MEXILETINE HYDROCHLORIDE capsule
Mexiletine Hydrochloride by
Drug Labeling and Warnings
Mexiletine Hydrochloride by is a Prescription medication manufactured, distributed, or labeled by AvKARE. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
DESCRIPTION
Mexiletine hydrochloride, USP is an orally active antiarrhythmic agent. It is a white to off-white crystalline powder with slightly bitter taste, freely soluble in water and in alcohol. Mexiletine hydrochloride, USP has a pKa of 9.2. The chemical name of mexiletine hydrochloride, USP is 1-methyl-2-(2,6-xylyloxy)ethylamine hydrochloride and its structural formula is:
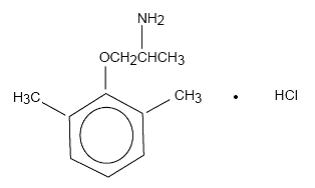
C 11H 17NO HCl M.W. 215.72
Each capsule for oral administration, contains 150 mg, 200 mg, or 250 mg of mexiletine hydrochloride, USP. 100 mg of mexiletine hydrochloride, USP is equivalent to 83.31 mg of mexiletine base. In addition, each capsule contains the following excipients: colloidal silicon dioxide, magnesium stearate and pregelatinized corn starch. The capsule shell contains: FD&C Yellow #6, gelatin and titanium dioxide. The 150 mg capsule also contains: D&C Red #28 and FD&C Blue #1 and the 250 mg capsule also contains: D&C Yellow #10 and FD&C Blue #1. The imprinting ink contains: ammonium hydroxide, black iron oxide, D&C Yellow #10, ethyl alcohol, FD&C Blue #1, FD&C Blue #2, FD&C Red #40, isopropyl alcohol, n-butyl alcohol, propylene glycol and shellac.
-
CLINICAL PHARMACOLOGY
Mechanism of Action
Mexiletine hydrochloride is a local anesthetic, antiarrhythmic agent, structurally similar to lidocaine, but orally active. In animal studies, mexiletine has been shown to be effective in the suppression of induced ventricular arrhythmias, including those induced by glycoside toxicity and coronary artery ligation. Mexiletine, like lidocaine, inhibits the inward sodium current, thus reducing the rate of rise of the action potential, Phase 0. Mexiletine decreased the effective refractory period (ERP) in Purkinje fibers. The decrease in ERP was of lesser magnitude than the decrease in action potential duration (APD), with a resulting increase in the ERP/APD ratio.
Electrophysiology in Man
Mexiletine is a Class 1B antiarrhythmic compound with electrophysiologic properties in man similar to those of lidocaine, but dissimilar from quinidine, procainamide, and disopyramide.
In patients with normal conduction systems, mexiletine has a minimal effect on cardiac impulse generation and propagation. In clinical trials, no development of second-degree or third degree AV block was observed. Mexiletine did not prolong ventricular depolarization (QRS duration) or repolarization (QT intervals) as measured by electrocardiography. Theoretically, therefore, mexiletine may be useful in the treatment of ventricular arrhythmias associated with a prolonged QT interval.
In patients with preexisting conduction defects, depression of the sinus rate, prolongation of sinus node recovery time, decreased conduction velocity and increased effective refractory period of the intraventricular conduction system have occasionally been observed.
The antiarrhythmic effect of mexiletine has been established in controlled comparative trials against placebo, quinidine, procainamide and disopyramide. Mexiletine hydrochloride, at doses of 200 to 400 mg q8h, produced a significant reduction of ventricular premature beats, paired beats, and episodes of non-sustained ventricular tachycardia compared to placebo and was similar in effectiveness to the active agents. Among all patients entered into the studies, about 30% in each treatment group had a 70% or greater reduction in PVC count and about 40% failed to complete the 3 month studies because of adverse effects. Follow-up of patients from the controlled trials has demonstrated continued effectiveness of mexiletine in long-term use.
Hemodynamics
Hemodynamic studies in a limited number of patients, with normal or abnormal myocardial function, following oral administration of mexiletine hydrochloride, have shown small, usually not statistically significant, decreases in cardiac output and increases in systemic vascular resistance, but no significant negative inotropic effect. Blood pressure and pulse rate remain essentially unchanged. Mild depression of myocardial function, similar to that produced by lidocaine, has occasionally been observed following intravenous mexiletine therapy in patients with cardiac disease.
