NAPRELAN- naproxen sodium tablet, film coated, extended release
NAPRELAN by
Drug Labeling and Warnings
NAPRELAN by is a Prescription medication manufactured, distributed, or labeled by STAT RX USA , STAT RX USA. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
- SPL UNCLASSIFIED SECTION
-
BOXED WARNING
(What is this?)
Cardiovascular Risk
- NSAIDs may cause an increased risk of serious cardiovascular thrombotic events, myocardial infarction, and stroke, which can be fatal. This risk may increase with duration of use. Patients with cardiovascular disease or risk factors for cardiovascular disease may be at greater risk. (See WARNINGS).
- Naproxen as NAPRELAN® is contraindicated for the treatment of peri-operative pain in the setting of coronary artery bypass graft (CABG) surgery (SeeWARNINGS).
Gastrointestinal Risk
- NSAIDs cause an increased risk of serious gastrointestinal adverse events including bleeding, ulceration, and perforation of the stomach or intestines, which can be fatal. These events can occur at any time during use and without warning symptoms. Elderly people are at greater risk for serous gastrointestinal events. (SeeWARNINGS).
-
DESCRIPTION
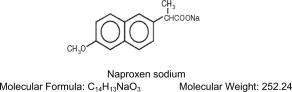
NAPRELAN®* Tablets contain naproxen sodium, a member of the arylacetic acid group of nonsteroidal anti-inflammatory drugs (NSAIDs). NAPRELAN® Tablets use the proprietary IPDAS®** (Intestinal Protective Drug Absorption System) technology. It is a rapidly disintegrating tablet system combining an immediate release component and a sustained release component of microparticles that are widely dispersed, allowing absorption of the active ingredient throughout the gastrointestinal (GI) tract, maintaining blood levels over 24 hours. The chemical name for naproxen sodium is 2-naphthaleneacetic acid, 6-methoxy-a-methyl-sodium salt, (S)- with the following structural formula:
Naproxen sodium is an odorless crystalline powder, white to creamy in color. It is soluble in methanol and water. NAPRELAN® Tablets contain 412.5 mg, 550 mg, or 825 mg of naproxen sodium, equivalent to 375 mg, 500 mg, and 750 mg of naproxen and 37.5, mg 50 mg, and 75 mg sodium respectively. Each NAPRELAN® Tablet also contains the following inactive ingredients: ammoniomethacrylate copolymer Type A, ammoniomethacrylate copolymer Type B, citric acid, crospovidone, magnesium stearate, methacrylic acid copolymer Type A, microcrystalline cellulose, povidone, and talc. The tablet coating contains hydroxypropyl methylcellulose, polyethylene glycol, and titanium dioxide.
-
CLINICAL PHARMACOLOGY
Naproxen is a nonsteroidal anti-inflammatory drug (NSAID), with analgesic and antipyretic properties. As with other NSAIDs, its mode of action is not fully understood; however, its ability to inhibit prostaglandin synthesis may be involved in the anti-inflammatory effect.
PHARMACOKINETICS
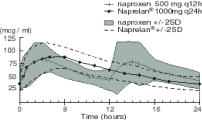
Although naproxen itself is well absorbed, the sodium salt form is more rapidly absorbed, resulting in higher peak plasma levels for a given dose. Approximately 30% of the total naproxen sodium dose in NAPRELAN® Tablets is present in the dosage form as an immediate release component. The remaining naproxen sodium is coated as microparticles to provide sustained release properties. After oral administration, plasma levels of naproxen are detected within 30 minutes of dosing, with peak plasma levels occurring approximately 5 hours after dosing. The observed terminal elimination half-life of naproxen from both immediate release naproxen sodium and NAPRELAN® Tablets is approximately 15 hours. Steady state levels of naproxen are achieved in 3 days and the degree of naproxen accumulation in the blood is consistent with this.
Pharmacokinetic Parameters at Steady State Day 5 (Mean of 24 Subjects) Parameter (units) naproxen 500 mg
Q12h/5 days
(1000 mg)NAPRELAN® 2 x 500 mg
tablets (1000 mg)
Q24h/5 daysMean SD Range Mean SD Range AUC 0-24
(mcgxh/mL)1446 168 1167 - 1858 1448 145 1173 - 1774 Cmax
(mcg/mL)95 13 71 - 117 94 13 74 - 127 Cavg
(mcg/mL)60 7 49 - 77 60 6 49 - 74 Cmin
(mcg/mL)36 9 13 - 51 33 7 23 - 48 Tmax
(hrs)3 1 1 - 4 5 2 2-10 Absorption
Naproxen itself is rapidly and completely absorbed from the GI tract with an in vivo bioavailability of 95%. Based on the pharmacokinetic profile, the absorption phase of NAPRELAN® Tablets occurs in the first 4-6 hours after administration. This coincides with disintegration of the tablet in the stomach, the transit of the sustained release microparticles through the small intestine and into the proximal large intestine. An in vivo imaging study has been performed in healthy volunteers that confirms rapid disintegration of the tablet matrix and dispersion of the microparticles.
The absorption rate from the sustained release particulate component of NAPRELAN® Tablets is slower than that for conventional naproxen sodium tablets. It is this prolongation of drug absorption processes that maintains plasma levels and allows for once daily dosing.
Food Effects
No significant food effects were observed when twenty-four subjects were given a single dose of NAPRELAN® Tablets 500 mg either after an overnight fast or 30 minutes after a meal. In common with conventional naproxen and naproxen sodium formulations, food causes a slight decrease in the rate of naproxen absorption following NAPRELAN® Tablets administration.
