VIVIMUSTA- bendamustine hydrochloride injection
VIVIMUSTA by
Drug Labeling and Warnings
VIVIMUSTA by is a Prescription medication manufactured, distributed, or labeled by Slayback Pharma LLC, Corden Pharma Latina S.p.A.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VIVIMUSTA safely and effectively. See full prescribing information for VIVIMUSTA.
VIVIMUSTA® (bendamustine hydrochloride injection), for intravenous use.
Initial U.S. Approval: 2008
INDICATIONS AND USAGE
VIVIMUSTA is an alkylating drug indicated for treatment of patients with:
Chronic lymphocytic leukemia (CLL). Efficacy relative to first line therapies other than chlorambucil has not been established. (1.1)
Indolent B-cell non-Hodgkin lymphoma (NHL) that has progressed during or within six months of treatment with rituximab or a rituximab-containing regimen. (1.2)DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Injection: 100 mg/4 mL (25 mg/mL) in a multiple-dose vial (3).
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
Myelosuppression: Delay or reduce dose, and restart treatment based on ANC and platelet count recovery. (5.1)
Infections: Monitor for fever and other signs of infection or reactivation of infections and treat promptly. (5.2)Progressive multifocal leukoencephalopathy (PML): Monitor for new or worsening neurological, cognitive or behavioral signs or symptoms suggestive of PML. (5.3)
Anaphylaxis and Infusion-Related Reactions: Severe anaphylactic reactions have occurred. Monitor clinically and discontinue drug for severe reactions. Pre-medicate in subsequent cycles for milder reactions. (5.4)
Tumor Lysis Syndrome: May lead to acute renal failure and death; anticipate and use supportive measures in patients at high risk. (5.5)
Skin Reactions: Discontinue for severe skin reactions. Cases of SJS, DRESS and TEN, some fatal, have been reported. (5.6).
Hepatotoxicity: Monitor liver chemistry tests prior to and during treatment. (5.7)
Other Malignancies: Pre-malignant and malignant diseases have been reported. (5.8)
Extravasation Injury: Take precautions to avoid extravasation, including monitoring intravenous infusion site during and after administration. (5.9)
Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential and males with female partners of reproductive potential of the potential risk to a fetus and to use an effective method of contraception. (5.10,8.1, 8.3)ADVERSE REACTIONS
Adverse reactions (> 5%) during infusion and within 24 hours post-infusion are nausea, and fatigue. (6.1)
Most common adverse reactions (≥15%) for CLL are anemia, thrombocytopenia, neutropenia, lymphopenia, leukopenia, hyperbilirubinemia, pyrexia, nausea, vomiting. (6.1)
Most common adverse reactions (≥15%) for NHL are lymphopenia, leukopenia, anemia neutropenia, thrombocytopenia, nausea, fatigue, vomiting, diarrhea, pyrexia, constipation, anorexia, cough, headache, weight decreased dyspnea, rash, and stomatitis. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Slayback Pharma at 1-844-566-2505 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatchDRUG INTERACTIONS
Consider alternative therapies that are not CYP1A2 inducers or inhibitors during treatment with VIVIMUSTA. (7.1)
USE IN SPECIFIC POPULATIONS
Lactation: Advise not to breastfeed. (8.2)
Infertility: May impair fertility. (8.3)
Renal Impairment: Do not use in patients with creatinine clearance < 30 mL/min. (8.6)
Hepatic Impairment: Do not use in patients with total bilirubin 1.5-3 x ULN and AST or ALT 2.5-1.0 x ULN, or total bilirubin > 3 x ULN. (8.7)See 17 for PATIENT COUNSELING INFORMATION.
Revised: 2/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS & USAGE
1.1 Chronic Lymphocytic Leukemia (CLL)
1.2 Non-Hodgkin Lymphoma (NHL)
2 DOSAGE & ADMINISTRATION
2.1 Dosing Instructions for CLL
2.2 Dosing Instructions for NHL
2.3 Preparation and Administration
2.4 Admixture Stability
2.5 Stability of Partially Used Vials (Needle Punched Vials)
3 DOSAGE FORMS & STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Myelosuppression
5.2 Infections
5.3 Progressive Multifocal Leukoencephalopathy (PML)
5.4 Anaphylaxis and Infusion-Related Reactions
5.5 Tumor Lysis Syndrome
5.6 Skin Reactions
5.7 Hepatotoxicity
5.8 Other Malignancies
5.9 Extravasation Injury
5.10 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on VIVIMUSTA
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis & Mutagenesis & Impairment Of Fertility
14 CLINICAL STUDIES
14.1 Chronic Lymphocytic Leukemia (CLL)
14.2 Non-Hodgkin Lymphoma (NHL)
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS & USAGE
-
2 DOSAGE & ADMINISTRATION
2.1 Dosing Instructions for CLL
Recommended Dosage
The recommended dosage is 100 mg/m2 administered intravenously over 20 minutes on Days 1 and 2 of a 28-day cycle for up to 6 cycles.
Dose Delays, Dose Modifications and Re-initiation of Therapy for CLL
Delay VIVIMUSTA for Grade 4 hematologic toxicity or clinically significant Grade 2 or greater non-hematologic toxicity.
Once non-hematologic toxicity has recovered to less than or equal to Grade 1 and/or the blood counts have improved [Absolute Neutrophil Count (ANC) greater than or equal to 1 x 109/L and platelets greater than or equal to 75 x 109/L], reinitiate VIVIMUSTA at the discretion of the healthcare provider. In addition, consider dose reduction [see Warnings and Precautions (5.1)]
Dose modifications for hematologic toxicity: for Grade 3 or greater toxicity, reduce the dose to 50 mg/m2 on Days 1 and 2 of each cycle; if Grade 3 or greater toxicity recurs, reduce the dose to 25 mg/m2 on Days 1 and 2 of each cycle.
Dose modifications for non-hematologic toxicity: for clinically significant Grade 3 or greater toxicity, reduce the dose to 50 mg/m2 on Days 1 and 2 of each cycle.
Consider dose re-escalation in subsequent cycles at the discretion of the healthcare provider.2.2 Dosing Instructions for NHL
Recommended Dosage
The recommended dosage is 120 mg/m2 administered intravenously over 20 minutes on Days 1 and 2 of a 21-day cycle for up to 8 cycles.Dose Delays, Dose Modifications and Re-initiation of Therapy for NHL
Delay VIVIMUSTA for Grade 4 hematologic toxicity or clinically significant Grade 2 or greater non-hematologic toxicity.
Once non-hematologic toxicity has recovered to less than or equal to Grade 1 and/or the blood counts have improved [Absolute Neutrophil Count (ANC) greater than or equal to 1 x 109/L and platelets greater than or equal to 75 x 109/L], reinitiate VIVIMUSTA at the discretion of the healthcare provider. In addition, consider dose reduction [see Warnings and Precautions (5.1)]
Dose modifications for hematologic toxicity: for Grade 4 toxicity, reduce the dose to 90 mg/m2 on Days 1 and 2 of each cycle; if Grade 4 toxicity recurs, reduce the dose to 60 mg/m2 on Days 1 and 2 of each cycle.
