Ezetimibe by AvPAK EZETIMIBE tablet
Ezetimibe by
Drug Labeling and Warnings
Ezetimibe by is a Prescription medication manufactured, distributed, or labeled by AvPAK. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
Ezetimibe Tablets, USP
Rx Only
These highlights do not include all the information needed to use EZETIMIBE TABLETS safely and effectively. See full prescribing information for EZETIMIBE TABLETS.
EZETIMIBE tablets, for oral use
Initial U.S. Approval: 2002INDICATIONS AND USAGE
Ezetimibe tablets are an inhibitor of intestinal cholesterol (and related phytosterol) absorption indicated as an adjunct to diet to:
- Reduce elevated total-C, LDL-C, Apo B, and non-HDL-C in patients with primary hyperlipidemia, alone or in combination with an HMG-CoA reductase inhibitor (statin) ( 1.1)
- Reduce elevated total-C, LDL-C, Apo B, and non-HDL-C in patients with mixed hyperlipidemia in combination with fenofibrate ( 1.1)
- Reduce elevated total-C and LDL-C in patients with homozygous familial hypercholesterolemia (HoFH), in combination with atorvastatin or simvastatin ( 1.2)
- Reduce elevated sitosterol and campesterol in patients with homozygous sitosterolemia (phytosterolemia) ( 1.3)
Limitations of Use ( 1.4)
- The effect of ezetimibe tablets on cardiovascular morbidity and mortality has not been determined.
- Ezetimibe tablets have not been studied in Fredrickson Type I, III, IV, and V dyslipidemias.
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
- Tablets: 10 mg ( 3)
CONTRAINDICATIONS
- Statin contraindications apply when ezetimibe is used with a statin:
- Active liver disease, which may include unexplained persistent elevations in hepatic transaminase levels (4, 5.2)
- Women who are pregnant or may become pregnant (4, 8.1)
- Nursing mothers (4, 8.3)
- Known hypersensitivity to product components (4, 6.2)
WARNINGS AND PRECAUTIONS
- Ezetimibe is not recommended in patients with moderate or severe hepatic impairment. ( 5.4, 8.7, 12.3)
- Liver enzyme abnormalities and monitoring: Persistent elevations in hepatic transaminase can occur when ezetimibe is added to a statin. Therefore, when ezetimibe is added to statin therapy, monitor hepatic transaminase levels before and during treatment according to the recommendations for the individual statin used. ( 5.2)
- Skeletal muscle effects (e.g., myopathy and rhabdomyolysis):
- Cases of myopathy and rhabdomyolysis have been reported in patients treated with ezetimibe co-administered with a statin and with ezetimibe administered alone. Risk for skeletal muscle toxicity increases with higher doses of statin, advanced age (>65), hypothyroidism, renal impairment, and depending on the statin used, concomitant use of other drugs. ( 5.3, 6.2)
ADVERSE REACTIONS
Common adverse reactions in clinical trials:
- Ezetimibe co-administered with a statin (incidence ≥2% and greater than statin alone)
- nasopharyngitis, myalgia, upper respiratory tract infection, arthralgia, and diarrhea ( 6)
- Ezetimibe administered alone (incidence ≥2% and greater than placebo):
- upper respiratory tract infection, diarrhea, arthralgia, sinusitis, and pain in extremity ( 6)
To report SUSPECTED ADVERSE REACTIONS, contact AvKARE, Inc. at 1-855-361-3993; email drugsafety@avkare.com; or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Cyclosporine: Combination increases exposure of ezetimibe and cyclosporine. Cyclosporine concentrations should be monitored in patients taking ezetimibe concomitantly. ( 7.1, 12.3)
- Fenofibrate: Combination increases exposure of ezetimibe. If cholelithiasis is suspected in a patient receiving ezetimibe and fenofibrate, gallbladder studies are indicated and alternative lipid-lowering therapy should be considered. ( 6.1, 7.3)
- Fibrates: Co-administration of ezetimibe with fibrates other than fenofibrate is not recommended until use in patients is adequately studied. ( 7.2)
- Cholestyramine: Combination decreases exposure of ezetimibe. ( 2.3, 7.4, 12.3)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 7/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Primary Hyperlipidemia
1.2 Homozygous Familial Hypercholesterolemia (HoFH)
1.3 Homozygous Sitosterolemia
1.4 Limitations of Use
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Information
2.2 Concomitant Lipid-Lowering Therapy
2.3 Co-Administration with Bile Acid Sequestrants
2.4 Patients with Hepatic Impairment
2.5 Patients with Renal Impairment
2.6 Geriatric Patients
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Use with Statins or Fenofibrate
5.2 Liver Enzymes
5.3 Myopathy/Rhabdomyolysis
5.4 Hepatic Impairment
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post-Marketing Experience
7 DRUG INTERACTIONS
7.1 Cyclosporine
7.2 Fibrates
7.3 Fenofibrate
7.4 Cholestyramine
7.5 Coumarin Anticoagulants
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Primary Hyperlipidemia
14.2 Homozygous Familial Hypercholesterolemia (HoFH)
14.3 Homozygous Sitosterolemia (Phytosterolemia)
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
17.1 Muscle Pain
17.2 Liver Enzymes
17.3 Pregnancy
17.4 Breast-feeding
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
Therapy with lipid-altering agents should be only one component of multiple risk factor intervention in individuals at significantly increased risk for atherosclerotic vascular disease due to hypercholesterolemia. Drug therapy is indicated as an adjunct to diet when the response to a diet restricted in saturated fat and cholesterol and other nonpharmacologic measures alone has been inadequate.
1.1 Primary Hyperlipidemia
Monotherapy
Ezetimibe tablets, administered alone, is indicated as adjunctive therapy to diet for the reduction of elevated total cholesterol (total-C), low-density lipoprotein cholesterol (LDL-C), apolipoprotein B (Apo B), and non-high-density lipoprotein cholesterol (non-HDL-C) in patients with primary (heterozygous familial and non-familial) hyperlipidemia.
Combination Therapy with HMG-CoA Reductase Inhibitors (Statins)
Ezetimibe tablets, administered in combination with a 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitor (statin), is indicated as adjunctive therapy to diet for the reduction of elevated total-C, LDL-C, Apo B, and non-HDL-C in patients with primary (heterozygous familial and non-familial) hyperlipidemia.
Combination Therapy with Fenofibrate
Ezetimibe tablets, administered in combination with fenofibrate, are indicated as adjunctive therapy to diet for the reduction of elevated total-C, LDL-C, Apo B, and non-HDL-C in adult patients with mixed hyperlipidemia.
1.2 Homozygous Familial Hypercholesterolemia (HoFH)
The combination of ezetimibe tablets and atorvastatin or simvastatin is indicated for the reduction of elevated total-C and LDL-C levels in patients with HoFH, as an adjunct to other lipid-lowering treatments (e.g., LDL apheresis) or if such treatments are unavailable.
-
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Information
The recommended dose of ezetimibe tablets is 10 mg once daily.
Ezetimibe tablets can be administered with or without food.
2.2 Concomitant Lipid-Lowering Therapy
Ezetimibe tablets may be administered with a statin (in patients with primary hyperlipidemia) or with fenofibrate (in patients with mixed hyperlipidemia) for incremental effect. For convenience, the daily dose of ezetimibe tablets may be taken at the same time as the statin or fenofibrate, according to the dosing recommendations for the respective medications.
2.3 Co-Administration with Bile Acid Sequestrants
Dosing of ezetimibe tablets should occur either ≥2 hours before or ≥4 hours after administration of a bile acid sequestrant [see Drug Interactions (7.4)] .
2.4 Patients with Hepatic Impairment
No dosage adjustment is necessary in patients with mild hepatic impairment [see Warnings and Precautions (5.4)] .
2.5 Patients with Renal Impairment
No dosage adjustment is necessary in patients with renal impairment [see Clinical Pharmacology (12.3)] . When given with simvastatin in patients with moderate to severe renal impairment (estimated glomerular filtration rate <60 mL/min/1.73 m 2), doses of simvastatin exceeding 20 mg should be used with caution and close monitoring [see Use in Specific Populations (8.6)] .
2.6 Geriatric Patients
No dosage adjustment is necessary in geriatric patients [see Clinical Pharmacology (12.3)] .
