ORIAHNN- elagolix and estradiol and norethisterone kit
Oriahnn by
Drug Labeling and Warnings
Oriahnn by is a Prescription medication manufactured, distributed, or labeled by AbbVie Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ORIAHNN safely and effectively. See full prescribing information for ORIAHNN.
ORIAHNN® (elagolix, estradiol, and norethindrone acetate capsules; elagolix capsules), co-packaged for oral use
Initial U.S. Approval: 2020
WARNING: THROMBOEMBOLIC DISORDERS AND VASCULAR EVENTS
See full prescribing information for complete boxed warning.
-
Estrogen and progestin combinations, including ORIAHNN, increase the risk of thrombotic or thromboembolic disorders, especially in women at increased risk for these events. (5.1)
- ORIAHNN is contraindicated in women with current or a history of thrombotic or thromboembolic disorders and in women at increased risk for these events including women over 35 years of age who smoke or women with uncontrolled hypertension. (4)
INDICATIONS AND USAGE
ORIAHNN is a combination of elagolix, a gonadotropin-releasing hormone (GnRH) receptor antagonist, estradiol, an estrogen, and norethindrone acetate, a progestin, indicated for the management of heavy menstrual bleeding associated with uterine leiomyomas (fibroids) in premenopausal women. (1)
Limitation of Use:
- Use of ORIAHNN should be limited to 24 months due to the risk of continued bone loss, which may not be reversible. (1)
DOSAGE AND ADMINISTRATION
One capsule (elagolix 300 mg, estradiol 1 mg, norethindrone acetate 0.5 mg) in the morning and one capsule (elagolix 300 mg) in the evening for up to 24 months. (2.1)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
- High risk of arterial, venous thrombotic, or thromboembolic disorder. (4)
- Pregnancy. (4)
- Known osteoporosis. (4)
- Current or history of breast cancer or other hormonally-sensitive malignancies. (4)
- Known liver impairment or disease. (4)
- Undiagnosed abnormal uterine bleeding. (4)
- Known hypersensitivity to ingredients of ORIAHNN. (4)
- Organic anion transporting polypeptide (OATP)1B1 inhibitors that are known or expected to significantly increase elagolix plasma concentrations. (4)
WARNINGS AND PRECAUTIONS
-
Thromboembolic Disorders and Vascular Events: Discontinue ORIAHNN if an arterial or venous thrombotic, cardiovascular, or cerebrovascular event occurs. Stop ORIAHNN if there is sudden unexplained partial or complete loss of vision, proptosis, diplopia, papilledema, or retinal vascular lesions and evaluate for retinal vein thrombosis immediately. (5.1)
-
Bone Loss: Duration-dependent decreases in bone mineral density (BMD) that may not be completely reversible. Baseline and periodic BMD assessments are recommended. Assess risk-benefit for women with additional risk factors for bone loss. (5.2)
-
Suicidal Ideation and Mood Disorders: Advise patients to seek medical attention for suicidal ideation, suicidal behavior, new onset or worsening depression, anxiety, or other mood changes. (5.4)
-
Hepatic Impairment and Transaminase Elevations: Counsel patients on signs and symptoms of liver injury. (5.5)
-
Elevated Blood Pressure: Do not use in women with uncontrolled hypertension. For women with well-controlled hypertension, continue to monitor blood pressure and stop ORIAHNN if blood pressure rises significantly. (5.6)
-
Change in Menstrual Bleeding Pattern and Reduced Ability to Recognize Pregnancy: Advise women to use non-hormonal contraception during treatment and for 28 days after discontinuing ORIAHNN. ORIAHNN may delay the ability to recognize the occurrence of a pregnancy because it alters menstrual bleeding. Perform pregnancy testing if pregnancy is suspected and discontinue ORIAHNN if pregnancy is confirmed. (5.8)
- Risk of Allergic Reactions Due to the Inactive Ingredient (FD&C Yellow No 5): This product contains FD&C Yellow No. 5 (tartrazine), which may cause allergic-type reactions (including bronchial asthma) in certain susceptible persons. (5.12)
ADVERSE REACTIONS
Most common adverse reaction (>5%) in clinical trials were hot flushes, headache, fatigue, metrorrhagia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AbbVie Inc. at 1–800–633–9110 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatchDRUG INTERACTIONS
See full prescribing information for a list of clinically important drug interactions. (7)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 12/2025
-
Estrogen and progestin combinations, including ORIAHNN, increase the risk of thrombotic or thromboembolic disorders, especially in women at increased risk for these events. (5.1)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: THROMBOEMBOLIC DISORDERS AND VASCULAR EVENTS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosing Information
2.2 Missed Dose
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Thromboembolic Disorders and Vascular Events
5.2 Bone Loss
5.3 Hormonally-Sensitive Malignancies
5.4 Suicidal Ideation, Suicidal Behavior, and Exacerbation of Mood Disorders
5.5 Hepatic Impairment and Transaminase Elevations
5.6 Elevated Blood Pressure
5.7 Gallbladder Disease or History of Cholestatic Jaundice
5.8 Change in Menstrual Bleeding Pattern and Reduced Ability to Recognize Pregnancy
5.9 Effects on Carbohydrate and Lipid Metabolism
5.10 Alopecia
5.11 Effect on Other Laboratory Results
5.12 Risk of Allergic Reactions Due to the Inactive Ingredient (FD&C Yellow No. 5)
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Potential for ORIAHNN to Affect Other Drugs
7.2 Potential for Other Drugs to Affect ORIAHNN
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.5 Pharmacogenomics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: THROMBOEMBOLIC DISORDERS AND VASCULAR EVENTS
-
Estrogen and progestin combinations, including ORIAHNN, increase the risk of thrombotic or thromboembolic disorders including pulmonary embolism, deep vein thrombosis, stroke and myocardial infarction, especially in women at increased risk for these events [see Warnings and Precautions (5.1)].
- ORIAHNN is contraindicated in women with current or a history of thrombotic or thromboembolic disorders and in women at increased risk for these events, including women over 35 years of age who smoke and women with uncontrolled hypertension [see Contraindications (4)].
-
Estrogen and progestin combinations, including ORIAHNN, increase the risk of thrombotic or thromboembolic disorders including pulmonary embolism, deep vein thrombosis, stroke and myocardial infarction, especially in women at increased risk for these events [see Warnings and Precautions (5.1)].
-
1 INDICATIONS AND USAGE
ORIAHNN is indicated for the management of heavy menstrual bleeding associated with uterine leiomyomas (fibroids) in premenopausal women.
Limitation of Use:
Use of ORIAHNN should be limited to 24 months due to the risk of continued bone loss, which may not be reversible [see Dosage and Administration (2.1) and Warnings and Precautions (5.2)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosing Information
- Exclude pregnancy before starting ORIAHNN or start ORIAHNN within 7 days from the onset of menses [see Use in Specific Populations (8.1) and (8.3)].
- The recommended dosage of ORIAHNN is:
○ One elagolix 300 mg, estradiol 1 mg, and norethindrone acetate 0.5 mg capsule in the morning (AM), and
○ One elagolix 300 mg capsule in the evening (PM).
- Take the morning and evening capsules at approximately the same time each day, with or without food.
- The recommended duration of treatment with ORIAHNN is 24 months [see Warnings and Precautions (5.2)].
2.2 Missed Dose
Instruct the patient to take the missed dose of ORIAHNN within 4 hours of the time that it was supposed to be taken and then the next dose at the usual time. If more than 4 hours have passed since a capsule is usually taken, instruct the patient not to take the missed dose and take the next dose at the usual time. Take only one morning capsule and one evening capsule per day.
- Exclude pregnancy before starting ORIAHNN or start ORIAHNN within 7 days from the onset of menses [see Use in Specific Populations (8.1) and (8.3)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
ORIAHNN is contraindicated in women:
- With a high risk of arterial, venous thrombotic, or thromboembolic disorders [see Boxed Warning and Warnings and Precautions (5.1)]. Examples include women over 35 years of age who smoke, and women who are known to have:
○ current or history of deep vein thrombosis or pulmonary embolism
○ vascular disease (e.g., cerebrovascular disease, coronary artery disease, peripheral vascular disease)
○ thrombogenic valvular or thrombogenic rhythm diseases of the heart (for example, subacute bacterial endocarditis with valvular disease, or atrial fibrillation)
○ inherited or acquired hypercoagulopathies
○ uncontrolled hypertension
○ headaches with focal neurological symptoms or have migraine headaches with aura if over age 35
- Who are pregnant. Exposure to ORIAHNN early in pregnancy may increase the risk of early pregnancy loss [see Use in Specific Populations (8.1)].
- With known osteoporosis because of the risk of further bone loss [see Warnings and Precautions (5.2)].
- With current or history of breast cancer or other hormonally-sensitive malignancies, and with increased risk for hormonally-sensitive malignancies [see Warnings and Precautions (5.3)].
- With known hepatic impairment or disease [see Warnings and Precautions (5.5)].
- With undiagnosed abnormal uterine bleeding.
- With known anaphylactic reaction, angioedema, or hypersensitivity to ORIAHNN or any of its components.
- Taking inhibitors of organic anion transporting polypeptide (OATP)1B1 (a hepatic uptake transporter) that are known or expected to significantly increase elagolix plasma concentrations [see Drug Interactions (7.2)].