Pharmacokinetics
Mexiletine is well absorbed (~90%) from the gastrointestinal tract. Unlike lidocaine, its first-pass metabolism is low. Peak blood levels are reached in two to three hours. In normal subjects, the plasma elimination half-life of mexiletine is approximately 10 to 12 hours. It is 50 to 60% bound to plasma protein, with a volume of distribution of 5 to 7 liters/kg. Mexiletine is mainly metabolized in the liver, the primary pathway being CYP2D6 metabolism, although it is also a substrate for CYP1A2. With involvement of CYP2D6, there can be either poor or extensive metabolizer phenotypes. Since approximately 90% of mexiletine hydrochloride is metabolized in the liver into inactive metabolites, pathological changes in the liver can restrict hepatic clearance of mexiletine hydrochloride and its metabolites. The metabolic degradation proceeds via various pathways including aromatic and aliphatic hydroxylation, dealkylation, deamination and N-oxidation. Several of the resulting metabolites are submitted to further conjugation with glucuronic acid (phase II metabolism); among these are the major metabolites p-hydroxymexiletine, hydroxy-methylmexiletine and N-hydroxy-mexiletine. Approximately 10% is excreted unchanged by the kidney. While urinary pH does not normally have much influence on elimination, marked changes in urinary pH influence the rate of excretion: acidification accelerates excretion, while alkalinization retards it.
Several metabolites of mexiletine have shown minimal antiarrhythmics activity in animal models. The most active is the minor metabolite N-methylmexiletine, which is less than 20% as potent as mexiletine. The urinary excretion of N-methylmexiletine in man is less than 0.5%. Thus the therapeutic activity of mexiletine is due to the parent compound.
Hepatic impairment prolongs the elimination half-life of mexiletine. In eight patients with moderate to severe liver disease, the mean half-life was approximately 25 hours.
Consistent with the limited renal elimination of mexiletine, little change in the half-life has been detected in patients with reduced renal function. In eight patients with creatinine clearance less than 10 mL/min, the mean plasma elimination half-life was 15.7 hours; in seven patients with creatinine clearance between 11 to 40 mL/min, the mean half-life was 13.4 hours.
The absorption rate of mexiletine is reduced in clinical situations such as acute myocardial infarction in which gastric emptying time is increased. Narcotics, atropine and magnesium-aluminum hydroxide have also been reported to slow the absorption of mexiletine. Metoclopramide has been reported to accelerate absorption.
Mexiletine plasma levels of at least 0.5 mcg/mL are generally required for therapeutic response. An increase in the frequency of central nervous system adverse effects has been observed when plasma levels exceed 2 mcg/mL. Thus the therapeutic range is approximately 0.5 to 2 mcg/mL. Plasma levels within the therapeutic range can be attained with either three times daily or twice daily dosing but peak to trough differences are greater with the latter regimen, creating the possibility of adverse effects at peak and arrhythmic escape at trough. Nevertheless, some patients may be transferred successfully to the twice daily regimen. (See DOSAGE AND ADMINISTRATION.)
-
INDICATIONS AND USAGE
Mexiletine hydrochloride capsules USP are indicated for the treatment of documented ventricular arrhythmias, such as sustained ventricular tachycardia, that, in the judgment of the physician, are life-threatening. Because of the proarrhythmic effects of mexiletine, its use with lesser arrhythmias is generally not recommended. Treatment of patients with asymptomatic ventricular premature contractions should be avoided.
Initiation of mexiletine treatment, as with other antiarrhythmic agents used to treat life-threatening arrhythmias, should be carried out in the hospital.
Antiarrhythmic drugs have not been shown to enhance survival in patients with ventricular arrhythmias.
- CONTRAINDICATIONS
-
BOXED WARNING
(What is this?)