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Ezetimibe is contraindicated in the following conditions:
- The combination of ezetimibe with a statin is contraindicated in patients with active liver disease or unexplained persistent elevations in hepatic transaminase levels.
- Women who are pregnant or may become pregnant. Because statins decrease cholesterol synthesis and possibly the synthesis of other biologically active substances derived from cholesterol, ezetimibe in combination with a statin may cause fetal harm when administered to pregnant women. Additionally, there is no apparent benefit to therapy during pregnancy, and safety in pregnant women has not been established. If the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus and the lack of known clinical benefit with continued use during pregnancy [see Use in Specific Populations (8.1)].
- Nursing mothers. Because statins may pass into breast milk, and because statins have the potential to cause serious adverse reactions in nursing infants, women who require ezetimibe treatment in combination with a statin should be advised not to nurse their infants [see Use in Specific Populations (8.3)] .
- Patients with a known hypersensitivity to any component of this product. Hypersensitivity reactions including anaphylaxis, angioedema, rash and urticaria have been reported with ezetimibe [see Adverse Reactions (6.2)] .
-
5 WARNINGS AND PRECAUTIONS
5.1 Use with Statins or Fenofibrate
Concurrent administration of ezetimibe with a specific statin or fenofibrate should be in accordance with the product labeling for that medication.
5.2 Liver Enzymes
In controlled clinical monotherapy studies, the incidence of consecutive elevations (≥3 X the upper limit of normal [ULN]) in hepatic transaminase levels was similar between ezetimibe (0.5%) and placebo (0.3%).
In controlled clinical combination studies of ezetimibe initiated concurrently with a statin, the incidence of consecutive elevations (≥3 X ULN) in hepatic transaminase levels was 1.3% for patients treated with ezetimibe administered with statins and 0.4% for patients treated with statins alone. These elevations in transaminases were generally asymptomatic, not associated with cholestasis, and returned to baseline after discontinuation of therapy or with continued treatment. When ezetimibe is co-administered with a statin, liver tests should be performed at initiation of therapy and according to the recommendations of the statin. Should an increase in ALT or AST ≥3 X ULN persist, consider withdrawal of ezetimibe and/or the statin.
5.3 Myopathy/Rhabdomyolysis
In clinical trials, there was no excess of myopathy or rhabdomyolysis associated with ezetimibe compared with the relevant control arm (placebo or statin alone). However, myopathy and rhabdomyolysis are known adverse reactions to statins and other lipid-lowering drugs. In clinical trials, the incidence of creatine phosphokinase (CPK) >10 X ULN was 0.2% for ezetimibe vs. 0.1% for placebo, and 0.1% for ezetimibe co-administered with a statin vs 0.4% for statins alone. Risk for skeletal muscle toxicity increases with higher doses of statin, advanced age (>65), hypothyroidism, renal impairment, and depending on the statin used, concomitant use of other drugs.
In post-marketing experience with ezetimibe, cases of myopathy and rhabdomyolysis have been reported. Most patients who developed rhabdomyolysis were taking a statin prior to initiating ezetimibe. However, rhabdomyolysis has been reported with ezetimibe monotherapy and with the addition of ezetimibe to agents known to be associated with increased risk of rhabdomyolysis, such as fibrates. Ezetimibe and any statin or fibrate that the patient is taking concomitantly should be immediately discontinued if myopathy is diagnosed or suspected. The presence of muscle symptoms and a CPK level >10 X the ULN indicates myopathy.
5.4 Hepatic Impairment
Due to the unknown effects of the increased exposure to ezetimibe in patients with moderate to severe hepatic impairment, ezetimibe is not recommended in these patients [see Clinical Pharmacology (12.3)].
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed in greater detail in other sections of the label:
- Liver enzyme abnormalities [see Warnings and Precautions (5.2)]
- Rhabdomyolysis and myopathy [see Warnings and Precautions (5.3)]
Monotherapy Studies:
In the ezetimibe controlled clinical trials database (placebo-controlled) of 2396 patients with a median treatment duration of 12 weeks (range 0 to 39 weeks), 3.3% of patients on ezetimibe and 2.9% of patients on placebo discontinued due to adverse reactions. The most common adverse reactions in the group of patients treated with ezetimibe that led to treatment discontinuation and occurred at a rate greater than placebo were:
- Arthralgia (0.3%)
- Dizziness (0.2%)
- Gamma-glutamyltransferase increased (0.2%)
The most commonly reported adverse reactions (incidence ≥2% and greater than placebo) in the ezetimibe monotherapy controlled clinical trial database of 2396 patients were: upper respiratory tract infection (4.3%), diarrhea (4.1%), arthralgia (3%), sinusitis (2.8%), and pain in extremity (2.7%).
Statin Co-Administration Studies:
In the ezetimibe + statin controlled clinical trials database of 11,308 patients with a median treatment duration of 8 weeks (range 0 to 112 weeks), 4% of patients on ezetimibe + statin and 3.3% of patients on statin alone discontinued due to adverse reactions. The most common adverse reactions in the group of patients treated with ezetimibe + statin that led to treatment discontinuation and occurred at a rate greater than statin alone were:
- Alanine aminotransferase increased (0.6%)
- Myalgia (0.5%)
- Fatigue, aspartate aminotransferase increased, headache, and pain in extremity (each at 0.2%)
The most commonly reported adverse reactions (incidence ≥2% and greater than statin alone) in the ezetimibe + statin controlled clinical trial database of 11,308 patients were: nasopharyngitis (3.7%), myalgia (3.2%), upper respiratory tract infection (2.9%), arthralgia (2.6%) and diarrhea (2.5%).
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in clinical practice.
Monotherapy
In 10 double-blind, placebo-controlled clinical trials, 2396 patients with primary hyperlipidemia (age range 9 to 86 years, 50% women, 90% Caucasians, 5% Blacks, 3% Hispanics, 2% Asians) and elevated
LDL-C were treated with ezetimibe 10 mg/day for a median treatment duration of 12 weeks (range 0 to 39 weeks).
Adverse reactions reported in ≥2% of patients treated with ezetimibe and at an incidence greater than placebo in placebo-controlled studies of ezetimibe, regardless of causality assessment, are shown in Table 1.
TABLE 1: Clinical Adverse Reactions Occurring in ≥2% of Patients Treated with Ezetimibe and at an Incidence Greater than Placebo, Regardless of Causality
Body System/Organ Class
Adverse Reaction
Ezetimibe 10 mg
(%)
n=2396Placebo
(%)
n=1159Gastrointestinal disorders
Diarrhea
4.1
3.7
General disorders and administration site conditions
Fatigue
2.4
1.5
Infections and infestations
Influenza
2
1.5
Sinusitis
2.8
2.2
Upper respiratory tract infection
4.3
2.5
Musculoskeletal and connective tissue disorders
Arthralgia
3
2.2
Pain in extremity
2.7
2.5
The frequency of less common adverse reactions was comparable between ezetimibe and placebo.
Combination with a Statin
In 28 double-blind, controlled (placebo or active-controlled) clinical trials, 11,308 patients with primary hyperlipidemia (age range 10 to 93 years, 48% women, 85% Caucasians, 7% Blacks, 4% Hispanics, 3% Asians) and elevated LDL-C were treated with ezetimibe 10 mg/day concurrently with or added to on-going statin therapy for a median treatment duration of 8 weeks (range 0 to 112 weeks).
The incidence of consecutive increased transaminases (≥3 X ULN) was higher in patients receiving ezetimibe administered with statins (1.3%) than in patients treated with statins alone (0.4%) [see Warnings and Precautions (5.2)].
Clinical adverse reactions reported in ≥2% of patients treated with ezetimibe + statin and at an incidence greater than statin, regardless of causality assessment, are shown in Table 2.