- With a high risk of arterial, venous thrombotic, or thromboembolic disorders [see Boxed Warning and Warnings and Precautions (5.1)]. Examples include women over 35 years of age who smoke, and women who are known to have:
-
5 WARNINGS AND PRECAUTIONS
5.1 Thromboembolic Disorders and Vascular Events
ORIAHNN is contraindicated in women with current or history of thrombotic or thromboembolic disorders and in women at increased risk for these events [see Contraindications (4)]. In the Phase 3 clinical trials (Studies UF-1, UF-2, and UF-3), two thrombotic events occurred in 453 ORIAHNN-treated women (thrombosis in the calf and pulmonary embolism) [see Adverse Reactions (6.1) and Clinical Studies (14)]. Estrogen and progestin combinations, including the estradiol/norethindrone acetate component of ORIAHNN, increase the risk of thrombotic or thromboembolic disorders, including pulmonary embolism, deep vein thrombosis, stroke, and myocardial infarction, especially in women at high risk for these events. In general, the risk is greatest among women over 35 years of age who smoke, and women with uncontrolled hypertension, dyslipidemia, vascular disease, or obesity.
Discontinue ORIAHNN if an arterial or venous thrombotic, cardiovascular, or cerebrovascular event occurs or is suspected. If feasible, discontinue ORIAHNN at least 4 to 6 weeks before surgery of the type associated with an increased risk of thromboembolism, or during periods of prolonged immobilization.
Stop ORIAHNN immediately if there is sudden unexplained partial or complete loss of vision, proptosis, diplopia, papilledema, or retinal vascular lesions and evaluate for retinal vein thrombosis as these have been reported in patients receiving estrogens and progestins.
5.2 Bone Loss
ORIAHNN is contraindicated in women with known osteoporosis [see Contraindications (4)]. ORIAHNN may cause a decrease in bone mineral density (BMD) in some patients. BMD loss is greater with increasing duration of use and may not be completely reversible after stopping treatment [see Adverse Reactions (6.1)].
In the Phase 3 clinical trials (Studies UF-1, UF-2, and UF-3) [see Clinical Studies (14)], seven out of 453 (1.5%) ORIAHNN-treated women experienced fractures, including one (0.2%) with a fragility fracture, compared to one out of 196 (0.5%) placebo-treated women (patient had a non-fragility fracture). Five of the seven ORIAHNN-treated women reported these fractures in the post-treatment follow-up period. The impact of BMD decreases on long-term bone health and future fracture risk in premenopausal women is unknown.
Consider the benefits and risks of ORIAHNN treatment in patients with a history of a low-trauma fracture or other risk factors for osteoporosis or bone loss, including taking medications that may decrease BMD (e.g., systemic or chronic inhaled corticosteroids, anticonvulsants, or proton pump inhibitors).
Assessment of BMD by dual-energy X-ray absorptiometry (DXA) is recommended at baseline and periodically thereafter. Consider discontinuing ORIAHNN if the risk associated with bone loss exceeds the potential benefit of treatment. Limit the duration of use to 24 months to reduce the extent of bone loss [see Indications and Usage (1) and Dosage and Administration (2.1)].
Although the effect of supplementation with calcium and vitamin D was not studied, such supplementation for patients with inadequate dietary intake may be beneficial.
5.3 Hormonally-Sensitive Malignancies
ORIAHNN is contraindicated in women with current or history of breast cancer and in women at increased risk for hormonally-sensitive malignancies, such as those with mutations in BRCA genes [see Contraindications (4)].
In the Phase 3 clinical trials (Studies UF-1, UF-2, and UF-3), two (0.4%) cases of breast cancer in 453 ORIAHNN-treated women were observed. No breast cancer cases were seen in placebo-treated women [see Adverse Reactions (6.1)].
The use of estrogen alone and estrogen plus progestin has been reported to result in an increase in abnormal mammograms requiring further evaluation. Surveillance measures, such as breast examinations and regular mammography, are recommended. Discontinue ORIAHNN if a hormonally-sensitive malignancy is diagnosed.
5.4 Suicidal Ideation, Suicidal Behavior, and Exacerbation of Mood Disorders
In Phase 3 placebo-controlled clinical trials (Studies UF-1 and UF-2), ORIAHNN-treated women had a higher incidence (3%) of depression, depressed mood, and/or tearfulness compared to placebo-treated women (1%) [see Adverse Reactions (6.1)]. Suicidal ideation and behavior, including a completed suicide, occurred in women treated with lower doses of elagolix in clinical trials conducted for a different indication.
Promptly evaluate patients with depressive symptoms to determine whether the risks of continued therapy outweigh the benefits. Patients with new or worsening depression, anxiety, or other mood changes should be referred to a mental health professional, as appropriate. Advise patients to seek immediate medical attention for suicidal ideation and behavior.
Reevaluate the benefits and risks of continuing ORIAHNN if such events occur.
5.5 Hepatic Impairment and Transaminase Elevations
Contraindication in Patients with Hepatic Impairment
ORIAHNN is contraindicated in women with known hepatic impairment or disease [see Contraindications (4), Use in Specific Populations (8.7), and Clinical Pharmacology (12.3)].
Transaminase Elevations
In Phase 3 placebo-controlled clinical trials (Studies UF-1 and UF-2), elevations (> 3 times the upper limit of the reference range) in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) occurred in 1.1% (4/379) and 1.3% (5/379) of ORIAHNN-treated patients, respectively, compared to no elevations in placebo. Transaminases peaked at 8 times the upper limit for ALT and 6 times the upper limit for AST. No pattern in time to onset of these liver transaminase elevations was identified. Transaminase levels returned to baseline within 4 months after peak values in these patients.
Instruct patients to promptly seek medical attention in case of symptoms or signs that may reflect liver injury, such as jaundice [see Adverse Reactions (6.1)].
5.6 Elevated Blood Pressure
ORIAHNN is contraindicated in women with uncontrolled hypertension [see Contraindications (4)]. In Studies UF-1 and UF-2, a maximum mean increase in systolic blood pressure of 5.1 mmHg [95% confidence interval (CI) 2.68, 7.59] occurred at Month 5, and a maximum mean increase in diastolic blood pressure of 2.1 mmHg (95% CI 0.43, 3.84) occurred at Month 4 in ORIAHNN-treated women, as compared to placebo-treated women [see Adverse Reactions (6.1)].
For women with well-controlled hypertension, continue to monitor blood pressure and stop ORIAHNN if blood pressure rises significantly. Monitor blood pressure in normotensive women treated with ORIAHNN.
5.7 Gallbladder Disease or History of Cholestatic Jaundice
Studies among estrogen users suggest a small increased relative risk of developing gallbladder disease. For women with a history of cholestatic jaundice associated with past estrogen use or with pregnancy, assess the risk-benefit of continuing therapy. Discontinue ORIAHNN if jaundice occurs.
5.8 Change in Menstrual Bleeding Pattern and Reduced Ability to Recognize Pregnancy
ORIAHNN may delay the ability to recognize the occurrence of a pregnancy because it may reduce the intensity, duration, and amount of menstrual bleeding [see Adverse Reactions (6.1)]. Perform pregnancy testing if pregnancy is suspected, and discontinue ORIAHNN if pregnancy is confirmed [see Use in Specific Populations (8.1, 8.3)].
The effect of hormonal contraceptives on the efficacy of ORIAHNN is unknown. Advise women to use non-hormonal contraception during treatment and for 28 days after discontinuing ORIAHNN [see Use in Specific Populations (8.1, 8.3)].
5.9 Effects on Carbohydrate and Lipid Metabolism
ORIAHNN may decrease glucose tolerance and result in increased glucose levels. More frequent monitoring in ORIAHNN-treated women with prediabetes and diabetes may be needed.
In women with pre-existing hypertriglyceridemia, estrogen therapy may be associated with elevations of plasma triglycerides leading to pancreatitis. Use of elagolix is associated with increases in total cholesterol, low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), and serum triglycerides. Monitor lipid levels and consider discontinuing ORIAHNN if hypercholesterolemia or hypertriglyceridemia worsens [see Adverse Reactions (6.1)].
5.10 Alopecia
In Phase 3 clinical trials (Studies UF-1 and UF-2), more women experienced alopecia, hair loss, and hair thinning with ORIAHNN (3.5%) compared to placebo (1.0%). In almost one-third (4/14) of affected ORIAHNN-treated women, alopecia was a reason for discontinuing treatment. No specific pattern was described. In the majority of affected women, hair loss was continuing when ORIAHNN was stopped. Whether the hair loss is reversible is unknown. Consider discontinuing ORIAHNN if hair loss becomes a concern [see Adverse Reactions (6.1)].
5.11 Effect on Other Laboratory Results
The use of estrogen and progestin combinations may raise serum concentrations of binding proteins (e.g., thyroid-binding globulin, corticosteroid-binding globulin), which may reduce the free thyroid or corticosteroid hormone levels. Patients with hypothyroidism and hypoadrenalism may require higher doses of thyroid hormone or cortisol replacement therapy, respectively.
The use of estrogen and progestin may also affect the levels of sex hormone-binding globulin, coagulation factors, lipids, and glucose [see Pharmacodynamics (12.2)].
