RELISTOR- methylnaltrexone bromide tablet RELISTOR- methylnaltrexone bromide injection, solution
Relistor by
Drug Labeling and Warnings
Relistor by is a Prescription medication manufactured, distributed, or labeled by Salix Pharmaceuticals, Inc., Valeant Pharmaceuticals North America LLC, Vetter Pharma Fertigung GmbH & Co. KG, Mallinckrodt, Inc., Patheon Inc., Carton Service, Incorporated. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use RELISTOR safely and effectively. See full prescribing information for RELISTOR.
RELISTOR® (methylnaltrexone bromide) tablets, for oral use
RELISTOR® (methylnaltrexone bromide) injection, for subcutaneous use
Initial U.S. Approval: 2008INDICATIONS AND USAGE
- RELISTOR is an opioid antagonist. RELISTOR tablets and RELISTOR injection are indicated for the treatment of opioid-induced constipation (OIC) in adults with chronic non-cancer pain, including patients with chronic pain related to prior cancer or its treatment who do not require frequent (e.g., weekly) opioid dosage escalation. (1.1)
- RELISTOR injection is indicated for the treatment of OIC in adults with advanced illness or pain caused by active cancer who require opioid dosage escalation for palliative care. (1.2)
DOSAGE AND ADMINISTRATION
Administration Instructions (2.1)
- Be within close proximity to toilet facilities once administered.
- Discontinue if treatment with opioid pain medication is also discontinued.
-
In adult patients with chronic non-cancer pain and OIC:
- Patients receiving opioids for less than 4 weeks may be less responsive to RELISTOR.
- Discontinue all maintenance laxative therapy before starting RELISTOR; may resume laxatives if there is a suboptimal response to RELISTOR after 3 days.
- Re-evaluate the continued need for RELISTOR when opioid regimen is changed to avoid adverse reactions.
- In patients with chronic non-cancer pain and OIC, take RELISTOR tablets with water on an empty stomach at least 30 minutes before the first meal of the day.
Dosing
-
For OIC in adult patients with chronic non-cancer pain (2.2):
- RELISTOR tablets: The recommended dosage is 450 mg once daily in the morning.
- RELISTOR injection: The recommended dosage is 12 mg subcutaneously once daily.
-
For OIC in adult patients with advanced illness (2.3):
- The pre-filled syringe is only for patients who require a RELISTOR injection dose of 8 mg or 12 mg. Use the vial for patients who require other doses of RELISTOR injection.
- RELISTOR injection: See Table 1 in the full prescribing information for the recommended dosage; administer one dose every other day, as needed, but no more frequently than one dose in a 24-hour period.
Dosage Adjustment
- See full prescribing information dosage adjustment in renal or hepatic impairment by indication. (2.4, 2.5)
Preparation and Administration of RELISTOR Injection (2.6)
- For subcutaneous use only.
- Inject in upper arm, abdomen or thigh. Rotate injection sites.
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Gastrointestinal Perforation: Consider the overall risk benefit in patients with known or suspected lesions of the GI tract. Monitor for severe, persistent or worsening abdominal pain; discontinue if development of symptoms. (5.1)
- Severe or Persistent Diarrhea: Discontinue if severe or persistent diarrhea occurs during treatment. (5.2)
- Opioid Withdrawal: Consider the overall risk benefit in patients with disruptions to the blood-brain barrier. Monitor closely for symptoms of opioid withdrawal. (5.3)
ADVERSE REACTIONS
The most common adverse reactions are:
OIC in adult patients with chronic non-cancer pain (6.1)
- RELISTOR tablets (≥ 2%): abdominal pain, diarrhea, headache, abdominal distention, vomiting, hyperhidrosis, anxiety, muscle spasms, rhinorrhea, and chills.
- RELISTOR injection (≥ 1%): abdominal pain, nausea, diarrhea, hyperhidrosis, hot flush, tremor, and chills.
OIC in adult patients with advanced illness (6.1)
- RELISTOR injection (≥ 5%): abdominal pain, flatulence, nausea, dizziness, and diarrhea.
To report SUSPECTED ADVERSE REACTIONS, contact Valeant Pharmaceuticals North America LLC at 1-800-321-4576 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Other Opioid Antagonists: Potential for additive effect and increased risk of opioid withdrawal; avoid concomitant use. (7.1)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 11/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Opioid-Induced Constipation in Adult Patients with Chronic Non-Cancer Pain
1.2 Opioid-Induced Constipation in Adult Patients with Advanced Illness
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Information
2.2 Opioid-Induced Constipation in Adult Patients with Chronic Non-Cancer Pain
2.3 Opioid-Induced Constipation in Adult Patients with Advanced Illness
2.4 Dosage in Patients with Renal Impairment
2.5 Dosage in Patients with Hepatic Impairment
2.6 Preparation and Administration of RELISTOR Injection
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Gastrointestinal Perforation
5.2 Severe or Persistent Diarrhea
5.3 Opioid Withdrawal
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Other Opioid Antagonists
7.2 Drugs Metabolized by Cytochrome P450 Isozymes
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Opioid-Induced Constipation in Adult Patients with Chronic Non-Cancer Pain
14.2 Opioid-Induced Constipation in Adult Patients with Advanced Illness
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Opioid-Induced Constipation in Adult Patients with Chronic Non-Cancer Pain
RELISTOR tablets and RELISTOR injection are indicated for the treatment of opioid-induced constipation (OIC) in adult patients with chronic non-cancer pain, including patients with chronic pain related to prior cancer or its treatment who do not require frequent (e.g., weekly) opioid dosage escalation.
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Information
- Be within close proximity to toilet facilities once RELISTOR is administered.
- Discontinue RELISTOR if treatment with the opioid pain medication is also discontinued.
-
In adult patients with chronic non-cancer pain and OIC:
- Patients receiving opioids for less than 4 weeks may be less responsive to RELISTOR [see Clinical Studies (14.1)].
- Discontinue all maintenance laxative therapy prior to initiation of RELISTOR. Laxative(s) can be used as needed if there is a suboptimal response to RELISTOR after three days.
- Re-evaluate the continued need for RELISTOR when the opioid regimen is changed to avoid adverse reactions.
- In patients with chronic non-cancer pain and OIC, take RELISTOR tablets with water on an empty stomach at least 30 minutes before the first meal of the day.
2.2 Opioid-Induced Constipation in Adult Patients with Chronic Non-Cancer Pain
- The recommended dosage of RELISTOR tablets is 450 mg taken orally once daily in the morning.
- The recommended dosage of RELISTOR injection is 12 mg administered subcutaneously once daily.
2.3 Opioid-Induced Constipation in Adult Patients with Advanced Illness
The pre-filled syringe is only for patients who require a RELISTOR injection dose of 8 mg or 12 mg. Use the vial for patients who require other doses of RELISTOR injection.
Table 1 below shows the recommended weight-based dose of RELISTOR injection and the corresponding injection volume. The recommended dosage regimen is one dose administered subcutaneously every other day, as needed. Do not administer more frequently than one dose per 24-hour period.
Table 1:Weight-Based Dosing of RELISTOR Injection and Corresponding Injection Volume for Adult Patients with OIC and Advanced Illness - * Calculate the injection volume for these patients by multiplying the patient weight in kilograms by 0.0075 and then rounding up the volume to the nearest 0.1 mL.
Weight of Adult Patient
Subcutaneous
Dose
Injection Volume
Less than 38 kg
0.15 mg/kg
See below*
38 kg to less than 62 kg
8 mg
0.4 mL
62 kg to 114 kg
12 mg
0.6 mL
More than 114 kg
0.15 mg/kg
See below*
2.4 Dosage in Patients with Renal Impairment
The recommended dosage of RELISTOR in patients with moderate and severe renal impairment (i.e., creatinine clearance less than 60 mL/minute as estimated by Cockcroft-Gault) [see Use in Specific Populations (8.6)], is shown below by indication:
OIC in Adult Patients with Chronic Non‑Cancer Pain
- RELISTOR tablets: 150 mg once daily in the morning.
- RELISTOR injection: 6 mg administered subcutaneously once daily
OIC in Adult Patients with Advanced Illness
RELISTOR injection: The recommended dosage regimen is one dose every other day, as needed is shown in Table 2.
Table 2: Weight-Based Dosing in Moderate and Severe Renal Impairment of RELISTOR Injection and Corresponding Injection Volume for Adult Patients with OIC and Advanced Illness - * Calculate the injection volume for these patients by multiplying the patient weight in kilograms by 0.00375 and then rounding up the volume to the nearest 0.1 mL.
Weight of Adult Patient
Subcutaneous Dose
Injection Volume
Less than 38 kg
0.075 mg/kg
See below*
38 kg to less than 62 kg
4 mg
0.2 mL
62 kg to 114 kg
6 mg
0.3 mL
More than 114 kg
0.075 mg/kg
See below*
2.5 Dosage in Patients with Hepatic Impairment
OIC in Adult Patients with Chronic Non-Cancer Pain
- RELISTOR tablets: The recommended dosage in adult patients with moderate or severe hepatic impairment (Child-Pugh Class B or C) is 150 mg, once daily in the morning [see Use in Specific Populations (8.7)].
- RELISTOR injection: If considering dose adjustment for patients with severe hepatic impairment, follow the recommendations in Table 3 [see Use in Specific Populations (8.7)].
Table 3: Weight-Based Dosing in Severe Hepatic Impairment of RELISTOR Injection and Corresponding Injection Volume for Adult Patients with OIC and Chronic Non-Cancer Pain - * Calculate the injection volume for these patients by multiplying the patient weight in kilograms by 0.00375 and then rounding up the volume to the nearest 0.1 mL.
Weight of Adult Patient
Subcutaneous
Dose
Injection Volume
Less than 38 kg
0.075 mg/kg
See below*
38 kg to less than 62 kg
4 mg
0.2 mL
62 kg to 114 kg
6 mg
0.3 mL
More than 114 kg
0.075 mg/kg
See below*
2.6 Preparation and Administration of RELISTOR Injection
- RELISTOR injection is for subcutaneous use only.
- Single-dose pre-filled syringes: Do not remove the pre-filled syringe from the tray until ready to administer.
- Inspect RELISTOR injection visually for particulate matter and discoloration prior to administration. RELISTOR injection is colorless to pale yellow. Discard the pre-filled syringe or vial if particulate matter or discoloration is present.
- Administer RELISTOR injection subcutaneously in the upper arm, abdomen or thigh. Do not inject at the same spot each time (rotate injection sites).
- RELISTOR single-dose vials: Once drawn into the syringe, if immediate administration is not possible, store at ambient room temperature and administer within 24 hours. Discard any unused portion that remains in the vial.
- For patient or caregiver instructions for preparation and administration of RELISTOR injection (including recommended specifications for the syringe and needle to be used with the single-dose vial), see Instructions for Use.
-
3 DOSAGE FORMS AND STRENGTHS
- Tablets:
- 1. 150 mg methylnaltrexone bromide supplied as film-coated, white, round, biconvex, debossed with “REL” on one side and plain on the other side.
Injection:
Single-dose Vial:
- 12 mg/0.6 mL methylnaltrexone bromide supplied as colorless to pale yellow solution.
Single-dose Pre-filled Syringe:
- 8 mg/0.4 mL methylnaltrexone bromide supplied as colorless to pale yellow solution.
- 12 mg/0.6 mL methylnaltrexone bromide supplied as colorless to pale yellow solution.
-
4 CONTRAINDICATIONS
RELISTOR is contraindicated in patients with known or suspected gastrointestinal obstruction and patients at increased risk of recurrent obstruction, due to the potential for gastrointestinal perforation [see Warnings and Precautions (5.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Gastrointestinal Perforation
Cases of gastrointestinal perforation have been reported in adult patients with OIC and advanced illness with conditions that may be associated with localized or diffuse reduction of structural integrity in the wall of the gastrointestinal tract (e.g., peptic ulcer disease, Ogilvie’s syndrome, diverticular disease, infiltrative gastrointestinal tract malignancies or peritoneal metastases). Take into account the overall risk-benefit profile when using RELISTOR in patients with these conditions or other conditions which might result in impaired integrity of the gastrointestinal tract wall (e.g., Crohn’s disease). Monitor for the development of severe, persistent, or worsening abdominal pain; discontinue RELISTOR in patients who develop this symptom [see Contraindications (4)].
5.2 Severe or Persistent Diarrhea
If severe or persistent diarrhea occurs during treatment, advise patients to discontinue therapy with RELISTOR and consult their healthcare provider.