TABLE 2: Clinical Adverse Reactions Occurring in ≥2% of Patients Treated with Ezetimibe
Co-Administered with a Statin and at an Incidence Greater than Statin,
Regardless of CausalityBody System/Organ Class
Adverse Reaction
All Statins *
(%)
n=9361Ezetimibe + All Statins *
(%)
n=11,308Gastrointestinal disorders
Diarrhea
2.2
2.5
General disorders and administration site conditions
Fatigue
1.6
2
Infections and infestations
Influenza
2.1
2.2
Nasopharyngitis
3.3
3.7
Upper respiratory tract infection
2.8
2.9
Musculoskeletal and connective tissue disorders
Arthralgia
2.4
2.6
Back pain
2.3
2.4
Myalgia
2.7
3.2
Pain in extremity
1.9
2.1
*All Statins = all doses of all statins
Combination with Fenofibrate
This clinical study involving 625 patients with mixed dyslipidemia (age range 20 to 76 years, 44% women, 79% Caucasians, 0.1% Blacks, 11% Hispanics, 5% Asians) treated for up to 12 weeks and 576 patients treated for up to an additional 48 weeks evaluated co-administration of ezetimibe and fenofibrate. This study was not designed to compare treatment groups for infrequent events. Incidence rates (95% CI) for clinically important elevations (≥3 X ULN, consecutive) in hepatic transaminase levels were 4.5% (1.9, 8.8) and 2.7% (1.2, 5.4) for fenofibrate monotherapy (n=188) and ezetimibe co-administered with fenofibrate (n=183), respectively, adjusted for treatment exposure. Corresponding incidence rates for cholecystectomy were 0.6% (95% CI: 0%, 3.1%) and 1.7% (95% CI: 0.6%, 4%) for fenofibrate monotherapy and ezetimibe co-administered with fenofibrate, respectively [see Drug Interactions (7.3)] . The numbers of patients exposed to co-administration therapy as well as fenofibrate and ezetimibe monotherapy were inadequate to assess gallbladder disease risk. There were no CPK elevations >10 X ULN in any of the treatment groups.
6.2 Post-Marketing Experience
Because the reactions below are reported voluntarily from a population of uncertain size, it is generally not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
The following additional adverse reactions have been identified during post-approval use of ezetimibe:
Hypersensitivity reactions, including anaphylaxis, angioedema, rash, and urticaria; erythema multiforme; arthralgia; myalgia; elevated creatine phosphokinase; myopathy/rhabdomyolysis [see Warnings and Precautions (5.3)] ; elevations in liver transaminases; hepatitis; abdominal pain; thrombocytopenia; pancreatitis; nausea; dizziness; paresthesia; depression; headache; cholelithiasis; cholecystitis.
To report SUSPECTED ADVERSE REACTIONS, contact AvKARE, Inc. at 1-855-361-3993; email drugsafety@avkare.com; or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
-
7 DRUG INTERACTIONS
[See Clinical Pharmacology (12.3).]
7.1 Cyclosporine
Caution should be exercised when using ezetimibe and cyclosporine concomitantly due to increased exposure to both ezetimibe and cyclosporine. Cyclosporine concentrations should be monitored in patients receiving ezetimibe and cyclosporine.
The degree of increase in ezetimibe exposure may be greater in patients with severe renal insufficiency. In patients treated with cyclosporine, the potential effects of the increased exposure to ezetimibe from concomitant use should be carefully weighed against the benefits of alterations in lipid levels provided by ezetimibe.
7.2 Fibrates
The efficacy and safety of co-administration of ezetimibe with fibrates other than fenofibrate have not been studied.
Fibrates may increase cholesterol excretion into the bile, leading to cholelithiasis. In a preclinical study in dogs, ezetimibe increased cholesterol in the gallbladder bile [see Nonclinical Toxicology (13.2)] . Co-administration of ezetimibe with fibrates other than fenofibrate is not recommended until use in patients is adequately studied.
7.3 Fenofibrate
If cholelithiasis is suspected in a patient receiving ezetimibe and fenofibrate, gallbladder studies are indicated and alternative lipid-lowering therapy should be considered [see Adverse Reactions (6.1) and the product labeling for fenofibrate] .
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C:
There are no adequate and well-controlled studies of ezetimibe in pregnant women. Ezetimibe should be used during pregnancy only if the potential benefit justifies the risk to the fetus.
In oral (gavage) embryo-fetal development studies of ezetimibe conducted in rats and rabbits during organogenesis, there was no evidence of embryolethal effects at the doses tested (250, 500, 1000 mg/kg/day). In rats, increased incidences of common fetal skeletal findings (extra pair of thoracic ribs, unossified cervical vertebral centra, shortened ribs) were observed at 1000 mg/kg/day (~10 X the human exposure at 10 mg daily based on AUC 0-24hr for total ezetimibe). In rabbits treated with ezetimibe, an increased incidence of extra thoracic ribs was observed at 1000 mg/kg/day (150 X the human exposure at 10 mg daily based on AUC 0-24hr for total ezetimibe). Ezetimibe crossed the placenta when pregnant rats and rabbits were given multiple oral doses.
Multiple-dose studies of ezetimibe given in combination with statins in rats and rabbits during organogenesis result in higher ezetimibe and statin exposures. Reproductive findings occur at lower doses in combination therapy compared to monotherapy.
All statins are contraindicated in pregnant and nursing women. When ezetimibe is administered with a statin in a woman of childbearing potential, refer to the pregnancy category and product labeling for the statin [see Contraindications (4)].
8.3 Nursing Mothers
It is not known whether ezetimibe is excreted into human breast milk. In rat studies, exposure to total ezetimibe in nursing pups was up to half of that observed in maternal plasma. Because many drugs are excreted in human milk, caution should be exercised when ezetimibe is administered to a nursing woman. Ezetimibe should not be used in nursing mothers unless the potential benefit justifies the potential risk to the infant.
8.4 Pediatric Use
The effects of ezetimibe co-administered with simvastatin (n=126) compared to simvastatin monotherapy (n=122) have been evaluated in adolescent boys and girls with heterozygous familial hypercholesterolemia (HeFH). In a multicenter, double-blind, controlled study followed by an open-label phase, 142 boys and 106 postmenarchal girls, 10 to 17 years of age (mean age 14.2 years, 43% females, 82% Caucasians, 4% Asian, 2% Blacks, 13% multi-racial) with HeFH were randomized to receive either ezetimibe co-administered with simvastatin or simvastatin monotherapy. Inclusion in the study required 1) a baseline LDL-C level between 160 and 400 mg/dL and 2) a medical history and clinical presentation consistent with HeFH. The mean baseline LDL-C value was 225 mg/dL (range: 161 to 351 mg/dL) in the ezetimibe co-administered with simvastatin group compared to 219 mg/dL (range: 149 to 336 mg/dL) in the simvastatin monotherapy group. The patients received co-administered ezetimibe and simvastatin (10 mg, 20 mg, or 40 mg) or simvastatin monotherapy (10 mg, 20 mg, or 40 mg) for 6 weeks, co-administered ezetimibe and 40 mg simvastatin or 40 mg simvastatin monotherapy for the next 27 weeks, and open-label co-administered ezetimibe and simvastatin (10 mg, 20 mg, or 40 mg) for 20 weeks thereafter.
The results of the study at Week 6 are summarized in Table 3. Results at Week 33 were consistent with those at Week 6.
TABLE 3: Mean Percent Difference at Week 6 Between the Pooled Ezetimibe Co-Administered with Simvastatin
Group and the Pooled Simvastatin Monotherapy Group in Adolescent Patients with Heterozygous Familial Hypercholesterolemia
Total-C
LDL-C
Apo B
Non-HDL-C
TG *
HDL-C
Mean percent difference between treatment groups
-12%
-15%
-12%
-14%
-2%
+0.1%
95% Confidence Interval
(-15%, -9%)
(-18%, -12%)
(-15%, -9%)
(-17%, -11%)
(-9%, +4%)
(-3%, +3%)
* For triglycerides, median % change from baseline
From the start of the trial to the end of Week 33, discontinuations due to an adverse reaction occurred in 7 (6%) patients in the ezetimibe co-administered with simvastatin group and in 2 (2%) patients in the simvastatin monotherapy group.
During the trial, hepatic transaminase elevations (two consecutive measurements for ALT and/or AST ≥3 X ULN) occurred in four (3%) individuals in the ezetimibe co-administered with simvastatin group and in two (2%) individuals in the simvastatin monotherapy group. Elevations of CPK (≥10 X ULN) occurred in two (2%) individuals in the ezetimibe co-administered with simvastatin group and in zero individuals in the simvastatin monotherapy group.
In this limited controlled study, there was no significant effect on growth or sexual maturation in the adolescent boys or girls, or on menstrual cycle length in girls.
Co-administration of ezetimibe with simvastatin at doses greater than 40 mg/day has not been studied in adolescents. Also, ezetimibe has not been studied in patients younger than 10 years of age or in pre-menarcheal girls.