5.12 Risk of Allergic Reactions Due to the Inactive Ingredient (FD&C Yellow No. 5)
ORIAHNN contains FD&C Yellow No. 5 (tartrazine), which may cause allergic-type reactions (including bronchial asthma) in certain susceptible persons. Although the overall incidence of FD&C Yellow No. 5 (tartrazine) sensitivity in the general population is low, it is frequently seen in patients who also have aspirin hypersensitivity.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed elsewhere in labeling:
- Thromboembolic Disorders and Vascular Events [see Warnings and Precautions (5.1)]
- Bone Loss [see Warnings and Precautions (5.2)]
- Suicidal Ideation, Suicidal Behavior, and Exacerbation of Mood Disorders [see Warnings and Precautions (5.4)]
- Hepatic Transaminase Elevations [see Warnings and Precautions (5.5)]
- Elevated Blood Pressure [see Warnings and Precautions (5.6)]
- Effects on Carbohydrate and Lipid Metabolism [see Warnings and Precautions (5.9)]
- Alopecia [see Warnings and Precautions (5.10)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety of ORIAHNN was evaluated in two 6-month, randomized, double-blind, placebo-controlled trials (Studies UF-1 and UF-2), in which 790 premenopausal women received at least 1 dose of ORIAHNN (n=395), elagolix 300 mg twice daily (n=199), or placebo (n=196) [see Clinical Studies (14)]. Women who completed 6-month treatment in either Study UF-1 or Study UF-2 and met eligibility criteria (n=433) entered a 6-month extension study (Study UF-3), receiving either ORIAHNN (n=276) or elagolix 300 mg twice daily (n=157). Elagolix 300 mg twice daily is not an approved dosage but was included as a reference arm. A total of 341 women received ORIAHNN for 6 months and 182 women received ORIAHNN for 12 months.
Serious Adverse Events
Serious adverse events were reported in three (0.8%) ORIAHNN-treated women in Studies UF-1 and UF-2. Two women had heavy menstrual bleeding and required blood transfusion due to anemia (0.5%) and one woman with history of bariatric surgery had a laparoscopic cholecystectomy due to cholelithiasis.
In Study UF-3, two women were diagnosed with breast cancer. One woman had completed 6 months of treatment with ORIAHNN in Study UF-1 and received 34 additional days of ORIAHNN in Study UF-3 when diagnosed. The second woman had received placebo in Study UF-2 and completed 6 months of ORIAHNN in Study UF-3 when diagnosed [see Warnings and Precautions (5.3)].
Adverse Reactions Leading to Study Discontinuation
In Studies UF-1 and UF-2, the discontinuation rate due to adverse reactions was 10% among ORIAHNN-treated women and 7% among placebo-treated women. The most common adverse reactions leading to study drug discontinuation in the ORIAHNN group were nausea (1%), headache (1%), alopecia (1%), metrorrhagia (1%), menorrhagia (1%), and hot flush (1%). One event each of the following adverse reactions led to study drug discontinuation: affect lability, angina pectoris, depression, hepatic enzyme increased, homicidal ideation, hypertension, irritability, thrombosis.
In women who received ORIAHNN in Studies UF-1 or UF-2 and then in Study UF-3, 4% discontinued treatment due to adverse reactions. Three women discontinued due to serious adverse events (one each for breast cancer, menorrhagia with pelvic pain, and hysterectomy).
Common Adverse Reactions
Adverse reactions reported in ≥5% of ORIAHNN-treated women in Studies UF-1 and UF-2 and at a greater frequency than placebo-treated women are presented in Table 1.
Table 1. Adverse Reactions that Occurred in at Least 5% of Women with Uterine Fibroids Who Received ORIAHNN in Studies UF-1 and UF-2 and at a Greater Incidence Than Placebo Adverse Reaction ORIAHNN
N=395Placebo
N=196Hot flush 22% 9% Headache 9% 7% Fatigue 6% 4% Metrorrhagia 5% 1% The most commonly reported adverse reactions in the blinded extension trial (Study UF-3) were consistent with those in the placebo-controlled trials.
Less Common Adverse Reactions
In Studies UF-1 and UF-2, adverse reactions reported in ≥3% and <5% in the ORIAHNN group and greater incidence than the placebo group included: libido decreased, arthralgia, hypertension, alopecia, mood swings, influenza, abdominal distension, upper respiratory tract infection, menorrhagia, vomiting, and weight increased.
Thromboembolic and Vascular Events
In the Studies UF-1, UF-2, and UF-3, two (0.4%) thrombotic events occurred in 453 ORIAHNN-treated patients (thrombosis in the calf and pulmonary embolism) [see Warnings and Precautions (5.1)]. One obese woman developed thrombosis in the left calf after 30 days of treatment with ORIAHNN. Another woman developed a pulmonary embolism after taking ORIAHNN for approximately 8 months.
Bone Loss
The effect of ORIAHNN on BMD was assessed by dual-energy X-ray absorptiometry (DXA).
In Studies UF-1 and UF-2, there was a greater decrease in BMD in women treated with ORIAHNN for 6 months compared to women treated with placebo. In Study UF-3, continued bone loss was observed in some women who received ORIAHNN for 12 consecutive months. The mean percent change from baseline in lumbar spine BMD at Month 6 (Studies UF-1 and UF-2) and Month 12 (Study UF-3) is presented in Table 2.
Table 2. Mean Percent Change (On-Treatment) from Baseline in Lumbar Spine BMD in Women with Fibroids at Month 6 in Studies UF-1 and UF-2 and Month 12 in Study UF-3 Studies UF-1 and UF-2
Treatment Month 6Study UF-3
Treatment Month 12Placebo ORIAHNN ORIAHNN Number of Subjects 150 305 175 Percent Change from Baseline -0.1 -0.7 -1.5 Treatment
Difference, %
(95% CI)-0.6
(-1.0, -0.1)CI: Confidence interval Following 12 months of ORIAHNN treatment in Study UF-3, a decline in lumbar spine BMD of >3% was seen in 27% (48/175) of women and a decline of ≥8% was seen in 1.7% (3/175) of women.
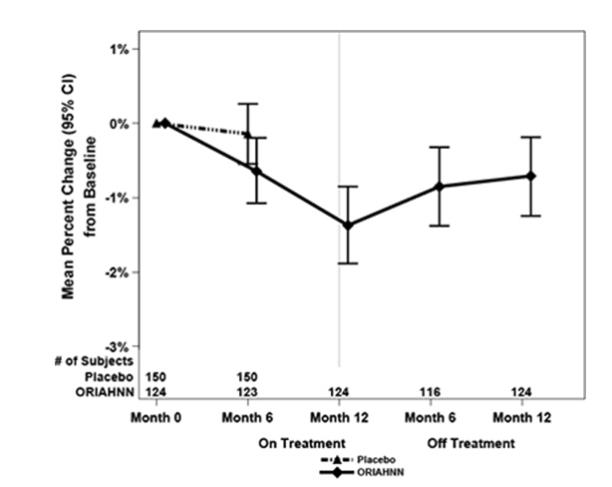
To assess for recovery, the change in BMD over time was analyzed for women who received continuous ORIAHNN treatment for up to 12 months and were then followed after cessation of therapy for an additional 12 months in Study UF-3 (Figure 1). The LS mean percent change from baseline in BMD 12 months after cessation of therapy was -0.72 (95% CI -1.2, -0.2), -0.59 (-1.0, -0.2), and -0.95 (-1.6, -0.3) at the lumbar spine, total hip, and femoral neck, respectively. Twelve months after cessation of ORIAHNN, continued bone loss was observed at the lumbar spine, total hip, and femoral neck in 24%, 32%, and 40% of women, respectively. Partial recovery was observed in 46%, 33%, and 38% and full recovery was observed in 30%, 35%, and 22% of women at these same sites. The time to maximum recovery in women who partially recovered is unknown.
Figure 1. Mean Percent Change From Baseline in Lumbar Spine BMD in Women Who Received 12 Months of ORIAHNN (On-Treatment) and 12 Months of Follow Up (Off Treatment)
Suicidal Ideation, Suicidal Behavior, and Exacerbation of Mood Disorders
In the placebo-controlled trials (Studies UF-1 and UF-2), ORIAHNN was associated with adverse mood changes. Depression, depressed mood, and/or tearfulness were reported in 3% of ORIAHNN-treated women compared to 1% of placebo-treated women. One woman treated with lower dose elagolix alone for another disease completed suicide 2 days after elagolix discontinuation.
Hepatic Transaminase Elevations
In Studies UF-1 and UF-2, elevations of serum ALT and AST with no concurrent elevations of bilirubin were reported.
- ALT elevations to at least 3 times the upper limit of normal (ULN) occurred in 1.1% (4/379) of ORIAHNN-treated women and no placebo-treated women. Peak elevation of ALT almost 8 times the ULN was reported in 1 ORIAHNN-treated woman.
- AST elevations to at least 3 times the ULN occurred in 5/379 (1.3%) in ORIAHNN-treated women and no placebo-treated women. Peak elevation of AST 6 times the ULN was reported in 1 ORIAHNN-treated woman.
Blood Pressure Elevations
There were more ORIAHNN-treated women with systolic blood pressure ≥ 160 mmHg (7.1%) and diastolic blood pressure ≥ 100 mmHg (11.3%) compared to placebo-treated women (3.7% and 6.3%, respectively). The incidence of hypertensive adverse reactions was 3.8% in ORIAHNN-treated women and 3.1% placebo-treated women. One ORIAHNN-treated woman in Study UF-1, with no prior history but with elevated cholesterol levels, had severe hypertension (BP 204/112) and chest pain. ECG was negative. Her hypertension was controlled with anti-hypertensives and she completed Study UF-3.
Changes in Lipid Parameters
Increases in total cholesterol, low-density lipoprotein cholesterol (LDL-C), serum triglycerides, and apolipoprotein B were noted during ORIAHNN treatment in Studies UF-1 and UF-2.
