 5 mg twice daily had greater improvement from baseline compared to placebo in Physical Component Summary (PCS) score, but not in Mental Component Summary (MCS) score at Month 3. Patients receiving XELJANZ 5 mg twice daily reported consistently greater improvement relative to placebo in the domains of Physical Functioning, Bodily Pain, Vitality, and Social Functioning, but not in Role Physical, General Health, Role Emotional, or Mental Health.
14.3 Ulcerative Colitis
Induction Trials (Study UC-I [NCT01465763] and Study UC-II [NCT01458951])
In two identical induction trials (UC-I and UC-II), 1139 patients were randomized (598 and 541 patients, respectively) to XELJANZ 10 mg twice daily or placebo with a 4:1 treatment allocation ratio. These trials included adult patients with moderately to severely active UC (total Mayo score of 6 to 12, with an endoscopy subscore of at least 2, and rectal bleeding subscore of at least 1) and who had failed or were intolerant to at least 1 of the following treatments: oral or intravenous corticosteroids, azathioprine, 6-MP or TNF blocker. XELJANZ is indicated for patients who have an inadequate response or who are intolerant to TNF blockers [see Indications and Usage (1)].
The disease activity was assessed by Mayo scoring index (0 to 12) which consists of four subscores (0 to 3 for each subscore): stool frequency, rectal bleeding, findings on endoscopy, and physician global assessment. An endoscopy subscore of 2 was defined by marked erythema, absent vascular pattern, any friability, and erosions; an endoscopy subscore of 3 was defined by spontaneous bleeding and ulceration.
Patients were permitted to use stable doses of oral aminosalicylates and corticosteroids (prednisone daily dose up to 25 mg equivalent). Concomitant immunosuppressants (oral immunomodulators or biologic therapies) were not permitted for UC patients during these studies.
A total of 52%, 73% and 72% of patients had previously failed or were intolerant to TNF blockers (51% in Study UC-1 and 52% in Study UC-II), corticosteroids (75% in Study UC-I and 71% in Study UC-II), and/or immunosuppressants (74% in Study UC-I and 70% in Study UC-II), respectively.
Oral corticosteroids were received as concomitant treatment for UC by 47% of patients (45% in Study UC-I and 48% in Study UC-II) and 71% were receiving concomitant aminosalicylates as treatment for UC (71% in Study UC-I, and 72% in Study UC-II). The baseline clinical characteristics were generally similar between the XELJANZ treated patients and patients receiving placebo.
The primary endpoint of Study UC-I and Study UC-II was the proportion of patients in remission at Week 8, and the key secondary endpoint was the proportion of patients with improvement of endoscopic appearance of the mucosa at Week 8.
The efficacy results of Study UC-I and Study UC-II based on the centrally read endoscopy results are shown in Table 16.
Table 16: Proportion of Patients Meeting Primary and Key Secondary Efficacy Endpoints at Week 8 (Induction Study UC-I and Study UC-II, Central Endoscopy Read) CI = Confidence interval; N = number of patients in the analysis set; TNF = tumor necrosis factor - * Remission was defined as clinical remission (a Mayo score ≤2 with no individual subscore >1) and rectal bleeding subscore of 0.
- † p-value <0.01,
- ‡ Prior TNF blocker failure was defined in this program as inadequate response, loss of response, or intolerance to TNF blocker therapy.
- § Patients in this group had failed one or more conventional therapies (corticosteroid, azathioprine, 6-mercaptopurine) but did not have history of prior failure of TNF blocker therapy.
- ¶ Improvement of endoscopic appearance of the mucosa was defined as Mayo endoscopy subscore of 0 (normal or inactive disease) or 1 (erythema, decreased vascular pattern).
- # p-value <0.001.
Study UC-I Endpoint Placebo XELJANZ
10 mg
Twice DailyTreatment Difference versus Placebo
(95% CI)Remission at Week 8* Total Population N=122 N=476 10%†
(4.3, 16.3)8% 18% With Prior TNF Blocker Failure‡ N=64 N=243 2% 11% Without Prior TNF Blocker Failure§ N=58 N=233 16% 26% Improvement of endoscopic appearance of the mucosa at Week 8¶ Total Population N=122 N=476 16%#
(8.1, 23.4)16% 31% With Prior TNF Blocker Failure‡ N=64 N=243 6% 23% Without Prior TNF Blocker Failure§ N=58 N=233 26% 40% Study UC-II Endpoint Placebo XELJANZ
10 mg
Twice DailyTreatment Difference
(95% CI)Remission at Week 8* Total Population N=112 N=429 13%#
(8.1, 17.9)4% 17% With Prior TNF Blocker Failure‡ N=60 N=222 0% 12% Without Prior TNF Blocker Failure§ N=52 N=207 8% 22% Improvement of endoscopic appearance of the mucosa at Week 8¶ Total Population N=112 N-429 17%#
(9.5, 24.1)12% 28% With Prior TNF Blocker Failure‡ N=60 N=222 7% 22% Without Prior TNF Blocker Failure§ N=52 N=207 17% 36% Clinical Response at Week 8
Clinical response was defined as a decrease from baseline in Mayo score of ≥3 points and ≥30%, with an accompanying decrease in the subscore for rectal bleeding of ≥1 point or absolute subscore for rectal bleeding of 0 or 1.