Based on total ezetimibe (ezetimibe + ezetimibe-glucuronide), there are no pharmacokinetic differences between adolescents and adults. Pharmacokinetic data in the pediatric population <10 years of age are not available.
8.5 Geriatric Use
Monotherapy Studies
Of the 2396 patients who received ezetimibe in clinical studies, 669 (28%) were 65 and older, and 111 (5%) were 75 and older.
Statin Co-Administration Studies
Of the 11,308 patients who received ezetimibe + statin in clinical studies, 3587 (32%) were 65 and older, and 924 (8%) were 75 and older.
No overall differences in safety and effectiveness were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
When used as monotherapy, no dosage adjustment of ezetimibe is necessary.
In the Study of Heart and Renal Protection (SHARP) trial of 9270 patients with moderate to severe renal impairment (6247 non-dialysis patients with median serum creatinine 2.5 mg/dL and median estimated glomerular filtration rate 25.6 mL/min/1.73 m 2, and 3023 dialysis patients), the incidence of serious adverse events, adverse events leading to discontinuation of study treatment, or adverse events of special interest (musculoskeletal adverse events, liver enzyme abnormalities, incident cancer) was similar between patients ever assigned to ezetimibe 10 mg plus simvastatin 20 mg (n=4650) or placebo (n=4620) during a median follow-up of 4.9 years. However, because renal impairment is a risk factor for statin-associated myopathy, doses of simvastatin exceeding 20 mg should be used with caution and close monitoring when administered concomitantly with ezetimibe in patients with moderate to severe renal impairment.
8.7 Hepatic Impairment
Ezetimibe is not recommended in patients with moderate to severe hepatic impairment [see Warnings and Precautions (5.4) and Clinical Pharmacology (12.3)] .
Ezetimibe given concomitantly with a statin is contraindicated in patients with active liver disease or unexplained persistent elevations of hepatic transaminase levels [see Contraindications (4); Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
In clinical studies, administration of ezetimibe, 50 mg/day to 15 healthy subjects for up to 14 days, 40 mg/day to 18 patients with primary hyperlipidemia for up to 56 days, and 40 mg/day to 27 patients with homozygous sitosterolemia for 26 weeks was generally well tolerated. One female patient with homozygous sitosterolemia took an accidental overdose of ezetimibe 120 mg/day for 28 days with no reported clinical or laboratory adverse events.
In the event of an overdose, symptomatic and supportive measures should be employed.
-
11 DESCRIPTION
Ezetimibe is in a class of lipid-lowering compounds that selectively inhibits the intestinal absorption of cholesterol and related phytosterols. The chemical name of ezetimibe is 1-(4-fluorophenyl)- 3(R)-[3-(4-fluorophenyl)-3(S)-hydroxypropyl]-4(S)-(4-hydroxyphenyl)-2-azetidinone. The empirical formula is C 24H 21F 2NO 3. Its molecular weight is 409.4 and its structural formula is:
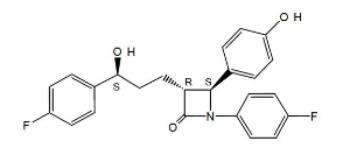
Ezetimibe, USP is a white, crystalline powder that is freely to very soluble in ethanol, methanol, and acetone and practically insoluble in water. Ezetimibe, USP has a melting point of about 163°C and is stable at ambient temperature. Ezetimibe tablets, USP are available as a tablet for oral administration containing 10 mg of ezetimibe, USP and the following inactive ingredients: croscarmellose sodium, lactose monohydrate, magnesium stearate, povidone, and sodium lauryl sulfate.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ezetimibe reduces blood cholesterol by inhibiting the absorption of cholesterol by the small intestine. In a 2-week clinical study in 18 hypercholesterolemic patients, ezetimibe inhibited intestinal cholesterol absorption by 54%, compared with placebo. Ezetimibe had no clinically meaningful effect on the plasma concentrations of the fat-soluble vitamins A, D, and E (in a study of 113 patients), and did not impair adrenocortical steroid hormone production (in a study of 118 patients).
The cholesterol content of the liver is derived predominantly from three sources. The liver can synthesize cholesterol, take up cholesterol from the blood from circulating lipoproteins, or take up cholesterol absorbed by the small intestine. Intestinal cholesterol is derived primarily from cholesterol secreted in the bile and from dietary cholesterol.
Ezetimibe has a mechanism of action that differs from those of other classes of cholesterol-reducing compounds (statins, bile acid sequestrants [resins], fibric acid derivatives, and plant stanols). The molecular target of ezetimibe has been shown to be the sterol transporter, Niemann-Pick C1-Like 1 (NPC1L1), which is involved in the intestinal uptake of cholesterol and phytosterols.
Ezetimibe does not inhibit cholesterol synthesis in the liver, or increase bile acid excretion. Instead, ezetimibe localizes at the brush border of the small intestine and inhibits the absorption of cholesterol, leading to a decrease in the delivery of intestinal cholesterol to the liver. This causes a reduction of hepatic cholesterol stores and an increase in clearance of cholesterol from the blood; this distinct mechanism is complementary to that of statins and of fenofibrate [see Clinical Studies (14.1)] .
12.2 Pharmacodynamics
Clinical studies have demonstrated that elevated levels of total-C, LDL-C and Apo B, the major protein constituent of LDL, promote human atherosclerosis. In addition, decreased levels of HDL-C are associated with the development of atherosclerosis. Epidemiologic studies have established that cardiovascular morbidity and mortality vary directly with the level of total-C and LDL-C and inversely with the level of HDL-C. Like LDL, cholesterol-enriched triglyceride-rich lipoproteins, including very-low-density lipoproteins (VLDL), intermediate-density lipoproteins (IDL), and remnants, can also promote atherosclerosis. The independent effect of raising HDL-C or lowering TG on the risk of coronary and cardiovascular morbidity and mortality has not been determined.
Ezetimibe reduces total-C, LDL-C, Apo B, non-HDL-C, and TG, and increases HDL-C in patients with hyperlipidemia. Administration of ezetimibe with a statin is effective in improving serum total-C, LDL-C, Apo B, non-HDL-C, TG, and HDL-C beyond either treatment alone. Administration of ezetimibe with fenofibrate is effective in improving serum total-C, LDL-C, Apo B, and non-HDL-C in patients with mixed hyperlipidemia as compared to either treatment alone. The effects of ezetimibe given either alone or in addition to a statin or fenofibrate on cardiovascular morbidity and mortality have not been established.
12.3 Pharmacokinetics
Absorption
After oral administration, ezetimibe is absorbed and extensively conjugated to a pharmacologically active phenolic glucuronide (ezetimibe-glucuronide). After a single 10-mg dose of ezetimibe to fasted adults, mean ezetimibe peak plasma concentrations (C max) of 3.4 to 5.5 ng/mL were attained within 4 to 12 hours (T max). Ezetimibe-glucuronide mean C max values of 45 to 71 ng/mL were achieved between 1 and 2 hours (T max). There was no substantial deviation from dose proportionality between 5 and 20 mg. The absolute bioavailability of ezetimibe cannot be determined, as the compound is virtually insoluble in aqueous media suitable for injection.
Effect of Food on Oral Absorption
Concomitant food administration (high-fat or non-fat meals) had no effect on the extent of absorption of ezetimibe when administered as ezetimibe 10-mg tablets. The C max value of ezetimibe was increased by 38% with consumption of high-fat meals. Ezetimibe can be administered with or without food.
Distribution
Ezetimibe and ezetimibe-glucuronide are highly bound (>90%) to human plasma proteins.
Metabolism and Excretion
Ezetimibe is primarily metabolized in the small intestine and liver via glucuronide conjugation (a phase II reaction) with subsequent biliary and renal excretion. Minimal oxidative metabolism (a phase I reaction) has been observed in all species evaluated.
In humans, ezetimibe is rapidly metabolized to ezetimibe-glucuronide. Ezetimibe and ezetimibe-glucuronide are the major drug-derived compounds detected in plasma, constituting approximately 10% to 20% and 80% to 90% of the total drug in plasma, respectively. Both ezetimibe and ezetimibe-glucuronide are eliminated from plasma with a half-life of approximately 22 hours for both ezetimibe and ezetimibe-glucuronide. Plasma concentration-time profiles exhibit multiple peaks, suggesting enterohepatic recycling.