BOXED WARNING
WARNINGS
Mortality
In the National Heart, Lung and Blood Institute’s Cardiac Arrhythmia Suppression Trial (CAST), a long-term, multicentered, randomized, double-blind study in patients with asymptomatic non-life-threatening ventricular arrhythmias who had a myocardial infarction more than six days but less than two years previously, an excessive mortality or non-fatal cardiac arrest rate (7.7%) was seen in patients treated with encainide or flecainide compared with that seen in patients assigned to carefully matched placebo-treated groups (3.0%). The average duration of treatment with encainide or flecainide in this study was ten months.
The applicability of the CAST results to other populations (e.g., those without recent myocardial infarction) is uncertain. Considering the known proarrhythmic properties of mexiletine and the lack of evidence of improved survival for any antiarrhythmic drug in patients without life-threatening arrhythmias, the use of mexiletine as well as other antiarrhythmic agents should be reserved for patients with life-threatening ventricular arrhythmia.
Acute Liver Injury
In postmarketing experience abnormal liver function tests have been reported, some in the first few weeks of therapy with mexiletine hydrochloride. Most of these have been observed in the setting of congestive heart failure or ischemia and their relationship to mexiletine hydrochloride has not been established.
Drug Reactions with Eosinophilia and Systemic Symptoms (DRESS)
Drug reactions with eosinophilia and systemic symptoms (DRESS) have been reported in patients taking mexiletine. DRESS typically presents with eosinophilia, fever, rash, and/or lymphadenopathy in association with other organ involvement, such as hepatitis, nephritis, hematologic abnormalities, myocarditis, or myositis, sometimes resembling an acute viral infection. Discontinue mexiletine if DRESS is suspected.
-
PRECAUTIONS
General
If a ventricular pacemaker is operative, patients with second or third degree heart block may be treated with mexiletine hydrochloride if continuously monitored. A limited number of patients (45 of 475 in controlled clinical trials) with preexisting first degree AV block were treated with mexiletine; none of these patients developed second or third degree AV block. Caution should be exercised when it is used in such patients or in patients with preexisting sinus node dysfunction or intraventricular conduction abnormalities.
Like other antiarrhythmics mexiletine hydrochloride can cause worsening of arrhythmias. This has been uncommon in patients with less serious arrhythmias (frequent premature beats or nonsustained ventricular tachycardia: see ADVERSE REACTIONS), but is of greater concern in patients with life-threatening arrhythmias such as sustained ventricular tachycardia. In patients with such arrhythmias subjected to programmed electrical stimulation or to exercise provocation, 10 to 15% of patients had exacerbation of the arrhythmia, a rate not greater than that of other agents.
Mexiletine should be used with caution in patients with hypotension and severe congestive heart failure because of the potential for aggravating these conditions.
Since mexiletine is metabolized in the liver, and hepatic impairment has been reported to prolong the elimination half-life of mexiletine, patients with liver disease should be followed carefully while receiving mexiletine. The same caution should be observed in patients with hepatic dysfunction secondary to congestive heart failure.
Concurrent drug therapy or dietary regimens which may markedly alter urinary pH should be avoided during mexiletine hydrochloride therapy. The minor fluctuations in urinary pH associated with normal diet do not affect the excretion of mexiletine.
SGOT Elevation and Liver Injury
In three month controlled trials, elevations of SGOT greater than three times the upper limit of normal occurred in about 1% of both mexiletine-treated and control patients. Approximately 2% of patients in the mexiletine compassionate use program had elevations of SGOT greater than or equal to three times the upper limit of normal. These elevations frequently occurred in association with identifiable clinical events and therapeutic measures such as congestive heart failure, acute myocardial infarction, blood transfusions and other medications. These elevations were often asymptomatic and transient, usually not associated with elevated bilirubin levels and usually did not require discontinuation of therapy. Marked elevations of SGOT (> 1000 U/L) were seen before death in four patients with end-stage cardiac disease (severe congestive heart failure, cardiogenic shock).
Rare instances of severe liver injury, including hepatic necrosis, have been reported in association with mexiletine treatment. It is recommended that patients in whom an abnormal liver test has occurred, or who have signs of symptoms suggesting liver dysfunction, be carefully evaluated. If persistent or worsening elevation of hepatic enzymes is detected, consideration should be given to discontinuing therapy.