Clinical response was observed in 60% of patients treated with XELJANZ 10 mg twice daily compared to 33% of placebo patients in Study UC-I and 55% compared to 29% in Study UC-II.
Normalization of the Endoscopic Appearance of the Mucosa at Week 8
Normalization of endoscopic appearance of the mucosa was defined as a Mayo endoscopic subscore of 0 and was observed in 7% of patients treated with XELJANZ 10 mg twice daily compared to 2% of placebo patients in both Studies UC-I and UC-II.
Maintenance Trial (Study UC-III [NCT01458574])
A total of 593 patients who completed the induction trials (UC-I or UC-II) and achieved clinical response were re-randomized with 1:1:1 treatment allocation ratio to XELJANZ 5 mg twice daily, XELJANZ 10 mg twice daily, or placebo for 52 weeks in Study UC-III. XELJANZ 5 mg twice daily is the recommended dosage for maintenance therapy; limit use of XELJANZ 10 mg twice daily beyond induction to those with loss of response and should be used for the shortest duration [see Dosage and Administration (2.3)]. As in the induction trials, patients were permitted to use stable doses of oral aminosalicylates; however, corticosteroid tapering was required upon entrance into this study for patients who were receiving corticosteroids at baseline. Concomitant immunosuppressants (oral immunomodulators or biologic therapies) were not permitted.
At baseline of Study UC-III:
- 179 (30%) patients were in remission
- 289 (49%) patients were receiving oral corticosteroids
- 265 (45%), 445 (75%), and 413 (70%) patients had previously failed or were intolerant to TNF blocker therapy, corticosteroids, and immunosuppressants, respectively.
The primary endpoint was the proportion of patients in remission at Week 52. There were 2 key secondary endpoints: the proportion of patients with improvement of endoscopic appearance at Week 52, and the proportion of patients with sustained corticosteroid-free remission at both Week 24 and Week 52 among patients in remission at baseline of Study UC-III.
The efficacy results of Study UC-III based on the centrally read endoscopy results are summarized in Table 17.
Table 17: Proportion of Patients Meeting Primary and Key Secondary Efficacy Endpoints in Maintenance Study UC-III (Central Endoscopy Read) Treatment Difference versus Placebo
(95% CI)Endpoint Placebo XELJANZ
5 mg
Twice DailyXELJANZ
10 mg
Twice DailyXELJANZ
5 mg
Twice DailyXELJANZ
10 mg
Twice DailyCI = Confidence interval; N = number of patients in the analysis set; TNF = tumor necrosis factor. - * Remission was defined as clinical remission (a Mayo score ≤2 with no individual subscore >1) and rectal bleeding subscore of 0.
- † p-value <0.0001.
- ‡ Prior TNF blocker failure was defined in this program as inadequate response, loss of response, or intolerance to TNF blocker therapy.
- § Patients in this group had failed one or more conventional therapies (corticosteroid, azathioprine, 6-mercaptopurine) but did not have history of prior failure of TNF blocker therapy.
- ¶ Improvement of endoscopic appearance of the mucosa was defined as Mayo endoscopy subscore of 0 (normal or inactive disease) or 1 (erythema, decreased vascular pattern).
- # Sustained corticosteroid-free remission was defined as being in remission and not taking corticosteroids for at least 4 weeks prior to the visit at both Week 24 and Week 52.