Following oral administration of 14C-ezetimibe (20 mg) to human subjects, total ezetimibe (ezetimibe + ezetimibe-glucuronide) accounted for approximately 93% of the total radioactivity in plasma. After 48 hours, there were no detectable levels of radioactivity in the plasma.
Approximately 78% and 11% of the administered radioactivity were recovered in the feces and urine, respectively, over a 10-day collection period. Ezetimibe was the major component in feces and accounted for 69% of the administered dose, while ezetimibe-glucuronide was the major component in urine and accounted for 9% of the administered dose.
Specific Populations
Geriatric Patients: In a multiple-dose study with ezetimibe given 10 mg once daily for 10 days, plasma concentrations for total ezetimibe were about 2-fold higher in older (≥65 years) healthy subjects compared to younger subjects.
Pediatric Patients: [see Use in Specific Populations (8.4)].
Gender: In a multiple-dose study with ezetimibe given 10 mg once daily for 10 days, plasma concentrations for total ezetimibe were slightly higher (<20%) in women than in men.
Race: Based on a meta-analysis of multiple-dose pharmacokinetic studies, there were no pharmacokinetic differences between Black and Caucasian subjects. Studies in Asian subjects indicated that the pharmacokinetics of ezetimibe were similar to those seen in Caucasian subjects.
Hepatic Impairment: After a single 10-mg dose of ezetimibe, the mean AUC for total ezetimibe was increased approximately 1.7-fold in patients with mild hepatic impairment (Child-Pugh score 5 to 6), compared to healthy subjects. The mean AUC values for total ezetimibe and ezetimibe were increased approximately 3- to 4-fold and 5- to 6-fold, respectively, in patients with moderate (Child-Pugh score 7 to 9) or severe hepatic impairment (Child-Pugh score 10 to 15). In a 14-day, multiple-dose study (10 mg daily) in patients with moderate hepatic impairment, the mean AUC values for total ezetimibe and ezetimibe were increased approximately 4-fold on Day 1 and Day 14 compared to healthy subjects. Due to the unknown effects of the increased exposure to ezetimibe in patients with moderate or severe hepatic impairment, ezetimibe is not recommended in these patients [see Warnings and Precautions (5.4)] .
Renal Impairment: After a single 10-mg dose of ezetimibe in patients with severe renal disease (n=8; mean CrCl ≤30 mL/min/1.73 m 2), the mean AUC values for total ezetimibe, ezetimibe-glucuronide, and ezetimibe were increased approximately 1.5-fold, compared to healthy subjects (n=9).
Drug Interactions [see also Drug Interactions (7)]
Ezetimibe had no significant effect on a series of probe drugs (caffeine, dextromethorphan, tolbutamide, and IV midazolam) known to be metabolized by cytochrome P450 (1A2, 2D6, 2C8/9 and 3A4) in a “cocktail” study of twelve healthy adult males. This indicates that ezetimibe is neither an inhibitor nor an inducer of these cytochrome P450 isozymes, and it is unlikely that ezetimibe will affect the metabolism of drugs that are metabolized by these enzymes.
TABLE 4: Effect of Co-Administered Drugs on Total Ezetimibe
Co-Administered Drug and Dosing Regimen
Total Ezetimibe *
Change in AUC
Change in C max
Cyclosporine-stable dose required (75-150 mg BID) †, ‡
↑240%
↑290%
Fenofibrate, 200 mg QD, 14 days ‡
↑48%
↑64%
Gemfibrozil, 600 mg BID, 7 days ‡
↑64%
↑91%
Cholestyramine, 4 g BID, 14 days ‡
↓55%
↓4%
Aluminum & magnesium hydroxide combination antacid, single dose §
↓4%
↓30%
Cimetidine, 400 mg BID, 7 days
↑6%
↑22%
Glipizide, 10 mg, single dose
↑4%
↓8%
Statins
Lovastatin 20 mg QD, 7 days
↑9%
↑3%
Pravastatin 20 mg QD, 14 days
↑7%
↑23%
Atorvastatin 10 mg QD, 14 days
↓2%
↑12%
Rosuvastatin 10 mg QD, 14 days
↑13%
↑18%
Fluvastatin 20 mg QD, 14 days
↓19%
↑7%
* Based on 10 mg dose of ezetimibe
† Post-renal transplant patients with mild impaired or normal renal function. In a different study, a renal transplant patient with severe renal insufficiency (creatinine clearance of 13.2 mL/min/1.73 m 2) who was receiving multiple medications, including cyclosporine, demonstrated a 12-fold greater exposure to total ezetimibe compared to healthy subjects.
‡ See Drug Interactions (7).
§ Supralox, 20 mL
TABLE 5: Effect of Ezetimibe Co-Administration on Systemic Exposure to Other Drugs
Co-Administered Drug and its Dosage Regimen
Ezetimibe Dosage Regimen
Change in AUC of Co-Administered Drug
Change in C max of Co-Administered Drug
Warfarin, 25 mg single dose on day 7
10 mg QD, 11 days
↓2% (R-warfarin)
↓4% (S-warfarin)
↑3% (R-warfarin)
↑1% (S-warfarin)
Digoxin, 0.5 mg single dose
10 mg QD, 8 days
↑2%
↓7%
Gemfibrozil, 600 mg BID, 7 days *
10 mg QD, 7 days
↓1%
↓11%
Ethinyl estradiol & Levonorgestrel, QD, 21 days
10 mg QD, days 8 to 14 of 21d
oral contraceptive cycleEthinyl estradiol
0%
Levonorgestrel
0%Ethinyl estradiol
↓9%
Levonorgestrel
↓5%Glipizide, 10 mg on days 1 and 9
10 mg QD, days 2 to 9
↓3%
↓5%
Fenofibrate, 200 mg QD, 14 days *
10 mg QD, 14 days
↑11%
↑7%
Cyclosporine, 100 mg single dose day 7 *
20 mg QD, 8 days
↑15%
↑10%
Statins
Lovastatin 20 mg QD, 7 days
10 mg QD, 7 days
↑19%
↑3%
Pravastatin 20 mg QD, 14 days
10 mg QD, 14 days
↓20%
↓24%
Atorvastatin 10 mg QD, 14 days
10 mg QD, 14 days
↓4%
↑7%
Rosuvastatin 10 mg QD, 14 days
10 mg QD, 14 days
↑19%
↑17%
Fluvastatin 20 mg QD, 14 days
10 mg QD, 14 days
↓39%
↓27%
* See Drug Interactions (7).
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
A 104-week dietary carcinogenicity study with ezetimibe was conducted in rats at doses up to 1500 mg/kg/day (males) and 500 mg/kg/day (females) (~20 X the human exposure at 10 mg daily based on AUC 0-24hr for total ezetimibe). A 104-week dietary carcinogenicity study with ezetimibe was also conducted in mice at doses up to 500 mg/kg/day (>150 X the human exposure at 10 mg daily based on AUC 0-24hr for total ezetimibe). There were no statistically significant increases in tumor incidences in drug-treated rats or mice.
No evidence of mutagenicity was observed in vitro in a microbial mutagenicity (Ames) test with Salmonella typhimurium and Escherichia coli with or without metabolic activation. No evidence of clastogenicity was observed in vitro in a chromosomal aberration assay in human peripheral blood lymphocytes with or without metabolic activation. In addition, there was no evidence of genotoxicity in the in vivo mouse micronucleus test.
In oral (gavage) fertility studies of ezetimibe conducted in rats, there was no evidence of reproductive toxicity at doses up to 1000 mg/kg/day in male or female rats (~7 X the human exposure at 10 mg daily based on AUC 0-24hr for total ezetimibe).
13.2 Animal Toxicology and/or Pharmacology
The hypocholesterolemic effect of ezetimibe was evaluated in cholesterol-fed Rhesus monkeys, dogs, rats, and mouse models of human cholesterol metabolism. Ezetimibe was found to have an ED 50 value of 0.5 mcg/kg/day for inhibiting the rise in plasma cholesterol levels in monkeys. The ED 50 values in dogs, rats, and mice were 7, 30, and 700 mcg/kg/day, respectively. These results are consistent with ezetimibe being a potent cholesterol absorption inhibitor.
In a rat model, where the glucuronide metabolite of ezetimibe (SCH 60663) was administered intraduodenally, the metabolite was as potent as the parent compound (SCH 58235) in inhibiting the absorption of cholesterol, suggesting that the glucuronide metabolite had activity similar to the parent drug.