5.3 Opioid Withdrawal
Symptoms consistent with opioid withdrawal, including hyperhidrosis, chills, diarrhea, abdominal pain, anxiety, and yawning have occurred in patients treated with RELISTOR [see Adverse Reactions (6.1)]. Patients having disruptions to the blood-brain barrier may be at increased risk for opioid withdrawal and/or reduced analgesia. Take into account the overall risk-benefit profile when using RELISTOR in such patients. Monitor for adequacy of analgesia and symptoms of opioid withdrawal in such patients.
-
6 ADVERSE REACTIONS
Serious and important adverse reactions described elsewhere in the labeling include:
- Gastrointestinal perforation [see Warnings and Precautions (5.1)]
- Severe or persistent diarrhea [see Warnings and Precautions (5.2)]
- Opioid withdrawal [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Opioid-Induced Constipation in Adult Patients with Chronic Non-Cancer Pain
The safety of RELISTOR tablets was evaluated in a double-blind, placebo-controlled trial in adult patients with OIC and chronic non-cancer pain receiving opioid analgesia. This study (Study 1) included a 12-week, double-blind, placebo-controlled period in which adult patients were randomized to receive RELISTOR tablets 450 mg orally (200 patients) or placebo (201 patients) [see Clinical Studies (14.1)]. After 4 weeks of double-blind treatment administered once daily, patients continued 8 weeks of double-blind treatment on an as needed basis (but not more than once daily).
The most common adverse reactions in adult patients with OIC and chronic non-cancer pain receiving RELISTOR tablets are shown in Table 4. Adverse reactions of abdominal pain, diarrhea, hyperhidrosis, anxiety, rhinorrhea, and chills may reflect symptoms of opioid withdrawal.
Table 4: Adverse Reactions* in 4-Week Double-Blind, Placebo-Controlled Period of Clinical Study of RELISTOR Tablets in Adult Patients with OIC and Chronic Non-Cancer Pain (Study 1) - * Adverse reactions occurring in at least 2% of patients receiving RELISTOR tablets 450 mg once daily and at an incidence greater than placebo.
- † Includes: abdominal pain, upper abdominal pain, lower abdominal pain, abdominal discomfort and abdominal tenderness
Adverse Reaction
RELISTOR Tablets
n = 200Placebo
n = 201Abdominal Pain†
14%
10%
Diarrhea
5%
2%
Headache
4%
3%
Abdominal Distention
4%
2%
Vomiting
3%
2%
Hyperhidrosis
3%
1%
Anxiety
2%
1%
Muscle Spasms
2%
1%
Rhinorrhea
2%
1%
Chills
2%
0%
The safety of RELISTOR injection was evaluated in a double-blind, placebo-controlled trial in adult patients with OIC and chronic non-cancer pain receiving opioid analgesia. This study (Study 2) included a 4-week, double-blind, placebo-controlled period in which adult patients were randomized to receive RELISTOR injection 12 mg subcutaneously once daily (150 patients) or placebo (162 patients) [see Clinical Studies (14.1)]. After 4 weeks of double-blind treatment, patients began an 8‑week open-label treatment period during which RELISTOR injection 12 mg subcutaneously was administered less frequently than the recommended dosage regimen of 12 mg once daily.
The most common adverse reactions in adult patients with OIC and chronic non-cancer pain receiving RELISTOR injection are shown in Table 5. The adverse reactions in the table below may reflect symptoms of opioid withdrawal.
Table 5: Adverse Reactions* in 4-Week Double-Blind, Placebo-Controlled Period of Clinical Study of RELISTOR Injection in Adult Patients with OIC and Chronic Non-Cancer Pain (Study 2) - * Adverse reactions occurring in at least 1% of patients receiving RELISTOR injection 12 mg subcutaneously once daily and at an incidence greater than placebo.
- † Includes: abdominal pain, upper abdominal pain, lower abdominal pain, abdominal discomfort and abdominal tenderness
Adverse Reaction
RELISTOR Injection
n = 150Placebo
n = 162Abdominal Pain†
21%
7%
Nausea
9%
6%
Diarrhea
6%
4%
Hyperhidrosis
6%
1%
Hot Flush
3%
2%
Tremor
1%
<1%
Chills
1%
0%
During the 4-week double-blind period, in patients with OIC and chronic non-cancer pain that received RELISTOR every other day, there was a higher incidence of adverse reactions, including nausea (12%), diarrhea (12%), vomiting (7%), tremor (3%), feeling of body temperature change (3%), piloerection (3%), and chills (2%) as compared to daily RELISTOR dosing. Use of RELISTOR injection 12 mg subcutaneously every other day is not recommended in patients with OIC and chronic non-cancer pain [see Dosage and Administration (2.2)]. The rates of discontinuation due to adverse reactions during the double-blind period (Study 2) were higher in the RELISTOR once daily (7%) than the placebo group (3%). Abdominal pain was the most common adverse reaction resulting in discontinuation from the double-blind period in the RELISTOR once daily group (2%).
The safety of RELISTOR injection was also evaluated in a 48-week, open-label, uncontrolled trial in 1034 adult patients with OIC and chronic non-cancer pain (Study 3). Patients were allowed to administer RELISTOR injection 12 mg subcutaneously less frequently than the recommended dosage regimen of 12 mg once daily, and took a median of 6 doses per week. A total of 624 patients (60%) completed at least 24 weeks of treatment and 477 (46%) completed the 48‑week study. The adverse reactions seen in this study were similar to those observed during the 4-week double-blind period of Study 2. Additionally, in Study 3, investigators reported 4 myocardial infarctions (1 fatal), 1 stroke (fatal), 1 fatal cardiac arrest and 1 sudden death. It is not possible to establish a relationship between these events and RELISTOR.
Opioid-Induced Constipation in Adult Patients with Advanced Illness
The safety of RELISTOR injection was evaluated in two, double-blind, placebo-controlled trials in adult patients with OIC and advanced illness receiving palliative care: Study 4 included a single‑dose, double‑blind, placebo-controlled period, whereas Study 5 included a 14-day multiple dose, double-blind, placebo-controlled period [see Clinical Studies (14.2)].
The most common adverse reactions in adult patients with OIC and advanced illness receiving RELISTOR injection are shown in Table 6 below.
Table 6: Adverse Reactions from All Doses in Double-Blind, Placebo-Controlled Clinical Studies of RELISTOR Injection in Adult Patients with OIC and Advanced Illness* (Studies 4 and 5) Adverse Reaction RELISTOR Injection
n = 165Placebo
n = 123- * Adverse reactions occurring in at least 5% of patients receiving all doses of RELISTOR injection (0.075, 0.15, and 0.3 mg/kg) and at an incidence greater than placebo
- † Includes: abdominal pain, upper abdominal pain, lower abdominal pain, abdominal discomfort and abdominal tenderness
Abdominal Pain†
29%
10%
Flatulence
13%
6%
Nausea
12%
5%
Dizziness
7%
2%
Diarrhea
6%
2%
The rates of discontinuation due to adverse reactions during the double-blind, placebo-controlled clinical trials (Study 4 and Study 5) were comparable between RELISTOR (1%) and placebo (2%).
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of RELISTOR injection. Because reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate the frequency or establish a causal relationship to drug exposure.
Gastrointestinal
Perforation, cramping, vomiting
General Disorders and Administration Site Disorders
Diaphoresis, flushing, malaise, pain. Cases of opioid withdrawal have been reported [see Warnings and Precautions (5.3)].
- 7 DRUG INTERACTIONS
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The limited available data with RELISTOR in pregnant women are not sufficient to inform a drug-associated risk for major birth defects and miscarriages. There are clinical considerations when RELISTOR is used by pregnant women [see Clinical Considerations]. In animal reproduction studies, no effects on embryofetal development were observed with the administration of intravenous methylnaltrexone bromide during organogenesis in rats and rabbits at doses up to 20 times and 26 times, respectively, the subcutaneous maximum recommended human dose (MRHD) of 12 mg RELISTOR injection per day. The intravenous doses in rats and rabbits are about 0.5 times and 0.7 times, respectively, the oral MRHD of 450 mg/day [see Data]. Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
The use of RELISTOR during pregnancy may precipitate opioid withdrawal in a fetus due to the immature fetal blood-brain barrier.
Data
Animal Data
Reproduction studies have been performed with methylnaltrexone bromide administered during the period of organogenesis to rats at intravenous doses up to 25 mg/kg/day (about 20 times the subcutaneous MRHD of 12 mg/day based on body surface area), and did not cause any adverse effects on embryofetal development. In rabbits, intravenous doses of methylnaltrexone bromide up to 16 mg/kg/day (about 26 times the subcutaneous MRHD of 12 mg/day) did not show any embryofetal toxicity. The intravenous doses in rats (25 mg/kg/day) and rabbits (16 mg/kg/day) are about 0.5 and 0.7 times, respectively, the oral MRHD of 450 mg/day based on body surface area. A pre- and postnatal development study in rats showed no evidence of any adverse effect on pre- and postnatal development at subcutaneous doses of methylnaltrexone bromide up to 100 mg/kg/day (about 81 times the subcutaneous MRHD of 12 mg/day; about 2.2 times the oral MRHD of 450 mg/day).
8.2 Lactation
Risk Summary
There is no information regarding the presence of methylnaltrexone in human milk, the effects on the breastfed infant, or the effects on milk production. Methylnaltrexone is present in rat milk [see Data]. Because of the potential for serious adverse reactions, including opioid withdrawal, in breastfed infants, advise women that breastfeeding is not recommended during treatment with RELISTOR.
Data
Radioactivity appeared in rat milk within 30 minutes of a single subcutaneous administration of radiolabeled methylnaltrexone bromide and was concentrated up to 24-fold at 8 hours after administration relative to plasma concentrations.
8.4 Pediatric Use
Safety and effectiveness of RELISTOR tablets and injection have not been established in pediatric patients.
Juvenile Animal Studies
In juvenile rats administered intravenous methylnaltrexone bromide for 13 weeks, adverse clinical signs such as convulsions, tremors and labored breathing were observed, and the juvenile rats were found to be more sensitive to the adverse effects of methylnaltrexone when compared to adult animals. Juvenile dogs administered intravenous methylnaltrexone bromide for 13 weeks had a toxicity profile similar to adult dogs [see Nonclinical Toxicology (13.2)].
8.5 Geriatric Use
Of the total number of patients in clinical studies of RELISTOR tablets, a total of 136 patients (10%) were aged 65 years and older, while 23 (2%) were aged 75 and older. In clinical studies of RELISTOR tablets, no overall differences in effectiveness were observed. Adverse reactions were similar; however, there was a higher incidence of diarrhea in elderly patients.
Of the total number of patients in clinical studies of RELISTOR injection, a total of 226 (28%) were aged 65 years and older, while 108 (13%) were aged 75 years and older. In clinical studies of RELISTOR injection, no overall differences in safety or effectiveness were observed between elderly patients and younger patients.
Based on pharmacokinetic data, and safety and efficacy data from controlled clinical trials, no dosage adjustment based on age is recommended. Monitor elderly patients for adverse reactions.
8.6 Renal Impairment
In a study of subjects with varying degrees of renal impairment receiving RELISTOR injection subcutaneously, there was a significant increase in the exposure to methylnaltrexone in subjects with moderate and severe renal impairment (creatinine clearance less than 60 mL/minute as estimated by Cockcroft-Gault) compared to healthy subjects [see Clinical Pharmacology (12.3)].
Therefore, a dosage reduction of RELISTOR tablets and RELISTOR injection is recommended in patients with moderate and severe renal impairment [see Dosage and Administration (2.4)]. No dosage adjustment of RELISTOR tablets or RELISTOR injection is needed in patients with mild renal impairment (creatinine clearance greater than 60 mL/minute as estimated by Cockcroft-Gault).
8.7 Hepatic Impairment
Tablets
In a study of subjects with varying degrees of hepatic impairment receiving a 450 mg dose of RELISTOR tablets, there was a significant increase in systemic exposure of methylnaltrexone for subjects with moderate (Child-Pugh Class B) and severe (Child-Pugh Class C) hepatic impairment compared to healthy subjects with normal hepatic function [see Clinical Pharmacology (12.3)]. Therefore, a dosage reduction of RELISTOR tablets is recommended in patients with moderate or severe hepatic impairment [see Dosage and Administration (2.5)]. No dosage adjustment of RELISTOR tablets is needed in patients with mild hepatic impairment (Child-Pugh Class A).