Dose modifications for non-hematologic toxicity: for Grade 3 or greater toxicity, reduce the dose to 90 mg/m2 on Days 1 and 2 of each cycle; if Grade 3 or greater toxicity recurs, reduce the dose to 60 mg/m2 on Days 1 and 2 of each cycle.2.3 Preparation and Administration
VIVIMUSTA is a hazardous drug. Follow applicable special handling and disposal procedures.1
VIVIMUSTA is a clear and colorless to yellow solution in a multiple-dose vial.
Store VIVIMUSTA refrigerated at 2°C to 8°C (36°F to 46°F). When refrigerated, the contents may partially freeze. Allow the vial to reach room temperature (15°C to 30°C or 59°F to 86°F) prior to use. Observe the contents of the vial for any visible solid or particulate matter. Do not use the product if solid or particulate matter is observed after reaching room temperature.
Intravenous Infusion
Aseptically withdraw the volume needed for the required dose from the 25 mg/mL solution as per Table 1 below and immediately transfer to a 250 mL infusion bag of one of the following diluents: 0.9% Sodium Chloride Injection, USP; or 2.5% Dextrose/0.45% Sodium Chloride Injection, USP.
The resulting final concentration of bendamustine hydrochloride in the infusion bag should be within 0.1 mg/mL to 1.36 mg/mL.
After transferring, thoroughly mix the contents of the infusion bag. The admixture should be a clear and colorless to slightly yellow solution.No other diluents have been shown to be compatible [see Dosage and Administration (2.4)].
Table 1: Volume of VIVIMUSTA required for Dilution into 250 mL of 0.9% Sodium Chloride Injection, USP, or 2.5% Dextrose/0.45% Sodium Chloride Injection, USP for a Given Dose and Body Surface Area
Body Surface Area (m2)
Volume of VIVIMUSTA to Withdraw (mL) from Vial
120 mg/m2
100 mg/m2
90 mg/m2
60 mg/m2
50 mg/m2
25 mg/m2
1
4.8
4
3.6
2.4
2
1
1.1
5.3
4.4
4
2.6
2.2
1.1
1.2
5.8
4.8
4.3
2.9
2.4
1.2
1.3
6.2
5.2
4.7
3.1
2.6
1.3
1.4
6.7
5.6
5
3.4
2.8
1.4
1.5
7.2
6
5.4
3.6
3
1.5
1.6
7.7
6.4
5.8
3.8
3.2
1.6
1.7
8.2
6.8
6.1
4.1
3.4
1.7
1.8
8.6
7.2
6.5
4.3
3.6
1.8
1.9
9.1
7.6
6.8
4.6
3.8
1.9
2
9.6
8
7.2
4.8
4
2
2.1
10.1
8.4
7.6
5
4.2
2.1
2.2
10.6
8.8
7.9
5.3
4.4
2.2
2.3
11
9.2
8.3
5.5
4.6
2.3
2.4
11.5
9.6
8.6
5.8
4.8
2.4
2.5
12
10
9
6
5
2.5
2.6
12.5
10.4
9.4
6.2
5.2
2.6
2.7
13
10.8
9.7
6.5
5.4
2.7
2.8
13.4
11.2
10.1
6.7
5.6
2.8
2.9
13.9
11.6
10.4
7
5.8
2.9
3
14.4
12
10.8
7.2
6
3
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever solution and container permit. Discard any unused solution according to institutional procedures for hazardous drugs.
2.4 Admixture Stability
VIVIMUSTA contains no antimicrobial preservative. Prepare the admixture as close as possible to the time of patient administration.
Once diluted with 0.9% Sodium Chloride Injection, USP, or 2.5% Dextrose/0.45% Sodium Chloride Injection, USP, the final admixture is stable for 24 hours when stored refrigerated (2°C to 8°C or 36°F to 46°F) or for 3 hours when stored at room temperature (15°C to 30°C or 59°F to 86°F) and room light. Complete administration of diluted VIVIMUSTA within this period of time.
VIVIMUSTA (bendamustine hydrochloride injection) is supplied in a multiple-dose vial. Retain the partially used vial in original package to protect from light and store refrigerated (2°C to 8°C or 36°F to 46°F) if additional dose withdrawal from the same vial is intended.2.5 Stability of Partially Used Vials (Needle Punched Vials)
VIVIMUSTA is supplied as a multiple-dose vial. Although it does not contain any antimicrobial preservative, VIVIMUSTA is bacteriostatic. The partially used vials are stable for up to 28 days when stored in its original carton under refrigeration (2°C to 8°C or 36°F to 46°F). Each vial is not recommended for more than a total of six (6) dose withdrawals.
After first use, store the partially used vial in the original carton at 2°C to 8°C (36°F to 46°F) and then discard after 28 days. - 3 DOSAGE FORMS & STRENGTHS
-
4 CONTRAINDICATIONS
VIVIMUSTA is contraindicated in patients with a known hypersensitivity (e.g., anaphylactic and anaphylactoid reactions) to bendamustine, polyethylene glycol 400, dehydrated alcohol, or monothioglycerol [see Warnings and Precautions (5.4)]
-
5 WARNINGS AND PRECAUTIONS
5.1 Myelosuppression
VIVIMUSTA causes myelosuppression. Bendamustine hydrochloride caused severe myelosuppression (Grade 3-4) in 98% of patients in the two NHL studies [see Adverse Reactions (6.1)]. Three patients (2%) died from myelosuppression-related adverse reactions; one each from neutropenic sepsis, diffuse alveolar hemorrhage with Grade 3 thrombocytopenia and pneumonia from an opportunistic infection (CMV).
Monitor complete blood counts, including leukocytes, platelets, hemoglobin (Hgb), and neutrophils frequently. In the clinical trials, blood counts were monitored every week initially. Hematologic nadirs were observed predominantly in the third week of therapy. Myelosuppression may require dose delays and/or subsequent dose reductions if recovery to the recommended values has not occurred by the first day of the next scheduled cycle. Delay the next cycle of therapy if ANC less than 1 x 109/L or platelet count less than 75 x 109/L [see Dosage and Administration (2.1, 2.2)].5.2 Infections
Infection, including pneumonia, sepsis, septic shock, hepatitis and death has occurred in adult and pediatric patients in clinical trials and in postmarketing reports for bendamustine hydrochloride [see Adverse Reactions (6.1, 6.2)]. Patients with myelosuppression following treatment with bendamustine hydrochloride are more susceptible to infections. Advise patients with myelosuppression following VIVIMUSTA treatment to contact healthcare provider immediately if they have symptoms or signs of infection.