In 1-month studies in dogs given ezetimibe (0.03 to 300 mg/kg/day), the concentration of cholesterol in gallbladder bile increased ~2- to 4-fold. However, a dose of 300 mg/kg/day administered to dogs for one year did not result in gallstone formation or any other adverse hepatobiliary effects. In a 14-day study in mice given ezetimibe (0.3 to 5 mg/kg/day) and fed a low-fat or cholesterol-rich diet, the concentration of cholesterol in gallbladder bile was either unaffected or reduced to normal levels, respectively.
A series of acute preclinical studies was performed to determine the selectivity of ezetimibe for inhibiting cholesterol absorption. Ezetimibe inhibited the absorption of 14C-cholesterol with no effect on the absorption of triglycerides, fatty acids, bile acids, progesterone, ethinyl estradiol, or the fat-soluble vitamins A and D.
In 4- to 12-week toxicity studies in mice, ezetimibe did not induce cytochrome P450 drug metabolizing enzymes. In toxicity studies, a pharmacokinetic interaction of ezetimibe with statins (parents or their active hydroxy acid metabolites) was seen in rats, dogs, and rabbits.
-
14 CLINICAL STUDIES
14.1 Primary Hyperlipidemia
Ezetimibe reduces total-C, LDL-C, Apo B, non-HDL-C, and TG, and increases HDL-C in patients with hyperlipidemia. Maximal to near maximal response is generally achieved within 2 weeks and maintained during chronic therapy.
Monotherapy
In two multicenter, double-blind, placebo-controlled, 12-week studies in 1719 patients with primary hyperlipidemia, ezetimibe significantly lowered total-C, LDL-C, Apo B, non-HDL-C, and TG, and increased HDL-C compared to placebo (see Table 6). Reduction in LDL-C was consistent across age, sex, and baseline LDL-C.
TABLE 6: Response to Ezetimibe in Patients with Primary Hyperlipidemia
(Mean* % Change from Untreated Baseline †)
Treatment Group
N
Total-C
LDL-C
Apo B
Non-HDL-C
TG *
HDL-C
Study 1 ‡
Placebo
205
+1
+1
-1
+1
-1
-1
Ezetimibe
622
-12
-18
-15
-16
-7
+1
Study 2 ‡
Placebo
226
+1
+1
-1
+2
+2
-2
Ezetimibe
666
-12
-18
-16
-16
-9
+1
Pooled Data ‡
(Studies 1 & 2)Placebo
431
0
+1
-2
+1
0
-2
Ezetimibe
1288
-13
-18
-16
-16
-8
+1
* For triglycerides, median % change from baseline
† Baseline - on no lipid-lowering drug
‡ Ezetimibe significantly reduced total-C, LDL-C, Apo B, non-HDL-C, and TG, and increased HDL-C compared to placebo.
Combination with Statins
Ezetimibe Added to On-going Statin Therapy
In a multicenter, double-blind, placebo-controlled, 8-week study, 769 patients with primary hyperlipidemia, known coronary heart disease or multiple cardiovascular risk factors who were already receiving statin monotherapy, but who had not met their NCEP ATP II target LDL-C goal were randomized to receive either ezetimibe or placebo in addition to their on-going statin.
Ezetimibe, added to on-going statin therapy, significantly lowered total-C, LDL-C, Apo B, non-HDL-C, and TG, and increased HDL-C compared with a statin administered alone (see Table 7). LDL-C reductions induced by Ezetimibe were generally consistent across all statins.
TABLE 7: Response to Addition of Ezetimibe to On-Going Statin Therapy* in Patients with
Hyperlipidemia
(Mean † % Change from Treated Baseline ‡)Treatment
(Daily Dose)N
Total-C
LDL-C
Apo B
Non-HDL-C
TG †
HDL-C
On-going Statin + Placebo §
390
-2
-4
-3
-3
-3
+1
On-going Statin + Ezetimibe §
379
-17
-25
-19
-23
-14
+3
* Patients receiving each statin: 40% atorvastatin, 31% simvastatin, 29% others (pravastatin, fluvastatin, cerivastatin, lovastatin)
† For triglycerides, median % change from baseline
‡ Baseline - on a statin alone.
§ Ezetimibe + statin significantly reduced total-C, LDL-C, Apo B, non-HDL-C, and TG, and increased HDL-C compared to statin alone.Ezetimibe Initiated Concurrently with a Statin
In four multicenter, double-blind, placebo-controlled, 12-week trials, in 2382 hyperlipidemic patients, ezetimibe or placebo was administered alone or with various doses of atorvastatin, simvastatin, pravastatin, or lovastatin.
When all patients receiving ezetimibe with a statin were compared to all those receiving the corresponding statin alone, ezetimibe significantly lowered total-C, LDL-C, Apo B, non-HDL-C, and TG, and, with the exception of pravastatin, increased HDL-C compared to the statin administered alone. LDL-C reductions induced by ezetimibe were generally consistent across all statins. (See footnote ‡, Tables 8 to 11.)
TABLE 8: Response to Ezetimibe and Atorvastatin Initiated Concurrently
in Patients with Primary Hyperlipidemia
(Mean * % Change from Untreated Baseline †)Treatment
(Daily Dose)N
Total-C
LDL-C
Apo B
Non-HDL-C
TG *
HDL-C
Placebo
60
+4
+4
+3
+4
-6
+4
Ezetimibe
65
-14
-20
-15
-18
-5
+4
Atorvastatin 10 mg
60
-26
-37
-28
-34
-21
+6
Ezetimibe +
Atorvastatin 10 mg65
-38
-53
-43
-49
-31
+9
Atorvastatin 20 mg
60
-30
-42
-34
-39
-23
+4
Ezetimibe +
Atorvastatin 20 mg62
-39
-54
-44
-50
-30
+9
Atorvastatin 40 mg
66
-32
-45
-37
-41
-24
+4
Ezetimibe +
Atorvastatin 40 mg65
-42
-56
-45
-52
-34
+5
Atorvastatin 80 mg
62
-40
-54
-46
-51
-31
+3
Ezetimibe +
Atorvastatin 80 mg63
-46
-61
-50
-58
-40
+7
Pooled data (All
Atorvastatin Dose) ‡248
-32
-44
-36
-41
-24
+4
Pooled data (All Ezetimibe +
Atorvastatin Doses) ‡255
-41
-56
-45
-52
-33
+7
* For triglycerides, median % change from baseline
† Baseline - on no lipid-lowering drug
‡ Ezetimibe + all doses of atorvastatin pooled (10 to 80 mg) significantly reduced total-C, LDL-C, Apo B, non-HDL-C, and TG, and increased HDL-C compared to all doses of atorvastatin pooled (10 to 80 mg).TABLE 9: Response to Ezetimibe and Simvastatin Initiated Concurrently
in Patients with Primary Hyperlipidemia
(Mean * % Change from Untreated Baseline †)Treatment
(Daily Dose)N
Total-C
LDL-C
Apo B
Non-HDL-C
TG *
HDL-C
Placebo
70
-1
-1
0
-1
+2
+1
Ezetimibe
61
-13
-19
-14
-17
-11
+5
Simvastatin 10 mg
70
-18
-27
-21
-25
-14
+8
Ezetimibe +
Simvastatin 10 mg67
-32
-46
-35
-42
-26
+9
Simvastatin 20 mg
61
-26
-36
-29
-33
-18
+6
Ezetimibe +
Simvastatin 20 mg69
-33
-46
-36
-42
-25
+9
Simvastatin 40 mg
65
-27
-38
-32
-35
-24
+6
Ezetimibe +
Simvastatin 40 mg73
-40
-56
-45
-51
-32
+11
Simvastatin 80 mg
67
-32
-45
-37
-41
-23
+8
Ezetimibe +
Simvastatin 80 mg65
-41
-58
-47
-53
-31
+8
Pooled data (All
Atorvastatin Dose) ‡263
-26
-36
-30
-34
-20
+7
Pooled data (All Ezetimibe +
Simvastatin Doses) ‡274
-37
-51
-41
-47
-29
+9
* For triglycerides, median % change from baseline
† Baseline - on no lipid-lowering drug
‡ Ezetimibe + all doses of simvastatin pooled (10 to 80 mg) significantly reduced total-C, LDL-C, Apo B, non-HDL-C, and TG, and increased HDL-C compared to all doses of simvastatin pooled (10 to 80 mg).TABLE 10: Response to Ezetimibe and Pravastatin Initiated Concurrently
in Patients with Primary Hyperlipidemia
(Mean * % Change from Untreated Baseline †)Treatment
(Daily Dose)N
Total-C
LDL-C
Apo B
Non-HDL-C
TG *
HDL-C
Placebo
65
0
-1
-2
0
-1
+2
Ezetimibe
64
-13
-20
-15
-17
-5
+4