Distribution
Naproxen has a volume of distribution of 0.16 L/kg. At therapeutic levels, naproxen is greater than 99% albumin-bound. At doses of naproxen greater than 500 mg/day, there is a less than proportional increase in plasma levels due to an increase in clearance caused by saturation of plasma protein binding at higher doses. However the concentration of unbound naproxen continues to increase proportionally to dose. NAPRELAN® Tablets exhibit similar dose proportional characteristics.
Metabolism
Naproxen is extensively metabolized to 6-0-desmethyl naproxen and both parent and metabolites do not induce metabolizing enzymes.
Elimination
The elimination half-life of NAPRELAN® Tablets and conventional naproxen is approximately 15 hours. Steady state conditions are attained after 2-3 doses of NAPRELAN® Tablets. Most of the drug is excreted in the urine, primarily as unchanged naproxen (less than 1%), 6-0-desmethyl naproxen (less than 1%) and their glucuronide or other conjugates (66-92%). A small amount (<5%) of the drug is excreted in the feces. The rate of excretion has been found to coincide closely with the rate of clearance from the plasma. In patients with renal failure, metabolites may accumulate.
Pediatric Use
No pediatric studies have been performed with NAPRELAN® Tablets, thus safety of NAPRELAN® Tablets in pediatric populations has not been established.
Renal Insufficiency
Naproxen pharmacokinetics have not been determined in subjects with renal insufficiency. Given that naproxen is metabolized and conjugates are primarily excreted by the kidneys, the potential exists for naproxen metabolites to accumulate in the presence of renal insufficiency. Elimination of naproxen is decreased in patients with severe renal impairment. Naproxen-containing products are not recommended for use in patients with moderate to severe and severe renal impairment (creatinine clearance <30mL/min)(see WARNINGS – Renal Effects).
-
CLINICAL STUDIES
RHEUMATOID ARTHRITIS
The use of NAPRELAN® Tablets for the management of the signs and symptoms of rheumatoid arthritis was assessed in a 12 week double-blind, randomized, placebo and active-controlled study in 348 patients. Two NAPRELAN® 500 mg tablets (1000 mg) once daily and naproxen 500 mg tablets twice daily (1000 mg) were more effective than placebo. Clinical effectiveness was demonstrated at one week and continued for the duration of the study.
OSTEOARTHRITIS
The use of NAPRELAN® Tablets for the management of the signs and symptoms of osteoarthritis of the knee was assessed in a 12 week double-blind, placebo and active-controlled study in 347 patients. Two NAPRELAN® 500 mg tablets (1000 mg) once daily and naproxen 500 mg tablets twice daily (1000 mg) were more effective than placebo. Clinical effectiveness was demonstrated at one week and continued for the duration of the study.
ANALGESIA
The onset of the analgesic effect of NAPRELAN® Tablets was seen within 30 minutes in a pharmacokinetic/pharmacodynamic study of patients with pain following oral surgery. In controlled clinical trials, naproxen has been used in combination with gold, D-penicillamine, methotrexate and corticosteroids. Its use in combination with salicylate is not recommended because there is evidence that aspirin increases the rate of excretion of naproxen and data are inadequate to demonstrate that naproxen and aspirin produce greater improvement over that achieved with aspirin alone. In addition, as with other NSAIDs the combination may result in higher frequency of adverse events than demonstrated for either product alone.
SPECIAL STUDIES
In a double-blind randomized, parallel group study, 19 subjects received either two NAPRELAN® 500 mg tablets (1000 mg) once daily or naproxen 500 mg tablets (1000 mg) twice daily for 7 days. Mucosal biopsy scores and endoscopic scores were lower in the subjects who received NAPRELAN® Tablets. In another double-blind, randomized, crossover study, 23 subjects received two NAPRELAN® 500 mg tablets (1000 mg) once daily, naproxen 500 mg tablets (1000 mg) twice daily and aspirin 650 mg four times daily (2600 mg) for 7 days each. There were significantly fewer duodenal erosions seen with NAPRELAN® Tablets than with either naproxen or aspirin. There were significantly fewer gastric erosions with both NAPRELAN® Tablets and naproxen than with aspirin. The clinical significance of these findings is unknown.
-
INDICATIONS AND USAGE
Carefully consider the potential benefits and risks of NAPRELAN® Tablets and other treatment options before deciding to use NAPRELAN® Tablets. Use the lowest effective dose for the shortest duration consistent with individual patient treatment goals (see WARNINGS).
NAPRELAN® Tablets are indicated for the treatment of rheumatoid arthritis, osteoarthritis, ankylosing spondylitis, tendinitis, bursitis and acute gout. It is also indicated in the relief of mild to moderate pain and the treatment of primary dysmenorrhea.
-
CONTRAINDICATIONS
NAPRELAN® is contraindicated in patients with known hypersensitivity to naproxen.
NAPRELAN® should not be given to patients who have experienced asthma, urticaria, or allergic-type reactions after taking aspirin or other NSAIDs. Severe, rarely fatal, anaphylactic-like reactions to NSAIDs have been reported in such patients (see WARNINGS- Anaphylactoid Reactions, and PRECAUTIONS- Preexisting Asthma).
NAPRELAN® is contraindicated for the treatment of peri-operative pain in the setting of coronary artery bypass graft (CABG) surgery (see WARNINGS).
-
WARNINGS
CARDIOVASCULAR EFFECTS
Cardiovascular Thrombotic Events
Clinical trials of several COX-2 selective and nonselective NSAIDs of up to three years duration have shown an increased risk of serious cardiovascular (CV) thrombotic events, myocardial infarction, and stroke, which can be fatal. All NSAIDs, both COX-2 selective and nonselective, may have a similar risk. Patients with known CV disease or risk factors for CV disease may be at greater risk. To minimize the potential risk for an adverse CV event in patients treated with an NSAID, the lowest effective dose should be used for the shortest duration possible. Physicians and patients should remain alert for the development of such events, even in the absence of previous CV symptoms. Patients should be informed about the signs and/or symptoms of serious CV events and the steps to take if they occur.