Of the women with Grade 0 LDL-C (<130 mg/dL) at baseline, 1/313 (0.3%) ORIAHNN-treated woman shifted to Grade 3 (≥ 190 mg/dL) compared to no placebo-treated woman. Of those with Grade 1 LDL-C (130 to <160 mg/dL) at baseline, 9/54 (16.7%) ORIAHNN-treated women shifted to Grade 3 compared to no placebo-treated woman. Of those with Grade 2 LDL-C (160 to <190 mg/dL) at baseline, 7/10 (70%) ORIAHNN-treated women shifted to Grade 3 compared to 1/5 (20%) placebo-treated woman.
Alopecia
In Phase 3 placebo-controlled clinical trials (Studies UF-1 and UF-2), 3.5% (14/395) of ORIAHNN-treated women experienced alopecia, hair loss, or hair thinning compared to 1.0% (2/196) of placebo-treated women. No specific pattern in hair loss was observed. In almost one-third (4/14) of affected ORIAHNN-treated women, alopecia was a reason for study drug discontinuation; no placebo-treated women discontinued because of alopecia. In ORIAHNN-treated women, 79% of the cases were mild and 21% were moderate in severity. Hair loss was ongoing at the end of the study for 4 out of 14 women (29%). Of these 4 women, one discontinued treatment due to hair loss, two had ongoing hair loss 12 months after discontinuing ORIAHNN, and one was lost to follow-up. In the remaining 10 women (71%), hair loss either resolved while on treatment or resolved within 24 days to approximately 9 months after discontinuing ORIAHNN.
Resumption of Menses after Discontinuation
After six months of ORIAHNN treatment, resumption of menses was reported by 39%, 68%, and 73% of women within 1, 2, and 6 months, respectively, in Study UF-1 and 39%, 85%, and 92% within 1, 2, and 6 months, respectively, in Study UF-2.
After 12 months of therapy with ORIAHNN (Study UF-1 or Study UF-2 then Study UF-3), resumption of menses was reported by 43%, 82%, and 90% of women within 1, 2, and 6 months after stopping treatment, respectively.
Whether those who did not resume having menses transitioned to a peri-postmenopausal status is unknown.
- Thromboembolic Disorders and Vascular Events [see Warnings and Precautions (5.1)]
-
7 DRUG INTERACTIONS
7.1 Potential for ORIAHNN to Affect Other Drugs
Elagolix (a component of ORIAHNN) is:
- A weak to moderate inducer of cytochrome P450 (CYP3A). Co-administration with ORIAHNN may decrease plasma concentrations of drugs that are substrates of CYP3A.
- A weak inhibitor of CYP2C19. Co-administration with ORIAHNN may increase plasma concentrations of drugs that are substrates of CYP2C19 (see Table 3).
- An inhibitor of efflux transporter P-glycoprotein (P-gp). Co-administration with ORIAHNN may increase plasma concentrations of drugs that are substrates of P-gp (see Table 3).
The effects of co-administration of ORIAHNN on concentrations of concomitant drugs and the clinical recommendations for these drug interactions are summarized in Table 3.
Table 3. Drug Interactions: Effects of ORIAHNN on Other Drugs Concomitant
Drug Class:
Drug NameEffect on
Plasma
Exposure of
Concomitant
DrugClinical Recommendations Cardiac
glycosides:
digoxin↑ digoxin Increase monitoring of digoxin concentrations and potential signs and symptoms of clinical toxicity when initiating ORIAHNN in patients who are taking digoxin. If ORIAHNN is discontinued, increase monitoring of digoxin concentrations. Benzodiazepines:
oral midazolam↓ midazolam Consider increasing the dose of midazolam by no more than 2-fold and individualize midazolam therapy based on the patient’s response. Statins:
rosuvastatin↓ rosuvastatin Monitor lipid levels and adjust the dose of rosuvastatin, if necessary. Proton pump
inhibitors:
omeprazole↑ omeprazole No dose adjustment needed for omeprazole 40 mg once daily when co-administered with ORIAHNN. When ORIAHNN is used concomitantly with higher doses of omeprazole, consider dosage reduction of omeprazole. See Tables 6 and 7 [see Clinical Pharmacology (12.3)].
The direction of the arrow indicates the direction of the change in the area under the curve (AUC) (↑= increase, ↓ = decrease).7.2 Potential for Other Drugs to Affect ORIAHNN
Elagolix (a component of ORIAHNN) is a substrate of CYP3A, P-gp, and OATP1B1; estradiol and norethindrone acetate are metabolized partially by CYP3A [see Clinical Pharmacology (12.3)]. Concomitant use of ORIAHNN with:
- Strong CYP3A inducers may decrease elagolix, estradiol, and norethindrone plasma concentrations and may result in a decrease of the therapeutic effects of ORIAHNN.
- Rifampin is not recommended. The concomitant use of rifampin increased plasma concentrations of elagolix [see Clinical Pharmacology (12.3)].
- Strong CYP3A inhibitors are not recommended. Concomitant use of ORIAHNN with strong CYP3A inhibitors may increase elagolix, estradiol, and norethindrone plasma concentrations and increase the risk of adverse reactions.
- OATP1B1 inhibitors that are known or expected to significantly increase elagolix plasma concentrations is contraindicated due to increased risk of elagolix-associated adverse reactions [see Contraindications (4)].
- A weak to moderate inducer of cytochrome P450 (CYP3A). Co-administration with ORIAHNN may decrease plasma concentrations of drugs that are substrates of CYP3A.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Use of ORIAHNN is contraindicated in pregnant women. Exposure to elagolix early in pregnancy may increase the risk of early pregnancy loss. Discontinue ORIAHNN if pregnancy occurs during treatment.
The limited human data with the use of elagolix in pregnant women are insufficient to determine whether there is a risk for major birth defects or miscarriage [see Data].
When pregnant rats and rabbits were orally dosed with elagolix during the period of organogenesis, postimplantation loss was observed in pregnant rats at doses 12 times the maximum recommended human dose (MRHD). Spontaneous abortion and total litter loss were observed in rabbits at doses 4 and 7 times the MRHD. There were no structural abnormalities in the fetuses at exposures up to 25 and 7 times the MRHD for the rat and rabbit, respectively [see Data].
Data
Human Data
There was one pregnancy reported in the 453 women who received ORIAHNN in the Phase 3 uterine fibroids clinical trials. The pregnancy resulted in a spontaneous abortion and the estimated fetal exposure to ORIAHNN occurred during the first 18 days of pregnancy.
Animal Data
Embryofetal development studies were conducted in the rat and rabbit. Elagolix was administered by oral gavage to pregnant rats (25 animals/dose) at doses of 0, 300, 600, and 1200 mg/kg/day and to rabbits (20 animals/dose) at doses of 0, 100, 150, and 200 mg/kg/day during the period of organogenesis (gestation day 6-17 in the rat and gestation day 7-20 in the rabbit).
In rats, maternal toxicity was present at all doses and included six deaths and decreases in body weight gain and food consumption. Increased postimplantation losses were present in the mid dose group, which was 12 times the MRHD based on AUC. In rabbits, three spontaneous abortions and a single total litter loss were observed at the highest maternally toxic dose, which was 7 times the MRHD based on AUC. A single total litter loss occurred at a lower non-maternally toxic dose of 150 mg/kg/day, which was 4 times the MRHD.
No fetal malformations were present at any dose level tested in either species even in the presence of maternal toxicity. At the highest doses tested, the exposure margins were 25 and 7 times the MRHD for the rat and rabbit, respectively. However, because elagolix binds poorly to the rat gonadotropin-releasing hormone (GnRH) receptor (~1000 fold less than to the human GnRH receptor), the rat study is unlikely to identify pharmacologically mediated effects of elagolix on embryofetal development. The rat study is still expected to provide information on potential non-target-related effects of elagolix.
In a pre- and postnatal development study in rats, elagolix was given in the diet to achieve doses of 0, 100, and 300 mg/kg/day (25 per dose group) from gestation day 6 to lactation day 20. There was no evidence of maternal toxicity. At the highest dose, two dams had total litter loss, and one failed to deliver. Pup survival was decreased from birth to postnatal day 4. Pups had lower birth weights, and lower body weight gains were observed throughout the pre-weaning period at 300 mg/kg/day. Smaller body size and effect on startle response were associated with lower pup weights at 300 mg/kg/day. Post-weaning growth, development, and behavioral endpoints were unaffected.
Maternal plasma concentrations in rats on lactation day 21 at 100 and 300 mg/kg/day (47 and 125 ng/mL) were 0.04-fold and 0.1-fold the maximal elagolix concentration (Cmax) in humans at the MRHD. Because the exposures achieved in rats were much lower than the human MRHD, this study is not predictive of potentially higher lactational exposure in humans.
8.2 Lactation
Risk Summary
There is no information on the presence of elagolix or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. When estrogen and progestins are administered to lactating women, these compounds and/or their metabolites are detected in human milk and can reduce milk production in breast-feeding females. This reduction can occur at any time but is less likely to occur once breast-feeding is well established. Advise the nursing female to use non-hormonal contraception until she discontinues breast-feeding. The developmental and health benefits of breast-feeding should be considered along with the mother’s clinical need for ORIAHNN and any potential adverse effects on the breast-fed child from ORIAHNN or from the underlying maternal condition [see Data].
Data
There is no information on the presence of elagolix or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. Estrogen administration to nursing women has been shown to decrease the quantity and quality of the breast milk. Detectable amounts of estrogen and progestin have been identified in the breast milk of women receiving estrogen and progestin combinations.
There are no adequate animal data on excretion of elagolix in milk.
8.3 Females and Males of Reproductive Potential
Based on the mechanism of action of elagolix, there is a risk of early pregnancy loss if ORIAHNN is administered to a pregnant woman [see Use in Specific Populations (8.1), Clinical Pharmacology (12.1)].