Blood Dyscrasias
Among 10,867 patients treated with mexiletine in the compassionate use program, marked leukopenia (neutrophils less than 1000/mm 3) or agranulocytosis were seen in 0.06% and milder depressions of leukocytes were seen in 0.08%, and thrombocytopenia was observed in 0.16%. Many of these patients were seriously ill and receiving concomitant medications with known hematologic adverse effects. Rechallenge with mexiletine in several cases was negative. Marked leukopenia or agranulocytosis did not occur in any patient receiving mexiletine alone; five of the six cases of agranulocytosis were associated with procainamide (sustained release preparations in four) and one with vinblastine. If significant hematologic changes are observed, the patient should be carefully evaluated, and, if warranted, mexiletine should be discontinued. Blood counts usually return to normal within a month of discontinuation (see ADVERSE REACTIONS).
Convulsions (seizures) did not occur in mexiletine controlled clinical trials. In the compassionate use program, convulsions were reported in about 2 of 1000 patients. Twenty-eight percent of these patients discontinued therapy. Convulsions were reported in patients with and without a prior history of seizures. Mexiletine should be used with caution in patients with known seizure disorder.
Drug Interactions
Since mexiletine hydrochloride is a substrate for the metabolic pathways involving CYP2D6 and CYP1A2 enzymes, inhibition or induction of either of these enzymes would be expected to alter mexiletine plasma concentrations. In a formal, single-dose interaction study (n = 6 males) the clearance of mexiletine was decreased by 38% following the coadministration of fluvoxamine, an inhibitor of CYP1A2. In another formal study (n = 8 extensive and n = 7 poor metabolizers of CYP2D6), coadministration of propafenone did not alter the kinetics of mexiletine in the poor CYP2D6 metabolizer group. However, the metabolic clearance of mexiletine in the extensive metabolizer phenotype decreased by about 70% making the poor and extensive metabolizer groups indistinguishable. In this crossover steady state study, the pharmacokinetics of propafenone were unaffected in either phenotype by the coadministration of mexiletine. Addition of mexiletine to propafenone did not lead to further electrocardiographic parameters changes of QRS, QT C, RR, and PR intervals than propafenone alone. When concomitant administration of either of these two drugs is initiated, the dose of mexiletine should be slowly titrated to desired effect.
In a large compassionate use program mexiletine has been used concurrently with commonly employed antianginal, antihypertensive, and anticoagulant drugs without observed interactions. A variety of antiarrhythmics such as quinidine or propranolol were also added, sometimes with improved control of ventricular ectopy. When phenytoin or other hepatic enzyme inducers such as rifampin and phenobarbital have been taken concurrently with mexiletine, lowered mexiletine plasma levels have been reported. Monitoring of mexiletine plasma levels is recommended during such concurrent use to avoid ineffective therapy.
In a formal study, benzodiazepines were shown not to affect mexiletine plasma concentrations. ECG intervals (PR, QRS, and QT) were not affected by concurrent mexiletine and digoxin, diuretics, or propranolol.
Concurrent administration of cimetidine and mexiletine has been reported to increase, decrease, or leave unchanged mexiletine plasma levels; therefore patients should be followed carefully during concurrent therapy.
Mexiletine does not alter serum digoxin levels but magnesium-aluminum hydroxide, when used to treat gastrointestinal symptoms due to mexiletine, has been reported to lower serum digoxin levels.
Concurrent use of mexiletine and theophylline may lead to increased plasma theophylline levels. One controlled study in eight normal subjects showed a 72% mean increase (range
35 to 136%) in plasma theophylline levels. This increase was observed at the first test point which was the second day after starting mexiletine. Theophylline plasma levels returned to pre-mexiletine values within 48 hours after discontinuing mexiletine. If mexiletine and theophylline are to be used concurrently, theophylline blood levels should be monitored, particularly when the mexiletine dose is changed. An appropriate adjustment in theophylline dose should be considered.
Additionally, in one controlled study in five normal subjects and seven patients, the clearance of caffeine was decreased 50% following the administration of mexiletine.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Studies of carcinogenesis in rats (24 months) and mice (18 months) did not demonstrate any tumorigenic potential. Mexiletine was found to be non-mutagenic in the Ames test. Mexiletine did not impair fertility in the rat.