Remission at Week 52* Total Population N=198 N=198 N=197 23%†
(15.3, 31.2)30%†
(21.4, 37.6)11% 34% 41% With Prior TNF Blocker Failure‡ N=89 N=83 N=93 11% 24% 37% Without Prior TNF Blocker Failure§ N=109 N=115 N=104 11% 42% 44% Improvement of endoscopic appearance of the mucosa at Week 52¶ Total Population N=198 N=198 N=197 24%†
(16.0, 32.5)33%†
(24.2, 41.0)13% 37% 46% With Prior TNF Blocker Failure‡ N=89 N=83 N=93 12% 30% 40% Without Prior TNF Blocker Failure§ N=109 N=115 N=104 14% 43% 51% Sustained corticosteroid-free remission at both Week 24 and Week 52 among patients in remission at baseline# Total Population N=59 N=65 N=55 30%†
(17.4, 43.2)42%†
(27.9, 56.5)5% 35% 47% With Prior TNF Blocker Failure‡ N=21 N=18 N=18 5% 22% 39% Without Prior TNF Blocker Failure§ N=38 N=47 N=37 5% 40% 51% Maintenance of Clinical Response
Maintenance of clinical response was defined as the proportion of patients who met the definition of clinical response (defined as a decrease from the induction study (UC-I, UC-II) baseline Mayo score of ≥3 points and ≥30%, with an accompanying decrease in the rectal bleeding subscore of ≥1 point or rectal bleeding subscore of 0 or 1) at both Baseline and Week 52 of Study UC-III.
Maintenance of clinical response was observed in 52% in the XELJANZ 5 mg twice daily group and 62% in the XELJANZ 10 mg twice daily group compared to 20% of placebo patients.
Maintenance of Remission (Among Patients in Remission at Baseline)
In the 179 patients who were in remission at baseline of Study UC-III (N = 59 for placebo, N = 65 for XELJANZ 5 mg twice daily, N = 55 for XELJANZ 10 mg twice daily), 46% in the XELJANZ 5 mg twice daily group and 56% in the XELJANZ 10 mg twice daily group maintained remission at Week 52 compared to 10% of placebo patients.
Normalization of the Endoscopic Appearance of the Mucosa
Normalization of endoscopic appearance of the mucosa was defined as a Mayo endoscopic subscore of 0 and was observed at Week 52 in 15% of patients in the XELJANZ 5 mg twice daily group and 17% of patients in the XELJANZ 10 mg twice daily group compared to 4% of placebo patients.
Open-label Extension Study (Study UC-IV [NCT01470612])
In Study UC-IV, 914 patients were treated of which 156 received 5 mg twice daily and 758 received 10 mg twice daily.
Of the 905 patients who were assigned to XELJANZ 10 mg twice daily in the 8-week induction studies (Study UC-I or Study UC-II), 322 patients completed the induction studies but did not achieve clinical response. Of these 322 patients, 291 continued to receive XELJANZ 10 mg twice daily (unblinded) and had available data after an additional 8 weeks in Study UC-IV. After 8 additional weeks (a total of 16 weeks treatment), 148 patients achieved clinical response, and 25 patients achieved remission (based on central endoscopy read). Among those 143 patients who achieved clinical response by 16 weeks and had available data at Week 52, 66 patients achieved remission (based on local endoscopy read) after continued treatment with XELJANZ 10 mg twice daily for 52 weeks.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Bottle Size
(number of tablets)NDC Number XELJANZ
5 mg tofacitinib tablets
White, round, immediate-release film-coated tablets, debossed with "Pfizer" on one side, and "JKI 5" on the other side28 NDC: 0069-1001-03 60 NDC: 0069-1001-01 XELJANZ
10 mg tofacitinib tablets
Blue, round, immediate-release film-coated tablets, debossed with "Pfizer" on one side, and "JKI 10" on the other side28 NDC: 0069-1002-03 60 NDC: 0069-1002-01 180 NDC: 0069-1002-02 XELJANZ XR
11 mg tofacitinib tablets
Pink, oval, extended-release film-coated tablets with a drilled hole at one end of the tablet band and "JKI 11" printed on one side of the tablet14 NDC: 0069-0501-14 30 NDC: 0069-0501-30 XELJANZ XR
22 mg tofacitinib tablets
Beige, oval, extended-release film-coated tablets with a drilled hole at one end of the tablet band and "JKI 22" printed on one side of the tablet30 NDC: 0069-0502-30 -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Serious Infections
Inform patients that XELJANZ/XELJANZ XR may lower the ability of their immune system to fight infections. Advise patients not to start taking XELJANZ/XELJANZ XR if they have an active infection. Instruct patients to contact their healthcare provider immediately during treatment if symptoms suggesting infection appear in order to ensure rapid evaluation and appropriate treatment [see Warnings and Precautions (5.1)].
Advise patients that the risk of herpes zoster, some cases of which can be serious, is increased in patients treated with XELJANZ/XELJANZ XR [see Warnings and Precautions (5.1)].
Malignancies and Lymphoproliferative Disorders
Inform patients that XELJANZ/XELJANZ XR may increase their risk of certain cancers, and that lymphoma and other cancers have been observed in patients taking XELJANZ. Instruct patients to inform their healthcare provider if they have ever had any type of cancer [see Warnings and Precautions (5.3)].