Pregnancy Testing
ORIAHNN may delay the ability to recognize the occurrence of a pregnancy because it may reduce the intensity, duration, and amount of menstrual bleeding [see Adverse Reactions (6.1)]. Exclude pregnancy before initiating treatment with ORIAHNN. Perform pregnancy testing if pregnancy is suspected during treatment with ORIAHNN and discontinue treatment if pregnancy is confirmed [see Contraindications (4) and Warnings and Precautions (5.8)].
Contraception
Advise women to use non-hormonal contraception during treatment with ORIAHNN and for 28 days after discontinuing ORIAHNN [see Warnings and Precautions (5.8)].
8.4 Pediatric Use
Safety and effectiveness of ORIAHNN in pediatric patients have not been established.
8.6 Renal Impairment
No dose adjustment of ORIAHNN is required in women with any degree of renal impairment or end-stage renal disease (including women on dialysis) [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
ORIAHNN is contraindicated in women with any hepatic impairment or disease [see Contraindications (4)]. The use of estradiol (a component of ORIAHNN) in patients with hepatic impairment, compared to patients with normal hepatic function, is expected to increase the blood levels of estradiol and increase the risk of estradiol-associated adverse reactions. Additionally, the use of elagolix (a component of ORIAHNN) in patients with moderate and severe hepatic impairment, compared to patients with normal hepatic function, increased elagolix exposures 3-fold and 7-fold, respectively, and this increases the risk of elagolix-associated adverse reactions [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
Overdosage of estrogen and progestin combination products may cause nausea, vomiting, breast tenderness, abdominal pain, drowsiness, fatigue, and withdrawal bleeding. In case of ORIAHNN overdose, monitor the patient for any signs or symptoms of adverse reactions and initiate appropriate symptomatic treatment, as needed.
-
11 DESCRIPTION
ORIAHNN consists of two capsules: one to be taken orally in the morning (AM) and one to be taken orally in the evening (PM). The AM capsule is white and yellow and contains 300 mg elagolix (equivalent to 310.4 mg of elagolix sodium), 1 mg estradiol, and 0.5 mg norethindrone acetate. The PM capsule is white and light blue and contains 300 mg of elagolix (equivalent to 310 mg of elagolix sodium).
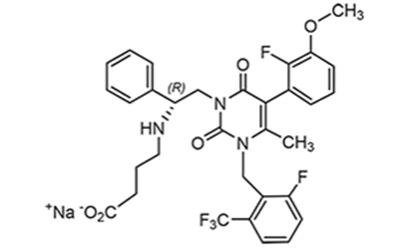
Elagolix
Elagolix sodium is the sodium salt of the active moiety elagolix, a nonpeptide small molecule, GnRH receptor antagonist. Elagolix sodium is chemically described as sodium 4-({(1R)-2-[5-(2-fluoro-3-methoxyphenyl)-3-{[2-fluoro-6-(trifluoromethyl)phenyl]methyl}-4-methyl-2,6-dioxo-3,6-dihydropyrimidin-1(2H)-yl]-1-phenylethyl}amino)butanoate. Elagolix sodium has a molecular formula of C32H29F5N3O5Na and a molecular weight of 653.58. Elagolix free acid has a molecular formula of C32H30F5N3O5 and a molecular weight of 631.60.
Elagolix sodium has the following structural formula:
Elagolix sodium is a white to off-white to light yellow powder and is freely soluble in water.
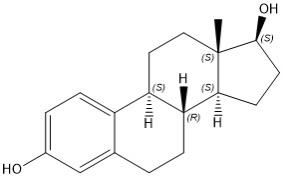
Estradiol
Estradiol (E2), an estrogen, is a white or almost white crystalline powder. Its chemical name is estra-1,3,5(10)-triene-3,17β-diol with the molecular formula of C18H24O2, and molecular weight of 272.38. The structural formula of E2 is as follows:
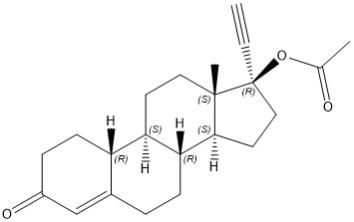
Norethindrone acetate
Norethindrone acetate (NETA), a progestin, is a white or yellowish white crystalline powder. Its chemical name is 17β-acetoxy-19-nor-17α-pregn-4-en-20-yn-3-one with the molecular formula of C22H28O3 and molecular weight of 340.46.
ORIAHNN morning (AM) capsules contain the following inactive ingredients: anhydrous sodium carbonate, polyethylene glycol 3350, crospovidone, colloidal silicon dioxide, magnesium stearate, polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, purified water, lactose monohydrate, starch (corn), copovidone, talc, hypromellose, triacetin, and gelatin capsule shell. The capsule shell contains the following ingredients: FD&C Red #40, FD&C Yellow #5 [see Warnings and Precautions (5.12)], FD&C Yellow #6, titanium dioxide, gelatin, and printing ink (shellac, dehydrated alcohol, isopropyl alcohol, butyl alcohol, propylene glycol, strong ammonia solution, black iron oxide, potassium hydroxide, and purified water).
ORIAHNN evening (PM) capsules contain the following inactive ingredients: anhydrous sodium carbonate, polyethylene glycol 3350, crospovidone, colloidal silicon dioxide, magnesium stearate, polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, purified water, and gelatin capsule shell. The capsule shell contains the following ingredients: FD&C Blue #2, FDA/E172 yellow iron oxide, titanium dioxide, gelatin, and printing ink (shellac, dehydrated alcohol, isopropyl alcohol, butyl alcohol, propylene glycol, strong ammonia solution, black iron oxide, potassium hydroxide, and purified water).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
ORIAHNN combines elagolix and estradiol/norethindrone acetate (E2/NETA), a combination of estrogen and progestin.
Elagolix is a GnRH receptor antagonist that inhibits endogenous GnRH signaling by binding competitively to GnRH receptors in the pituitary gland. Administration of elagolix results in dose-dependent suppression of luteinizing hormone (LH) and follicle-stimulating hormone (FSH), leading to decreased blood concentrations of the ovarian sex hormones estradiol and progesterone and reduces bleeding associated with uterine fibroids.
E2 acts by binding to nuclear receptors that are expressed in estrogen-responsive tissues. As a component of ORIAHNN, the addition of exogenous estradiol may reduce the increase in bone resorption and resultant bone loss that can occur due to a decrease in circulating estrogen from elagolix alone.
Progestins such as NETA act by binding to nuclear receptors that are expressed in progesterone-responsive tissues. As a component of ORIAHNN, NETA may protect the uterus from the potential adverse endometrial effects of unopposed estrogen.
12.2 Pharmacodynamics
Estradiol and norethindrone acetate (components of ORIAHNN) may have the following effects:
- Increased thyroxin-binding globulin levels leading to [see Warnings and Precautions (5.11)]:
○ Increased circulating total thyroid hormone levels as measured by protein-bound iodine (PBI), thyroxine (T4) levels (by column or by radioimmunoassay), or triiodothyronine (T3) levels by radioimmunoassay
○ Decreased T3 resin uptake
○ Unaltered free T4 and free T3 concentrations in women with normal thyroid function [see Warnings and Precautions (5.11)].
- Elevated corticosteroid-binding globulin (CBG) and sex hormone-binding globulin (SHBG) leading to increased total circulating corticosteroids and sex steroids, respectively [see Warnings and Precautions (5.11)].
- Possible decreased free testosterone concentrations.
- Possible increased other plasma proteins concentrations (angiotensinogen/renin substrate, alpha-1 antitrypsin, ceruloplasmin).
- Increased plasma high-density lipoprotein (HDL) and HDL2 cholesterol subfraction concentration, reduced low-density lipoprotein concentration, increased triglyceride levels [see Warnings and Precautions (5.9)].
- Accelerated prothrombin time, partial thromboplastin time, and platelet aggregation time; increased platelet count; increased factors II, VII antigen, VIII coagulant activity, IX, X, XII, VII-X complex, and beta-thromboglobulin; decreased levels of anti-factor Xa and antithrombin III, decreased antithrombin III activity, increased levels of fibrinogen and fibrinogen activity; increased plasminogen antigen and activity.
Cardiac Electrophysiology
The effect of elagolix (a component of ORIAHNN) on the QTc interval was evaluated in a randomized, placebo- and positive-controlled, open-label, single-dose, crossover thorough QTc study in 48 healthy adult premenopausal women. Elagolix concentrations in women given a single dose of 1200 mg were 9 times higher than the concentration in women given elagolix 300 mg twice daily. There was no clinically relevant prolongation of the QTc interval.
The effect of estradiol and norethindrone acetate (two components of ORIAHNN) on the QTc interval has not been studied.
12.3 Pharmacokinetics
The pharmacokinetic properties of ORIAHNN in healthy subjects are summarized in Table 4.
The pharmacokinetic parameters under fasting conditions are summarized in Table 5.