Patients treated with VIVIMUSTA are at risk for reactivation of infections including (but not limited to) hepatitis B, cytomegalovirus, Mycobacterium tuberculosis, and herpes zoster. Implement appropriate measures (including clinical and laboratory monitoring, prophylaxis, and treatment) for infection and infection reactivation prior to administration.5.3 Progressive Multifocal Leukoencephalopathy (PML)
Progressive multifocal leukoencephalopathy (PML), including fatal cases, have occurred following treatment with bendamustine, primarily in combination with rituximab or obinutuzumab [see Adverse Reactions (6.2)]. Consider PML in the differential diagnosis in patients with new or worsening neurological, cognitive or behavioral signs or symptoms. If PML is suspected, withhold VIVIMUSTA treatment and perform appropriate diagnostic evaluations. Consider discontinuation or reduction of any concomitant chemotherapy or immunosuppressive therapy in patients who develop PML.
5.4 Anaphylaxis and Infusion-Related Reactions
Infusion-related reactions to bendamustine hydrochloride have occurred commonly in clinical trials. Symptoms include fever, chills, pruritus and rash. In rare instances, severe anaphylactic and anaphylactoid reactions have occurred, particularly in the second and subsequent cycles of therapy.
Monitor clinically and discontinue drug for severe reactions. Ask patients about symptoms suggestive of infusion-related reactions after their first cycle of therapy. Do not rechallenge patients who experienced Grade 3 or worse allergic-type reactions. Consider measures to prevent severe reactions, including antihistamines, antipyretics and corticosteroids in subsequent cycles in patients who have experienced Grade 1 or 2 infusion-related reactions. Discontinue VIVIMUSTA for patients with Grade 4 infusion-related reactions. Consider discontinuation for Grade 3 infusion-related reactions as clinically appropriate considering individual benefits, risks, and supportive care.5.5 Tumor Lysis Syndrome
Tumor lysis syndrome associated with bendamustine hydrochloride has occurred in patients in clinical trials and in postmarketing reports. The onset tends to be within the first treatment cycle of bendamustine hydrochloride and, without intervention, may lead to acute renal failure and death.
Administer vigorous hydration and monitor blood chemistry, particularly potassium and uric acid levels at baseline and closely during treatment with VIVIMUSTA. Allopurinol has also been used during the beginning of bendamustine hydrochloride therapy. However, there may be an increased risk of severe skin toxicity when bendamustine hydrochloride and allopurinol are administered concomitantly [see Warnings and Precautions (5.6)].5.6 Skin Reactions
Fatal and serious skin reactions have been reported with bendamustine hydrochloride treatment in clinical trials and postmarketing safety reports, including toxic skin reactions [Stevens-Johnson Syndrome (SJS), toxic epidermal necrolysis (TEN), and drug reaction with eosinophilia and systemic symptoms (DRESS)], bullous exanthema, and rash. Events occurred when bendamustine hydrochloride was given as a single agent and in combination with other anticancer agents or allopurinol.
Where skin reactions occur, they may be progressive and increase in severity with further treatment. Monitor patients with skin reactions closely. If skin reactions are severe or progressive, withhold or discontinue VIVIMUSTA.5.7 Hepatotoxicity
Fatal and serious cases of liver injury have been reported with Bendamustine Hydrochloride Injection [see Adverse Reactions (6.1)]. Combination therapy, progressive disease or reactivation of hepatitis B were confounding factors in some patients [see Warnings and Precautions (5.2)]. Most cases were reported within the first three months of starting therapy.
Monitor liver chemistry tests prior to and during treatment with VIVIMUSTA.5.8 Other Malignancies
Pre-malignant and malignant diseases have developed in patients who have been treated with bendamustine hydrochloride, including myelodysplastic syndrome, myeloproliferative disorders, acute myeloid leukemia, bronchial carcinoma, and non-melanoma skin cancer including basal cell carcinoma and squamous cell carcinoma [see Adverse Reactions (6.2)].
Monitor patients for the development of secondary malignancies. Perform dermatologic evaluations during and after treatment with VIVIMUSTA.5.9 Extravasation Injury
Bendamustine hydrochloride extravasations have been reported in postmarketing resulting in hospitalizations from erythema, marked swelling, and pain.
Assure good venous access prior to starting VIVIMUSTA infusion and monitor the intravenous infusion site for redness, swelling, pain, infection, and necrosis during and after administration of VIVIMUSTA.5.10 Embryo-Fetal Toxicity
Based on findings from animal reproduction studies and the drug's mechanism of action, VIVIMUSTA can cause fetal harm when administered to a pregnant woman. Single intraperitoneal doses of bendamustine (that approximated the maximum recommended human dose based on body surface area) to pregnant mice and rats during organogenesis caused adverse developmental outcomes, including an increase in resorptions, skeletal and visceral malformations, and decreased fetal body weights.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use an effective method of contraception during treatment with VIVIMUSTA and for 6 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with VIVIMUSTA and for 3 months after the last dose [see Use in Specific Populations (8.1, 8.3) and Clinical Pharmacology (12.1)]. -
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labelling:
Myelosuppression [see Warnings and Precautions (5.1)]
Infections [see Warnings and Precautions (5.2)]Progressive Multifocal Leukoencephalopathy [see Warnings and Precautions (5.3)]
Anaphylaxis and Infusion-Related Reactions [see Warnings and Precautions (5.4)]
Tumor Lysis Syndrome [see Warnings and Precautions (5.5)]
Skin Reactions [see Warnings and Precautions (5.6)]
Hepatotoxicity [see Warnings and Precautions (5.7)]
Other Malignancies [see Warnings and Precautions (5.8)]
Extravasation Injury [see Warnings and Precautions (5.9)]6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Chronic Lymphocytic Leukemia (CLL)
The data described below reflect exposure to bendamustine hydrochloride in 153 patients. Bendamustine hydrochloride was studied in an active-controlled, randomized trial. The population was 45-77 years of age, 63% were male, 100% were White, and had treatment naïve CLL. All patients started the study at a dose of 100 mg/m2 intravenously over 30 minutes on Days 1 and 2 every 28 days.
Adverse reactions were reported according to NCI CTC v.2.0. In the randomized CLL clinical study, non-hematologic adverse reactions (any grade) in the bendamustine hydrochloride group that occurred with a frequency greater than 15% were pyrexia (24%), nausea (20%), and vomiting (16%).
Other adverse reactions seen frequently in one or more studies included asthenia, fatigue, malaise, and weakness; dry mouth; somnolence; cough; constipation; headache; mucosal inflammation and stomatitis.Worsening hypertension was reported in 4 patients treated with bendamustine hydrochloride and in none treated with chlorambucil. Three of these 4 adverse reactions were described as a hypertensive crisis and were managed with oral medications and resolved.
The most frequent adverse reactions leading to study withdrawal for patients receiving bendamustine hydrochloride were hypersensitivity (2%) and pyrexia (1%).
Table 2 summarizes the adverse reactions that were reported in ≥ 5% of patients in either treatment group in the randomized CLL clinical study.