Thrombosis
Advise patients to stop taking XELJANZ/XELJANZ XR and to call their healthcare provider right away if they experience any symptoms of thrombosis (sudden shortness of breath, chest pain worsened with breathing, swelling of leg or arm, leg pain or tenderness, red or discolored skin in the affected leg or arm) [see Warnings and Precautions (5.4)].
Hypersensitivity
Advise patients to stop taking XELJANZ/XELJANZ XR and to call their healthcare provider right away if they experience any symptoms of allergic reactions while taking XELJANZ/XELJANZ XR [see Warnings and Precautions (5.6)].
Important Information on Laboratory Abnormalities
Inform patients that XELJANZ/XELJANZ XR may affect certain lab test results, and that blood tests are required before and during XELJANZ/XELJANZ XR treatment [see Warnings and Precautions (5.7)].
Pregnancy
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females to inform their prescriber of a known or suspected pregnancy. Inform patients that Pfizer has a registry for pregnant women who have taken XELJANZ/XELJANZ XR during pregnancy. Advise patients to contact the registry at 1-877-311-8972 to enroll [see Use in Specific Populations (8.1)].
Lactation
Advise women not to breastfeed during treatment with XELJANZ/XELJANZ XR and for at least 18 hours after the last dose of XELJANZ or 36 hours after the last dose of XELJANZ XR [see Use in Specific Populations (8.2)].
Infertility
Advise females of reproductive potential that XELJANZ/XELJANZ XR may impair fertility [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)]. It is not known if this effect is reversible.
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Revised: Dec 2019
MEDICATION GUIDE
XELJANZ (ZEL' JANS')
(tofacitinib)
tablets, for oral useXELJANZ XR (ZEL' JANS' EKS-AHR)
(tofacitinib)
extended-release tablets, for oral useWhat is the most important information I should know about XELJANZ/XELJANZ XR?
XELJANZ/XELJANZ XR may cause serious side effects including:
1. Serious infections. XELJANZ/XELJANZ XR is a medicine that affects your immune system. XELJANZ/XELJANZ XR can lower the ability of your immune system to fight infections. Some people can have serious infections while taking XELJANZ/XELJANZ XR, including tuberculosis (TB), and infections caused by bacteria, fungi, or viruses that can spread throughout the body. Some people have died from these infections.
- Your healthcare provider should test you for TB before starting XELJANZ/XELJANZ XR and during treatment.
- Your healthcare provider should monitor you closely for signs and symptoms of TB infection during treatment with XELJANZ/XELJANZ XR.
You should not start taking XELJANZ/XELJANZ XR if you have any kind of infection unless your healthcare provider tells you it is okay. You may be at a higher risk of developing shingles (herpes zoster).
People taking the higher dose of XELJANZ (10 mg twice daily) or XELJANZ XR (22 mg one time each day) have a higher risk of serious infections and shingles.
Before starting XELJANZ/XELJANZ XR, tell your healthcare provider if you:
- think you have an infection or have symptoms of an infection such as:
- fever, sweating, or chills
- cough
- blood in phlegm
- warm, red, or painful skin or sores on your body
- burning when you urinate or urinating more often than normal
- muscle aches
- shortness of breath
- weight loss
- diarrhea or stomach pain
- feeling very tired
- are being treated for an infection.
- get a lot of infections or have infections that keep coming back.
- have diabetes, chronic lung disease, HIV, or a weak immune system. People with these conditions have a higher chance for infections.
- have TB, or have been in close contact with someone with TB.
- live or have lived, or have traveled to certain parts of the country (such as the Ohio and Mississippi River valleys and the Southwest) where there is an increased chance for getting certain kinds of fungal infections (histoplasmosis, coccidioidomycosis, or blastomycosis). These infections may happen or become more severe if you use XELJANZ/XELJANZ XR. Ask your healthcare provider if you do not know if you have lived in an area where these infections are common.
- have or have had hepatitis B or C.
After starting XELJANZ/XELJANZ XR, call your healthcare provider right away if you have any symptoms of an infection. XELJANZ/XELJANZ XR can make you more likely to get infections or make worse any infection that you have.
2. Increased risk of death in people 50 years of age and older with rheumatoid arthritis who have at least 1 heart disease (cardiovascular) risk factor and who are taking a higher than recommended dose of XELJANZ/XELJANZ XR. The recommended dose in patients with rheumatoid arthritis and psoriatic arthritis is XELJANZ 5 mg twice daily or XELJANZ XR 11 mg one time each day.
3. Cancer and immune system problems. XELJANZ/XELJANZ XR may increase your risk of certain cancers by changing the way your immune system works.
- Lymphoma and other cancers including skin cancers can happen in patients taking XELJANZ/XELJANZ XR. People taking the higher dose of XELJANZ (10 mg twice daily) or XELJANZ XR (22 mg one time each day) have a higher risk of skin cancers. Tell your healthcare provider if you have ever had any type of cancer.