There is no consistent evidence that concurrent use of aspirin mitigates the increased risk of serious CV thrombotic events associated with NSAID use. The concurrent use of aspirin and an NSAID does increase the risk of serious GI events (see GI Effects – Risk of Ulceration, Bleeding, and Perforation).
Two large, controlled, clinical trials of a COX-2 selective NSAID for the treatment of pain in the first 10-14 days following CABG surgery found an increased incidence of myocardial infarction and stroke (see CONTRAINDICATIONS).
Hypertension
NSAIDs, including NAPRELAN® can lead to onset of new hypertension or worsening of preexisting hypertension, either of which may contribute to the increased incidence of CV events. Patients taking thiazides or loop diuretics may have impaired response to these therapies when taking NSAIDs. NSAIDs, including NAPRELAN®, should be used with caution in patients with hypertension. Blood pressure (BP) should be monitored closely during the initiation of NSAID treatment and throughout the course of therapy.
GASTROINTESTINAL EFFECTS - Risk of Ulceration, Bleeding, and Perforation
NSAIDs, including NAPRELAN®, can cause serious gastrointestinal (GI) adverse events including inflammation, bleeding, ulceration, and perforation of the stomach, small intestine, or large intestine, which can be fatal. These serious adverse events can occur at any time, with or without warning symptoms, in patients treated with NSAIDs. Only one in five patients who develop a serious upper GI adverse event on NSAID therapy is symptomatic. Upper GI ulcers, gross bleeding, or perforation caused by NSAIDs occur in approximately 1% of patients treated for 3-6 months, and in about 2-4% of patients treated for one year. These trends continue with longer duration of use, increasing the likelihood of developing a serious GI event at some time during the course of therapy. However, even short-term therapy is not without risk.
NSAIDs should be prescribed with extreme caution in those with a prior history of ulcer disease or gastrointestinal bleeding. Patients with a prior history of peptic ulcer disease and/or gastrointestinal bleeding who use NSAIDs have a greater than 10-fold increased risk for developing a GI bleed compared to patients with neither of these risk factors. Other factors that increase the risk for GI bleeding in patients treated with NSAIDs include concomitant use of oral corticosteroids or anticoagulants, longer duration of NSAID therapy, smoking, use of alcohol, older age, and poor general health status. Most spontaneous reports of fatal GI events are in elderly or debilitated patients and therefore, special care should be taken in treating this population.
To minimize the potential risk for an adverse GI event in patients treated with an NSAID, the lowest effective dose should be used for the shortest possible duration. Patients and physicians should remain alert for signs and symptoms of GI ulceration and bleeding during NSAID therapy and promptly initiate additional evaluation and treatment if a serious GI adverse event is suspected. This should include discontinuation of the NSAID until a serious GI adverse event is ruled out. For high risk patients, alternate therapies that do not involve NSAIDs should be considered.
RENAL EFFECTS
Long-term administration of NSAIDs has resulted in renal papillary necrosis and other renal injury. Renal toxicity has also been seen in patients in whom renal prostaglandins have a compensatory role in the maintenance of renal perfusion. In these patients, administration of a nonsteroidal anti-inflammatory drug may cause a dose-dependent reduction in prostaglandin formation and, secondarily, in renal blood flow, which may precipitate overt renal decompensation. Patients at greatest risk of this reaction are those with impaired renal function, heart failure, liver dysfunction, those taking diuretics and ACE inhibitors, and the elderly. Discontinuation of NSAID therapy is usually followed by recovery to the pretreatment state.
Advanced Renal Disease
No information is available from from controlled clinical studies regarding the use of NAPRELAN® in patients with advanced renal disease. Therefore, treatment with NAPRELAN® is not recommended in those patients with advanced renal disease. If NAPRELAN® therapy must be initiated, close monitoring of the patient's renal function is advisable.
ANAPHYLACTOID REACTIONS
As with other NSAIDs, anaphylactoid reactions may occur in patients without known prior exposure to NAPRELAN®. NAPRELAN® should not be given to patients with the aspirin triad. This symptom complex typically occurs in asthmatic patients who experience rhinitis with or without nasal polyps, or who exhibit severe, potentially fatal bronchospasm after taking aspirin or other NSAIDs (see CONTRAINDICATIONS and PRECAUTIONS- Preexisting Asthma). Emergency help should be sought in cases where an anaphylactoid reaction occurs.
SKIN REACTIONS
NSAIDs, including NAPRELAN®, can cause serious skin adverse events such as exfoliative dermatitis, Stevens-Johnson Syndrome (SJS), and toxic epidermal necrolysis (TEN), which can be fatal. These serious events may occur without warning. Patients should be informed about the signs and symptoms of serious skin manifestations and use of the drug should be discontinued at the first appearance of skin rash or any other sign of hypersensitivity.
-
PRECAUTIONS
General
NAPRELAN® cannot be expected to substitute for corticosteroids or to treat corticosteroid insufficiency. Abrupt discontinuation of corticosteroids may lead to disease exacerbation. Patients on prolonged corticosteroid therapy should have their therapy tapered slowly if a decision is made to discontinue corticosteroids. The pharmacological activity of NAPRELAN® in reducing fever and inflammation may diminish the utility of these diagnostic signs in detecting complications of presumed noninfectious, painful conditions.