Table 4. Pharmacokinetic Properties of ORIAHNN in Healthy Subjects Absorption Elagolix Estradiola Norethindrone Tmax (h)b,c 1.5 (1.0 – 4.0) 2.0 (0.0 - 10.0) 1.0 (0.5 - 2.0) Effect of Food High-fat
meald (relative to
fasting)AUC: ↓25%,
Cmax: ↓36%AUC: no change,
Cmax: ↓23%AUC: ↑23%,
Cmax: ↓50%Distribution % Bound to
human plasma
proteins80 98 97 Blood-to-plasma ratio 0.6 NA NA Metabolism Metabolism CYP3A (major)
Minor pathways include:
CYP2D6, CYP2C8, and
uridine glucuronosyl transferases (UGTs)CYP3A (partial)
Other pathways
include: sulfation and
glucuronidationCYP3A (partial) Elimination Major route of
eliminationHepatic metabolism Hepatic metabolism Hepatic metabolism Terminal phase
elimination
half-life (t1/2) (h)c, e5.9 ± 2.1 14.5 ± 6.6 9.2 ± 4.0 % of dose
excreted in urine<3 NA NA % of dose
excreted in feces90 NA NA NA=not available
aBaseline adjusted unconjugated estradiol
bMedian and range
cFollowing administration of a single dose under fasting conditions
dHigh-fat meal is approximately 826 kcal, 52% fat.
eMean ± SDTable 5. Mean (%CV) Pharmacokinetic Parameters of ORIAHNN Pharmacokinetic Parameter
(Units)Elagolix 300 mg
Twice Dailya
N = 8Estradiolb 1 mg
N = 163Norethindroneb
0.5 mg
N = 163Cmax (ng/mL) 1200 (45) 0.06 (52) 6.1 (35) AUCτ (ng●hr/mL) 2826 (44) 0.86 (38) 23.8 (48) aData obtained at steady state (Day 21); AUCτ represents the area under the plasma concentration-time curve from 0 to 12 hours post dose.
bData obtained following single dose administration; AUCτ represents AUC from 0 to 24 hours post dose; estradiol: baseline adjusted unconjugated estradiol.
CV: Coefficient of variation
Cmax: Plasma peak concentrationSpecific Populations
Patients with Renal Impairment
Elagolix exposures (Cmax and AUC) were not altered by renal impairment. The elagolix mean plasma exposures were similar for women with moderate to severe or end-stage renal disease (including women on dialysis) compared to women with normal renal function.
The effect of renal impairment on the pharmacokinetics of E2/NETA has not been studied.
Patients with Hepatic Impairment
Elagolix exposures (Cmax and AUC) were similar between women with normal hepatic function and women with mild hepatic impairment. Elagolix exposures in women with moderate and severe hepatic impairment were approximately 3-fold and 7-fold, respectively, higher than exposures from women with normal hepatic function.
The use of estradiol in patients with hepatic impairment, compared to patients with normal hepatic function, is expected to increase the estradiol blood levels [see Warnings and Precautions (5.5) and Use in Specific Populations (8.7)].
Racial or Ethnic Groups
No clinically meaningful difference in the pharmacokinetics of elagolix between White and Black subjects or between Hispanics and others was observed. There is no clinically meaningful difference in the pharmacokinetics of elagolix between Japanese and Han Chinese subjects. The effect of race/ethnicity on the pharmacokinetics of E2/NETA has not been studied.
Body Weight/Body Mass Index
Body weight or body mass index does not affect the pharmacokinetics of elagolix.
The effect of body weight/body mass index on the pharmacokinetics of E2/NETA has not been studied.
Drug Interaction Studies
Drug interaction studies were performed with elagolix and other drugs likely to be co-administered and with drugs commonly used as probes for pharmacokinetic interactions. Tables 6 and 7 summarize the pharmacokinetic effects when elagolix was co-administered with these drugs.
Table 6. Drug Interactions: Change in Pharmacokinetics of Elagolix in the Presence of Co-administered Drugs Co-administered
DrugCo-administered
Drug RegimenElagolix Regimen N Ratio (90% CI)* Ketoconazole 400 mg once daily 150 mg single dose& 11 Cmax AUC 1.77
(1.48 - 2.12)2.20
(1.98 - 2.44)Rifampin** 600 mg
single dose150 mg single dose& 12 4.37
(3.62 - 5.28)5.58
(4.88 - 6.37)600 mg
once daily2.00
(1.66 - 2.41)1.65
(1.45 - 1.89)CI: Confidence interval
& The elagolix dose in these studies was 0.5 times the approved dose in ORIAHNN (0.25 times the total approved daily dosage of elagolix in ORIAHNN)
* ratios for Cmax and AUC compare co-administration of the medication with elagolix vs. administration of elagolix alone.
**A single dose of 600 mg rifampin inhibits OATP1B1; 600 mg once daily dose of rifampin inhibits OATP1B1 and induces CYP3A.No clinically significant changes in elagolix exposures were observed when elagolix 300 mg twice daily was co-administered with rosuvastatin (20 mg once daily), sertraline (25 mg once daily), or fluconazole (200 mg single dose). The effect of co-administered rosuvastatin, sertraline, or fluconazole on E2/NETA has not been studied.
Table 7. Drug Interactions: Change in Pharmacokinetics of Co-administered Drug in the Presence of Elagolix Co-administered
DrugCo-administered
Drug RegimenElagolix
RegimenN Ratio (90% CI)* Digoxin 0.5 mg
single dose200 mg
twice daily
x 10 days11 Cmax AUC 1.71
(1.53 - 1.91)1.26
(1.17 - 1.35)Rosuvastatin 20 mg
once daily300 mg
twice daily
x 7 days10 0.99
(0.73 - 1.35)0.60
(0.50 - 0.71)Midazolam 2 mg
single dose300 mg
twice daily
x 11 days20 0.56
(0.51 - 0.62)0.46
(0.41 - 0.50)2 mg
single dose150 mg
once daily
x 13 days11 0.81
(0.74 - 0.89)0.65
(0.58 - 0.72)Omeprazole 40 mg single dose 300 mg
twice daily
x 9 days20 1.95
(1.50 - 2.53)1.78
(1.39 - 2.27)CI: Confidence interval
*ratios for Cmax and AUC compare co-administration of the medication with elagolix vs.
administration of the medication alone.No clinically significant changes in sertraline, fluconazole, bupropion, or transdermal patch E2/NETA 0.51/4.8 mg exposures were observed when co-administered with elagolix 300 mg twice daily.
12.5 Pharmacogenomics
Hepatic uptake of elagolix (a component of ORIAHNN) involves the OATP1B1 transporter protein. Higher plasma concentrations of elagolix have been observed in patients who have two reduced function alleles of the gene that encodes OATP1B1 (SLCO1B1 521T>C) (these patients are likely to have reduced hepatic uptake of elagolix and thus, higher plasma elagolix concentrations). The frequency of the SLCO1B1 521C/C genotype is generally less than 5% in most racial/ethnic groups. Women with this genotype are expected to have approximately 2-fold higher elagolix mean concentrations compared to women with normal transporter function (i.e., SLCO1B1 521T/T genotype). Adverse effects of elagolix have not been fully evaluated in subjects who have two reduced function alleles of the gene that encodes OATP1B1 (SLCO1B1 521T>C).
- Increased thyroxin-binding globulin levels leading to [see Warnings and Precautions (5.11)]:
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Elagolix
Two-year carcinogenicity studies conducted in mice (50, 150, or 500 mg/kg/day) and rats (150, 300, or 800 mg/kg/day) that administered elagolix by the dietary route revealed no increase in tumors in mice at up to 11.9-fold the MRHD based on AUC. In the rat, there was an increase in thyroid (male and female) and liver (males only) tumors at the high dose (7.7- to 8.1-fold the MRHD). The rat tumors were likely species-specific and of negligible relevance to humans.
Elagolix was not genotoxic or mutagenic in a battery of tests, including the in vitro bacterial reverse mutation assay, the in vitro mammalian cell forward mutation assay at the thymidine kinase (TK+/-) locus in L5178Y mouse lymphoma cells, and the in vivo mouse micronucleus assay.
In a fertility study conducted in the rat, there was no effect of elagolix on fertility at any dose (50, 150, or 300 mg/kg/day). Based on AUC, the exposure multiple for the MRHD in women compared to the highest dose of 300 mg/kg/day in female rats is approximately 2.9-fold. However, because elagolix has low affinity for the GnRH receptor in the rat [see Use in Specific Populations (8.1)], and because effects on fertility are most likely to be mediated via the GnRH receptor, these data have low relevance to humans.
E2/NETA
Long-term continuous administration of natural and synthetic estrogens in certain animal species increases the frequency of carcinomas of the breast, uterus, cervix, vagina, testis, and liver [see Warnings and Precautions (5.3)].
-
14 CLINICAL STUDIES
The efficacy of ORIAHNN in the management of heavy menstrual bleeding (HMB) associated with uterine fibroids was demonstrated in two randomized, double-blind, placebo-controlled studies [Study UF-1 (NCT02654054) and Study UF-2 (NCT02691494)] in which 790 premenopausal women with heavy menstrual bleeding received ORIAHNN (elagolix 300 mg, estradiol 1 mg, and norethindrone acetate 0.5 mg in the morning and elagolix 300 mg in the evening) or placebo for 6 months. Heavy menstrual bleeding at baseline was defined as having at least two menstrual cycles with greater than 80 mL of menstrual blood loss (MBL) as assessed by alkaline hematin (AH) method (an objective, validated measure to quantify MBL volume on sanitary products).
In Studies UF-1 and UF-2, the median age of enrolled women was 43 years (ranging from 25 to 53 years); 68% of the women were Black or African American, 29% were White, and 3% were other races.
Menstrual Blood Loss
The primary endpoint in both studies was the proportion of responders, defined as women who achieved both 1) MBL volume less than 80 mL at the Final Month and 2) 50% or greater reduction in MBL volume from Baseline to the Final Month. Final Month was defined as the last 28 days before and including the last treatment visit date or the last dose date. A higher proportion of ORIAHNN-treated women were responders compared to placebo-treated women (Table 8).