Pravastatin 10 mg
66
-15
-21
-16
-20
-14
+6
Ezetimibe +
Pravastatin 10 mg71
-24
-34
-27
-32
-23
+8
Pravastatin 20 mg
69
-15
-23
-18
-20
-8
+8
Ezetimibe +
Pravastatin 20 mg66
-27
-40
-31
-36
-21
+8
Pravastatin 40 mg
70
-22
-31
-26
-28
-19
+6
Ezetimibe +
Pravastatin 40 mg67
-30
-42
-32
-39
-21
+8
Pooled data (All
Pravastatin Dose) ‡205
-17
-25
-20
-23
-14
+7
Pooled data (All Ezetimibe +
Pravastatin Doses) ‡204
-27
-39
-30
-36
-21
+8
* For triglycerides, median % change from baseline
† Baseline - on no lipid-lowering drug
‡ Ezetimibe + all doses of pravastatin pooled (10 to 40 mg) significantly reduced total-C, LDL-C, Apo B, non-HDL-C, and TG compared to all doses of pravastatin pooled (10 to 40 mg).TABLE 11: Response to Ezetimibe and Lovastatin Initiated Concurrently
in Patients with Primary Hyperlipidemia
(Mean * % Change from Untreated Baseline †)Treatment
(Daily Dose)N
Total-C
LDL-C
Apo B
Non-HDL-C
TG *
HDL-C
Placebo
64
+1
0
+1
+1
+6
0
Ezetimibe
72
-13
-19
-14
-16
-5
+3
Lovastatin 10 mg
73
-15
-20
-17
-19
-11
+5
Ezetimibe +
Lovastatin 10 mg65
-24
-34
-27
-31
-19
+8
Lovastatin 20 mg
74
-19
-26
-21
-24
-12
+3
Ezetimibe +
Lovastatin 20 mg62
-29
-41
-34
-39
-27
+9
Lovastatin 40 mg
73
-21
-30
-25
-27
-15
+5
Ezetimibe +
Lovastatin 40 mg65
-33
-46
-38
-43
-27
+9
Pooled data (All
Lovastatin Dose) ‡220
-18
-25
-21
-23
-12
+4
Pooled data (All Ezetimibe +
Lovastatin Doses) ‡192
-29
-40
-33
-38
-25
+9
* For triglycerides, median % change from baseline
† Baseline - on no lipid-lowering drug
‡ Ezetimibe + all doses of lovastatin pooled (10 to 40 mg) significantly reduced total-C, LDL-C, Apo B, non-HDL-C, and TG, and increased HDL-C compared to all doses of lovastatin pooled (10 to 40 mg).Combination with Fenofibrate
In a multicenter, double-blind, placebo-controlled, clinical study in patients with mixed hyperlipidemia, 625 patients were treated for up to 12 weeks and 576 for up to an additional 48 weeks. Patients were randomized to receive placebo, ezetimibe alone, 160 mg fenofibrate alone, or ezetimibe and 160 mg fenofibrate in the 12-week study. After completing the 12-week study, eligible patients were assigned to ezetimibe co-administered with fenofibrate or fenofibrate monotherapy for an additional 48 weeks.
Ezetimibe co-administered with fenofibrate significantly lowered total-C, LDL-C, Apo B, and non-HDL-C compared to fenofibrate administered alone. The percent decrease in TG and percent increase in HDL-C for Ezetimibe co-administered with fenofibrate were comparable to those for fenofibrate administered alone (see Table 12).
TABLE 12: Response to Ezetimibe and Fenofibrate Initiated Concurrently
in Patients with Mixed Hyperlipidemia
(Mean * % Change from Untreated Baseline †)Treatment
(Daily Dose)N
Total-C
LDL-C
Apo B
Non-HDL-C
TG *
Non-HDL-C
Placebo
63
0
0
-1
-9
+3
0
Ezetimibe
185
-12
-13
-11
-11
+4
-15
Fenofibrate 160 mg
188
-11
-6
-15
-43
+19
-16
Ezetimibe + Fenofibrate 160 mg
183
-22
-20
-26
-44
+19
-30
* For triglycerides, median % change from baseline
† Baseline - on no lipid-lowering drug
The changes in lipid endpoints after an additional 48 weeks of treatment with ezetimibe co-administered with fenofibrate or with fenofibrate alone were consistent with the 12-week data displayed above.
14.2 Homozygous Familial Hypercholesterolemia (HoFH)
A study was conducted to assess the efficacy of ezetimibe in the treatment of HoFH. This double-blind, randomized, 12-week study enrolled 50 patients with a clinical and/or genotypic diagnosis of HoFH, with or without concomitant LDL apheresis, already receiving atorvastatin or simvastatin (40 mg). Patients were randomized to one of three treatment groups, atorvastatin or simvastatin (80 mg), ezetimibe administered with atorvastatin or simvastatin (40 mg), or ezetimibe administered with atorvastatin or simvastatin (80 mg). Due to decreased bioavailability of ezetimibe in patients concomitantly receiving cholestyramine [see Drug Interactions (7.4)] , ezetimibe was dosed at least 4 hours before or after administration of resins. Mean baseline LDL-C was 341 mg/dL in those patients randomized to atorvastatin 80 mg or simvastatin 80 mg alone and 316 mg/dL in the group randomized to ezetimibe plus atorvastatin 40 or 80 mg or simvastatin 40 or 80 mg. Ezetimibe, administered with atorvastatin or simvastatin (40 and 80 mg statin groups, pooled), significantly reduced LDL-C (21%) compared with increasing the dose of simvastatin or atorvastatin monotherapy from 40 to 80 mg (7%). In those treated with ezetimibe plus 80 mg atorvastatin or with ezetimibe plus 80 mg simvastatin, LDL-C was reduced by 27%.
14.3 Homozygous Sitosterolemia (Phytosterolemia)
A study was conducted to assess the efficacy of ezetimibe in the treatment of homozygous sitosterolemia. In this multicenter, double-blind, placebo-controlled, 8-week trial, 37 patients with homozygous sitosterolemia with elevated plasma sitosterol levels (>5 mg/dL) on their current therapeutic regimen (diet, bile-acid-binding resins, statins, ileal bypass surgery and/or LDL apheresis), were randomized to receive ezetimibe (n=30) or placebo (n=7). Due to decreased bioavailability of ezetimibe in patients concomitantly receiving cholestyramine [see Drug Interactions (7.4)] , ezetimibe was dosed at least 2 hours before or 4 hours after resins were administered. Excluding the one subject receiving LDL apheresis, ezetimibe significantly lowered plasma sitosterol and campesterol, by 21% and 24% from baseline, respectively. In contrast, patients who received placebo had increases in sitosterol and campesterol of 4% and 3% from baseline, respectively. For patients treated with ezetimibe, mean plasma levels of plant sterols were reduced progressively over the course of the study. The effects of reducing plasma sitosterol and campesterol on reducing the risks of cardiovascular morbidity and mortality have not been established.
Reductions in sitosterol and campesterol were consistent between patients taking ezetimibe concomitantly with bile acid sequestrants (n=8) and patients not on concomitant bile acid sequestrant therapy (n=21).
Limitations of Use
The effect of ezetimibe on cardiovascular morbidity and mortality has not been determined.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Ezetimibe tablets USP, 10 mg are supplied as white to off-white, capsule-shaped tablets debossed with “AA69” on one side and plain on other side.
They are supplied as follows:NDC: 50268-298-12 (10 tablets per card, 2 cards per carton)
Dispensed in Unit Dose Package. For Institutional Use Only.
Storage
Store at 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature]. Protect from moisture.
-
17 PATIENT COUNSELING INFORMATION
See FDA-Approved Patient Labeling ( Patient Information).
Patients should be advised to adhere to their National Cholesterol Education Program (NCEP)-recommended diet, a regular exercise program, and periodic testing of a fasting lipid panel.