Hepatic Effects
Borderline elevations of one or more liver tests may occur in up to 15% of patients taking NSAIDs including NAPRELAN®. These laboratory abnormalities may progress, may remain unchanged, or may be transient with continuing therapy. Notable elevations of ALT or AST (approximately three or more times the upper limit of normal) have been reported in approximately 1% of patients in clinical trials with NSAIDs. In addition, rare cases of severe hepatic reactions, including jaundice and fatal fulminant hepatitis, liver necrosis and hepatic failure, some of them with fatal outcomes have been reported.
A patient with symptoms and/or signs suggesting liver dysfunction, or in whom an abnormal liver test has occurred, should be evaluated for evidence of the development of a more severe hepatic reaction while on therapy with NAPRELAN®. If clinical signs and symptoms consistent with liver disease develop, or if systemic manifestations occur (e.g., eosinophilia, rash, etc.), NAPRELAN® should be discontinued.
Hematological Effects
Anemia is sometimes seen in patients receiving NSAIDs, including NAPRELAN®. This may be due to fluid retention, occult or gross GI blood loss, or an incompletely described effect upon erythropoiesis. Patients on long-term treatment with NSAIDs, including NAPRELAN®, should have their hemoglobin or hematocrit checked if they exhibit any signs or symptoms of anemia.
NSAIDs inhibit platelet aggregation and have been shown to prolong bleeding time in some patients. Unlike aspirin, their effect on platelet function is quantitatively less, of shorter duration, and reversible. Patients receiving NAPRELAN® who may be adversely affected by alterations in platelet function, such as those with coagulation disorders or patients receiving anticoagulants, should be carefully monitored.
Preexisting Asthma
Patients with asthma may have aspirin-sensitive asthma. The use of aspirin in patients with aspirin-sensitive asthma has been associated with severe bronchospasm which can be fatal. Since cross reactivity, including bronchospasm, between aspirin and other nonsteroidal anti-inflammatory drugs has been reported in such aspirin-sensitive patients, NAPRELAN® should not be administered to patients with this form of aspirin sensitivity and should be used with caution in patients with preexisting asthma.
-
INFORMATION FOR PATIENTS
Patients should be informed of the following information before initiating therapy with an NSAID and periodically during the course of ongoing therapy. Patients should also be encouraged to read the NSAID Medication Guide that accompanies each prescription dispensed.
- NAPRELAN®, like other NSAIDs, may cause serious CV side effects, such as MI or stroke, which may result in hospitalization and even death. Although serious CV events can occur without warning symptoms, patients should be alert for the signs and symptoms of chest pan, shortness of breath, weakness, slurring of speech, and should ask for medical advice when observing any indicative sign or symptoms. Patients should be apprised of the importance of this follow-up (see WARNINGS, Cardiovascular Effects).
- NAPRELAN®, like other NSAIDs, can cause GI discomfort and, rarely, serious GI side effects, such as ulcers and bleeding, which may result in hospitalization and even death. Although serious GI tract ulcerations and bleeding can occur without warning symptoms, patients should be alert for the signs and symptoms of ulcerations and bleeding, and should ask for medical advice when observing any indicative sign or symptoms including epigastric pain, dyspepsia, melena, and hematemesis. Patients should be apprised of the importance of this follow-up (see WARNINGS, Gastrointestinal Effects: Risk of Ulceration, Bleeding and Perforation).
- NAPRELAN®, like other NSAIDs, can cause serious skin side effects such as exfoliative dermatitis, SJS, and TEN, which may result in hospitalization and even death. Although serious skin reactions may occur without warning, patients should be alert for the signs and symptoms of skin rash and blisters, fever, or other signs of hypersensitivity such as itching, and should ask for medical advice when observing any indicative signs or symptoms. Patients should be advised to stop the drug immediately if they develop any type of rash and contact their physicians as soon as possible.
- Patients should promptly report signs or symptoms of unexplained weight gain or edema to their physicians.
- Patients should be informed of the warning signs and symptoms of hepatotoxicity (e.g., nausea, fatigue, lethargy, pruritus, jaundice, right upper quadrant tenderness, and “flu-like” symptoms). If these occur, patients should be instructed to stop therapy and seek immediate medical therapy.
- Patients should be informed of the signs of an anaphylactoid reaction (e.g., difficulty breathing, swelling of the face or throat). If these occur, patients should be instructed to seek immediate emergency help (see WARNINGS).
- In late pregnancy, as with other NSAIDs, NAPRELAN® should be avoided because it may cause premature closure of the ductus arteriosus.
Laboratory Tests
Because serious GI tract ulcerations and bleeding can occur without warning symptoms, physicians should monitor for signs or symptoms of GI bleeding. Patients on long-term treatment with NSAIDs, should have their CBC and a chemistry profile checked periodically. If clinical signs and symptoms consistent with liver or renal disease develop, systemic manifestations occur (e.g., eosinophilia, rash, etc.) or if abnormal liver tests persist or worsen, NAPRELAN® should be discontinued.
-
DRUG INTERACTIONS
ACE-inhibitors
Reports suggest that NSAIDs may diminish the antihypertensive effect of ACE-inhibitors. This interaction should be given consideration in patients taking NSAIDs concomitantly with ACE-inhibitors.
Aspirin
When NAPRELAN® is administered with aspirin, its protein binding is reduced, although the clearance of free NAPRELAN® is not altered. The clinical significance of this interaction is not known; however, as with other NSAIDs, concomitant administration of naproxen and aspirin is not generally recommended because of the potential of increased adverse effects.
Diuretics
Clinical studies, as well as post-marketing observations, have shown that NAPRELAN® can reduce the natriuretic effect of furosemide and thiazides in some patients. This response has been attributed to inhibition of renal prostaglandin synthesis. During concomitant therapy with NSAIDs, the patient should be observed closely for signs of renal failure (see WARNINGS, Renal Effects), as well as to assure diuretic efficacy.