Table 2: Non-Hematologic Adverse Reactions that Occurred in at Least 5% of Patients Who Received Bendamustine Hydrochloride or Chlorambucil in the Randomized CLL Clinical Study
Adverse Reaction
Bendamustine Hydrochloride
(N=153)
Chlorambucil
(N=143)
All Grades
n (%)
Grade 3 or 4
n (%)
All Grades
n (%)
Grade 3 or 4
n (%)
Total number of patients with at least 1 adverse reaction
121 (79)
52 (34)
96 (67)
25 (17)
Gastrointestinal disorders
Nausea
31 (20)
1 (<1)
21 (15)
1 (<1)
Vomiting
24 (16)
1 (<1)
9 (6)
0
Diarrhea
14 (9)
2 (1)
5 (3)
0
General disorders and administration site conditions
Pyrexia
36 (24)
6 (4)
8 (6)
2 (1)
Fatigue
14 (9)
2 (1)
8 (6)
0
Asthenia
13 (8)
0
6 (4)
0
Chills
9 (6)
0
1 (<1)
0
Immune system disorders
Hypersensitivity
7 (5)
2 (1)
3 (2)
0
Infections and infestations
Nasopharyngitis
10 (7)
0
12 (8)
0
Infection
9 (6)
3 (2)
1 (<1)
1 (<1)
Herpes simplex
5 (3)
0
7 (5)
0
Investigations
Weight decreased
11 (7)
0
5 (3)
0
Metabolism and nutrition disorders
Hyperuricemia
11 (7)
3 (2)
2 (1)
0
Respiratory, thoracic and mediastinal disorders
Cough
6 (4)
1 (<1)
7 (5)
1 (<1)
Skin and subcutaneous tissue disorders
Rash
12 (8)
4 (3)
7 (5)
3 (2)
Pruritus
8 (5)
0
2 (1)
0
Hematology laboratory abnormalities are described in Table 3. Red blood cell transfusions were administered to 20% of patients receiving bendamustine hydrochloride compared with 6% of patients receiving chlorambucil. Bilirubin elevation occurred in 34% of patients, some without associated significant elevations in AST and ALT. Grade 3 or 4 increased bilirubin occurred in 3% of patients. Increases in AST and ALT of Grade 3 or 4 were limited to 1% and 3% of patients, respectively. Patients treated with bendamustine hydrochloride may also have changes in their creatinine levels.
Table 3: Hematology Laboratory Abnormalities in Patients Who Received Bendamustine Hydrochloride or Chlorambucil in the Randomized CLL Clinical Study
Laboratory
Abnormality
Bendamustine Hydrochloride
(N=150)
Chlorambucil
(N=141)
All Grades
n (%)
Grade 3 or 4
n (%)
All Grades
n (%)
Grade 3 or 4
n (%)
Hemoglobin Decreased
134 (89)
20 (13)
115 (82)
12 (9)
Platelets Decreased
116 (77)
16 (11)
110 (78)
14 (10)
Neutrophils Decreased
113 (75)
65 (43)
86 (61)
30 (21)
Lymphocytes Decreased
102 (68)
70 (47)
27 (19)
6 (4)
Leukocytes Decreased
92 (61)
42 (28)
26 (18)
4 (3)
Non-Hodgkin Lymphoma (NHL)
The data described below reflect exposure to bendamustine hydrochloride in 176 patients with indolent B-cell NHL treated in two single-arm studies. The population was 31-84 years of age; 60% were male; 89% were White, 7% were Black, 3% were Hispanic, 1% were other, and <1% were Asian. These patients received bendamustine hydrochloride at a dose of 120 mg/m2 intravenously on Days 1 and 2 for up to eight 21-day cycles.
In both studies, serious adverse reactions, were reported in 37% of patients receiving bendamustine hydrochloride. The most frequent serious adverse reactions occurring in ≥5% of patients were febrile neutropenia and pneumonia. Other important serious adverse reactions reported in clinical trials and/or postmarketing experience were acute renal failure, cardiac failure, hypersensitivity, skin reactions, pulmonary fibrosis, and myelodysplastic syndrome.
Serious adverse reactions reported in clinical trials included myelosuppression, infection, pneumonia, tumor lysis syndrome and infusion-related reactions [see Warningsand Precautions (5)]. Adverse reactions occurring less frequently but possibly related to bendamustine hydrochloride treatment were hemolysis, dysgeusia/taste disorder, atypical pneumonia, sepsis, herpes zoster, erythema, dermatitis, and skin necrosis.The most common non-hematologic adverse reactions (≥30%) were nausea (75%), fatigue (57%), vomiting (40%), diarrhea (37%) and pyrexia (34%). The most common non-hematologic Grade 3 or 4 adverse reactions (≥5%) were fatigue (11%), febrile neutropenia (6%), and pneumonia, hypokalemia and dehydration, each reported in 5% of patients.
Non-hematologic adverse reactions are shown in Table 4.Table 4: Non-Hematologic Adverse Reactions that Occurred in at Least 5% of Patients who Received Bendamustine Hydrochloride in the NHL Studies
Bendamustine Hydrochloride
(N=176*)
Adverse Reaction
All Grades
n(%)
Grade3 or 4
n(%)
Total number of patients with at least 1 adverse reaction
176 (100)
94 (53)
Cardiac Disorders
Tachycardia
13 (7)
0
Gastrointestinal disorders
Nausea
132 (75)
7 (4)
Vomiting
71 (40)
5 (3)
Diarrhea
65 (37)
6 (3)
Constipation
51 (29)
1 (<1)
Stomatitis
27 (15)
1 (<1)
Abdominal pain
22 (13)
2 (1)
Dyspepsia
20 (11)
0
Gastroesophageal reflux disease
18 (10)
0
Dry mouth
15 (9)
1 (<1)
Abdominal pain upper
8 (5)
0
Abdominal distension
8 (5)
0
General disorders and administration site conditions
Fatigue
101 (57)
19 (11)
Pyrexia
59 (34)
3 (2)
Chills
24 (14)
0
Edema peripheral
23 (13)
1 (<1)
Asthenia
19 (11)
4 (2)
Chest pain
11 (6)
1 (<1)
Infusion site pain
11 (6)
0
Pain
10 (6)
0
Catheter site pain
8 (5)
0
Infections and infestations
Herpes zoster
18 (10)
5 (3)
Upper respiratory tract infection
18 (10)
0
Urinary tract infection
17 (10)
4 (2)
Sinusitis
15 (9)
0
Pneumonia
14 (8)
9 (5)
Febrile neutropenia
11 (6)
11 (6)
Oral candidiasis
11 (6)
2 (1)
Nasopharyngitis
11 (6)
0
Investigations
Weight decreased
31 (18)
3 (2)
Metabolism and nutrition disorders
Anorexia
40 (23)
3 (2)
Dehydration
24 (14)
8 (5)
Decreased appetite
22 (13)
1 (<1)
Hypokalemia
15 (9)
9 (5)
Musculoskeletal and connective tissue disorders
Back pain
25 (14)
5 (3)
Arthralgia
11 (6)
0
Pain in extremity
8 (5)
2 (1)
Bone pain
8 (5)
0
Nervous system disorders
Headache
36 (21)
0
Dizziness
25 (14)
0
Dysgeusia
13 (7)
0
Psychiatric disorder
Insomnia
23 (13)
0
Anxiety
14 (8)
1 (<1)
Depression
10 (6)
0
Respiratory, thoracic and mediastinal disorders
Cough
38 (22)
1 (<1)
Dyspnea
28 (16)
3 (2)
Pharyngolaryngeal pain
14 (8)
1 (<1)
Wheezing
8 (5)
0
Nasal congestion
8 (5)
0
Skin and subcutaneous tissue disorders
Rash
28 (16)
1 (<1)
Pruritus
11 (6)
0
Dry skin
9 (5)
0
Night sweats
9 (5)
0
Hyperhidrosis
8 (5)
0
Vascular disorders
Hypotension
10 (6)
2 (1)
*Patients may have reported more than 1 adverse reaction.