Injection
In a study of subjects with mild or moderate hepatic impairment, there was no clinically meaningful change in systemic exposure of methylnaltrexone compared to healthy subjects with normal hepatic function [see Clinical Pharmacology (12.3)]. Therefore, no dosage adjustment of RELISTOR injection is needed for patients with mild or moderate hepatic impairment [see Clinical Pharmacology (12.3)].
Patients with severe hepatic impairment were not studied. In patients with severe hepatic impairment, monitor for methylnaltrexone-related adverse reactions. If considering dosage adjustment, follow the recommendations in Table 3 [see Dosage and Administration (2.5)].
-
10 OVERDOSAGE
During clinical trials of RELISTOR administered orally and subcutaneously, one accidental case of methylnaltrexone bromide overdose was reported and no adverse events were reported as a result of the overdosage.
A study of healthy subjects noted orthostatic hypotension associated with a dose of 0.64 mg/kg administered as an intravenous bolus. Monitor for signs or symptoms of orthostatic hypotension and initiate treatment as appropriate.
If a patient on opioid therapy receives an overdose of RELISTOR, the patient should be monitored closely for potential evidence of opioid withdrawal symptoms such as chills, rhinorrhea, diaphoresis or reversal of central analgesic effect. Base treatment on the degree of opioid withdrawal symptoms, including changes in blood pressure and heart rate, and on the need for analgesia.
-
11 DESCRIPTION
RELISTOR® (methylnaltrexone bromide) is a mu-opioid receptor antagonist. The chemical name for methylnaltrexone bromide is (R)-N-(cyclopropylmethyl) noroxymorphone methobromide. The molecular formula is C21H26NO4Br, and the molecular weight is 436.36.
The structural formula is:
RELISTOR tablets for oral administration are film-coated and contain 150 mg of methylnaltrexone bromide (equivalent to 122.5 mg methylnaltrexone). Inactive ingredients are silicified microcrystalline cellulose, microcrystalline cellulose, sodium lauryl sulfate, croscarmellose sodium, crospovidone, poloxamer 407, stearic acid (vegetable source), colloidal silicon dioxide, edetate calcium disodium, polyvinyl alcohol, titanium dioxide, polyethylene glycol and talc.
RELISTOR for subcutaneous administration is a sterile, clear and colorless to pale yellow aqueous solution. Each 3 mL vial contains 12 mg of methylnaltrexone bromide (equivalent to 9.8 mg of methylnaltrexone) in 0.6 mL of water. The excipients are 3.9 mg sodium chloride USP, 0.24 mg edetate calcium disodium USP, and 0.18 mg glycine hydrochloride. During manufacture, the pH may have been adjusted with hydrochloric acid and/or sodium hydroxide.
Each 8 mg/0.4 mL pre-filled syringe (1 mL syringe) contains 8 mg of methylnaltrexone bromide (equivalent to 6.5 mg of methylnaltrexone) in 0.4 mL of water. The excipients are 2.6 mg sodium chloride USP, 0.16 mg edetate calcium disodium USP, and 0.12 mg glycine hydrochloride.
Each 12 mg/0.6 mL pre-filled syringe (1 mL syringe) contains 12 mg of methylnaltrexone bromide (equivalent to 9.8 mg of methylnaltrexone) in 0.6 mL of water. The excipients are 3.9 mg sodium chloride USP, 0.24 mg edetate calcium disodium USP, and 0.18 mg glycine hydrochloride.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Methylnaltrexone is a selective antagonist of opioid binding at the mu-opioid receptor. As a quaternary amine, the ability of methylnaltrexone to cross the blood-brain barrier is restricted. This allows methylnaltrexone to function as a peripherally-acting mu‑opioid receptor antagonist in tissues such as the gastrointestinal tract, thereby decreasing the constipating effects of opioids without impacting opioid-mediated analgesic effects on the central nervous system (CNS).
12.2 Pharmacodynamics
Cardiac Electrophysiology
In a randomized, double-blind placebo- and (open-label) moxifloxacin-controlled 4‑period crossover study, 56 healthy subjects were administered methylnaltrexone bromide 0.3 mg/kg and methylnaltrexone bromide 0.64 mg/kg by intravenous infusion over 20 minutes (RELISTOR is not approved for intravenous use), placebo, and a single oral dose of moxifloxacin. At a dose approximately 4.3 times the maximum recommended dose (7.5 times the mean peak plasma concentration for RELISTOR injection and 22 times the peak plasma concentration for RELISTOR tablets), methylnaltrexone does not prolong the QTc interval to any clinically relevant extent.
12.3 Pharmacokinetics
Between the oral dosage range of 150 mg to 450 mg for RELISTOR tablets and the subcutaneous dosage range of 0.15 mg/kg to 0.50 mg/kg for RELISTOR injection, the mean Cmax and area under the plasma concentration-time curve (AUC) of methylnaltrexone increased in a dose-proportional manner. There was no significant accumulation of methylnaltrexone following once-daily oral dosing of 450 mg RELISTOR tablets or subcutaneous dosing of 12 mg RELISTOR injection for seven consecutive days in healthy subjects.
Absorption
- Tablets
Following administration of a single 450 mg dose of RELISTOR tablets in OIC patients or healthy subjects, peak concentrations (Cmax) of methylnaltrexone were observed at approximately 1.5 hours. The absolute bioavailability of oral methylnaltrexone bromide has not been determined. The Cmax and AUC in healthy subjects were 48.1 ng/mL and 382 ng·hr/mL, respectively, following a single 450 mg dose of RELISTOR tablets. Exposure in the OIC patient population was approximately 27% lower than in healthy subjects.
Food Effect
Administration of a single 450 mg dose of RELISTOR tablets to healthy subjects with a high-fat breakfast (containing approximately 800 to 1000 total calories, with 60%, 25% and 15% of calories derived from fat, carbohydrate and protein, respectively) resulted in a decrease in the Cmax of methylnaltrexone by 60%, the AUC by 43% and delayed the Tmax by 2 hours [see Dosage and Administration (2.1)].
Injection
Following administration of RELISTOR injection subcutaneously, methylnaltrexone achieved peak concentrations (Cmax) at approximately 0.5 hours (see Table 7).
Table 7: Pharmacokinetic Parameters of Methylnaltrexone Following Subcutaneous Doses - * Expressed as mean (SD).
- † Expressed as median (range).
Parameter
0.15 mg/kg
Single Dose
12 mg
Single Dose
12 mg
at Steady-State
Cmax (ng/mL)*
117 (32.7)
140 (35.6)
119 (27.2)
Tmax (hr)†
0.5 (0.25 to 0.75)
0.25 (0.25 to 0.5)
0.25 (0.25 to 0.5)
AUC24 (ng·hr/mL) *
175 (36.6)
218 (28.3)
223 (28.2)
Distribution
The steady-state volume of distribution (Vss) of methylnaltrexone is approximately 1.1 L/kg. The fraction of methylnaltrexone bound to human plasma proteins is 11% to 15%, as determined by equilibrium dialysis.
Elimination
Following oral administration of a single 450 mg dose of RELISTOR tablets, concentrations of methylnaltrexone declined in multiphasic manner with a terminal half-life (t1/2) of approximately 15 hours.
Metabolism
In an intravenous mass balance study, approximately 44% of the administered radioactivity was recovered in the urine over 24 hours with 5 distinct metabolites. None of the detected metabolites was in amounts over 6% of administered radioactivity. Conversion to methyl-6-naltrexol isomers (5% of total) and methylnaltrexone sulfate (1% of total) appear to be the primary pathways of metabolism. N‑demethylation of methylnaltrexone to produce naltrexone is not significant.
Systemic exposure of methylnaltrexone metabolites after oral administration of a single 450 mg dose of RELISTOR tablets are greater than the systemic exposure of methylnaltrexone metabolites after subcutaneous administration of a single 12 mg dose of RELISTOR injection. Subcutaneous administration is not subject to first-pass hepatic metabolism prior to appearance in the systemic circulation. After 12 mg subcutaneous once daily dosing the mean AUC0-24 ratio of metabolites to methylnaltrexone at steady-state was 30%, 19%, and 9% for methylnaltrexone sulfate, methyl-6α-naltrexol, and methyl-6ß-naltrexol, respectively. After 450 mg oral once daily dosing, the ratio of the mean AUC0-24 of metabolites to methylnaltrexone at steady-state was 79%, 38%, and 21% for methylnaltrexone sulfate, methyl-6α-naltrexol, and methyl-6ß-naltrexol, respectively. Methylnaltrexone sulfate is a weak mu-opioid receptor antagonist; methyl-6α-naltrexol, and methyl-6ß-naltrexol are active mu-opioid receptor antagonists.
Methylnaltrexone is conjugated by sulfotransferase SULT1E1 and SULT2A1 isoforms to methylnaltrexone sulfate. Conversion to methyl-6-naltrexol isomers is mediated by aldo-keto reductase 1C enzymes.
Excretion
In an intravenous mass balance study, approximately half of the dose was excreted in the urine (54%), and 17% of administered dose was excreted in the feces up to 168 hours postdose; however, radiolabeled recovery in this study was only 71% after 7 days. Methylnaltrexone is excreted primarily as the unchanged drug in the urine and feces. Active renal secretion of methylnaltrexone is suggested by renal clearance of methylnaltrexone that is approximately 4- to 5-fold higher than creatinine clearance.
No mass balance clinical studies were conducted with oral administration of methylnaltrexone bromide. However, following once daily dosing of 450 mg RELISTOR tablets for 1 week, the percentage of dose recovered in the urine as the parent methylnaltrexone was low (approximately 1% on both Day 1 and Day 7).
Specific Populations
Age: Geriatric Population
A study was conducted to characterize the pharmacokinetics of methylnaltrexone after a single dose of 24 mg methylnaltrexone bromide via intravenous infusion over 20 min in healthy adults between 18 and 45 years of age and in healthy adults aged 65 years and older. In elderly subjects (mean age 72 years old), mean clearance was about 20% lower (56 L/h versus 70 L/h) and AUC∞ was 26% higher than in subjects between 18 and 45 years of age (mean age 30 years old) [see Use in Specific Populations (8.5)].
Renal Impairment
Administration of a single subcutaneous dose of 0.3 mg/kg of RELISTOR injection in subjects with varying degrees of renal impairment (8 subjects each cohort) resulted in a 1.3-, 1.7- and 1.9-fold higher AUC0-∞ of methylnaltrexone, respectively, compared to 8 subjects with normal renal function [see Dosage and Administration (2.4)]. The mean Cmax did not change significantly with renal impairment. No studies were performed in patients with end-stage renal impairment requiring dialysis.
Hepatic Impairment
Administration of a single 450 mg dose of RELISTOR tablets in subjects with mild, moderate, and severe hepatic impairment (6 subjects each of Child-Pugh Class A, B, and C) resulted in a 1.7-, 4.8- and 3.8-fold higher Cmax of methylnaltrexone, respectively, compared to 6 subjects with normal liver function. The AUC0-∞, was comparable between healthy subjects and subjects with mild hepatic impairment, but increased approximately 2.1-fold in subjects with moderate and severe hepatic impairment [see Dosage and Administration (2.5)].
Administration of a single dose of 0.3 mg/kg of RELISTOR injection subcutaneously in 8 subjects with mild hepatic impairment (Child-Pugh Class A) and 8 subjects with moderate hepatic impairment (Child-Pugh Class B) did not result in any meaningful change in the AUC or Cmax when compared to 8 healthy subjects with normal hepatic function. The effect of severe (Child-Pugh Class C) hepatic impairment on the pharmacokinetics of RELISTOR injection has not been studied [see Dosage and Administration (2.5), Use in Specific Populations (8.7)].
Drug Interaction Studies
Effect of Methylnaltrexone and its Metabolites on CYP Enzymes
In vitro, methylnaltrexone did not significantly inhibit or induce the activity of cytochrome P450 (CYP) isozymes CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, or CYP3A4. In addition, methylnaltrexone did not induce CYP2E1.
In vitro, the methylnaltrexone metabolites, methylnaltrexone sulfate, methyl-6α-naltrexol and methyl-6β-naltrexol did not inhibit CYP isozymes CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A4. The metabolites of methylnaltrexone did not induce activity of CYP1A2, CYP2B6, or CYP3A4.