- Some people who have taken XELJANZ with certain other medicines to prevent kidney transplant rejection have had a problem with certain white blood cells growing out of control (Epstein Barr Virus-associated post-transplant lymphoproliferative disorder).
4. Blood clots in the lungs, veins of the legs or arms, and arteries. Blood clots in the lungs (pulmonary embolism, PE), veins of the legs (deep vein thrombosis, DVT) and arteries (arterial thrombosis) have happened more often in patients with rheumatoid arthritis who are 50 years of age and older and with at least 1 heart disease (cardiovascular) risk factor taking a higher than recommended dose of XELJANZ/XELJANZ XR. The recommended dose in patients with rheumatoid arthritis and psoriatic arthritis is XELJANZ 5 mg twice daily or XELJANZ XR 11 mg one time each day. Blood clots in the lungs have also happened in patients with ulcerative colitis. Some people have died from these blood clots.
- Stop taking XELJANZ/XELJANZ XR and tell your healthcare provider right away if you develop signs and symptoms of a blood clot, such as sudden shortness of breath or difficulty breathing, chest pain, swelling of the leg or arm, leg pain or tenderness, or redness or discoloration in the leg or arm.
5. Tears (perforation) in the stomach or intestines.
- Tell your healthcare provider if you have had diverticulitis (inflammation in parts of the large intestine) or ulcers in your stomach or intestines. Some people taking XELJANZ/XELJANZ XR can get tears in their stomach or intestines. This happens most often in people who also take nonsteroidal anti-inflammatory drugs (NSAIDs), corticosteroids, or methotrexate. Tell your healthcare provider right away if you have fever and stomach-area pain that does not go away, and a change in your bowel habits.
6. Allergic reactions.
- Symptoms such as swelling of your lips, tongue, or throat, or hives (raised, red patches of skin that are often very itchy) that may mean you are having an allergic reaction have been seen in patients taking XELJANZ/XELJANZ XR. Some of these reactions were serious. If any of these symptoms occur while you are taking XELJANZ/XELJANZ XR, stop XELJANZ/XELJANZ XR and call your healthcare provider right away.
7. Changes in certain laboratory test results. Your healthcare provider should do blood tests before you start receiving XELJANZ/XELJANZ XR and while you take XELJANZ/XELJANZ XR to check for the following side effects:
- changes in lymphocyte counts. Lymphocytes are white blood cells that help the body fight off infections.
- low neutrophil counts. Neutrophils are white blood cells that help the body fight off infections.
- low red blood cell count. This may mean that you have anemia, which may make you feel weak and tired.
Your healthcare provider should routinely check certain liver tests.
You should not receive XELJANZ/XELJANZ XR if your lymphocyte count, neutrophil count, or red blood cell count is too low or your liver tests are too high.
Your healthcare provider may stop your XELJANZ/XELJANZ XR treatment for a period of time if needed because of changes in these blood test results.
You may also have changes in other laboratory tests, such as your blood cholesterol levels. Your healthcare provider should do blood tests to check your cholesterol levels 4 to 8 weeks after you start receiving XELJANZ/XELJANZ XR, and as needed after that. Normal cholesterol levels are important to good heart health.
See "What are the possible side effects of XELJANZ/XELJANZ XR?" for more information about side effects.
What is XELJANZ/XELJANZ XR?
XELJANZ/XELJANZ XR is a prescription medicine called a Janus kinase (JAK) inhibitor. XELJANZ/XELJANZ XR is used to treat adults with moderately to severely active rheumatoid arthritis in whom methotrexate did not work well or cannot be tolerated.
XELJANZ/XELJANZ XR is used to treat adults with active psoriatic arthritis in which methotrexate or other similar medicines called nonbiologic disease-modifying antirheumatic drugs (DMARDs) did not work well or cannot be tolerated.
XELJANZ/XELJANZ XR is used to treat adults with moderately to severely active ulcerative colitis when medicines called tumor necrosis factor (TNF) blockers did not work well or cannot be tolerated.
It is not known if XELJANZ/XELJANZ XR is safe and effective in people with Hepatitis B or C.
XELJANZ/XELJANZ XR is not recommended for people with severe liver problems.
It is not known if XELJANZ/XELJANZ XR is safe and effective in children.
What should I tell my healthcare provider before taking XELJANZ/XELJANZ XR?
Before taking XELJANZ/XELJANZ XR, tell your healthcare provider about all of your medical conditions, including if you:
- have an infection. See "What is the most important information I should know about XELJANZ/XELJANZ XR?"