Table 8. Proportion of Responders for Reduction in MBL Volume at Final Month in Women with Uterine Fibroids (Studies UF-1 and UF-2) Study UF-1 Study UF-2 ORIAHNN
N=206Placebo
N=102ORIAHNN
N=189Placebo
N=94Women with MBL volume
< 80 mL and ≥ 50% reduction
in MBL volume from Baseline
to the Final Month68.5% 8.7% 76.5% 10.5% Difference from placebo %
95% CI
P-value59.8%
(51.1, 68.5)
< 0.00166.0%
(57.1, 75.0)
< 0.001CI: confidence interval Changes in MBL Volume
Treatment with ORIAHNN resulted in a reduction in mean MBL volume from Baseline at Months 1, 3, and 6 compared to placebo (see Figures 2 and 3).
Figure 2. Monthly Change from Baseline in MBL Volume in Women with Uterine Fibroids (Study UF-1)
Figure 3. Monthly Change from Baseline in MBL Volume in Women with Uterine Fibroids (Study UF-2)
In Study UF-1, mean baseline MBL was 238 mL for ORIAHNN and 255 mL for placebo. In Study UF-2, mean baseline MBL was 228 mL for ORIAHNN and 254 mL for placebo. Women taking ORIAHNN had a mean reduction of MBL volume from Baseline to Final Month in both Studies UF-1 and UF-2 compared to women taking placebo (Study UF-1: -177 mL for ORIAHNN and 1 mL for placebo; Study UF-2: -169 mL for ORIAHNN and -4 mL for placebo).
Suppression of Bleeding
In Studies UF-1 and UF-2, a greater proportion (57% and 61%, respectively) of women receiving ORIAHNN experienced suppression of bleeding, defined as no bleeding (but spotting allowed), at Final Month, compared to 4% and 5%, respectively, of women receiving placebo.
Hemoglobin (Hgb)
In Studies UF-1 and UF-2, a greater proportion of ORIAHNN-treated women who were anemic with baseline Hgb ≤ 10.5 g/dL achieved an increase > 2 g/dL in Hgb from Baseline to Month 6 compared to placebo-treated women (see Table 9). Over 90% of women with baseline Hgb ≤ 10.5 g/dL took supplemental iron.
Table 9. Proportion of Women with Uterine Fibroids with Baseline Hgb ≤ 10.5 g/dL and Increase > 2 g/dL in Hgb at Month 6 UF-1 UF-2 ORIAHNN
n=52
(N=206)Placebo
n=31
(N=102)ORIAHNN
n=48
(N=189)Placebo
n=24
(N=94)(%) at Month 6 62% 16% 50% 21% Difference from placebo %
95% CI
p-value45%
(27, 64)
< 0.00129%
(8, 51)
0.02CI: confidence interval
n: number of subjects with Hgb ≤10.5 g/dL at Baseline and had Hgb measurements at Month 6
N: number of subjects in each treatment arm -
16 HOW SUPPLIED/STORAGE AND HANDLING
ORIAHNN consists of two capsules: one to be taken in the morning (AM) and one to be taken in the evening (PM).
- morning (AM) capsules are white and yellow, printed with “EL300 AM” and contain elagolix 300 mg, estradiol 1 mg, and norethindrone acetate 0.5 mg.
- evening (PM) capsules are white and light blue, printed with “EL300 PM” and contain elagolix 300 mg.
ORIAHNN is packaged in weekly blister packs. Each blister pack contains seven AM capsules and seven PM capsules. Four blisters are packaged into a carton (NDC: 0074-1017-56).
Store at 20°C to 25°C (68°F to 77°F), excursions permitted to 15˚C to 30˚C (59˚F to 86˚F). [See USP Controlled Room Temperature].
Dispose unused medication via a take-back option if available. Otherwise, follow FDA instructions for disposing medication in the household trash, www.fda.gov/drugdisposal. Do NOT flush down the toilet.
- morning (AM) capsules are white and yellow, printed with “EL300 AM” and contain elagolix 300 mg, estradiol 1 mg, and norethindrone acetate 0.5 mg.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Thromboembolic Disorders and Vascular Events
Advise patients that use of estrogen and progestin combinations may increase the risk of thromboembolic disorders and vascular events, especially in women at high risk for these events [see Boxed Warning, Contraindications (4), Warnings and Precautions (5.1), and Adverse Reactions (6.1)].
Bone Loss
Advise patients about the risk of bone loss. Advise patients that supplementary calcium and vitamin D may be beneficial if dietary intake of calcium and vitamin D is not adequate. Advise patients that oral iron supplement should not be taken at the same time as calcium and vitamin D [see Warnings and Precautions (5.2) and Adverse Reactions (6.1)].
Suicidal Ideation and Exacerbation of Mood Disorders
Advise patients that suicidal ideation and exacerbation of mood disorders may occur with ORIAHNN use. Instruct patients with new onset or worsening depression, anxiety, or other mood changes to promptly seek medical attention [see Warnings and Precautions (5.3) and Adverse Reactions (6.1)].
Liver Injury
Advise patients to promptly seek medical attention in case of signs or symptoms that may reflect liver injury, such as jaundice [see Warnings and Precautions (5.5) and Adverse Reactions (6.1)].
Change in Menstrual Bleeding Pattern
Advise patients that ORIAHNN may delay the recognition of pregnancy because it may reduce the duration and amount of menstrual bleeding. Advise patients to use effective non-hormonal contraception while taking ORIAHNN, for 28 days after discontinuing ORIAHNN, and to discontinue ORIAHNN if pregnancy is diagnosed [see Warnings and Precautions (5.8) and Use in Specific Populations (8.1, 8.3)].
Alopecia
Advise patients that alopecia, hair loss, and hair thinning in no specific pattern, may occur with ORIAHNN use. Advise patients that hair loss and hair thinning may not resolve completely after stopping ORIAHNN. Advise patients to contact their healthcare provider if they have concerns about changes to their hair [see Warnings and Precautions (5.10) and Adverse Reactions (6.1)].
Drug Interactions
Advise patients to inform their healthcare providers of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products. Advise patients to avoid grapefruit juice while taking ORIAHNN [see Drug Interactions (7)].
ORIAHNN Missed Dose Instructions
Instruct patients about what to do in the event a dose is missed. See “If you miss a dose of ORIAHNN” section in FDA-approved Medication Guide.
ORIAHNN Disposal Instructions
Instruct patients to dispose of unused medication via a take-back option if available or to otherwise follow FDA instructions for disposing of medication in the household trash, www.fda.gov/drugdisposal, and not to flush down the toilet.
Manufactured by AbbVie Inc. North Chicago, IL 60064
ORIAHNN is a trademark of AbbVie Inc.
© 2025 AbbVie Inc. All rights reserved.
20096646
-
MEDICATION GUIDE
MEDICATION GUIDE
ORIAHNN® (or-ee-ahn)
(elagolix, estradiol, and norethindrone acetate capsules; elagolix capsules)
co-packaged for oral useWhat is the most important information I should know about ORIAHNN?
ORIAHNN may cause serious side effects, including:-
cardiovascular conditions
○ ORIAHNN may increase your chances of heart attack, stroke, or blood clots, especially if you are over 35 years of age and smoke, have uncontrolled high blood pressure, high cholesterol, diabetes, or are obese. Stop taking ORIAHNN and call your healthcare provider right away or go to the nearest hospital emergency room right away if you have:
■ leg pain or swelling that will not go away
■ sudden shortness of breath
■ double vision, bulging of the eyes, sudden blindness, partial or complete
■ pain or pressure in your chest, arm, or jaw
■ sudden, severe headache unlike your usual headaches
■ weakness or numbness in an arm or leg, or trouble speaking
-
bone loss (decreased bone mineral density)
○ While you are taking ORIAHNN, your estrogen levels may be low. Low estrogen levels can lead to bone mineral density loss.
○ If you have bone loss on ORIAHNN, your bone density may improve after you stop taking ORIAHNN, but complete recovery may not occur. It is unknown if these bone changes could increase your risk for broken bones as you age. For this reason, you should not take ORIAHNN for more than 24 months.
○ Your healthcare provider may order an X-ray test called a DXA scan to check your bone mineral density when you start taking ORIAHNN and periodically after you start.
○ Your healthcare provider may advise you to take vitamin D and calcium supplements as part of a healthy lifestyle that promotes bone health. Iron supplements should not be taken at the same time that you take vitamin D and calcium supplements.
-
effects on pregnancy
○ Do not take ORIAHNN if you are trying to become pregnant or are pregnant. It may increase the risk of early pregnancy loss.
○ If you think you may be pregnant, stop taking ORIAHNN right away and call your healthcare provider.
○ ORIAHNN can decrease your menstrual bleeding or result in no menstrual bleeding at all, making it hard to know if you are pregnant. Watch for other signs of pregnancy such as breast tenderness, weight gain, and nausea.
○ ORIAHNN does not prevent pregnancy. You will need to use effective methods of birth control while taking ORIAHNN and for 28 days after you stop taking ORIAHNN. Examples of effective methods can include condoms or spermicide, which do not contain hormones.
○ Talk to your healthcare provider about which birth control to use during treatment with ORIAHNN. Your healthcare provider may change the birth control you were on before you start taking ORIAHNN.
What is ORIAHNN?
ORIAHNN is a prescription medicine used to control heavy menstrual bleeding in premenopausal women (before “change of life” or menopause) with uterine fibroids.