Pregnancy
Teratogenic Effects
Pregnancy category C
Reproduction studies performed with mexiletine in rats, mice and rabbits at doses up to four times the maximum human oral dose (24 mg/kg in a 50 kg patient) revealed no evidence of teratogenicity or impaired fertility but did show an increase in fetal resorption. There are no adequate and well-controlled studies in pregnant women; this drug should be used in pregnancy only if the potential benefit justifies the potential risk to the fetus.
-
ADVERSE REACTIONS
Mexiletine hydrochloride commonly produces reversible gastrointestinal and nervous system adverse reactions but is otherwise well tolerated. Mexiletine has been evaluated in 483 patients in one month and three month controlled studies and in over 10,000 patients in a large compassionate use program. Dosages in the controlled studies ranged from 600 to 1200 mg/day; some patients (8%) in the compassionate use program were treated with higher daily doses (1600 to 3200 mg/day). In the three month controlled trials comparing mexiletine to quinidine, procainamide and disopyramide, the most frequent adverse reactions were upper gastrointestinal distress (41%), lightheadedness (10.5%), tremor (12.6%) and coordination difficulties (10.2%). Similar frequency and incidence were observed in the one month placebo-controlled trial. Although these reactions were generally not serious, and were dose-related and reversible with a reduction in dosage, by taking the drug with food or antacid or by therapy discontinuation, they led to therapy discontinuation in 40% of patients in the controlled trials. Table 1 presents the adverse events reported in the one-month placebo-controlled trial.
Table 1: Comparative Incidence (%) of Adverse Events Among Patients Treated with Mexiletine and Placebo in the 4 Week, Double-Blind Crossover Trial
Mexiletine
N = 53Placebo
N = 49Cardiovascular Palpitations 7.5 10.2 Chest Pain 7.5 4.1 Increased Ventricular Arrythmia/PVCs 1.9 - Digestive Nausea/Vomiting/Heartburn 39.6 6.1 Central Nervous System Dizziness/Lightheadedness 26.4 14.3 Tremor 13.2 - Nervousness 11.3 6.1 Coordination Difficulties 9.4 - Changes in Sleep Habits 7.5 16.3 Paresthesias/Numbness 3.8 2 Weakness 1.9 4.1 Fatigue 1.9 2 Tinnitus 1.9 4.1 Confusion/Clouded Sensorium 1.9 2 Other Headache 7.5 6.1 Blurred Vision/Visual Disturbances 7.5 2 Dyspnea/Respiratory 5.7 10.2 Rash 3.8 2 Non-specific Edema 3.8 - Table 2 presents the adverse reactions occurring in one percent or more of patients in the three month controlled studies.
Table 2: Comparative Incidence (%) of Adverse Events Among Patients Treated with Mexiletine or Control Drugs in the 12 Week Double-Blind Trials
Mexiletine
N = 430Quinidine
N = 262Procainamide
N = 78Disopyramide
N = 69Cardiovascular Palpitations 4.3 4.6 1.3 5.8 Chest Pain 2.6 3.4 1.3 2.9 Angina/Angina-like Pain 1.7 1.9 2.6 2.9 Increased Ventricular Arrhythmias/PVCs 1 2.7 2.6 - Digestive Nausea/Vomiting/ Heartburn 39.3 21.4 33.3 14.5 Diarrhea 5.2 33.2 2.6 8.7 Constipation 4 - 6.4 11.6 Changes in Appetite 2.6 1.9 - - Abdominal Pain/Cramps/ Discomfort 1.2 1.5 - 1.4 Central Nervous System Dizziness/Lightheadedness 18.9 14.1 14.1 2.9 Tremor 13.2 2.3 3.8 1.4 Coordination Difficulties 9.7 1.1 1.3 - Changes in Sleep Habits 7.1 2.7 11.5 8.7 Weakness 5 5.3 7.7 2.9 Nervousness 5 1.9 6.4 5.8 Fatigue 3.8 5.7 5.1 1.4 Speech Difficulties 2.6 0.4 - - Confusion/Clouded Sensorium 2.6 - 3.8 - Paresthesias/Numbness 2.4 2.3 2.6 - Tinnitus 2.4 1.5 - - Depression 2.4 1.1 1.3 1.4 Other Blurred Vision/Visual Disturbances 5.7 3.1 5.1 7.2 Headache 5.7 6.9 7.7 4.3 Rash 4.2 3.8 10.3 1.4 Dyspnea/Respiratory 3.3 3.1 5.1 2.9 Dry Mouth 2.8 1.9 5.1 14.5 Arthralgia 1.7 2.3 5.1 1.4 Fever 1.2 3.1 2.6 - Less than 1%: Syncope, edema, hot flashes, hypertension, short-term memory loss, loss of consciousness, other psychological changes, diaphoresis, urinary hesitancy/retention, malaise, impotence/decreased libido, pharyngitis, congestive heart failure.