- have had blood clots in the veins of your legs, arms, or lungs, or clots in the arteries in the past.
- have liver problems.
- have kidney problems.
- have any stomach area (abdominal) pain or been diagnosed with diverticulitis or ulcers in your stomach or intestines.
- have had a reaction to tofacitinib or any of the ingredients in XELJANZ/XELJANZ XR.
- have recently received or are scheduled to receive a vaccine. People who take XELJANZ/XELJANZ XR should not receive live vaccines. People taking XELJANZ/XELJANZ XR can receive non-live vaccines.
- plan to become pregnant or are pregnant. XELJANZ/XELJANZ XR may affect the ability of females to get pregnant. It is not known if this will change after stopping XELJANZ/XELJANZ XR. It is not known if XELJANZ/XELJANZ XR will harm an unborn baby.
- Pregnancy Registry: Pfizer has a registry for pregnant women who take XELJANZ/XELJANZ XR. The purpose of this registry is to check the health of the pregnant mother and her baby. If you are pregnant or become pregnant while taking XELJANZ/XELJANZ XR, talk to your healthcare provider about how you can join this pregnancy registry or you may contact the registry at 1-877-311-8972 to enroll.
- plan to breastfeed or are breastfeeding. You and your healthcare provider should decide if you will take XELJANZ/XELJANZ XR or breastfeed. You should not do both. After you stop your treatment with XELJANZ/XELJANZ XR do not start breastfeeding again until:
- 18 hours after your last dose of XELJANZ or
- 36 hours after your last dose of XELJANZ XR
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. XELJANZ/XELJANZ XR and other medicines may affect each other causing side effects.
Especially tell your healthcare provider if you take:
- any other medicines to treat your rheumatoid arthritis, psoriatic arthritis, or ulcerative colitis. You should not take tocilizumab (Actemra®), etanercept (Enbrel®), adalimumab (Humira®), infliximab (Remicade®), rituximab (Rituxan®), abatacept (Orencia®), anakinra (Kineret®), certolizumab (Cimzia®), golimumab (Simponi®), ustekinumab (Stelara®), secukinumab (Cosentyx®), vedolizumab (Entyvio®), azathioprine, cyclosporine, or other immunosuppressive drugs while you are taking XELJANZ or XELJANZ XR. Taking XELJANZ or XELJANZ XR with these medicines may increase your risk of infection.
- medicines that affect the way certain liver enzymes work. Ask your healthcare provider if you are not sure if your medicine is one of these.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.
How should I take XELJANZ/XELJANZ XR? Take XELJANZ/XELJANZ XR exactly as your healthcare provider tells you to take it.
- Take XELJANZ 2 times a day with or without food.
- Take XELJANZ XR 1 time a day with or without food.
- Swallow XELJANZ XR tablets whole and intact. Do not crush, split, or chew.
- When you take XELJANZ XR, you may see something in your stool that looks like a tablet. This is the empty shell from the tablet after the medicine has been absorbed by your body.
- If you take too much XELJANZ/XELJANZ XR, call your healthcare provider or go to the nearest hospital emergency room right away.
- For the treatment of psoriatic arthritis, take XELJANZ/XELJANZ XR in combination with methotrexate, sulfasalazine or leflunomide as instructed by your healthcare provider.
What are possible side effects of XELJANZ/XELJANZ XR?
XELJANZ/XELJANZ XR may cause serious side effects, including:
- See "What is the most important information I should know about XELJANZ/XELJANZ XR?"
- Hepatitis B or C activation infection in people who carry the virus in their blood. If you are a carrier of the hepatitis B or C virus (viruses that affect the liver), the virus may become active while you use XELJANZ/XELJANZ XR. Your healthcare provider may do blood tests before you start treatment with XELJANZ/XELJANZ XR and while you are using XELJANZ/XELJANZ XR. Tell your healthcare provider if you have any of the following symptoms of a possible hepatitis B or C infection:
- feel very tired
- little or no appetite
- clay-colored bowel movements
- chills
- muscle aches
- skin rash
- skin or eyes look yellow
- vomiting
- fevers
- stomach discomfort
- dark urine
Common side effects of XELJANZ/XELJANZ XR in rheumatoid arthritis patients and psoriatic arthritis patients include:
- upper respiratory tract infections (common cold, sinus infections)
- headache
- diarrhea
- nasal congestion, sore throat, and runny nose (nasopharyngitis)
- high blood pressure (hypertension)
Common side effects of XELJANZ/XELJANZ XR in ulcerative colitis patients include:
- nasal congestion, sore throat, and runny nose (nasopharyngitis)
- increased cholesterol levels
- headache
- upper respiratory tract infections (common cold, sinus infections)
- increased muscle enzyme levels
- rash
- diarrhea
- shingles (herpes zoster)
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of XELJANZ/XELJANZ XR. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
You may also report side effects to Pfizer at 1-800-438-1985.