Effect of Methylnaltrexone and its Metabolites on Transporters
Based on the in vitro studies, methylnaltrexone and its three major metabolites, methylnaltrexone sulfate, methyl-6α-naltrexol and methyl-6β-naltrexol, are unlikely to have clinically meaningful in vivo drug-drug interactions via inhibition of P-glycoprotein (P-gp), Breast Cancer Resistance Protein (BCRP), Multidrug Resistance Protein 2 (MRP2), Organic Anion-Transporting Polypeptide (OATP)1B1, OATP1B3, Organic Cation Transporter (OCT)1, OCT2, Organic Anion Transporter (OAT)1, OAT3, Multidrug and Toxic Extrusion Transporter (MATE)1 and MATE2-K at the recommended dosage of 450 mg orally or 12 mg subcutaneously once daily.
Methylnaltrexone: Methylnaltrexone was a substrate of OCT1, OCT2, MATE1, and MATE2-K, but not a substrate for P-gp, BCRP, MRP2, OATP1B1, OATP1B3, OAT1 and OAT3.
Methylnaltrexone sulfate: Methylnaltrexone sulfate was a substrate for MATE2-K and a potential substrate of BCRP, but was not a substrate of P-gp, MRP2, OATP1B1, OATP1B3, OCT1, OCT2, OAT1, OAT3, and MATE1.
Methyl-6α-naltrexol: Methyl-6α-naltrexol was a substrate of BCRP, OCT1, OCT2, MATE1, and MATE2-K, but was not a substrate of P-gp, MRP2, OATP1B1, OATP1B3, OAT1 and OAT3.
Methyl-6β-naltrexol: Methyl-6β-naltrexol was a substrate of OCT1, OCT2, MATE1, and MATE2-K, but was not a substrate of P-gp, BCRP, MRP2, OATP1B1, OATP1B3, OAT1 and OAT3.
Cimetidine
A clinical drug interaction study in healthy adult subjects evaluated the effects of cimetidine, a drug that inhibits the active renal secretion of organic cations, on the pharmacokinetics of methylnaltrexone (24 mg administered as an intravenous infusion over 20 minutes). A single dose of methylnaltrexone bromide was administered before cimetidine dosing and with the last dose of cimetidine (400 mg every 8 hours for 6 days). Mean Cmax and AUC of methylnaltrexone increased by 10% with concomitant cimetidine administration. The renal clearance of methylnaltrexone decreased about 40%. This change is not considered to be clinically meaningful.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Two-year oral carcinogenicity studies have been conducted with methylnaltrexone bromide in CD-1 mice at doses up to 200 mg/kg/day (about 81 times the subcutaneous maximum recommended human dose (MRHD) of 12 mg/day based on body surface area) in males and 400 mg/kg/day (about 162 times the subcutaneous MRHD of 12 mg/day) in females and in Sprague Dawley rats at oral doses up to 300 mg/kg/day (about 243 times the subcutaneous MRHD of 12 mg/day). The 200 mg/kg/day and 400 mg/kg/day doses in male and female mice are about 2.2 and 4.4 times, respectively, the oral MRHD of 450 mg/day, and the 300 mg/kg/day dose in rats is about 6.5 times the oral MRHD of 450 mg/day, based on body surface area. Oral administration of methylnaltrexone bromide for 104 weeks did not produce tumors in mice and rats.
Mutagenesis
Methylnaltrexone bromide was negative in the Ames test, chromosome aberration tests in Chinese hamster ovary cells and human lymphocytes, in the mouse lymphoma cell forward mutation tests and in the in vivo mouse micronucleus test.
Impairment of Fertility
Methylnaltrexone bromide at subcutaneous doses up to 150 mg/kg/day (about 122 times the subcutaneous MRHD of 12 mg/day; about 3.3 times the oral MRHD of 450 mg/day) was found to have no adverse effect on fertility and reproductive performance of male and female rats.
13.2 Animal Toxicology and/or Pharmacology
In an in vitro human cardiac potassium ion channel (hERG) assay, methylnaltrexone caused concentration-dependent inhibition of hERG current (1%, 12%, 13% and 40% inhibition at 30, 100, 300 and 1000 micromolar concentrations, respectively). Methylnaltrexone had a hERG IC50 of more than 1000 micromolar. In isolated dog Purkinje fibers, methylnaltrexone caused prolongations in action potential duration (APD). The highest tested concentration (10 micromolar) in the dog Purkinje fiber study was about 18 and 37 times the Cmax at human subcutaneous doses of 0.3 and 0.15 mg/kg, respectively. In isolated rabbit Purkinje fibers, methylnaltrexone (up to 100 micromolar) did not have an effect on APD, compared to vehicle control. The highest methylnaltrexone concentration (100 micromolar) tested was about 186 and 373 times the human Cmax at subcutaneous doses of 0.3 and 0.15 mg/kg, respectively. In anesthetized dogs, methylnaltrexone bromide caused decreases in blood pressure, heart rate, cardiac output, left ventricular pressure, left ventricular end diastolic pressure, and +dP/dt at 1 mg/kg or more. In conscious dogs, methylnaltrexone bromide caused a dose-related increase in QTc interval. After a single intravenous dosage of 20 mg/kg to beagle dogs, predicted Cmax and AUC values were approximately 482 and 144 times, respectively, the exposure at human subcutaneous dose of 0.15 mg/kg and 241 times and 66 times, respectively, the exposure at a human subcutaneous dose of 0.3 mg/kg. In conscious guinea pigs, methylnaltrexone bromide caused mild prolongation of QTc (4% over baseline) at 20 mg/kg, intravenous. A thorough QTc assessment was conducted in humans [see Clinical Pharmacology (12.2)].
In juvenile rats administered intravenous methylnaltrexone bromide for 13 weeks, adverse clinical signs such as convulsions, tremors and labored breathing occurred at dosages of 3 and 10 mg/kg/day (about 2.4 and 8 times, respectively, the subcutaneous MRHD of 12 mg/day; about 0.06 and 0.22 times, respectively, the oral MRHD of 450 mg/day). Similar adverse clinical signs were seen in adult rats at 20 mg/kg/day (about 16 times the subcutaneous MRHD of 12 mg/day; about 0.43 times the oral MRHD of 450 mg/day). Juvenile rats were found to be more sensitive to the toxicity of methylnaltrexone bromide when compared to adults. The no observed adverse effect levels (NOAELs) in juvenile and adult rats were 1 and 5 mg/kg/day, respectively (about 0.8 and 4 times, respectively, the subcutaneous MRHD of 12 mg/day; about 0.02 and 0.11 times, respectively, the oral MRHD of 450 mg/day).
Juvenile dogs administered intravenous methylnaltrexone bromide for 13 weeks had a toxicity profile similar to adult dogs. Following intravenous administration of methylnaltrexone bromide for 13 weeks, decreased heart rate (13.2% reduction compared to pre-dose) in juvenile dogs and prolonged QTc interval in juvenile (9.6% compared to control) and adult (up to 15% compared to control) dogs occurred at 20 mg/kg/day (about 54 times the subcutaneous MRHD of 12 mg/day; about 1.5 times the oral MRHD of 450 mg/day). Clinical signs consistent with effects on the CNS (including tremors and decreased activity) occurred in both juvenile and adult dogs. The NOAELs in juvenile and adult dogs were 5 mg/kg/day (about 14 times the subcutaneous MRHD of 12 mg/day; about 0.4 times the oral MRHD of 450 mg/day).
-
14 CLINICAL STUDIES
14.1 Opioid-Induced Constipation in Adult Patients with Chronic Non-Cancer Pain
RELISTOR Tablets
The efficacy of RELISTOR tablets in the treatment of OIC in patients with chronic non-cancer pain was evaluated in a randomized, double‑blind, placebo‑controlled study (Study 1). This study compared 4‑week treatment of RELISTOR tablets 450 mg orally once daily with placebo.
A total of 401 patients (200 RELISTOR, 201 placebo) were enrolled and treated in the double-blind period. Patients had a history of chronic non-cancer pain for which they were taking opioids. The most common pain condition requiring opioid use was back pain. Other frequently reported primary pain conditions were arthritis, neurologic/neuropathic pain, joint/extremity pain, and fibromyalgia. Prior to screening, patients were receiving opioid therapy for pain for 1 month or longer (median daily baseline oral morphine equivalent dose of 156 mg) and had OIC (less than 3 spontaneous bowel movements per week during the screening period). Constipation due to opioid use had to be associated with 1 or more of the following: A Bristol Stool Form Scale score of 1 or 2 for at least 25% of the bowel movements (BM), straining during at least 25% of the BMs or a sensation of incomplete evacuation after at least 25% of the BMs.
Patients were required to be on a stable opioid regimen (daily dose 50 mg or more of oral morphine equivalents per day) a minimum of 2 weeks prior to the screening visit and received their opioid medication during the study as clinically needed. The median duration of OIC at baseline was 53 months (4 years). The mean patient age was 52 years (range 23 to 78 years), 64% were female, and 84% of patients were Caucasian.
Eligible patients were required to discontinue all previous laxative therapy and use only the study-permitted rescue laxative (bisacodyl tablets). If patients did not have a bowel movement for 3 consecutive days during the study, they were permitted to use rescue medication (up to 3 bisacodyl tablets taken orally once during a 24-hour period). Bisacodyl tablets were taken 5 hours or longer and up to 8 hours after study drug administration. If rescue treatment with bisacodyl tablets did not result in a bowel movement, a second dose of bisacodyl or an enema 24 hours after rescue was permitted. Enema use was permitted after rescue with bisacodyl tablets had failed at least once.
A responder analysis was performed which defined the proportion of patients with 3 or more spontaneous bowel movements (SBMs)/week, with an increase of 1 or more SBM/week over baseline, for 3 or more out of the first 4 weeks of the treatment period. A SBM was defined as a bowel movement that occurred without laxative use during the previous 24 hours. Table 8 presents the proportion of patients who responded during the double-blind treatment period in the intent-to-treat (ITT) population, which included all randomized patients who received at least one dose of double-blind study medication.
Table 8: Proportion of Responders* in the ITT Population in Study 1 of RELISTOR Tablets for the Treatment of OIC in Patients with Chronic Non-Cancer Pain - * A responder is defined as a patient with 3 or more SBMs/week, with an increase of 1 or more SBM/week over baseline, for 3 or more out of the first 4 weeks of the treatment period.
- † Difference for active treatment vs. placebo;
Treatment
N
n (%)
Percent Difference†
(2-sided 95% CI)
RELISTOR Tablets 450 mg Once Daily
- 200
103 (52%)
13%
(3%, 23%)
Placebo
- 201
77 (38%)
CI = confidence interval; ITT = intent-to-treat
RELISTOR Injection
The efficacy of RELISTOR injection in the treatment of OIC in patients with chronic non-cancer pain were evaluated in a randomized, double‑blind, placebo‑controlled study (Study 2). This study compared 4‑week treatment of RELISTOR injection 12 mg administered subcutaneously once daily with placebo.
A total of 312 patients (150 RELISTOR, 162 placebo) were enrolled and treated in the double-blind period. Patients had a history of chronic non-cancer pain for which they were taking opioids. The majority of patients had a primary diagnosis of back pain; other primary diagnoses included joint/extremity pain, fibromyalgia, neurologic/neuropathic pain, and rheumatoid arthritis. Prior to screening, patients had been receiving opioid therapy for pain for 1 month or longer (median daily baseline oral morphine equivalent dose of 161 mg) and had OIC (less than 3 spontaneous bowel movements per week during the screening period). Constipation due to opioid use had to be associated with 1 or more of the following: A Bristol Stool Form Scale score of 1 or 2 for at least 25% of the bowel movements (BM), straining during at least 25% of the BMs or a sensation of incomplete evacuation after at least 25% of the BMs.
Patients were required to be on a stable opioid regimen (daily dose 50 mg or more of oral morphine equivalents per day) for at least 2 weeks before the screening visit and received their opioid medication during the study as clinically needed. The median duration of OIC at baseline was 59 months (5 years). The median patient age at baseline was 49 years, 62% were females and 90% were Caucasian.
Eligible patients were required to discontinue all previous laxative therapy and use only the study-permitted rescue laxative (bisacodyl tablets). If patients did not have a bowel movement for 3 consecutive days during the study, they were permitted to use rescue medication (up to 4 bisacodyl tablets taken orally once during a 24-hour period). Rescue laxatives were prohibited until at least 4 hours after taking an injection of study medication.
A responder analysis was performed which defined the proportion of patients with 3 or more (SBMs) per week for each of the 4 weeks of the double-blind period. A SBM was defined as a bowel movement that occurred without laxative use during the previous 24 hours. Table 9 presents the proportion of patients who responded during the double-blind treatment period in the modified intent-to-treat (mITT) population, which included all randomized subjects who received at least one dose of double-blind study medication.