Lithium
NSAIDs have produced an elevation of plasma lithium levels and a reduction in renal lithium clearance. The mean minimum lithium concentration increased 15% and the renal clearance was decreased by approximately 20%. These effects have been attributed to inhibition of renal prostaglandin synthesis by the NSAID. Thus, when NSAIDs and lithium are administered concurrently, subjects should be observed carefully for signs of lithium toxicity.
-
DRUG/LABORATORY TEST INTERACTIONS
Naproxen may decrease platelet aggregation and prolong bleeding time. This effect should be kept in mind when bleeding times are determined.
The administration of naproxen may result in increased urinary values for 17-ketogenic steroids because of an interaction between the drug and/or its metabolites with m-dinitrobenzene used in this assay. Although 17-hydroxy-corticosteroid measurements (Porter-Silber test) do not appear to be artificially altered, it is suggested that therapy with naproxen be temporarily discontinued 72 hours before adrenal function tests are performed if the Porter-Silber test is to be used.
Naproxen may interfere with some urinary assays of 5-hydroxy indoleacetic acid (5HIAA).
-
CARCINOGENESIS, MUTAGENESIS, IMPAIRMENT OF FERTILITY
A two year study was performed in rats to evaluate the carcinogenic potential of naproxen at doses of 8 mg/kg/day, 16 mg/kg/day, and 24 mg/kg/day (50 mg/m2, 100 mg/m2, and 150 mg/m2). The maximum dose used was 0.28 times the systemic exposure to humans at the recommended dose. No evidence of tumorigenicity was found.
-
PREGNANCY
Teratogenic Effects. Pregnancy Category C.
Reproduction studies have been performed in rats at 20mg/kg/day (125 mg/m2/day, 0.23 times the human systemic exposure) rabbits at 20mg/kg/day (220 mg/m2/day, 0.27 times the human systemic exposure) and mice at 170mg/kg/day (510 mg/m2/day, 0.28 times the human systemic exposure) with no evidence of impaired fertility or harm to the fetus due to the drug. There are no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predicitve of human response, NAPRELAN® Tablets should be used during pregnancy only if the potential benefits justify the potential risks to the fetus.
Nonteratogenic effects
There is some evidence to suggest that when inhibitors of prostaglandin synthesis are used to delay preterm labor, there is an increased risk of neonatal complications such as necrotizing enterocolitis, patent ductus arteriosus and intracranial hemorrhage. Naproxen treatment given in late pregnancy to delay parturition has been associated with persistent pulmonary hypertension, renal dysfunction, and abnormal prostaglandin E levels in preterm infants. Because of the known effect of drugs of this class on the human fetal cardiovascular system (closure of the ductus arteriosus), use during third trimester should be avoided.
LABOR AND DELIVERY
In rat studies with NSAIDs, as with other drugs known to inhibit prostaglandin synthesis, an increased incidence of dystocia, delayed parturition, and decreased pup survival occurred. Naproxen-containing products are not recommended in labor and delivery because, through its prostaglandin synthesis inhibitory effect, naproxen may adversely affect fetal circulation and inhibit uterine contractions, thus increasing the risk of uterine hemorrhage. The effects of NAPRELAN® on labor and delivery in pregnant women are unknown.
- PEDIATRIC USE
-
GERIATRIC USE
Clinical studies of NAPRELAN® Tablets did not include a sufficient number of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for the elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Aging may affect the pharmacokinetics of naproxen. Studies indicate that although total plasma concentration of naproxen is unchanged, the unbound plasma fraction of naproxen is increased in the elderly. Caution is advised when high doses are required and some adjustment of dosage may be required in elderly patients. Elderly or debilitated patients seem to tolerate GI ulcerations and bleeding less well than other individuals taking NSAIDs (see WARNINGS, RISK of GI Ulceration, Bleeding And Perforation With NSAID Therapy). Additionally, elderly patients may be more sensitive to dose-dependent reduction in renal prostaglandin formation while taking NSAIDs (see WARNINGS, Renal Effects).
-
ADVERSE REACTIONS
As with all drugs in this class, the frequency and severity of adverse events depends on several factors: the dose of the drug and duration of treatment; the age, the sex, physical condition of the patient; any concurrent medical diagnoses or individual risk factors. The following adverse reactions are divided into three parts based on frequency and whether or not the possibility exists of a causal relationship between drug usage and these adverse events. In those reactions listed as “Probable Causal Relationship” there is at least one case for each adverse reaction where there is evidence to suggest that there is a causal relationship between drug usage and the reported event. The adverse reactions reported were based on the results from two double-blind controlled clinical trials of three months duration with an additional nine month open-label extension. A total of 542 patients received NAPRELAN® Tablets either in the double-blind period or in the nine month open-label extension. Of these 542 patients, 232 received NAPRELAN® Tablets, 167 were initially treated with Naprosyn®*** and 143 were initially treated with placebo. Adverse reactions reported by patients who received NAPRELAN® Tablets are shown by body system. Those adverse reactions observed with naproxen but not reported in controlled trials with NAPRELAN® Tablets are italicized.
The most frequent adverse events from the double-blind and open-label clinical trials were headache (15%), followed by dyspepsia (14%), and flu syndrome (10%). The incidence of other adverse events occurring in 3% - 9% of the patients are marked with an asterisk.
Those reactions occurring in less than 3% of the patients are unmarked.
INCIDENCE GREATER THAN 1% (PROBABLE CAUSAL RELATIONSHIP)
- Body as a Whole—Pain (back)*, pain*, infection*, fever, injury (accident), asthenia, pain chest, headache (15%), flu syndrome (10%).
- Gastrointestinal—Nausea*, diarrhea*, constipation*, abdominal pain*, flatulence, gastritis, vomiting, dysphagia, dyspepsia (14%), heartburn*, stomatitis.