NOTE: Patients counted only once in each preferred term category and once in each body system category.
Hematologic toxicities, based on laboratory values and CTC grade, in patients with NHL treated in both single arm studies combined are described in Table 5. Clinically important chemistry laboratory values that were new or worsened from baseline and occurred in >1% of patients at grade 3 or 4, in patients with NHL who were treated in both single arm studies combined were hyperglycemia (3%), elevated creatinine (2%), hyponatremia (2%), and hypocalcemia (2%).
Table 5: Hematology Laboratory Abnormalities in Patients Who Received Bendamustine Hydrochloride in the NHL Studies
Hematology Variable
Bendamustine Hydrochloride
All Grades
(%)
Grade 3 or 4
(%)
Lymphocytes Decreased
99
94
Leukocytes Decreased
94
56
Hemoglobin Decreased
88
11
Neutrophils Decreased
86
60
Platelets Decreased
86
25
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of bendamustine hydrochloride. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and lymphatic systems disorders: Pancytopenia.
Cardiovascular disorders: Atrial fibrillation, congestive heart failure (some fatal), myocardial infarction (some fatal), palpitation.
General disorders and administration site conditions: Injection site reactions (including phlebitis, pruritus, irritation, pain, swelling), infusion site reactions (including phlebitis, pruritus, irritation, pain, swelling).
Immune system disorders: Anaphylaxis.
Infections and infestations: Pneumocystis jiroveci pneumonia, progressive multifocal leukoencephalopathy (PML).
Renal and urinary disorders: Nephrogenic diabetes insipidus (NDI)
Respiratory, thoracic and mediastinal disorders: Pneumonitis.
Skin and subcutaneous tissue disorders: Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), drug reaction with eosinophilia and systemic symptoms (DRESS), and non-melanoma skin cancer (NMSC).
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on VIVIMUSTA
CYP1A2 Inhibitors
The coadministration of VIVIMUSTA with CYP1A2 inhibitors may increase bendamustine plasma concentrations and may result in increased incidence of adverse reactions with VIVIMUSTA [see Clinical Pharmacology (12.3)]. Consider alternative therapies that are not CYP1A2 inhibitors during treatment with VIVIMUSTA.CYP1A2 Inducers
The coadministration of VIVIMUSTA with CYP1A2 inducers may decrease bendamustine plasma concentrations and may result in decreased efficacy of VIVIMUSTA [seeClinical Pharmacology (12.3)]. Consider alternative therapies that are not CYP1A2 inducers during treatment with VIVIMUSTA. -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
In animal reproduction studies, intraperitoneal administration of bendamustine to pregnant mice and rats during organogenesis at doses 0.6 to 1.8 times the maximum recommended human dose (MRHD) resulted in embryo-fetal and/or infant mortality, structural abnormalities, and alterations to growth (see Data). There are no available data on bendamustine hydrochloride use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Bendamustine hydrochloride was intraperitoneally administered once to mice from 210 mg/m2 (approximately 1.8 times the MRHD) during organogenesis and caused an increase in resorptions, skeletal and visceral malformations (exencephaly, cleft palates, accessory rib, and spinal deformities) and decreased fetal body weights. This dose did not appear to be maternally toxic and lower doses were not evaluated. Repeat intraperitoneal administration of bendamustine hydrochloride in mice on gestation days 7 to 11 resulted in an increase in resorptions from 75 mg/m2 (approximately 0.6 times the MRHD) and an increase in abnormalities from 112.5 mg/m2 (approximately 0.9 times the MRHD), similar to those seen after a single intraperitoneal administration.
Bendamustine hydrochloride was intraperitoneally administered once to rats from 120 mg/m2 (approximately the MRHD) on gestation days 4, 7, 9, 11, or 13 and caused embryo and fetal lethality as indicated by increased resorptions and a decrease in live fetuses. A significant increase in external (effect on tail, head, and herniation of external organs [exomphalos]) and internal (hydronephrosis and hydrocephalus) malformations were seen in dosed rats.8.2 Lactation
Risk Summary
There are no data on the presence of bendamustine hydrochloride or its metabolites in either human or animal milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in the breastfed child, advise women not to breastfeed during treatment with VIVIMUSTA and for 1 week after the last dose.8.3 Females and Males of Reproductive Potential
VIVIMUSTA can cause embryo-fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Pregnancy testing is recommended for females of reproductive potential prior to initiation of treatment with VIVIMUSTA.
Contraception
Females
Advise female patients of reproductive potential to use effective contraception during treatment with VIVIMUSTA and for 6 months after the last dose.
Males
Based on genotoxicity findings, advise males with female partners of reproductive potential to use effective contraception during treatment with VIVIMUSTA and for 3 months after the last dose [see Nonclinical Toxicology (13.1)].
Infertility
Based on findings from clinical studies, VIVIMUSTA may impair male fertility. Impaired spermatogenesis, azoospermia, and total germinal aplasia have been reported in male patients treated with alkylating agents, especially in combination with other drugs. In some instances spermatogenesis may return in patients in remission, but this may occur only several years after intensive chemotherapy has been discontinued. Advise patients of the potential risk to their reproductive capacities.
Based on findings from animal studies, VIVIMUSTA may impair male fertility due to an increase in morphologically abnormal spermatozoa. The long-term effects of VIVIMUSTA on male fertility, including the reversibility of adverse effects, have not been studied [see Nonclinical Toxicology (13.1)].8.4 Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
Safety, pharmacokinetics and efficacy were assessed in a single open-label trial (NCT01088984) in patients aged 1-19 years with relapsed or refractory acute leukemia, including 27 patients with acute lymphocytic leukemia (ALL) and 16 patients with acute myeloid leukemia (AML).
Bendamustine hydrochloride was administered as an intravenous infusion over 60 minutes on Days 1 and 2 of each 21-day cycle. There was no treatment response (CR+ CRp) in any patient. The safety profile in these patients was consistent with that seen in adults and no new safety signals were identified.