Table 9: Proportion of Responders* in the mITT Population in Study 2 of RELISTOR Injection for the Treatment of OIC in Patients with Chronic Non-Cancer Pain - * A responder is defined as a patient with 3 or more SBMs per week for each of the 4 weeks in the double-blind period.
- † Difference for active treatment vs. placebo;
- ‡ P-value for active treatment vs. placebo based on 2-sided Chi-square test.
Treatment
N
n (%)
Percent Difference†
(2-sided 95% CI)
P-value‡
RELISTOR Injection 12 mg Once Daily
- 150
88 (59%)
20%
(10%, 31%)
<0.001
Placebo
- 162
62 (38%)
CI = confidence interval; mITT = modified intent-to-treat
Following the first dose, 33% of patients in the RELISTOR treatment group had a SBM within 4 hours and approximately half of patients had a SBM prior to the second dose of RELISTOR injection.
14.2 Opioid-Induced Constipation in Adult Patients with Advanced Illness
The efficacy of RELISTOR injection in the treatment of OIC in advanced illness patients receiving palliative care was demonstrated in two randomized, double‑blind, placebo‑controlled studies. In these studies, the median age was 68 years (range 21 to 100 years); 51% were females. In both studies, patients had advanced illness and received care to control their symptoms. The majority of patients had a primary diagnosis of incurable cancer; other primary diagnoses included end-stage COPD/emphysema, cardiovascular disease/heart failure, Alzheimer's disease/dementia, HIV/AIDS, or other advanced illnesses. Prior to screening, patients had been receiving palliative opioid therapy (median daily baseline oral morphine equivalent dose of 172 mg), and had OIC (either less than 3 bowel movements in the preceding week or no bowel movement for 2 or more days). Patients were on a stable opioid regimen 3 or more days prior to randomization (not including PRN or rescue pain medication) and received their opioid medication during the study as clinically needed. Patients maintained their regular laxative regimen for at least 3 days prior to study entry, and throughout the study. Rescue laxatives were prohibited from 4 hours before to 4 hours after taking an injection of study medication.
Study 4 was a double-blind, placebo-controlled study which compared a single, subcutaneous dose of RELISTOR injection 0.15 mg/kg, and RELISTOR injection 0.3 mg/kg versus placebo. The double-blind dose was followed by an open-label 4‑week dosing period, where RELISTOR injection could be used as needed, no more frequently than one dose in a 24-hour period. Throughout both study periods, patients maintained their regular laxative regimen. A total of 154 patients (47 RELISTOR 0.15 mg/kg, 55 RELISTOR 0.3 mg/kg, 52 placebo) were enrolled and treated in the double-blind period. The primary endpoint was the proportion of patients with a rescue-free laxation within 4 hours of the double-blind dose of study medication. RELISTOR-treated patients had a significantly higher rate of laxation within 4 hours of the double-blind dose (62% for 0.15 mg/kg and 58% for 0.3 mg/kg) than did placebo‑treated patients (14%); p <0.0001 for each dose versus placebo (Figure 1).
Study 5 was a double-blind, placebo-controlled study which compared subcutaneous doses of RELISTOR injection given every other day for 2 weeks versus placebo. Patients received opioid medication for at least 2 weeks prior to receiving study medication. During the first week (Days 1, 3, 5, 7) patients received either 0.15 mg/kg RELISTOR injection or placebo. In the second week the patient's assigned dose could be increased to 0.3 mg/kg if the patient had 2 or fewer rescue-free laxations up to Day 8. At any time, the patient's assigned dose could be reduced based on tolerability. Data from 133 (62 RELISTOR, 71 placebo) patients were analyzed. There were 2 primary endpoints: proportion of patients with a rescue-free laxation within 4 hours of the first dose of study medication and proportion of patients with a rescue-free laxation within 4 hours after at least 2 of the first 4 doses of study medication. RELISTOR-treated patients had a higher rate of laxation within 4 hours of the first dose (48%) than placebo-treated patients (16%); p <0.0001 (Figure 1). RELISTOR-treated patients also had significantly higher rates of laxation within 4 hours after at least 2 of the first 4 doses (52%) than did placebo-treated patients (9%); p <0.0001. In both studies, in approximately 30% of patients, laxation was reported within 30 minutes of a dose of RELISTOR.
In both studies, there was no evidence of differential effects of age or gender on safety or efficacy. No meaningful subgroup analysis could be conducted on race because the study population was predominantly Caucasian (88%).
Durability of Response
Durability of response was explored in Study 5, and the laxation response rate was consistent from dose 1 through dose 7 over the course of the 2-week, double-blind period.
The efficacy of RELISTOR injection was also demonstrated in open-label treatment administered from Day 2 through Week 4 in Study 4, and in two open-label extension studies (Study 4 EXT and Study 5 EXT) in which RELISTOR was given as needed for up to 4 months. During open-label treatment, patients maintained their regular laxative regimen. A total of 136, 21, and 82 patients received at least one open-label dose in Studies 4, 4 EXT, and 5 EXT, respectively. Laxation response was also explored in this open-label setting and appeared to be maintained over the course of 3 to 4 months of open-label treatment.
Opioid Use and Pain Scores
No relationship between baseline opioid dose and laxation response in RELISTOR-treated patients was identified in exploratory analyses of these studies. In addition, median daily opioid dose did not vary meaningfully from baseline in either RELISTOR-treated patients or in placebo‑treated patients. There were no clinically relevant changes in pain scores from baseline in either the RELISTOR or placebo-treated patients.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
NDC Number
Pack Size
Contents65649-150-90
90-count bottle
100‑mL bottle containing 90 tablets and 2 silica gel desiccant canisters. Each 150 mg film-coated tablet is white, round, biconvex, and debossed with “REL” on one side and plain on the other side.
65649-551-02
1 vial per carton
One 12 mg/0.6 mL single-dose vial containing a colorless to pale yellow solution.
65649-552-04
7 pre-filled syringes per carton
Seven 8 mg/0.4 mL single-dose pre-filled syringes with needle guard system containing a colorless to pale yellow solution.
65649-551-03
7 pre-filled syringes per carton
Seven 12 mg/0.6 mL single-dose pre-filled syringes with needle guard system containing a colorless to pale yellow solution.
65649-551-07
1 pre-filled syringe per carton
One 12 mg/0.6 mL single-dose pre-filled syringe with needle guard system containing a colorless to pale yellow solution.
Storage
Tablets
Store at up to 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].
Injection
Store at 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].
Do not freeze. Protect from light.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Administration
- Be within close proximity to toilet facilities once RELISTOR is administered.
- Discontinue RELISTOR if treatment with the opioid pain medication is also discontinued.
-
Advise chronic non-cancer pain patients receiving RELISTOR for OIC to:
- Discontinue all maintenance laxative therapy prior to initiation of RELISTOR. Laxative(s) can be used as needed if there is a suboptimal response to RELISTOR after 3 days.
- Inform their healthcare provider if their opioid regimen is changed, to avoid adverse reactions, such as diarrhea.
Tablets
Advise patients with chronic non-cancer pain receiving RELISTOR tablets for OIC to take RELISTOR tablets once daily with water on an empty stomach at least 30 minutes before the first meal of the day.
Injection
-
Advise all patients receiving RELISTOR injection to:
- Inject RELISTOR subcutaneously in the upper arm, abdomen or thigh. Do not inject at the same spot each time (rotate injection sites).
- Safely dispose of needles by following the sharps disposal recommendations described in the Instructions for Use.
- Advise chronic non-cancer pain patients receiving RELISTOR injection for OIC to inject one dose every day.
- Advise patients with advanced illness receiving RELISTOR injection for OIC to inject one dose every other day, as needed, but no more frequently than one dose in a 24-hour period.
Gastrointestinal Perforation
Advise patients to discontinue RELISTOR and to promptly seek medical attention if they develop unusually severe, persistent, or worsening abdominal pain [see Warnings and Precautions (5.1)].
Severe or Persistent Diarrhea
Advise patients to discontinue RELISTOR if they experience severe or persistent diarrhea [see Warnings and Precautions 5.2].
Opioid Withdrawal
Advise patients that symptoms consistent with opioid withdrawal may occur while taking RELISTOR, including sweating, chills, diarrhea, abdominal pain, anxiety, and yawning [see Warnings and Precautions (5.3), Adverse Reactions (6.1)].
Pregnancy
Fetal Opioid Withdrawal
Advise females of reproductive potential, who become pregnant or are planning to become pregnant, that the use of RELISTOR during pregnancy may precipitate opioid withdrawal in a fetus due to the undeveloped blood-brain barrier [see Use in Specific Populations (8.1)].
Lactation
Advise patients that breastfeeding is not recommended during treatment with RELISTOR [see Use in Specific Populations (8.2)].
Manufactured for:
Salix Pharmaceuticals, a division of
Valeant Pharmaceuticals North America LLC
Bridgewater, NJ 08807 USAUnder license from:
Progenics Pharmaceuticals, Inc.
Tarrytown, NY 10591 USAFor Injection: U.S. Patent Numbers: 8,247,425; 8,420,663; 8,552,025; 8,822,490; 9,180,125; 9,492,445 and 9,669,096
For Tablets: U.S. Patent Numbers: 8,420,663; 8,524,276; 8,956,651; 9,180,125; 9,314,461; 9,492,445 and 9,724,343
For more information, go to www.RELISTOR.com or call 1-800-321-4576.RELISTOR is a trademark of Valeant Pharmaceuticals International, Inc. or its affiliates.
All other product/brand names are trademarks of the respective owners.
© 2018 Valeant Pharmaceuticals North America LLC9493104
70013976 -
MEDICATION GUIDE
RELISTOR® (rel-i –store)
(methylnaltrexone bromide)
tablets
and
RELISTOR (rel-i –store)
(methylnaltrexone bromide)
injection, for subcutaneous useWhat is the most important information I should know about RELISTOR?
RELISTOR can cause serious side effects, including:
- Tear in your stomach or intestinal wall (perforation). Stomach pain that is severe can be a sign of a serious medical condition. If you get stomach pain that is severe, does not go away, or gets worse, stop taking RELISTOR and get emergency medical help right away.
- Diarrhea that is severe or that will not go away. Stop taking RELISTOR and call your healthcare provider if you get diarrhea that is severe or that does not go away during treatment with RELISTOR.
- Opioid withdrawal. You may have symptoms of opioid withdrawal during treatment with RELISTOR including sweating, chills, diarrhea, stomach pain, anxiety, and yawning. Tell your healthcare provider if you have any of these symptoms.
What is RELISTOR?
RELISTOR is a prescription medicine used to treat constipation in adults that is caused by prescription pain medicines called opioids:
- RELISTOR tablets and RELISTOR injection are used to treat constipation caused by opioids in adults with long-lasting (chronic) pain that is not caused by active cancer.
- RELISTOR injection is used to treat constipation caused by opioids in adults with advanced illness or pain caused by active cancer and who need increases in their opioid dose for comfort care.
It is not known if RELISTOR is safe and effective in children.
Who should not use RELISTOR?
Do not use RELISTOR if you have a bowel blockage (intestinal obstruction) or have a history of bowel blockage.
What should I tell my healthcare provider before taking RELISTOR?
Before you start taking RELISTOR, tell your healthcare provider about all of your medical conditions, including if you:
- have kidney problems.
- have liver problems.
- have any stomach or bowel (intestines) problems, including stomach ulcer, Crohn’s disease, diverticulitis, cancer of the stomach or bowel, or Ogilvie’s syndrome.
- are pregnant or plan to become pregnant. Taking RELISTOR during pregnancy may cause opioid withdrawal symptoms in your unborn baby. Tell your healthcare provider right away if you become pregnant during treatment with RELISTOR.
- are breastfeeding or plan to breastfeed. It is not known if RELISTOR passes into your breast milk. Taking RELISTOR while you are breastfeeding may cause opioid withdrawal in your baby. You should not breastfeed during treatment with RELISTOR.
Tell your healthcare provider about all of the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
How should I take RELISTOR?
- Stay close to a toilet after taking RELISTOR.
- Stop taking RELISTOR if you stop taking your prescription opioid pain medicine. Tell your healthcare provider if your pain medicine changes.
- If you take too much RELISTOR, call your healthcare provider or go to the nearest emergency room right away.
- If you take RELISTOR for long-lasting (chronic) pain that is not caused by cancer:
- RELISTOR has been shown to be effective in people who have taken opioid pain medicines for at least 4 weeks to treat long-lasting (chronic) pain not caused by cancer.