- Hematologic—Anemia, ecchymosis.
- Respiratory—Pharyngitis*, rhinitis*, sinusitis*, bronchitis, cough increased.
- Renal—Urinary tract infection*, cystitis.
- Dermatologic—Skin rash*, skin eruptions*, ecchymoses*, purpura.
- Metabolic and Nutrition—Peripheral edema, hyperglycemia.
- Central Nervous System—Dizziness, paresthesia, insomnia, drowsiness*, lightheadedness.
- Cardiovascular—Hypertension, edema*, dyspnea*, palpitations.
- Musculoskeletal—Cramps (leg), myalgia, arthralgia, joint disorder, tendon disorder.
- Special Senses—Tinnitus*, hearing disturbances, visual disturbances.
- General—Thirst.
INCIDENCE LESS THAN 1% (PROBABLE CAUSAL RELATIONSHIP)
- Body as a Whole—Abscess, monilia, neck rigid, pain neck, abdomen enlarged, carcinoma, cellulitis, edema general, LE syndrome, malaise, mucous membrane disorder, allergic reaction, pain pelvic.
- Gastrointestinal—Anorexia, cholecystitis, cholelithiasis, eructation, GI hemorrhage, rectal hemorrhage, stomatitis aphthous, stomatitis ulcer, ulcer mouth, ulcer stomach, periodontal abscess, cardiospasm, colitis, esophagitis, gastroenteritis, GI disorder, rectal disorder, tooth disorder, hepatosplenomegaly, liver function abnormality, melena, ulcer esophagus, hematemesis, jaundice, pancreatitis, necrosis.
- Renal—Dysmenorrhea, dysuria, kidney function abnormality, nocturia, prostate disorder, pyelonephritis, carcinoma breast, urinary incontinence, kidney calculus, kidney failure, menorrhagia, metrorrhagia, neoplasm breast, nephrosclerosis, hematuria, pain kidney, pyuria, urine abnormal, urinary frequency, urinary retention, uterine spasm, vaginitis, glomerular nephritis, hyperkalemia, interstitial nephritis, nephrotic syndrome, renal disease, renal failure, renal papillary necrosis.
- Hematologic—Leukopenia, bleeding time increased, eosinophilia, abnormal RBC, abnormal WBC, thrombocytopenia, agranulocytosis, granulocytopenia.
- Central Nervous System—Depression, anxiety, hypertonia, nervousness, neuralgia, neuritis, vertigo, amnesia, confusion, co-ordination, abnormal diplopia, emotional lability, hematoma subdural, paralysis, dream abnormalities, inability to concentrate, muscle weakness.
- Dermatologic: Angiodermatitis, herpes simplex, dry skin, sweating, ulcer skin, acne, alopecia, dermatitis contact, eczema, herpes zoster, nail disorder, skin necrosis, subcutaneous nodule, pruritus, urticaria, neoplasm skin, photosensitive dermatitis, photosensitivity reactions resembling porphyria cutaneous tarda, epidermolysis bullosa.
- Special Senses—Amblyopia, scleritis, cataract, conjunctivitis, deaf, ear disorder, keratoconjunctivitis, lacrimation disorder, otitis media, pain eye.
- Cardiovascular—Angina pectoris, coronary artery disease, myocardial infarction, deep thrombophlebitis, vasodilation, vascular anomaly, arrhythmia, bundle branch block, abnormal ECG, heart failure right, hemorrhage, migraine, aortic stenosis, syncope, tachycardia, congestive heart failure.
- Respiratory—Asthma, dyspnea, lung edema, laryngitis, lung disorder, epistaxis, pneumonia, respiratory distress, respiratory disorder, eosinophilic pneumonitis.
- Musculoskeletal—Myasthenia, bone disorder, spontaneous bone fracture, fibrotendinitis, bone pain, ptosis, spasm general, bursitis.
- Metabolic and Nutrition—Creatinine increase, glucosuria, hypercholesteremia, albuminuria, alkalosis, BUN increased, dehydration, edema, glucose tolerance decrease, hyperuricemia, hypokalemia, SGOT increase, SGPT increase, weight decrease.
- General—Anaphylactoid reactions, angioneurotic edema, menstrual disorders, hypoglycemia, pyrexia (chills and fevers).
INCIDENCE LESS THAN 1% (CAUSAL RELATIONSHIP UNKNOWN)
Other adverse reactions listed in the naproxen package label, but not reported by those who received NAPRELAN® Tablets are shown in italics. These observations are being listed as alerting information to the physician.
- Hematologic—Aplastic anemia, hemolytic anemia.
- Central Nervous System—Aseptic meningitis, cognitive dysfunction.
- Dermatologic—Epidermal necrolysis, erythema multiforme, Stevens-Johnson syndrome.
- Gastrointestinal—Non-peptic GI ulceration, ulcerative stomatitis.
- Cardiovascular—Vasculitis.
-
OVERDOSAGE
Significant naproxen overdosage may be characterized by drowsiness, heartburn, indigestion, nausea or vomiting. Because naproxen sodium may be rapidly absorbed, high and early blood levels should be anticipated. A few patients have experienced seizures, but it is not clear whether or not these were drug-related. It is not known what dose of the drug would be life threatening. The oral LD50 of the drug is 500 mg/kg in rats, 1200 mg/kg in mice, 4000 mg/kg in hamsters and greater than 1000 mg/kg in dogs. In animals 0.5 g/kg of activated charcoal was effective in reducing plasma levels of naproxen.