How should I store XELJANZ/XELJANZ XR?
- Store XELJANZ/XELJANZ XR at room temperature between 68°F to 77°F (20°C to 25°C).
- Safely throw away medicine that is out of date or no longer needed.
Keep XELJANZ/XELJANZ XR and all medicines out of the reach of children.
General information about the safe and effective use of XELJANZ/XELJANZ XR.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use XELJANZ/XELJANZ XR for a condition for which it was not prescribed. Do not give XELJANZ/XELJANZ XR to other people, even if they have the same symptoms you have. It may harm them.
This Medication Guide summarizes the most important information about XELJANZ/XELJANZ XR. If you would like more information, talk to your healthcare provider. You can ask your pharmacist or healthcare provider for information about XELJANZ/XELJANZ XR that is written for health professionals.
What are the ingredients in XELJANZ 5 mg?
Active ingredient: tofacitinib citrate
Inactive ingredients: croscarmellose sodium, HPMC 2910/Hypromellose 6cP, lactose monohydrate, macrogol/PEG3350, magnesium stearate, microcrystalline cellulose, titanium dioxide, and triacetin.
What are the ingredients in XELJANZ 10 mg?
Active ingredient: tofacitinib citrate
Inactive ingredients: croscarmellose sodium, FD&C Blue #1/Brilliant Blue FCF Aluminum Lake, FD&C Blue #2/Indigo Carmine Aluminum Lake, HPMC 2910/Hypromellose 6cP, lactose monohydrate, macrogol/PEG3350, magnesium stearate, microcrystalline cellulose, titanium dioxide, and triacetin.
What are the ingredients in XELJANZ XR 11 mg?
Active ingredient: tofacitinib citrate
Inactive ingredients: cellulose acetate, copovidone, hydroxyethyl cellulose, hydroxypropyl cellulose, HPMC 2910/Hypromellose, magnesium stearate, red iron oxide, sorbitol, titanium dioxide, and triacetin. Printing ink contains ammonium hydroxide, ferrosoferric oxide/black iron, propylene glycol, and shellac glaze.
What are the ingredients in XELJANZ XR 22 mg?
Active ingredient: tofacitinib citrate
Inactive ingredients: cellulose acetate, copovidone, FD&C Blue #2 Aluminum Lake, hydroxyethyl cellulose, hydroxypropyl cellulose, HPMC 2910/Hypromellose, magnesium stearate, red iron oxide, sorbitol, titanium dioxide, triacetin, and yellow iron oxide. Printing ink contains ammonium hydroxide, ferrosoferric oxide/black iron oxide, propylene glycol, and shellac glaze.

LAB-0535-10.0
-
PRINCIPAL DISPLAY PANEL - 5 mg Tablet Bottle Label
PROFESSIONAL SAMPLE-NOT FOR SALE
ALWAYS DISPENSE WITH MEDICATION GUIDENDC: 63539-012-02
Pfizer
Xeljanz™
(tofacitinib tablets)5 mg*
60 Tablets
Rx only
-
PRINCIPAL DISPLAY PANEL - 11 mg Tablet Bottle Label
PROFESSIONAL SAMPLE-NOT FOR SALE
ALWAYS DISPENSE WITH MEDICATION GUIDENDC: 63539-501-30
Pfizer
Xeljanz® XR
(tofacitinib) tabletsExtended Release Tablets
11 mg*
30 Tablets
Rx only
-
PRINCIPAL DISPLAY PANEL - 10 mg Tablet Bottle Label
PROFESSIONAL SAMPLE – NOT FOR SALE
ALWAYS DISPENSE WITH MEDICATION GUIDENDC: 63539-016-02
Pfizer
Xeljanz®
(tofacitinib) tablets10 mg*
10mg Is Recommended Only In
Ulcerative Colitis60 Tablets
Rx only
-
PRINCIPAL DISPLAY PANEL - 22 mg Tablet Bottle Label
PROFESSIONAL SAMPLE-NOT FOR SALE
ALWAYS DISPENSE WITH MEDICATION GUIDEPfizer
NDC: 63539-502-30
Xeljanz® XR
(tofacitinib) tabletsExtended Release Tablets
22 mg*
22mg Is Recommended Only In
Ulcerative Colitis30 Tablets
Rx only
-
INGREDIENTS AND APPEARANCE
XELJANZ
tofacitinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 63539-012 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TOFACITINIB CITRATE (UNII: O1FF4DIV0D) (TOFACITINIB - UNII:87LA6FU830) TOFACITINIB 5 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TRIACETIN (UNII: XHX3C3X673) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) Product Characteristics Color WHITE (white to off-white) Score no score Shape ROUND Size 8mm Flavor Imprint Code PFIZER;JK15 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 63539-012-02 60 in 1 BOTTLE; Type 0: Not a Combination Product 11/08/2012 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203214 11/08/2012 XELJANZ XR