The pharmacokinetics of bendamustine in 43 patients, aged 1 to 19 years (median age of 10 years) were within range of values previously observed in adults given the same dose based on body surface area.8.5 Geriatric Use
No overall differences in safety were observed between patients ≥65 years of age and younger patients. Efficacy was lower in patients 65 and over with CLL receiving bendamustine hydrochloride based upon an overall response rate of 47% for patients 65 and over and 70% for younger patients. Progression free survival was also longer in younger patients with CLL receiving bendamustine (19 months vs. 12 months). No overall differences in efficacy in patients with non-Hodgkin Lymphoma were observed between geriatric patients and younger patients.
8.6 Renal Impairment
Do not use VIVIMUSTA in patients with creatinine clearance (CLcr) less than 30 mL/min [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Do not use VIVIMUSTA in patients with AST or ALT 2.5 to 10 times upper limit of normal (ULN) and total bilirubin 1.5 to 3 times ULN or total bilirubin greater than 3 times ULN [see Clinical Pharmacology (12.3)]
-
10 OVERDOSAGE
The intravenous lethal dose 50 (LD50) of bendamustine hydrochloride is 240 mg/m2 in the mouse and rat. Toxicities included sedation, tremor, ataxia, convulsions and respiratory distress.
Across all clinical experience, the reported maximum single dose received was 280 mg/m2. Three of four patients who received this dose showed ECG changes considered dose-limiting at 7 and 21 days post-dosing. These changes included QT prolongation (one patient), sinus tachycardia (one patient), ST and T wave deviations (two patients) and left anterior fascicular block (one patient). Cardiac enzymes and ejection fractions remained normal in all patients.
No specific antidote for bendamustine hydrochloride overdose is known. Management of overdosage should include general supportive measures, including monitoring of hematologic parameters and ECGs. -
11 DESCRIPTION
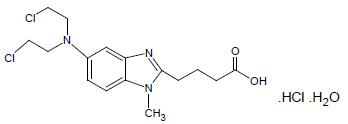
Bendamustine hydrochloride is an alkylating agent. The chemical name of bendamustine hydrochloride monohydrate is 5-[Bis(2-chloroethyl)-amino]-1-methyl-1H-benzimidazole-2- butanoic acid hydrochloride monohydrate. Its empirical molecular formula is C16H21Cl2N3O2 ∙ HCl.H2O and the molecular weight is 412.74. Bendamustine hydrochloride monohydrate contains a mechlorethamine group and a benzimidazole heterocyclic ring with a butyric acid substituent, and has the following structural formula:
VIVIMUSTA (bendamustine hydrochloride injection) is intended for intravenous use after dilution with either 0.9% Sodium Chloride Injection, USP or 2.5% Dextrose/0.45% Sodium Chloride Injection, USP. It is supplied as a sterile, clear, and colorless to yellow solution in a clear glass multiple-dose vial. Each milliliter contains 25 mg of bendamustine hydrochloride equivalent to 22.7 mg of bendamustine, 5 mg of monothioglycerol, 39.45 mg (5% v/v) of absolute alcohol, and q.s. to 1 mL polyethylene glycol 400. Sodium hydroxide is used to adjust pH of polyethylene glycol 400.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Bendamustine is a bifunctional mechlorethamine derivative containing a purine-like benzimidazole ring. Mechlorethamine and its derivatives form electrophilic alkyl groups. These groups form covalent bonds with electron-rich nucleophilic moieties, resulting in interstrand DNA crosslinks. The bifunctional covalent linkage can lead to cell death via several pathways. Bendamustine is active against both quiescent and dividing cells. The exact mechanism of action of bendamustine remains unknown.
12.2 Pharmacodynamics
Based on the pharmacokinetics/pharmacodynamics analyses of data from adult patients with NHL, nausea increased with increasing bendamustine maximum concentrations (Cmax).
Cardiac Electrophysiology
The effect of bendamustine on the QTc interval was evaluated in 53 patients with indolent NHL and mantle cell lymphoma on Day 1 of Cycle 1 after administration of rituximab at 375 mg/m2 intravenous infusion followed by a 30-minute intravenous infusion of bendamustine at 90 mg/m2/day. No mean changes greater than 20 milliseconds were detected up to one hour post infusion. The potential for delayed effects on the QT interval after one hour was not evaluated.12.3 Pharmacokinetics
Absorption
Following a single IV dose of bendamustine hydrochloride Cmax typically occurred at the end of infusion. The dose proportionality of bendamustine has not been studied.
Distribution
The protein binding of bendamustine ranged from 94-96% and was concentration independent from 1 to 50 μg/mL. The blood to plasma concentration ratios in human blood ranged from 0.84 to 0.86 over a concentration range of 10 to 100 μg/mL.
The mean steady-state volume of distribution (Vss) of bendamustine was approximately 20 to 25 L.
Elimination
After a single intravenous dose of 120 mg/m2 of bendamustine over 1 hour, the intermediate half-life (t½) of the parent compound was approximately 40 minutes. The mean terminal elimination t½ of two active metabolites, γ-hydroxybendamustine (M3) and N desmethylbendamustine (M4) were approximately 3 hours and 30 minutes, respectively. Bendamustine clearance in humans was approximately 700 mL/min.
Metabolism
Bendamustine is extensively metabolized via hydrolytic, oxidative, and conjugative pathways. Bendamustine is primarily metabolized via hydrolysis to monohydroxy (HP1) and dihydroxy-bendamustine (HP2) metabolites with low cytotoxic activity in vitro. Two active minor metabolites, M3 and M4, are primarily formed via CYP1A2 in vitro. M3 and M4 concentrations of these metabolites in plasma are 1/10th and 1/100th that of the parent compound, respectively.
Excretion
Following intravenous infusion of radiolabeled bendamustine hydrochloride in patients with cancer, approximately 76% of the dose was recovered. Approximately 50% of the dose was recovered in the urine (3.3% unchanged) and approximately 25% of the dose was recovered in the feces. Less than 1% of the dose was recovered in the urine as M3 and M4, and less than 5% of the dose was recovered in the urine as HP2.
Specific Populations
No clinically meaningful effects on the pharmacokinetics of bendamustine were observed based on age (31 to 84 years), sex, mild to moderate renal impairment (CLcr ≥ 30 mL/min), or hepatic impairment with total bilirubin 1.5 < ULN and AST or ALT < 2.5 × ULN. The effects of severe renal impairment (CLcr < 30 mL/min), or hepatic impairment with total bilirubin 1.5 to 3 × ULN and AST or ALT 2.5 to 10 × ULN or total bilirubin > 3 × ULN on the pharmacokinetics of bendamustine is unknown.
Race/Ethnicity
Exposures in Japanese subjects (n=6) were 40% higher than non-Japanese subjects receiving the same dose. The clinical importance of this difference on the safety and efficacy of bendamustine hydrochloride in Japanese subjects has not been established.
Drug Interaction Studies
In Vitro Studies
Effect of Bendamustine Hydrochloride on CYP Substrates: Bendamustine hydrochloride did not inhibit CYP1A2, 2C9/10, 2D6, 2E1, or 3A4/5. Bendamustine hydrochloride did not induce metabolism of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2E1, or CYP3A4/5.