Patients should be managed by symptomatic and supportive care following an NSAID overdose. There are no specific antidotes. Hemodialysis does not decrease the plasma concentration of naproxen because of the high degree of its protein binding. Emesis and/or activated charcoal (60 to 100 g in adults, 1 to 2 g/kg in children) and/or osmotic carthartic may be indicated in patients seen within 4 hours of ingestion with symptoms or following a large overdose. Forced diuresis, alkalinization of urine or hemoperfusion may not be useful due to high protein binding.
-
DOSAGE AND ADMINISTRATION
Carefully consider the potential benefits and risks of NAPRELAN® and other treatment options before deciding to use NAPRELAN®. Use the lowest effective dose for the shortest duration consistent with individual patient treatment goals (see WARNINGS).
After observing the response to initial therapy with NAPRELAN®, the dose and frequency should be adjusted to suit an individual patient's needs.
For the relief of:
Rheumatoid Arthritis, Osteoarthritis, and Ankylosing Spondylitis
The recommended starting dose of NAPRELAN® Tablets in adults is two NAPRELAN® 375 mg tablets (750 mg) once daily, one NAPRELAN® 750 mg (750 mg) once daily, or two NAPRELAN® 500 mg tablets (1000 mg) once a daily. Patients already taking naproxen 250 mg, 375 mg, or 500mg twice daily (morning and evening) may have their total daily dose replaced with NAPRELAN® Tablets as a single daily dose.
During long-term administration, the dose of NAPRELAN® Tablets may be adjusted up or down depending on the clinical response of the patient. In patients who tolerate lower doses of NAPRELAN® Tablets well, the dose may be increased to two NAPRELAN® 750 mg tablets (1500 mg), or three NAPRELAN® 500 mg tablets (1500 mg) once daily for limited periods when a higher level of anti-inflammatory/analgesic activity is required. When treating patients, especially at the higher dose levels, the physician should observe sufficient increased clinical benefit to offset the potential increased risk (see CLINICAL PHARMACOLOGY). The lowest effective dose should be sought and used in every patient. Symptomatic improvement in arthritis usually begins within one week; however, treatment for two weeks may be required to achieve a therapeutic benefit.
A lower dose should be considered in patients with renal or hepatic impairment or in elderly patients (see PRECAUTIONS). Studies indicate that although total plasma contcentration of naproxen is unchanged, the unbound plasma fraction of naproxen is increased in the elderly. Caution is advised when high doses are required and some adjustment of dosage may be required in elderly patients. As with other drugs used in the elderly it is prudent to use the lowest effective dose.
Management of Pain, Primary Dysmenorrhea, and Acute Tendinitis and Bursitis
The recommended starting dose is two NAPRELAN® 500 mg tablets (1000 mg) once daily. For patients requiring greater analgesic benefit, two NAPRELAN® 750 mg tablets (1500 mg) or three NAPRELAN® 500 mg tablets (1500 mg) may be used for a limited period. Thereafter, the total daily dose should not exceed two NAPRELAN® 500 mg tablets (1000 mg). The NAPRELAN® DOSE CARD provides a 10 day tapered dose regimen contained in a single blister dose pack that provides 1500 mg given once daily (two NAPRELAN® 750 mg tablets) for 3 days, with a taper to 1000 mg given once daily (two NAPRELAN® 500 mg tablets) for the remaining 7 days.
-
HOW SUPPLIED
NAPRELAN® (naproxen sodium) Controlled-Release Tablets are available as follows:
- NAPRELAN® 375: white, capsule-shaped tablet with “N” on one side and “375” on the reverse; in bottles of 100; NDC: 68453-375-10. Each tablet contains 412.5 mg naproxen sodium equivalent to 375 mg naproxen.
- NAPRELAN® 500: white, capsule-shaped tablet with “N” on one side and “500” on the reverse; in bottles of 75; NDC: 68453-850-75. Each tablet contains 550 mg naproxen sodium equivalent to 500 mg naproxen.
- NAPRELAN® 750: white-capsule-shaped tablet with “N” on one side and “750” on the reverse; in bottles of 30; 68453-777-03; in bottles of 60; NDC: 68453-777-06. Each tablet contains 825 mg naproxen sodium equivalent to 750 mg naproxen.
- NAPRELAN® DOSE CARD: contains six 750 mg white-capsule-shaped tablets with “N” on one side and “750” on the reverse (3 days of therapy) and fourteen 500 mg white, capsule-shaped tablets with "N" on one side and "500" on the reverse (7 days of therapy) in a single blister card; NDC: 68453-900-02.
Rx Only
US Patent 5,637,320
Store at controlled room temperature, 20° - 25° C (68° - 77° F).
Dispense in a well-closed container.
______________________________________________________________
*Registered Trademark of Elan Corporation plc
**Registered Trademark of Elan Pharma Int. Ltd.
***Naprosyn® is a registered trademark of Syntex Puerto Rico, Inc.
Manufactured for: Victory Pharma, Inc., San Diego, CA 92130
Manufactured by: Elan Pharma International Ltd., Athlone, Ireland
PI3750107
Rev 8/09
-
MEDICATION GUIDE
Medication Guide for Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
(See the end of this Medication Guide for a list of prescription NSAID medicines.)
NAPRELAN®
(naproxen sodium)
CONTROLLED-RELEASE TABLETSWhat is the most important information I should know about medicines called Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)?
NSAID medicines may increase the chance of a heart attack or stroke that can lead to death._
This chance increases:
- with longer use of NSAID medicines
- in people who have heart disease
NSAID medicines should never be used right before or after a heart surgery called a “coronary artery bypass graft (CABG).”