An additional group of over 10,000 patients has been treated in a program allowing administration of mexiletine hydrochloride under compassionate use circumstances. These patients were seriously ill with the large majority on multiple drug therapy. Twenty-four percent of the patients continued in the program for one year or longer. Adverse reactions leading to therapy discontinuation occurred in 15 percent of patients (usually upper gastrointestinal system or nervous system effects). In general, the more common adverse reactions were similar to those in the controlled trials. Less common adverse events possibly related to mexiletine use include:
Cardiovascular System
Syncope and hypotension, each about 6 in 1000; bradycardia, about 4 in 1000; angina/angina-like pain, about 3 in 1000; edema, atrioventricular block/conduction disturbances and hot flashes, each about 2 in 1000; atrial arrhythmias, hypertension and cardiogenic shock, each about 1 in 1000.
Central Nervous System
Short-term memory loss, about 9 in 1000 patients; hallucinations and other psychological changes, each about 3 in 1000; psychosis and convulsions/seizures, each about 2 in 1000; loss of consciousness, about 6 in 10,000.
Digestive
Dysphagia, about 2 in 1000; peptic ulcer, about 8 in 10,000; upper gastrointestinal bleeding, about 7 in 10,000; esophageal ulceration, about 1 in 10,000. Rare cases of severe hepatitis/acute hepatic necrosis.
Skin
Rare cases of exfoliative dermatitis and Stevens-Johnson syndrome with mexiletine treatment have been reported.
Laboratory
Abnormal liver function tests, about 5 in 1000; positive ANA and thrombocytopenia, each about 2 in 1000; leukopenia (including neutropenia and agranulocytosis), about 1 in 1000; myelofibrosis, about 2 in 10,000 patients.
Other
Diaphoresis, about 6 in 1000; altered taste, about 5 in 1000; salivary changes, hair loss and impotence/decreased libido, each about 4 in 1000; malaise, about 3 in 1000; urinary hesitancy/retention, each about 2 in 1000; hiccups, dry skin, laryngeal and pharyngeal changes and changes in oral mucous membranes, each about 1 in 1000; SLE syndrome, about 4 in 10,000.
Hematology
Blood dyscrasias were not seen in the controlled trials but did occur among 10,867 patients treated with mexiletine in the compassionate use program (see PRECAUTIONS).
Myelofibrosis was reported in two patients in the compassionate use program; one was receiving long-term thiotepa therapy and the other had pretreatment myeloid abnormalities.
In postmarketing experience, there have been isolated, spontaneous reports of pulmonary changes including pulmonary infiltration and pulmonary fibrosis during mexiletine therapy with or without other drugs or diseases that are known to produce pulmonary toxicity. A causal relationship to mexiletine therapy has not been established. In addition, there have been isolated reports of drowsiness, nystagmus, ataxia, dyspepsia, hypersensitivity reaction, and exacerbation of congestive heart failure in patients with preexisting compromised ventricular function. There have been rare reports of pancreatitis associated with mexiletine treatment.