Effect of Transporters on Bendamustine Hydrochloride:Bendamustine hydrochloride is a substrate of P-glycoprotein and breast cancer resistance protein (BCRP).
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis & Mutagenesis & Impairment Of Fertility
Bendamustine was carcinogenic in mice. After intraperitoneal injections at 37.5 mg/m2/day (the lowest dose tested, approximately 0.3 times the maximum recommended human dose [MRHD]) and 75 mg/m2/day (approximately 0.6 times the MRHD) for 4 days, peritoneal sarcomas in female AB/jena mice were produced. Oral administration at 187.5 mg/m2/day (the only dose tested, approximately 1.6 times the MRHD) for 4 days induced mammary carcinomas and pulmonary adenomas.
Bendamustine is a mutagen and clastogen. In a reverse bacterial mutation assay (Ames assay), bendamustine was shown to increase revertant frequency in the absence and presence of metabolic activation. Bendamustine was clastogenic in human lymphocytes in vitro, and in rat bone marrow cells in vivo (increase in micronucleated polychromatic erythrocytes) from 37.5 mg/m2, (the lowest dose tested, approximately 0.3 times the MRHD).
Bendamustine induced morphologic abnormalities in spermatozoa in mice. Following tail vein injection of bendamustine at 120 mg/m2 or a saline control on days 1 and 2 for a total of three weeks, the number of spermatozoa with morphologic abnormalities was 16% higher in the bendamustine-treated group as compared to the saline control group.
-
14 CLINICAL STUDIES
14.1 Chronic Lymphocytic Leukemia (CLL)
The efficacy of bendamustine hydrochloride was evaluated in an open-label, randomized, controlled multicenter trial comparing bendamustine hydrochloride to chlorambucil. The trial was conducted in 301 previously-untreated patients with Binet Stage B or C (Rai Stages I - IV) CLL requiring treatment. Need-to-treat criteria included hematopoietic insufficiency, B-symptoms, rapidly progressive disease or risk of complications from bulky lymphadenopathy. Patients with autoimmune hemolytic anemia or autoimmune thrombocytopenia, Richter’s syndrome, or transformation to prolymphocytic leukemia were excluded from the study. Patients were randomly assigned to receive either bendamustine hydrochloride 100 mg/m2 intravenously over 30 minutes on Days 1 and 2 of each 28-day cycle or chlorambucil 0.8 mg/kg (Broca’s normal weight) orally on Days 1 and 15 of each 28-day cycle.
The patient populations in the bendamustine hydrochloride and chlorambucil treatment groups were balanced with regard to the following baseline characteristics: age (median 63 vs. 66 years), sex (63% vs. 61% male), Binet stage (71% vs. 69% Binet B), lymphadenopathy (79% vs. 82%), enlarged spleen (76% vs. 80%), enlarged liver (48% vs. 46%), hypercellular bone marrow (79% vs. 73%), “B” symptoms (51% vs. 53%), lymphocyte count (mean 65.7 x 109/L vs. 65.1 x 109/L), and serum lactate dehydrogenase concentration (mean 370.2 vs. 388.4 U/L). Ninety percent of patients in both treatment groups had immuno-phenotypic confirmation of CLL (CD5, CD23 and either CD19 or CD20 or both).
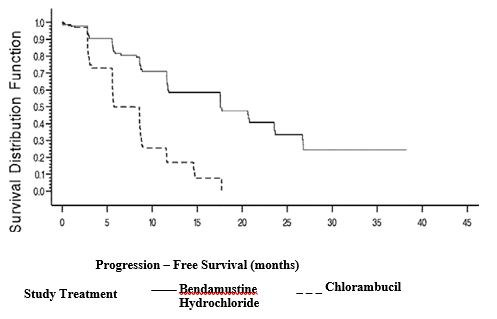
Efficacy endpoints of objective response rate and progression-free survival were calculated using a pre-specified algorithm based on NCI working group criteria for CLL. The results of this open-label randomized study demonstrated a higher rate of overall response and a longer progression-free survival for bendamustine hydrochloride compared to chlorambucil (see Table 6). Survival data are not mature.
Table 6: Efficacy Data for CLL
Bendamustine Hydrochloride (N=153)
Chlorambucil
(N=148)
p-value
Response Rate n (%)
Overall response rate
90 (59)
38 (26)
<0.0001
(95% CI)
(51, 66.6)
(18.6, 32.7)
Complete response (CR)*
13 (8)
1 (<1)
Nodular partial response (nPR)**
4 (3)
0
Partial response (PR) †
73 (48)
37 (25)
Progression-Free Survival††
Median, months (95% CI)
18 (11.7, 23.5)
6 (5.6, 8.6)
Hazard ratio (95% CI)
0.27 (0.17, 0.43)
<0.0001
CI = confidence interval
* CR was defined as peripheral lymphocyte count ≤ 4 x 109/L, neutrophils ≥ 1.5 x 109/L, platelets >100 x 109/L,
hemoglobin >110 g/L, without transfusions, absence of palpable hepatosplenomegaly, lymph nodes ≤ 1.5 cm,
< 30% lymphocytes without nodularity in at least a normocellular bone marrow and absence of “B” symptoms.
The clinical and laboratory criteria were required to be maintained for a period of at least 56 days.** nPR was defined as described for CR with the exception that the bone marrow biopsy shows persistent nodules.
† PR was defined as ≥50% decrease in peripheral lymphocyte count from the pre-treatment baseline
value, and either ≥50% reduction in lymphadenopathy, or ≥50% reduction in the size of spleen or liver,
as well as one of the following hematologic improvements: neutrophils ≥ 1.5 x 109/L or 50% improvement over
baseline, platelets >100 x 109/L or 50% improvement over baseline, hemoglobin >110 g/L or 50% improvement
over baseline without transfusions, for a period of at least 56 days.
†† PFS was defined as time from randomization to progression or death from any cause.
Kaplan-Meier estimates of progression-free survival comparing bendamustine hydrochloride with chlorambucil are shown in Figure 1.
Figure 1. Progression-Free Survival
14.2 Non-Hodgkin Lymphoma (NHL)
The efficacy of bendamustine hydrochloride was evaluated in a single arm study of 100 patients with indolent B-cell NHL that had progressed during or within six months of treatment with rituximab or a rituximab-containing regimen. Patients were included if they relapsed within 6 months of either the first dose (monotherapy) or last dose (maintenance regimen or combination therapy) of rituximab. All patients received bendamustine hydrochloride 120 mg/m2 intravenously on Days 1 and 2 of a 21-day treatment cycle for up to 8 cycles.
The median age was 60 years, 65% were male, and 95% had a baseline WHO performance status of 0 or 1. Major tumor subtypes were follicular lymphoma (62%), diffuse small lymphocytic lymphoma (21%), and marginal zone lymphoma (16%). Ninety-nine percent of patients had received previous chemotherapy, 91% of patients had received previous alkylator therapy, and 97% of patients had relapsed within 6 months of either the first dose (monotherapy) or last dose (maintenance regimen or combination therapy) of rituximab.