- Stop taking other laxatives before you start treatment with RELISTOR. You may use other laxatives if RELISTOR does not work after 3 days of treatment.
Tablets:
- Take RELISTOR tablets 1 time each day with water. Take RELISTOR tablets on an empty stomach at least 30 minutes before your first meal of the day.
Injection (Vials and Pre-filled Syringes):
See the detailed “Instructions for Use” that comes with RELISTOR injection for information about how to prepare and inject RELISTOR injection, and properly throw away (dispose of) used needles and syringes the right way.
-
RELISTOR injection is injected under the skin (subcutaneous injection) of the upper arm, stomach-area (abdomen), or thigh.
- Inject RELISTOR injection exactly as your healthcare provider tells you.
-
If you use RELISTOR injection for long-lasting (chronic) pain that is not caused by cancer:
- Inject 1 dose of RELISTOR injection each day.
-
If you use RELISTOR injection and are receiving treatment for advanced illness:
- Inject 1 dose of RELISTOR injection every other day, as needed. You should not inject more than 1 dose of RELISTOR injection in a 24-hour period.
What are the possible side effects of RELISTOR?
See “What is the most important information I should know about RELISTOR?”
- The most common side effects of RELISTOR tablets in people with long-lasting (chronic) pain that is not caused by cancer include: stomach-area (abdomen) pain, diarrhea, headache, swelling or a feeling of fullness or pressure in your abdomen, sweating, anxiety, muscle spasms, runny nose, and chills.
- The most common side effects of RELISTOR injection in people with long-lasting (chronic) pain that is not caused by cancer include: stomach-area (abdomen) pain, nausea, diarrhea, sweating, hot flush, tremor, and chills.
- The most common side effects of RELISTOR injection in people receiving treatment for their advanced illness include: stomach-area (abdomen) pain, gas, nausea, dizziness, and diarrhea.
Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all of the possible side effects of RELISTOR.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1‑800‑FDA‑1088.
You may also report side effects to Valeant Pharmaceuticals North America LLC at 1-800-321-4576.
How should I store RELISTOR?
RELISTOR tablets:
- Store RELISTOR tablets at room temperature between 68° to 77°F (20° to 25°C).
- The bottle of RELISTOR tablets contains 2 desiccant canisters to help keep your medicine dry. Do not remove the desiccant canisters from the bottle.
RELISTOR injection (Vials and Pre-filled Syringes):
- Store RELISTOR vials and pre-filled syringes at room temperature between 68° to 77°F (20° to 25°C).
- Do not freeze RELISTOR vials or pre-filled syringes.
- Keep RELISTOR vials and pre-filled syringes away from light until you are ready to use them.
- If the contents of a RELISTOR vial have been drawn into a syringe and you are not able to use the medicine right away, keep the syringe at room temperature for up to 24 hours.
Keep RELISTOR and all medicines, needles and syringes out of the reach of children.
General information about the safe and effective use of RELISTOR
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use RELISTOR for a condition for which it was not prescribed. Do not give RELISTOR to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about RELISTOR that is written for health professionals.
What are the ingredients in RELISTOR tablets and RELISTOR injection?
Active ingredient: methylnaltrexone bromide
Inactive ingredients (tablets): silicified microcrystalline cellulose, microcrystalline cellulose, sodium lauryl sulfate, croscarmellose sodium, crospovidone, poloxamer 407, stearic acid (vegetable source), colloidal silicon dioxide, edetate calcium disodium, polyvinyl alcohol, titanium dioxide, polyethylene glycol and talcInactive ingredients (injection: vials and pre-filled syringes): sodium chloride USP, edetate calcium disodium USP, glycine hydrochloride. During manufacture, the pH may have been adjusted with hydrochloric acid and/or sodium hydroxide.
Manufactured for:
Salix Pharmaceuticals, a division of
Valeant Pharmaceuticals North America LLC
Bridgewater, NJ 08807 USAUnder license from:
Progenics Pharmaceuticals, Inc.
Tarrytown, NY 10591 USAFor Injection: U.S. Patent Numbers: 8,247,425; 8,420,663; 8,552,025; 8,822,490; 9,180,125; 9,492,445 and 9,669,096
For Tablets: U.S. Patent Numbers: 8,420,663; 8,524,276; 8,956,651; 9,180,125; 9,314,461; 9,492,445 and 9,724,343For more information, go to www.RELISTOR.com or call 1-800-321-4576.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Revised: November 2018
9493104
70013976 -
Instructions for Use – Pre-filled Syringe
Instructions for Use
RELISTOR® (rel-i-store)
(methylnaltrexone bromide)
injection, for subcutaneous usePre-filled Syringe
Read this Instructions for Use before you start using RELISTOR and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
The following instructions explain how to prepare and give an injection of RELISTOR the right way, when using a pre-filled syringe of RELISTOR.
Important information:
- Do not use a RELISTOR pre-filled syringe and attached needle more than 1 time, even if there is medicine left in the syringe. See Step 4 “Dispose of used pre-filled syringes and needles.”
- Safely throw away RELISTOR pre-filled syringes and attached needle after use.
- To avoid needle-stick injuries, do not recap used needles.
- Avoid touching the trigger fingers of the RELISTOR pre-filled syringe to keep from activating the needle guard (safety device) too soon. The needle guard is activated by pressure from the plunger on the trigger fingers (See Figure A).
Gather the supplies you will need for your injection (See Figure A). These include:
- 1 RELISTOR pre-filled syringe with attached needle
- 1 alcohol swab
- 1 cotton ball or gauze
- 1 adhesive bandage
- a puncture resistant container, such as an FDA-cleared sharps disposal container to dispose of used pre-filled syringes and needles. See Step 4: “Dispose of used pre-filled syringes and needles.”
Step 1: Choose and prepare the injection site
-
Choose an injection site on your stomach-area (abdomen), thighs, or upper arms. See the shaded areas in Figures B and C below. Do not inject at the exact same spot each time (rotate injection sites). Do not inject into areas where the skin is tender, bruised, red or hard. Avoid areas with scars or stretch marks.
Figure B Abdomen or thigh – use these sites when injecting yourself or another person.
Figure C Upper arm – use this site only when injecting another person.
- Clean the injection site with an alcohol swab and let it air dry. Do not touch this area again before giving the injection (See Figure D).
Step 2: Prepare the pre-filled syringe
- Choose a flat, clean, well-lit work surface.
- Wash your hands with soap and water before preparing for the injection.
- Look at the pre-filled syringe of RELISTOR (See Figure E). Make sure that the dose prescribed by your healthcare provider matches the dose on the pre-filled syringe label. Look at the plunger rod of the syringe. If the dose prescribed by your healthcare provider is 8 mg, the plunger rod will be yellow; if the prescribed dose is 12 mg, the plunger rod of the syringe will be dark blue (See Figure E).
- The liquid in the pre-filled syringe should be colorless to pale yellow, and should not have any particles in it. Do not use the pre-filled syringe if it looks discolored, cloudy, or has any particles.
- Use one hand to firmly hold the barrel of the pre-filled syringe. Use your other hand to pull the needle cap straight off (See Figure F). Do not touch the needle or allow it to touch anything.
Step 3: Inject RELISTOR
- Use one hand to pinch the skin around the injection site (See Figure G).
- Use your other hand to hold the pre-filled syringe. Insert the full length of the needle into the skin at a 45-degree angle with a quick “dart‑like” motion (See Figure H).
- Let go of the skin and slowly push the plunger in with your thumb until the pre-filled syringe is empty (See Figure I). This will release the needle guard (safety device).
- Continue to hold pressure on the plunger with your thumb and quickly pull the needle out of the skin. Be careful to keep the needle at the same angle as it was inserted. Remove your thumb from the plunger to allow the protective sleeve to cover the needle (See Figure J). There may be a little bleeding at the injection site.
- Hold a cotton ball or gauze over the injection site (See Figure K). Do not rub the injection site. Apply an adhesive bandage to the injection site if needed.
Step 4: Dispose of used pre-filled syringes and needles
- Do not reuse the pre-filled syringe and attached needle.
- To avoid needle-stick injuries, do not recap used needles.
- Put your used pre-filled syringes and attached needles in an FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) loose needles and syringes in your household trash.
-
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA’s website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
- If you have any questions, talk to your healthcare provider or pharmacist.
- How should I store RELISTOR?
- Store pre-filled syringes at room temperature between 68° to 77°F (20° to 25°C).
- Do not freeze RELISTOR.
- Keep RELISTOR away from light until you are ready to use it.
-
Keep RELISTOR and all medicines, needles and syringes out of the reach of children.
For more information, go to www.Relistor.com or call 1-800-321-4576.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured for:
Salix Pharmaceuticals, a division of
Valeant Pharmaceuticals North America LLC
Bridgewater, NJ 08807 USAUnder license from:
Progenics Pharmaceuticals, Inc.
Tarrytown, NY 10591 USAFor Injection: U.S. Patent Numbers: 8,247,425; 8,420,663; 8,552,025; 8,822,490; 9,180,125; 9,492,445 and 9,669,096
For Tablets: U.S. Patent Numbers: 8,420,663; 8,524,276; 8,956,651; 9,180,125; 9,314,461; 9,492,445 and 9,724,343RELISTOR is a trademark of Valeant Pharmaceuticals International, Inc. or its affiliates.
All other product/brand names are trademarks of the respective owners.
© 2018 Valeant Pharmaceuticals North America LLCRevised: 11/2018
9493104
70013976 -
Instructions for Use - Vial and Syringe with Retractable Needle in Tray
Instructions for Use
RELISTOR® (rel-i-store)
(methylnaltrexone bromide)
injection, for subcutaneous use
Vial and Syringe with Retractable Needle in TrayRead this Instructions for Use before you start using RELISTOR and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
The following instructions explain how to prepare and give an injection of RELISTOR the right way, when using a RELISTOR tray containing a syringe with a retractable needle. A retractable needle is one that is pulled back so that it is covered after use, to prevent needle-stick injury.
Important information:
- Do not use a RELISTOR vial more than 1 time, even if there is medicine left in the vial.
- If RELISTOR has been drawn into a syringe and you are unable to use the medicine right away, carefully recap the needle and keep the syringe at room temperature for up to 24 hours. For more information about how to store RELISTOR, see the section called “How should I store RELISTOR?” at the end of this Instructions for Use.
- Safely throw away RELISTOR vials after use.
- Do not reuse syringes and needles. See Step 5: “Dispose of used syringes and needles” for information about how to safely throw away used needles and syringes.
- To avoid needle-stick injuries, do not recap used needles.
Your tray should include (See Figure A):
- 1 RELISTOR vial
- 1 1 mL syringe with retractable needle (VanishPoint)
- 2 alcohol swabs
- You will also need:
- 1 cotton ball or gauze
- 1 adhesive bandage
- a puncture resistant container, such as an FDA-cleared sharps disposal container to dispose of your used syringes and needles. See Step 5: “Dispose of used syringes and needles.”
Step 1: Choose and prepare the injection site
- Choose an injection site on your stomach-area (abdomen), thighs, or upper arms. See the shaded areas in Figures B and C below. Do not inject at the exact same spot each time (rotate injection sites). Do not inject into areas where the skin is tender, bruised, red, or hard. Avoid areas with scars or stretch marks.
-
Figure B Abdomen or thigh – use these sites when injecting yourself or another person.
Figure C Upper arm – use this site only when injecting another person.
- Clean the injection site with an alcohol swab and let it air dry. Do not touch this area again before giving the injection (See Figure D).
Step 2: Prepare the injection
- Choose a flat, clean, well-lit work surface.
- Wash your hands with soap and water before preparing for the injection.
- Look at the vial of RELISTOR (See Figure E). The liquid in the vial should be colorless to pale yellow, and should not have any particles in it. Do not use the vial if it looks discolored, cloudy, or has any particles.
Step 3: Prepare the syringe
- Remove the cap from the vial containing RELISTOR (See Figure F).
- Wipe the rubber stopper with an alcohol swab (See Figure G).
- Firmly hold the barrel of the syringe with one hand. With your other hand, pull the needle cap straight off (See Figure H). Do not touch the needle or allow it to touch anything.
- Carefully pull back on the plunger to the line that matches the dose prescribed by your healthcare provider (See Figures I and J). For most people, this will be the 0.4 mL mark which is an 8 mg dose or the 0.6 mL mark which is a 12 mg dose.
- Use one hand to hold the vial steady. Use your other hand to insert the needle straight down into the rubber top of the RELISTOR vial (See Figure K). Do not insert it at an angle. This may cause the needle to bend or break. You will feel some resistance as the needle passes through the rubber top.