-
OVERDOSAGE
Clinical findings associated with mexiletine overdosage have included drowsiness, confusion, nausea, hypotension, sinus bradycardia, paresthesia, seizures, bundle branch block, AV heart block, asystole, ventricular tachyarrythmia, including ventricular fibrillation, cardiovascular collapse and coma. The lowest known dose in a fatality case was 4.4 g with postmortem serum mexiletine level of 34 to 37 mcg/mL (Jequier P. et. al., Lancet 1976: 1 (7956): 429). Patients have recovered from ingestion of 4 g to 18 g of mexiletine (Frank S. E. et. al., Am J Emerg Med 1991: 9:43-48).
There is no specific antidote for mexiletine. Management of mexiletine overdosage includes general supportive measures, close observation and monitoring of vital signs. In addition, the use of pharmacologic interventions (e.g., pressor agents, atropine or anticonvulsants) or transvenous cardiac pacing is suggested, depending on the patient’s clinical condition.
-
DOSAGE AND ADMINISTRATION
The dosage of mexiletine hydrochloride must be individualized on the basis of response and tolerance, both of which are dose-related. Administration with food or antacid is recommended. Initiate mexiletine therapy with 200 mg every eight hours when rapid control of arrhythmia is not essential. A minimum of two to three days between dose adjustments is recommended. Dose may be adjusted in 50 or 100 mg increments up or down.
As with any antiarrhythmic drug, clinical and electrocardiographic evaluation (including Holter monitoring if necessary for evaluation) are needed to determine whether the desired antiarrhythmic effect has been obtained and to guide titration and dose adjustment.
Satisfactory control can be achieved in most patients by 200 to 300 mg given every eight hours with food or antacid. If satisfactory response has not been achieved at 300 mg q8h, and the patient tolerates mexiletine well, a dose of 400 mg q8h may be tried. As the severity of CNS side effects increases with total daily dose, the dose should not exceed 1200 mg/day.
In general, patients with renal failure will require the usual doses of mexiletine hydrochloride. Patients with severe liver disease, however, may require lower doses and must be monitored closely. Similarly, marked right-sided congestive heart failure can reduce hepatic metabolism and reduce the needed dose. Plasma level may also be affected by certain concomitant drugs (see PRECAUTIONS, Drug Interactions).
Loading Dose
When rapid control of ventricular arrhythmia is essential, an initial loading dose of 400 mg of mexiletine hydrochloride may be administered, followed by a 200 mg dose in eight hours. Onset of therapeutic effect is usually observed within 30 minutes to two hours.
Q12H Dosage Schedule
Some patients responding to mexiletine may be transferred to a 12 hour dosage schedule to improve convenience and compliance. If adequate suppression is achieved on a mexiletine hydrochloride dose of 300 mg or less every eight hours, the same total daily dose may be given in divided doses every 12 hours while carefully monitoring the degree of suppression of ventricular ectopy. This dose may be adjusted up to a maximum of 450 mg every 12 hours to achieve the desired response.
Transferring to Mexiletine Hydrochloride
The following dosage schedule, based on theoretical considerations rather than experimental data, is suggested for transferring patients from other Class I oral antiarrhythmic agents to mexiletine: mexiletine hydrochloride treatment may be initiated with a 200 mg dose, and titrated to response as described above, 6 to 12 hours after the last dose of quinidine sulfate, 3 to 6 hours after the last dose of procainamide, 6 to 12 hours after the last dose of disopryramide or 8 to 12 hours after the last dose of tocainide.
In patients in whom withdrawal of the previous antiarrhythmic agent is likely to produce life-threatening arrhythmias, hospitalization of the patient is recommended.
When transferring from lidocaine to mexiletine, the lidocaine infusion should be stopped when the first oral dose of mexiletine hydrochloride is administered. The infusion line should be left open until suppression of the arrhythmia appears to be satisfactorily maintained. Consideration should be given to the similarity of the adverse effects of lidocaine and mexiletine and the possibility that they may be additive.
-
HOW SUPPLIED
Mexiletine hydrochloride capsules USP, 150 mg are white granular powder in a hard gelatin capsule with an opaque tan cap and an opaque orange body, imprinted with "N" on one side and "739" and "150" on the other in black ink. They are supplied as follows: NDC 42291-624-01 bottles of 100.
Mexiletine hydrochloride capsules USP, 200 mg are white granular powder in a hard gelatin capsule with an opaque orange cap and an opaque orange body, imprinted with "N" one one side and "740" and "200" on the other in black ink. They are supplied as follows: NDC 42291-625-01 bottles of 100.