Efficacy was based on the assessments by a blinded independent review committee (IRC) and included overall response rate (complete response + complete response unconfirmed + partial response) and duration of response (DR) as summarized in Table 7.
Table 7: Efficacy Data for NHL*
Bendamustine Hydrochloride (N=100)
Response Rate (%)
Overall response rate (CR+CRu+PR)
74
(95% CI)
(64.3, 82.3)
Complete response (CR)
13
Complete response unconfirmed (CRu)
4
Partial response (PR)
57
Duration of Response (DR)
Median, months (95% CI)
9.2 months (7.1, 10.8)
CI = confidence interval
*IRC assessment was based on modified International Working Group response criteria (IWG-RC). Modifications to IWG-RC specified that a persistently positive bone marrow in patients who met all other criteria for CR would be scored as PR. Bone marrow sample lengths were not required to be ≥20 mm. - 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
VIVIMUSTA (bendamustine hydrochloride injection) is a sterile clear, colorless to yellow solution for intravenous use supplied in individual cartons of multiple-dose vials providing 100 mg bendamustine hydrochloride per 4 mL.100 mg/4 mL (25 mg/mL) NDC: 71225-120-01
Safe Handling and DisposalVIVIMUSTA (bendamustine hydrochloride injection) is a hazardous drug. Follow applicable special handling and disposal procedures1. Care should be exercised in the handling and preparation of solutions prepared from VIVIMUSTA. The use of gloves and safety glasses is recommended to avoid exposure in case of breakage of the vial or other accidental spillage. If gloves come in contact with VIVIMUSTA prior to dilution, remove gloves and follow disposal procedures. If a solution of VIVIMUSTA (bendamustine hydrochloride injection) contacts the skin, wash the skin immediately and thoroughly with soap and water. If VIVIMUSTA (bendamustine hydrochloride injection) contacts the mucous membranes, flush thoroughly with water.
StorageStore VIVIMUSTA (bendamustine hydrochloride injection) refrigerated at 2°C to 8°C (36°F to 46°F). Retain in original carton until time of use to protect from light.
-
17 PATIENT COUNSELING INFORMATION
Myelosuppression
Inform patients of the likelihood that bendamustine hydrochloride will cause a decrease in white blood cells, platelets, and red blood cells and the need for frequent monitoring of blood counts. Advise patients to report shortness of breath, significant fatigue, bleeding, fever, or other signs of infection [see Warnings and Precautions (5.1)].
Progressive Multifocal Leukoencephalopathy (PML)
Inform patients to immediately contact their healthcare provider if they experience confusion, memory loss, trouble thinking, difficulty talking or walking, vision loss or other neurological or cognitive symptoms [see Warnings and Precautions (5.3)].
Anaphylaxis and Infusion-Related Reactions
Inform patients of the possibility of serious or mild allergic reactions and to immediately report rash, facial swelling, or difficulty breathing during or soon after infusion [see Warnings and Precautions (5.4)].
Skin Reactions
Advise patients that a rash or itching may occur during treatment with VIVIMUSTA. Advise patients to immediately report severe or worsening rash or itching [see Warnings and Precautions (5.6)].
Hepatotoxicity
Inform patients of the possibility of developing liver function abnormalities and serious hepatic toxicity. Advise patients to immediately contact their healthcare provider if signs of liver failure occur, including jaundice, anorexia, bleeding or bruising [see Warnings and Precautions (5.7)].
Fatigue
Advise patients that VIVIMUSTA may cause tiredness and to avoid driving any vehicle or operating any dangerous tools or machinery if they experience this side effect [see Adverse Reactions (6.1)].
Nausea and Vomiting
Advise patients that VIVIMUSTA may cause nausea and/or vomiting. Patients should report nausea and vomiting so that symptomatic treatment may be provided [see Adverse Reactions (6.1)].
Diarrhea
Advise patients that VIVIMUSTA may cause diarrhea. Patients should report diarrhea to the healthcare provider so that symptomatic treatment may be provided [see Adverse Reactions (6.1)].
Non-melanoma Skin Cancer (NMSC)
Advise patients to undergo regular skin cancer screenings, and to report any suspicious skin changes to their healthcare provider [see Warnings and Precautions (5.8)].
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.10), Use inSpecific Populations (8.1, 8.3), and Nonclinical Toxicology (13.1)].
Advise female patients of reproductive potential to use effective contraception during treatment with VIVIMUSTA and for 6 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
Advise males with female partners of reproductive potential to use effective contraception during treatment with VIVIMUSTA and for 3 months after the last dose [see Use in Specific Populations (8.3) and Nonclinical Toxicology (13.1)].
Lactation
Advise females not to breastfeed during treatment with VIVIMUSTA and for 1 week after the last dose [see Use in Specific Populations (8.2)].Infertility
Advise males of reproductive potential that VIVIMUSTA may impair fertility [see Use in Specific Populations (8.3)].
Manufactured for:
Slayback Pharma LLC
Princeton, NJ 08540.Manufactured at:
Latina Pharma S.p.A.
04013 Sermoneta (LT), Italy. -
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
Principal Display Panel - Carton Label
NDC: 71225-120-01
VIVIMUSTA® (bendamustine hydrochloride injection)100 mg/4mL (25 mg/mL)
Principal Display Panel - Vial Label
NDC: 71225-120-01
VIVIMUSTA® (bendamustine hydrochloride injection)
100 mg/4mL (25 mg/mL)

-
INGREDIENTS AND APPEARANCE
VIVIMUSTA
bendamustine hydrochloride injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 71225-120 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BENDAMUSTINE HYDROCHLORIDE (UNII: 981Y8SX18M) (BENDAMUSTINE - UNII:9266D9P3PQ) BENDAMUSTINE HYDROCHLORIDE 25 mg in 1 mL Inactive Ingredients Ingredient Name Strength MONOTHIOGLYCEROL (UNII: AAO1P0WSXJ) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) SODIUM HYDROXIDE (UNII: 55X04QC32I) ALCOHOL (UNII: 3K9958V90M) Product Characteristics Color YELLOW (colorless to yellow) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 71225-120-01 1 in 1 CARTON 12/07/2022 1 4 mL in 1 VIAL, MULTI-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212209 12/07/2022 Labeler - Slayback Pharma LLC (967770848) Establishment Name Address ID/FEI Business Operations Corden Pharma Latina S.p.A. 339062883 ANALYSIS(71225-120) , MANUFACTURE(71225-120) , PACK(71225-120)
Trademark Results [VIVIMUSTA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 VIVIMUSTA 97791611 not registered Live/Pending |
Slayback Pharma Limited Liability Company 2023-02-13 |
 VIVIMUSTA 97326086 not registered Live/Pending |
Slayback Pharma Limited Liability Company 2022-03-23 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.