- Gently push down on the plunger until you feel resistance, and most of the air has gone from the syringe into the vial (See Figure L). Stop pushing down on the plunger when you feel resistance. If you continue to push down on the plunger when you feel resistance, the needle will pull back (retract) into the syringe barrel.
- With the needle still in the vial, turn the vial and syringe upside down. Hold the syringe at eye level. Make sure the tip of the needle is in the fluid. Slowly pull back on the plunger (See Figure M) until the top of the plunger lines up with the mark that matches your prescribed dose. For most people, this will be the 0.4 mL mark which is an 8 mg dose or the 0.6 mL mark which is a 12 mg dose.
- You may see some fluid or bubbles inside the vial when the syringe is filled. This is normal.
- With the needle still in the vial, gently tap the syringe to make any air bubbles rise to the top (See Figure N).
- Gently push the plunger up until all air bubbles are out of the syringe (See Figure O). A small air bubble may stay in the syringe. This is okay and it will not affect the dose of medicine in the syringe.
- Make sure the tip of the needle is in the fluid. Slowly pull back the plunger to draw the right amount of liquid back into the syringe (See Figure P).
- Check to be sure that you have the right dose of RELISTOR in the syringe.
- Slowly withdraw the needle from the vial. Do not touch the needle or allow it to touch anything. Safely throw away the vial with any unused medicine.
Step 4: Inject RELISTOR
- Use one hand to pinch the skin around the injection site (See Figure Q).
- Use your other hand to hold the syringe. Insert the full length of the needle into the skin at a 45-degree angle with a quick “dart-like” motion (See Figure R).
- Let go of the skin and slowly push in on the plunger past the resistance point, until the syringe is empty and you hear a click (See Figure S).
- The click sound means that the needle (See Figure T) has been pulled back (retracted) into the syringe barrel (See Figure U). You can now remove the syringe from your skin.
- Hold a cotton ball or gauze over the injection site (See Figure V). Do not rub the injection site. Apply an adhesive bandage to the injection site if needed.
Step 5: Dispose of used syringes and needles
- Do not re-use syringes or needles.
- To avoid needle-stick injuries, do not recap used needles.
- Put your used needles and syringes in an FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) loose needles and syringes in your household trash.
-
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA’s website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
- If you have any questions, talk to your healthcare provider or pharmacist.
How should I store RELISTOR?
- Store RELISTOR vials at room temperature between 68° to 77°F (20° to 25°C).
- Do not freeze RELISTOR.
- Keep RELISTOR away from light until you are ready to use it.
- If RELISTOR has been drawn into a syringe and you are unable to use the medicine right away, keep the syringe at room temperature for up to 24 hours.
Keep RELISTOR and all medicines, needles and syringes out of the reach of children.
For more information, go to www.Relistor.com or call 1-800-321-4576.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured for:
Salix Pharmaceuticals, a division of
Valeant Pharmaceuticals North America LLC
Bridgewater, NJ 08807 USAUnder license from:
Progenics Pharmaceuticals, Inc.
Tarrytown, NY 10591 USAFor Injection: U.S. Patent Numbers: 8,247,425; 8,420,663; 8,552,025; 8,822,490; 9,180,125; 9,492,445 and 9,669,096
For Tablets: U.S. Patent Numbers: 8,420,663; 8,524,276; 8,956,651; 9,180,125; 9,314,461; 9,492,445 and 9,724,343RELISTOR is a trademark of Valeant Pharmaceuticals International, Inc. or its affiliates.
All other product/brand names are trademarks of the respective owners.
© 2018 Valeant Pharmaceuticals North America LLCRevised: 11/2018
9493104
70013976 -
Instructions for Use - Vial
Instructions for Use
RELISTOR® (rel-i-store)
(methylnaltrexone bromide)
injection, for subcutaneous useVial
Read this Instructions for Use before you start using RELISTOR and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
The following instructions explain how to prepare and give an injection of RELISTOR the right way, when using a vial of RELISTOR.
Important information:
- Use the syringes and needles prescribed by your healthcare provider.
- Do not use a RELISTOR vial more than 1 time, even if there is medicine left in the vial.
- If RELISTOR has been drawn into a syringe and you are unable to use the medicine right away, carefully recap the needle and keep the syringe at room temperature for up to 24 hours. For more information about how to store RELISTOR, see the section “How should I store RELISTOR?” at the end of this Instructions for Use.
- Safely throw away RELISTOR vials after use.
- Do not re-use syringes or needles. See Step 5 “Dispose of used syringes and needles” for information about how to safely throw away used needles and syringes.
- To avoid needle-stick injuries, do not recap used needles.
Gather the supplies you will need for your injection (See Figure A.). These include:
- 1 RELISTOR vial
- 1 1 mL syringe with a 27-gauge, ½ inch needle for subcutaneous use
- 2 alcohol swabs
- 1 cotton ball or gauze
- 1 adhesive bandage
- a puncture resistant container, such as an FDA-cleared sharps disposal container to dispose of used syringes and needles. See Step 5: “Dispose of used syringes and needles.”
Step 1: Choose and prepare the injection site
- Choose an injection site on your stomach-area (abdomen), thighs, or upper arms. See the shaded areas in Figures B and C below. Do not inject at the exact same spot each time (rotate injection sites). Do not inject into areas where the skin is tender, bruised, red or hard. Avoid areas with scars or stretch marks.
- Figure B Abdomen or thigh – use these sites when injecting yourself or another person.
- Figure C Upper arm – use this site only when injecting another person.
- Clean the injection site with an alcohol swab and let it air dry. Do not touch this area again before giving the injection (See Figure D).
Step 2: Prepare the injection
- Choose a flat, clean, well-lit work surface.
- Wash your hands with soap and water before preparing for the injection.
- Look at the vial of RELISTOR (See Figure E). The liquid in the vial should be colorless to pale yellow, and should not have any particles in it. Do not use the vial if it looks discolored, cloudy, or has any particles.
Step 3: Prepare the syringe
- Remove the cap from the RELISTOR vial (See Figure F).
- Wipe the rubber stopper with an alcohol swab (See Figure G).
- Firmly hold the barrel of the syringe with one hand. With your other hand, pull the needle cap straight off (See Figure H). Do not touch the needle or allow it to touch anything.
- Carefully pull back on the plunger to the line that matches the dose prescribed by your healthcare provider (See Figures I and J). For most people, this will be the 0.4 mL mark which is an 8 mg dose or the 0.6 mL mark which is a 12 mg dose.
- Use one hand to hold the vial steady. Use your other hand to insert the needle straight down into the rubber top of the vial (See Figure K). Do not insert it at an angle. This may cause the needle to bend or break. You will feel some resistance as the needle passes through the rubber top.
- Gently push down the plunger until all of the air has gone from the syringe into the vial (See Figure L).
- With the needle still in the vial, turn the vial and syringe upside down. Hold the syringe at eye level. Make sure the tip of the needle is in the fluid. Slowly pull back on the plunger (See Figure M) until the top of the plunger lines up with the mark that matches your prescribed dose. For most people, this will be the 0.4 mL mark which is an 8 mg dose or the 0.6 mL mark which is a 12 mg dose.
- You may see some fluid or bubbles inside the vial when the syringe is filled. This is normal.
- With the needle still in the vial, gently tap the side of the syringe to make any air bubbles rise to the top (See Figure N).
- Slowly push the plunger up until all air bubbles are out of the syringe (See Figure O). A small air bubble may stay in the syringe. This is okay and it will not affect the dose of medicine in the syringe.
- Make sure the tip of the needle is in the fluid. Slowly pull back the plunger to draw the right amount of liquid back into the syringe (See Figure P).
- Check to be sure that you have the right dose of RELISTOR in the syringe.
- Slowly withdraw the needle from the vial. Do not touch the needle or allow it to touch anything. Safely throw away the vial with any unused medicine.
Step 4: Inject RELISTOR
- Use one hand to pinch the skin around the injection site (See Figure Q).
- Use your other hand to hold the syringe. Insert the full length of the needle into the skin at a 45-degree angle with a quick “dart‑like” motion (See Figure R).
- Let go of the skin and slowly push in on the plunger until the syringe is empty (Figure S).
- When the syringe is empty, quickly pull the needle out of the skin, being careful to keep it at the same angle as it was inserted. There may be a little bleeding at the injection site.
- Hold a cotton ball or gauze over the injection site (Figure T). Do not rub the injection site. Apply an adhesive bandage to the injection site if needed.
Step 5: Dispose of used syringes and needles
- Do not re-use a syringe or needle.
- To avoid needle-stick injuries, do not recap a used needle.
- Put your used needles and syringes in an FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) loose needles and syringes in your household trash.
-
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA’s website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
- If you have any questions, talk to your healthcare provider or pharmacist.
How should I store RELISTOR?
- Store RELISTOR vials at room temperature between 68° to 77°F (20° to 25°C).
- Do not freeze RELISTOR.
- Keep RELISTOR away from light until you are ready to use it.
- If RELISTOR has been drawn into a syringe and you are unable to use the medicine right away, keep the syringe at room temperature for up to 24 hours.
Keep RELISTOR and all medicines, needles and syringes out of the reach of children.
For more information, go to www.Relistor.com or call 1-800-321-4576.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured for:
Salix Pharmaceuticals, a division of
Valeant Pharmaceuticals North America LLC
Bridgewater, NJ 08807 USAUnder license from:
Progenics Pharmaceuticals, Inc.
Tarrytown, NY 10591 USAFor Injection: U.S. Patent Numbers: 8,247,425; 8,420,663; 8,552,025; 8,822,490; 9,180,125; 9,492,445 and 9,669,096
For Tablets: U.S. Patent Numbers: 8,420,663; 8,524,276; 8,956,651; 9,180,125; 9,314,461; 9,492,445 and 9,724,343RELISTOR is a trademark of Valeant Pharmaceuticals International, Inc. or its affiliates.
All other product/brand names are trademarks of the respective owners.
© 2018 Valeant Pharmaceuticals North America LLCRevised: 11/2018
9493104
70013976 -
PRINCIPAL DISPLAY PANEL - 12 mg/0.6 mL
NDC 65649-551-03
Rx only
RELISTOR®
(methylnaltrexone bromide)
Subcutaneous Injection12 mg/0.6 ml per syringe
PHARMACIST: Dispense the enclosed
Medication Guide to each patient.For Subcutaneous Injection Only
Contains 7 Pre-filled Syringes with Needle Guard
Single Use Only. Discard after use.Protect syringe from light.
Salix
Pharmaceuticals -
PRINCIPAL DISPLAY PANEL - 8 mg/0.4 mL
NDC 65649-552-04
Rx only
RELISTOR®
(methylnaltrexone bromide)
Subcutaneous Injection8 mg/0.4 mL per syringe
PHARMACIST: Dispense the enclosed
Medication Guide to each patient.For Subcutaneous Injection Only
Contains 7 Pre-filled Syringes with Needle Guard
Single Use Only. Discard after use.Protect syringe from light.