tofacitinib tablet, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 63539-501 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TOFACITINIB CITRATE (UNII: O1FF4DIV0D) (TOFACITINIB - UNII:87LA6FU830) TOFACITINIB 11 mg Inactive Ingredients Ingredient Name Strength SORBITOL (UNII: 506T60A25R) HYDROXYETHYL CELLULOSE (140 MPA.S AT 5%) (UNII: 8136Y38GY5) COPOVIDONE K25-31 (UNII: D9C330MD8B) MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE ACETATE (UNII: 3J2P07GVB6) HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) TRIACETIN (UNII: XHX3C3X673) FERRIC OXIDE RED (UNII: 1K09F3G675) SHELLAC (UNII: 46N107B71O) AMMONIA (UNII: 5138Q19F1X) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) FERROSOFERRIC OXIDE (UNII: XM0M87F357) Product Characteristics Color PINK Score no score Shape OVAL Size 8mm Flavor Imprint Code JKI11 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 63539-501-14 14 in 1 BOTTLE; Type 0: Not a Combination Product 03/07/2016 2 NDC: 63539-501-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 03/07/2016 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208246 03/07/2016 XELJANZ
tofacitinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 63539-016 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TOFACITINIB CITRATE (UNII: O1FF4DIV0D) (TOFACITINIB - UNII:87LA6FU830) TOFACITINIB 10 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TRIACETIN (UNII: XHX3C3X673) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) Product Characteristics Color BLUE Score no score Shape ROUND Size 10mm Flavor Imprint Code PFIZER;JK110 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 63539-016-02 60 in 1 BOTTLE; Type 0: Not a Combination Product 07/02/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203214 07/02/2018 XELJANZ XR
tofacitinib tablet, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 63539-502 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TOFACITINIB CITRATE (UNII: O1FF4DIV0D) (TOFACITINIB - UNII:87LA6FU830) TOFACITINIB 22 mg Inactive Ingredients Ingredient Name Strength SORBITOL (UNII: 506T60A25R) HYDROXYETHYL CELLULOSE (140 MPA.S AT 5%) (UNII: 8136Y38GY5) COPOVIDONE K25-31 (UNII: D9C330MD8B) MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE ACETATE (UNII: 3J2P07GVB6) HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) TRIACETIN (UNII: XHX3C3X673) FERRIC OXIDE RED (UNII: 1K09F3G675) SHELLAC (UNII: 46N107B71O) AMMONIA (UNII: 5138Q19F1X) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) FERROSOFERRIC OXIDE (UNII: XM0M87F357) FD&C BLUE NO. 2--ALUMINUM LAKE (UNII: 4AQJ3LG584) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color BROWN (beige) Score no score Shape OVAL Size 8mm Flavor Imprint Code JKI22 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 63539-502-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 01/21/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208246 01/21/2020 Labeler - U.S. Pharmaceuticals (829076905) Registrant - Pfizer Inc (113480771) Establishment Name Address ID/FEI Business Operations Pfizer Pharmaceuticals LLC 829084545 ANALYSIS(63539-012, 63539-016, 63539-501, 63539-502) , MANUFACTURE(63539-012, 63539-016, 63539-501, 63539-502) Establishment Name Address ID/FEI Business Operations Pfizer Pharmaceuticals LLC 829084552 LABEL(63539-012, 63539-016, 63539-501, 63539-502) , PACK(63539-012, 63539-016, 63539-501, 63539-502) Establishment Name Address ID/FEI Business Operations Pfizer Ireland Pharmaceuticals 985104227 ANALYSIS(63539-012, 63539-016, 63539-501, 63539-502) , API MANUFACTURE(63539-012, 63539-016, 63539-501, 63539-502) Establishment Name Address ID/FEI Business Operations Pfizer Manufacturing Deutschland GmbH 341970073 ANALYSIS(63539-012, 63539-016) , LABEL(63539-012, 63539-016) , MANUFACTURE(63539-012, 63539-016) , PACK(63539-012, 63539-016)

Trademark Results [XELJANZ]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 XELJANZ 86126490 4563705 Live/Registered |
Pfizer Inc. 2013-11-22 |
 XELJANZ 85678862 not registered Dead/Abandoned |
Pfizer Inc. 2012-07-17 |
 XELJANZ 77688128 4099367 Live/Registered |
Pfizer Inc. 2009-03-11 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.