17.1 Muscle Pain
All patients starting therapy with ezetimibe should be advised of the risk of myopathy and told to report promptly any unexplained muscle pain, tenderness or weakness. The risk of this occurring is increased when taking certain types of medication. Patients should discuss all medication, both prescription and over-the-counter, with their physician.
17.2 Liver Enzymes
Liver tests should be performed when ezetimibe is added to statin therapy and according to statin recommendations.
17.3 Pregnancy
Women of childbearing age should be advised to use an effective method of birth control to prevent pregnancy while using ezetimibe added to statin therapy. Discuss future pregnancy plans with your patients, and discuss when to stop combination ezetimibe and statin therapy if they are trying to conceive. Patients should be advised that if they become pregnant they should stop taking combination ezetimibe and statin therapy and call their healthcare professional.
17.4 Breast-feeding
Women who are breast-feeding should be advised to not use ezetimibe added to statin therapy. Patients who have a lipid disorder and are breastfeeding should be advised to discuss the options with their healthcare professionals.
Manufactured for:
AvKARE, Inc.
Pulaski, TN 38478Mfg. Rev. 05-2017-00
AV 07/18 (P)
AvPAK -
Patient Information
Ezetimibe (ez-ET-i-mibe) Tablets
Read this information carefully before you start taking ezetimibe tablets and each time you get more ezetimibe tablets. There may be new information. This information does not take the place of talking with your doctor about your medical condition or your treatment. If you have any questions about ezetimibe tablets, ask your doctor. Only your doctor can determine if ezetimibe tablets are right for you.
What are ezetimibe tablets?
Ezetimibe tablets are a medicine used to lower levels of total cholesterol and LDL (bad) cholesterol in the blood. Ezetimibe tablets are for patients who cannot control their cholesterol levels by diet and exercise alone. It can be used by itself or with other medicines to treat high cholesterol. You should stay on a cholesterol-lowering diet while taking this medicine.
Ezetimibe tablets work to reduce the amount of cholesterol your body absorbs. Ezetimibe tablets do not help you lose weight. Ezetimibe tablets have not been shown to prevent heart disease or heart attacks.
For more information about cholesterol, see the “What should I know about high cholesterol?” section that follows.
Who should not take ezetimibe tablets?
- Do not take ezetimibe tablets if you are allergic to ezetimibe, the active ingredient in ezetimibe tablets, or to the inactive ingredients. For a list of inactive ingredients, see the “Inactive ingredients” section that follows.
- If you have active liver disease, do not take ezetimibe tablets while taking cholesterol-lowering medicines called statins.
- If you are pregnant or breast-feeding, do not take ezetimibe tablets while taking a statin.
- If you are a woman of childbearing age, you should use an effective method of birth control to prevent pregnancy while using ezetimibe tablets added to statin therapy.
Ezetimibe tablets have not been studied in children under age 10.
What should I tell my doctor before and while taking ezetimibe tablets?
Tell your doctor about any prescription and non-prescription medicines you are taking or plan to take, including natural or herbal remedies.
Tell your doctor about all your medical conditions including allergies.
Tell your doctor if you:
- ever had liver problems. Ezetimibe tablets may not be right for you.
- are pregnant or plan to become pregnant. Your doctor will discuss with you whether ezetimibe tablets are right for you.
- are breast-feeding. We do not know if ezetimibe can pass to your baby through your milk. Your doctor will discuss with you whether ezetimibe tablets are right for you.
- experience unexplained muscle pain, tenderness, or weakness.
How should I take ezetimibe tablets?
- Take ezetimibe tablets once a day, with or without food. It may be easier to remember to take your dose if you do it at the same time every day, such as with breakfast, dinner, or at bedtime. If you also take another medicine to reduce your cholesterol, ask your doctor if you can take them at the same time.
- If you forget to take ezetimibe tablets, take it as soon as you remember. However, do not take more than one dose of ezetimibe tablets a day.
- Continue to follow a cholesterol-lowering diet while taking ezetimibe tablets. Ask your doctor if you need diet information.
- Keep taking ezetimibe tablets unless your doctor tells you to stop. It is important that you keep taking ezetimibe tablets even if you do not feel sick.
See your doctor regularly to check your cholesterol level and to check for side effects. Your doctor may do blood tests to check your liver before you start taking ezetimibe tablets with a statin and during treatment.
What are the possible side effects of ezetimibe tablets?
In clinical studies, patients reported few side effects while taking ezetimibe tablets. These included diarrhea, joint pains, and feeling tired.
Patients have experienced severe muscle problems while taking ezetimibe tablets, usually when ezetimibe tablets were added to a statin drug. If you experience unexplained muscle pain, tenderness, or weakness while taking ezetimibe tablets, contact your doctor immediately. You need to do this promptly, because on rare occasions, these muscle problems can be serious, with muscle breakdown resulting in kidney damage.
Additionally, the following side effects have been reported in general use: allergic reactions (which may require treatment right away) including swelling of the face, lips, tongue, and/or throat that may cause difficulty in breathing or swallowing, rash, and hives; raised red rash, sometimes with target-shaped lesions; joint pain; muscle aches; alterations in some laboratory blood tests; liver problems; stomach pain; inflammation of the pancreas; nausea; dizziness; tingling sensation; depression; headache; gallstones; inflammation of the gallbladder.
Tell your doctor if you are having these or any other medical problems while on ezetimibe tablets. For a complete list of side effects, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA -1088
What should I know about high cholesterol?
Cholesterol is a type of fat found in your blood. Your total cholesterol is made up of LDL and HDL cholesterol.
LDL cholesterol is called “bad” cholesterol because it can build up in the wall of your arteries and form plaque. Over time, plaque build-up can cause a narrowing of the arteries. This narrowing can slow or block blood flow to your heart, brain, and other organs. High LDL cholesterol is a major cause of heart disease and one of the causes for stroke.
HDL cholesterol is called “good” cholesterol because it keeps the bad cholesterol from building up in the arteries.
Triglycerides also are fats found in your blood.
General information about ezetimibe tablets
Medicines are sometimes prescribed for conditions that are not mentioned in patient information leaflets. Do not use ezetimibe tablets for a condition for which it was not prescribed. Do not give ezetimibe tablets to other people, even if they have the same condition you have. It may harm them.
This summarizes the most important information about ezetimibe tablets. If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about ezetimibe tablets that is written for health professionals.
Inactive ingredients:
Croscarmellose sodium, lactose monohydrate, magnesium stearate, povidone, and sodium lauryl sulfate.
*All trademarks are property of their respective owners.
This Patient Information has been approved by the U.S. Food and Drug Administration.
Manufactured for:
AvKARE, Inc.
Pulaski, TN 38478Mfg. Rev. 05-2017-00
AV 07/18 (P)
AvPAK -
PRINCIPAL DISPLAY PANEL
NDC: 50268-298-12
Ezetimibe
Tablets, USP10 mg
Rx Only
20 Tablets (2 x 10) Unit Dose
5026829812
NDC: 50268-298-12
Ezetimibe
Tablets, USP10 mg
Rx Only
20 Tablets (2 x 10) Unit Dose
5026829812
Each tablet contains 10 mg ezetimibe, USP.
USUAL DOSAGE: One tablet daily. See Insert.
Store at 20° to 25°C (68° to 77°F); excursions
permitted between 15° to 30°C (59° to 86°F)
[see USP Controlled Room Temperature].
Protect from moisture.Keep this and all drugs out of the reach of children.
Manufactured for:
AvKARE, Inc.
Pulaski, TN 38478AvPAK TM
A PRODUCT OF AvKARE
Mfg. Rev. 05-2017-00 AV 06/18
-
INGREDIENTS AND APPEARANCE
EZETIMIBE
ezetimibe tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 50268-298(NDC:69238-1154) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EZETIMIBE (UNII: EOR26LQQ24) (EZETIMIBE - UNII:EOR26LQQ24) EZETIMIBE 10 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) POVIDONE (UNII: FZ989GH94E) SODIUM LAURETH-12 SULFATE (UNII: 8M492LDU23) Product Characteristics Color white (TO OFF WHITE) Score no score Shape CAPSULE Size 8mm Flavor Imprint Code AA;69 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 50268-298-12 20 in 1 BOX, UNIT-DOSE 07/02/2017 1 NDC: 50268-298-11 1 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA208803 07/02/2017 Labeler - AvPAK (832926666)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.