Mexiletine hydrochloride capsules USP, 250 mg are white granular powder in a hard gelatin capsule with an opaque green cap and an opaque orange body, imprinted with "N" on one side and "741" and "250" on the other in black ink. They are supplied as follows: NDC 42291-626-01 bottles of 100.
Store at 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature].
Dispense in a tight, light-resistant container as defined in the USP, with a child-resistant closure (as required).
Manufactured For:
AvKARE, Inc.
Pulaski, TN 38478
Mfg. Rev. 03/15
AV Rev. 08/18 (P) - Label - 150mg
- Label - 200mg
- Label - 250mg
-
INGREDIENTS AND APPEARANCE
MEXILETINE HYDROCHLORIDE
mexiletine hydrochloride capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 42291-624(NDC:0093-8739) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength MEXILETINE HYDROCHLORIDE (UNII: 606D60IS38) (MEXILETINE - UNII:1U511HHV4Z) MEXILETINE HYDROCHLORIDE 150 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) STARCH, CORN (UNII: O8232NY3SJ) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) GELATIN (UNII: 2G86QN327L) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) D&C RED NO. 28 (UNII: 767IP0Y5NH) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) AMMONIA (UNII: 5138Q19F1X) FERROSOFERRIC OXIDE (UNII: XM0M87F357) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) ALCOHOL (UNII: 3K9958V90M) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) FD&C RED NO. 40 (UNII: WZB9127XOA) ISOPROPYL ALCOHOL (UNII: ND2M416302) BUTYL ALCOHOL (UNII: 8PJ61P6TS3) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SHELLAC (UNII: 46N107B71O) Product Characteristics Color orange, brown (Tan) Score no score Shape CAPSULE Size 18mm Flavor Imprint Code N;739;150 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 42291-624-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 07/09/2013 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA074377 07/09/2013 MEXILETINE HYDROCHLORIDE
mexiletine hydrochloride capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 42291-625(NDC:0093-8740) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength MEXILETINE HYDROCHLORIDE (UNII: 606D60IS38) (MEXILETINE - UNII:1U511HHV4Z) MEXILETINE HYDROCHLORIDE 200 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) STARCH, CORN (UNII: O8232NY3SJ) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) GELATIN (UNII: 2G86QN327L) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) AMMONIA (UNII: 5138Q19F1X) FERROSOFERRIC OXIDE (UNII: XM0M87F357) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) ALCOHOL (UNII: 3K9958V90M) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) FD&C RED NO. 40 (UNII: WZB9127XOA) ISOPROPYL ALCOHOL (UNII: ND2M416302) BUTYL ALCOHOL (UNII: 8PJ61P6TS3) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SHELLAC (UNII: 46N107B71O) Product Characteristics Color orange Score no score Shape CAPSULE Size 19mm Flavor Imprint Code N;740;200 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 42291-625-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 07/09/2013 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA074377 07/09/2013 MEXILETINE HYDROCHLORIDE
mexiletine hydrochloride capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 42291-626(NDC:0093-8741) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength MEXILETINE HYDROCHLORIDE (UNII: 606D60IS38) (MEXILETINE - UNII:1U511HHV4Z) MEXILETINE HYDROCHLORIDE 250 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) STARCH, CORN (UNII: O8232NY3SJ) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) GELATIN (UNII: 2G86QN327L) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) AMMONIA (UNII: 5138Q19F1X) FERROSOFERRIC OXIDE (UNII: XM0M87F357) ALCOHOL (UNII: 3K9958V90M) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) FD&C RED NO. 40 (UNII: WZB9127XOA) ISOPROPYL ALCOHOL (UNII: ND2M416302) BUTYL ALCOHOL (UNII: 8PJ61P6TS3) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SHELLAC (UNII: 46N107B71O) Product Characteristics Color orange, green Score no score Shape CAPSULE Size 22mm Flavor Imprint Code N;741;250 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 42291-626-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 07/09/2013 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA074377 07/09/2013 Labeler - AvKARE, Inc. (796560394)



© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.