NSAID medicines can cause ulcers and bleeding in the stomach and intestines at any time during treatment. Ulcers and bleeding:
- can happen without warning symptoms
- may cause death
The chance of a person getting an ulcer or bleeding increases with:
- taking medicines called “corticosteroids” and “anti-coagulants”
- longer use
- smoking
- drinking alcohol
- older age
- having poor health
NSAID medicines should only be used:
- exactly as prescribed
- at the lowest dose possible for your treatment
- for the shortest time needed
________________________________________________
What are Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)?
NSAID medicines are used to treat pain and redness, swelling, and heat (inflammation) from medical conditions such as:
- different types of arthritis
- menstrual cramps and other types of short-term pain
Who should not take a Non-Steroidal Anti-Inflammatory Drug (NSAID)?
Do not take an NSAID medicine:
- if you had an asthma attack, hives, or other allergic reaction with aspirin or any other NSAID medicine.
- for pain right before or after heart bypass surgery.
Tell your healthcare provider:
- about all your medical conditions
- about all of the medicines you take. NSAIDs and some other medicines can interact with each other and cause serious side effects. Keep a list of your medicines to show to your healthcare provider and pharmacist.
- if you are pregnant. NSAID medicines should not be used by pregnant women late in their pregnancy.
- if you are breastfeeding. Talk to your doctor.
What are the possible side effects of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)?
Serious side effects include:
- heart attack
- stroke
- high blood pressure
- heart failure from body swelling (fluid retention)
- kidney problems including kidney failure
- bleeding and ulcers in the stomach and intestine
- low red blood cells (anemia)
- life-threatening skin reactions
- life-threatening allergic reactions
- liver problems including liver failure
- asthma attacks in people who have asthma
Other side effects include:
- stomach pain
- constipation
- diarrhea
- gas
- heartburn
- nausea
- vomiting
- dizziness
Get emergency help right away if you have any of the following symptoms:
- shortness of breath or trouble breathing
- chest pain
- weakness in one part or side of your body
- slurred speach
- swelling of the face or throat
Stop your NSAID medicine and call your healthcare provider right away if you have any of the following symptoms:
- nausea
- more tired or weaker than usual
- itching
- your skin or eyes look yellow
- stomach pain
- flu-like symptoms
- vomit blood
- there is blood in your bowel movement or it is black and sticky like tar
- unusual weight gain
- skin rash or blisters with fever
- swelling of the arms and legs, hands and feet
These are not all the side effects with NSAID medicines. Talk to your healthcare provider or pharmacist for more information about NSAID medicines. Call your doctor for medial advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
Other information about Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
- Aspirin is an NSAID medicine but it does not increase the chance of a heart attack. Aspirin can cause bleeding in the brain, stomach, and intestines. Aspirin can also cause ulcers in the stomach and intestines.
- Some of these NSAID medicines are sold in lower doses without a prescription (over-the-counter). Talk to your healthcare provider before using over-the-counter NSAIDs for more than 10 days.
NSAID medicines that need a prescription *Vicoprofen contains the same dose of ibuprofen as over-the-counter (OTC) NSAIDs, and is usually used for less than 10 days to treat pain. The OTC NSAID label warns that long term continuous use may increase the risk of heart attack or stroke.
Generic Name Tradename Celecoxib Celebrex Diclofenac Cataflam, Voltaren, Arthrotec (combined with misoprostol) Diflunisal Dolobid Etodolac Lodine, Lodine XL Fenoprofen Nalfon, Nalfon 200 Flurbiprofen Ansaid Ibuprofen Motrin, Tab-Profen, Vicoprofen* (combined with hydrocodone), Combunox (combined with oxycodone) Indomethacin Indocin, Indocin SR, Indo-Lemmon, Indomethagan Ketoprofen Oruvail Ketorolac Toradol Mefenamic Acid Ponstel Meloxicam Mobic Nabumetone Relafen Naproxen Naprosyn, Anaprox, Anaprox DS, EC-Naproxyn, NAPRELAN, Naprapac (copackaged with lansoprazole) Oxaprozin Daypro Piroxicam Feldene Sulindac Clinoril Tolmetin Tolectin, Tolectin DS, Tolectin 600 This Medication Guide has been approved by the U.S. Food and Drug Administration.
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
NAPRELAN
naproxen sodium tablet, film coated, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 16590-242(NDC: 68453-777) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength NAPROXEN SODIUM (UNII: 9TN87S3A3C) (NAPROXEN - UNII:57Y76R9ATQ) NAPROXEN SODIUM 750 mg Inactive Ingredients Ingredient Name Strength ANHYDROUS CITRIC ACID (UNII: XF417D3PSL) CROSPOVIDONE (UNII: 68401960MK) MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POVIDONE (UNII: FZ989GH94E) TALC (UNII: 7SEV7J4R1U) HYPROMELLOSE (UNII: 3NXW29V3WO) POLYETHYLENE GLYCOL (UNII: 3WJQ0SDW1A) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color white Score no score Shape CAPSULE Size 22mm Flavor Imprint Code N;750 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 16590-242-30 30 in 1 BOTTLE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020353 12/19/2008 Labeler - STAT RX USA (786036330) Establishment Name Address ID/FEI Business Operations STAT RX USA 786036330 repack, relabel
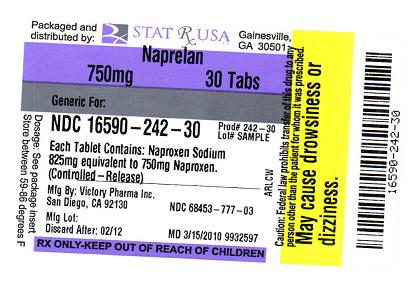 NAPRELAN 750MG LABEL IMAGE
NAPRELAN 750MG LABEL IMAGETrademark Results [NAPRELAN]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 NAPRELAN 73659443 1955461 Live/Registered |
ELAN CORPORATION, PLC. 1987-05-07 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.