- PRINCIPAL DISPLAY PANEL – 150 mg Tablets
-
INGREDIENTS AND APPEARANCE
RELISTOR
methylnaltrexone bromide tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 65649-150 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength METHYLNALTREXONE BROMIDE (UNII: RFO6IL3D3M) (METHYLNALTREXONE - UNII:0RK7M7IABE) METHYLNALTREXONE BROMIDE 150 mg Product Characteristics Color WHITE Score 2 pieces Shape ROUND Size 6mm Flavor Imprint Code REL;PLAIN Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 65649-150-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 07/19/2016 2 NDC: 65649-150-60 60 in 1 BOTTLE; Type 0: Not a Combination Product 07/19/2016 06/30/2017 3 NDC: 65649-150-06 1 in 1 CARTON 07/19/2016 3 6 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208271 08/01/2008 RELISTOR
methylnaltrexone bromide injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 65649-552 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength METHYLNALTREXONE BROMIDE (UNII: RFO6IL3D3M) (METHYLNALTREXONE - UNII:0RK7M7IABE) METHYLNALTREXONE BROMIDE 8 mg in 0.4 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) 2.6 mg in 0.4 mL EDETATE CALCIUM DISODIUM (UNII: 25IH6R4SGF) 0.16 mg in 0.4 mL GLYCINE HYDROCHLORIDE (UNII: 225ZLC74HX) 0.12 mg in 0.4 mL WATER (UNII: 059QF0KO0R) 0.4 mL in 0.4 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 65649-552-04 7 in 1 CARTON 04/24/2008 1 1 in 1 BLISTER PACK 1 0.4 mL in 1 SYRINGE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021964 04/24/2008 RELISTOR
methylnaltrexone bromide injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 65649-551 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength METHYLNALTREXONE BROMIDE (UNII: RFO6IL3D3M) (METHYLNALTREXONE - UNII:0RK7M7IABE) METHYLNALTREXONE BROMIDE 12 mg in 0.6 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) 3.9 mg in 0.6 mL EDETATE CALCIUM DISODIUM (UNII: 25IH6R4SGF) 0.24 mg in 0.6 mL GLYCINE HYDROCHLORIDE (UNII: 225ZLC74HX) 0.18 mg in 0.6 mL WATER (UNII: 059QF0KO0R) 0.6 mL in 0.6 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 65649-551-07 1 in 1 CARTON 04/24/2008 1 1 in 1 BLISTER PACK 1 0.6 mL in 1 SYRINGE; Type 0: Not a Combination Product 2 NDC: 65649-551-03 7 in 1 CARTON 04/24/2008 2 1 in 1 BLISTER PACK 2 0.6 mL in 1 SYRINGE; Type 0: Not a Combination Product 3 NDC: 65649-551-02 1 in 1 CARTON 04/24/2008 3 0.6 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021964 04/24/2008 Labeler - Salix Pharmaceuticals, Inc. (793108036) Registrant - Valeant Pharmaceuticals North America LLC (042230623) Establishment Name Address ID/FEI Business Operations Vetter Pharma Fertigung GmbH & Co. KG 316126754 MANUFACTURE(65649-150, 65649-552, 65649-551) Establishment Name Address ID/FEI Business Operations Mallinckrodt, Inc. 163205300 MANUFACTURE(65649-150) Establishment Name Address ID/FEI Business Operations Patheon Inc. 205475333 MANUFACTURE(65649-551) Establishment Name Address ID/FEI Business Operations Carton Service, Incorporated 928861723 REPACK(65649-150) , RELABEL(65649-150)
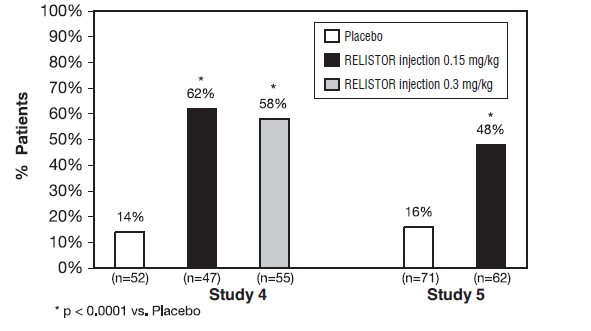
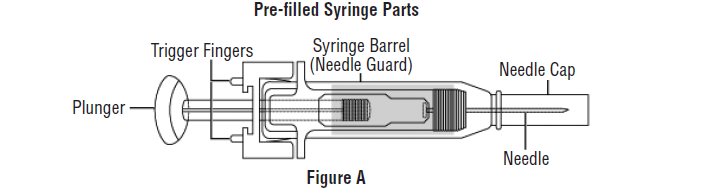
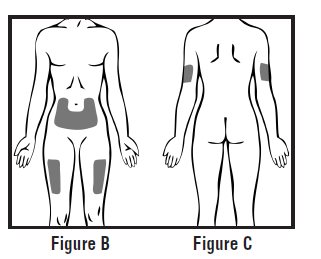
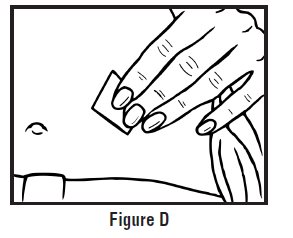
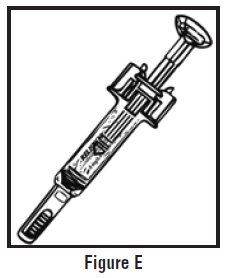
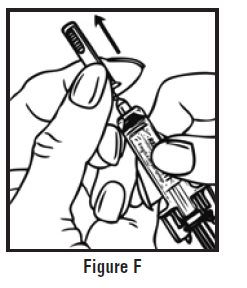
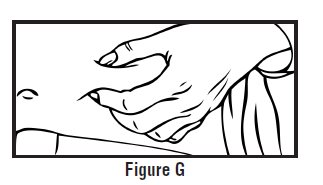
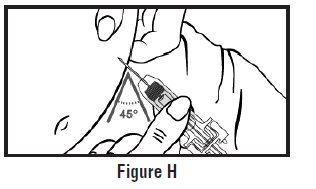
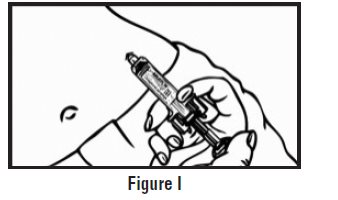
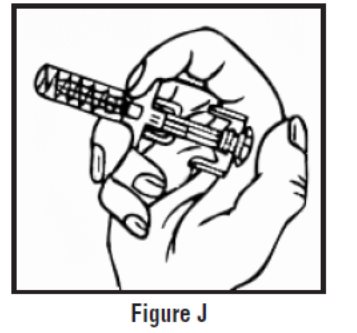
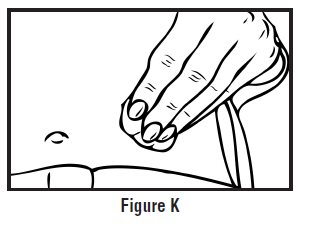
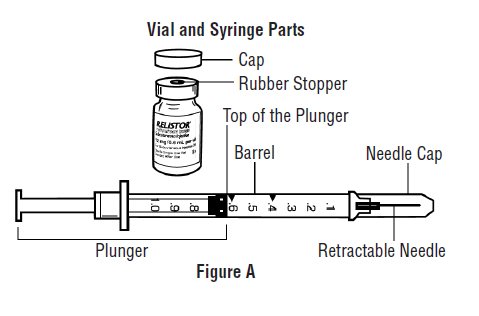
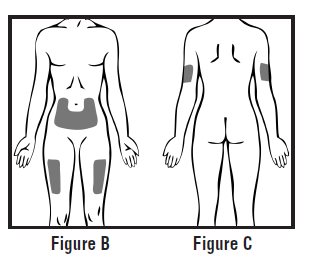
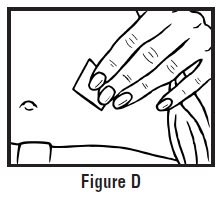

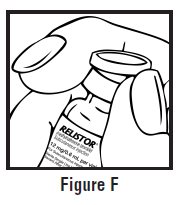
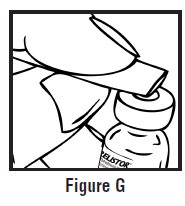
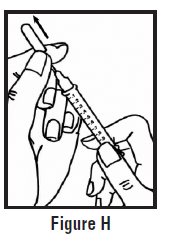

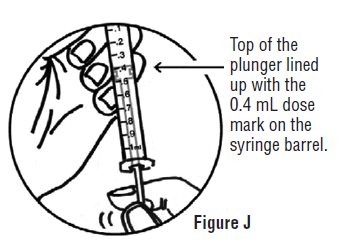
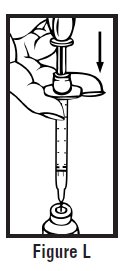
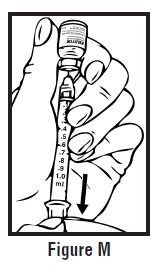
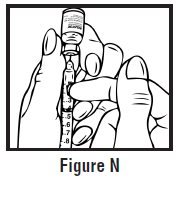
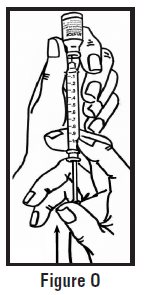
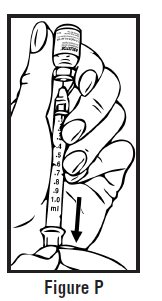
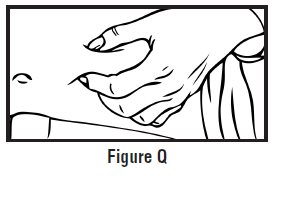

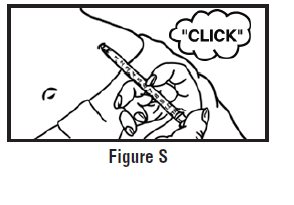
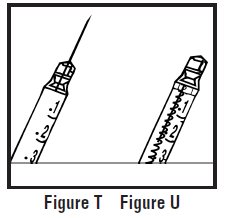
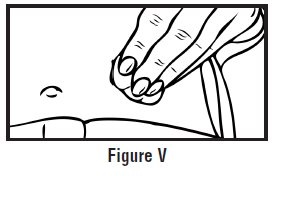
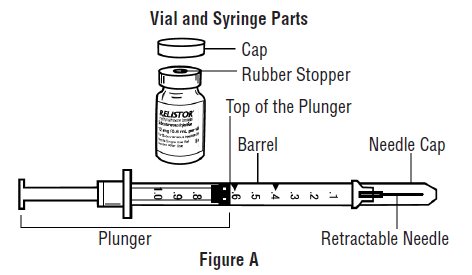
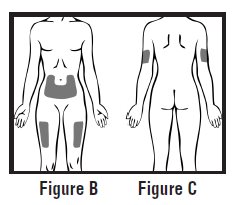
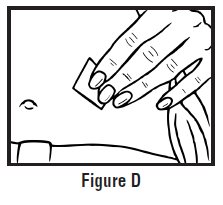
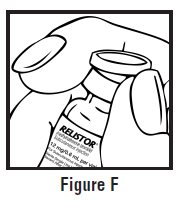
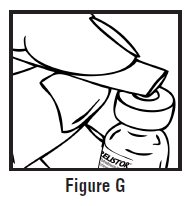
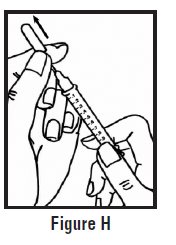

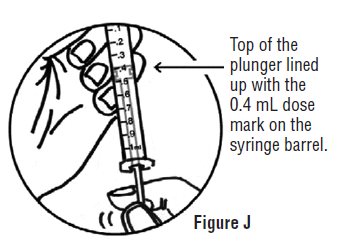
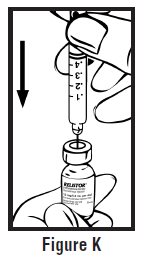
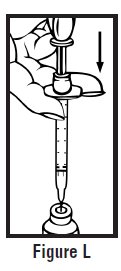
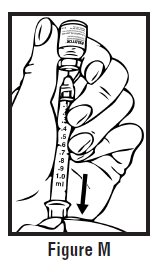
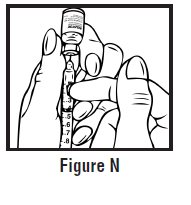
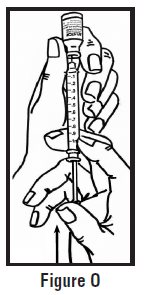
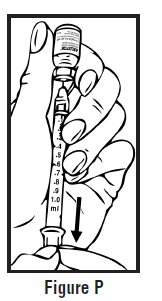
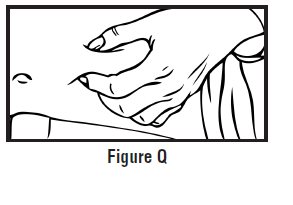
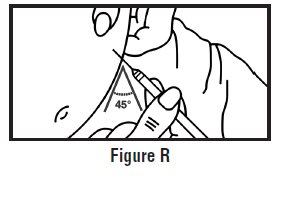
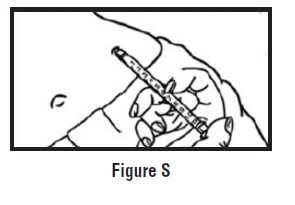
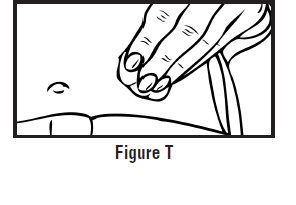



Trademark Results [Relistor]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 RELISTOR 78939607 3535582 Live/Registered |
SALIX PHARMACEUTICALS, INC. 2006-07-28 |
 RELISTOR 77324836 3592407 Dead/Cancelled |
SALIX PHARMACEUTICALS, INC. 2007-11-08 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.