It is not known if ORIAHNN is safe and effective in children under 18 years of age.Do not take ORIAHNN if you: - have or have had:
○ stroke or a heart attack
○ a problem that makes your blood clot more than normal
○ blood circulation disorder
○ certain heart valve problems or heart rhythm abnormalities that can cause blood clots to form in the heart
○ blood clots in your legs (deep vein thrombosis), lungs (pulmonary embolism), or eyes (retinal thrombosis)
○ high blood pressure not well controlled by medicine
○ diabetes with kidney, eye, nerve, or blood vessel damage
○ certain kinds of headaches with numbness, weakness, or changes in vision or have migraine headaches with aura if you are over age 35
○ breast cancer or any cancer that is sensitive to female hormones
○ osteoporosis
○ unexplained vaginal bleeding that has not been diagnosed. Your healthcare provider should check any unexplained vaginal bleeding to find out the cause.
○ liver problems including liver disease
○ smoke and are over 35 years old
- are taking medicines known as OATP1B1 inhibitors that are known or expected to significantly increase the blood levels of elagolix (an ingredient in ORIAHNN). Ask your healthcare provider if you are not sure if you are taking this type of medicine.
- have had a serious allergic reaction to elagolix, estradiol, norethindrone acetate, or any of the ingredients in ORIAHNN. Ask your healthcare provider if you are not sure.
- FD&C Yellow No.5 (tartrazine) is an ingredient in ORIAHNN which may cause an allergic type reaction such as bronchial asthma in some patients who are also allergic to aspirin. See the end of this Medication Guide for a complete list of ingredients in ORIAHNN.
Before you take ORIAHNN, tell your healthcare provider about all of your medical conditions, including if you:
- have or have had:
○ broken bones or other conditions that may cause bone problems.
○ depression, mood swings, or suicidal thoughts or behavior.
○ yellowing of the skin or eyes (jaundice) or jaundice caused by pregnancy (cholestasis of pregnancy).
- are scheduled for surgery. ORIAHNN may increase your risk of blood clots after surgery. Your healthcare provider may advise you to stop taking ORIAHNN before you have surgery. If this happens, talk to your healthcare provider about when to restart ORIAHNN after surgery.
- are pregnant or think you may be pregnant.
- are breastfeeding. It is not known if ORIAHNN can pass into your breastmilk. Talk to your healthcare provider about the best way to feed your baby if you take ORIAHNN.
Women on thyroid or cortisol replacement therapy may need increased doses of the hormone.
Know the medicines you take. Keep a list of your medicines with you to show to your healthcare provider and pharmacist when you get a new medicine.How should I take ORIAHNN?
- Take ORIAHNN exactly as your healthcare provider tells you to take it.
- Your healthcare provider will give you a pregnancy test before you start taking ORIAHNN or will have you start taking ORIAHNN within 7 days after you start your period.
- Take 1 white and yellow ORIAHNN capsule in the morning and 1 white and light blue ORIAHNN capsule in the evening each day.
- Take ORIAHNN at about the same time each morning and evening with or without food.
- If you take too much ORIAHNN, call your healthcare provider or go to the nearest hospital emergency room right away.
- Take the missed dose within 4 hours of the time that it was supposed to be taken. Then take the next dose at the usual time.
- If more than 4 hours have passed since you usually take the morning or evening dose, skip the missed dose. Take your next dose at the usual time.
- Do not take 2 doses to make up for the missed dose.
What should I avoid while taking ORIAHNN? - Avoid grapefruit and grapefruit juice during treatment with ORIAHNN since they may affect the level of ORIAHNN in your blood, which may increase side effects.
What are the possible side effects of ORIAHNN?
ORIAHNN may cause serious side effects including:
- See “What is the most important information I should know about ORIAHNN?”
-
suicidal thoughts, suicidal behavior, and worsening of mood. ORIAHNN may cause suicidal thoughts or actions. Call your healthcare provider or get emergency medical help right away if you have any of these symptoms, especially if they are new, worse, or bother you:
○ thoughts about suicide or dying
○ attempts to commit suicide
○ new or worse depression
○ new or worse anxiety
○ other unusual changes in behavior or mood
-
abnormal liver tests. Call your healthcare provider right away if you have any of these signs and symptoms of liver problems:
○ jaundice
○ dark amber-colored urine
○ feeling tired (fatigue or exhaustion)
○ nausea and vomiting
○ generalized swelling
○ right upper stomach area (abdomen) pain
○ bruising easily
-
high blood pressure. You should see your healthcare provider to check your blood pressure regularly.
-
gallbladder problems (cholestasis), especially if you had cholestasis of pregnancy.
-
increases in blood sugar, cholesterol and fat (triglyceride) levels.
-
hair loss (alopecia). Hair loss and hair thinning can happen while taking ORIAHNN and it can continue even after you stop taking ORIAHNN. It is not known if this hair loss or hair thinning is reversible. Talk to your healthcare provider if this is a concern for you.
- changes in laboratory tests including thyroid and other hormone, cholesterol, and blood clotting tests.
These are not all the possible side effects of ORIAHNN. For more information, ask your healthcare provider or pharmacist.
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store ORIAHNN?
- Store ORIAHNN at room temperature between 68°F to 77°F (20°C to 25°C).
-
Do not keep medicine that is out of date or that you no longer need.
- Dispose of unused medicines through community take-back disposal programs when available. If no community take-back disposal program is available go to www.fda.gov/drugdisposal for information on how to dispose of ORIAHNN the right way.
- Do not flush ORIAHNN down the toilet.
General information about the safe and effective use of ORIAHNN.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use ORIAHNN for a condition for which it was not prescribed. Do not give ORIAHNN to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about ORIAHNN that is written for health professionals.What are the ingredients in ORIAHNN?
Yellow/White AM Capsule:
Active ingredient: elagolix, estradiol, norethindrone acetate.
Inactive ingredients: anhydrous sodium carbonate, polyethylene glycol 3350, crospovidone, colloidal silicon dioxide, magnesium stearate, polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, purified water, lactose monohydrate, starch (corn), copovidone, talc, hypromellose, triacetin, and a gelatin capsule shell. The capsule shell contains the following ingredients: FD&C Red #40, FD&C Yellow #5, FD&C Yellow #6, titanium dioxide, gelatin, and printing ink (shellac, dehydrated alcohol, isopropyl alcohol, butyl alcohol, propylene glycol, strong ammonia solution, black iron oxide, potassium hydroxide, and purified water).
Light Blue/White PM Capsule:
Active ingredient: elagolix.
Inactive ingredients: anhydrous sodium carbonate, polyethylene glycol 3350, crospovidone, colloidal silicon dioxide, magnesium stearate, polyvinyl alcohol, titanium dioxide, polyethylene glycol, and talc, purified water, and a gelatin capsule shell. The capsule shell contains the following ingredients: FD&C Blue #2, FDA/E172 yellow iron oxide, titanium dioxide, gelatin, and printing ink (shellac, dehydrated alcohol, isopropyl alcohol, butyl alcohol, propylene glycol, strong ammonia solution, black iron oxide, potassium hydroxide, and purified water).Manufactured by AbbVie Inc. North Chicago, IL 60064
ORIAHNN is a trademark of AbbVie Inc.
For more information, go to www.ORIAHNN.com or call 1-844-674-2466.This Medication Guide has been approved by the U.S. Food and Drug Administration.
12/2025
20096646
-
cardiovascular conditions
-
PRINCIPAL DISPLAY PANEL
NDC 0074-1017-56
Rx only
56 CAPSULES
FOR 28 DAYS IN 4 WEEKLY BLISTER PACKS
Oriahnn®
elagolix, estadiol and norethindrone acetate capsules and elagolix capsules 300 mg/1 mg/0.5 mg and 300 mg
Co-Packaged for Oral Use
300 mg / 1 mg / 0.5 mg 300mg
*Elagolix 300 mg (equivalent to 310 mg of elagolix sodium)
Contains FD&C Yellow No. 5 (Tartrazine) as a color additive
Each weekly blister pack contains 7 capsules to be taken in the morning
Each capsule contains elagolix* (300 mg), estradiol (1 mg) and norethindrone acetate (0.5 mg)
Each weekly blister pack contains 7 capsules to be taken in the evening
Each capsule contains elagolix* (300 mg)

-
PRINCIPAL DISPLAY PANEL
NDC 0074-1017-14
Rx only
14 CAPSULES
FOR 7 DAYS
Oriahnn®
elagolix, estadiol and norethindrone acetate capsules and elagolix capsules 300 mg/1 mg/0.5 mg and 300 mg
Co-Packaged for Oral Use
Each pack contains 7 capsules of elagolix* (300 mg),
estradiol (1mg), and norethindrone acetate (0.5 mg),
and 7 capsules of elagolix* 300 mg in a weekly blister pack for 1 week of treatmentContains FD&C Yellow No. 5 (Tartrazine) as a color additive
PROFESSIONAL SAMPLE. NOT FOR SALE.

-
INGREDIENTS AND APPEARANCE
ORIAHNN
elagolix and estradiol and norethisterone kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0074-1017 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0074-1017-56 4 in 1 CARTON 05/29/2020 1 1 in 1 BLISTER PACK; Type 0: Not a Combination Product 2 NDC: 0074-1017-14 1 in 1 CARTON 09/24/2020 2 1 in 1 BLISTER PACK; Type 0: Not a Combination Product Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 BLISTER PACK 7 Part 2 1 BLISTER PACK 7 Part 1 of 2 ORIAHNN
elagolix and estradiol and norethisterone capsuleProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ELAGOLIX SODIUM (UNII: 5948VUI423) (ELAGOLIX - UNII:5B2546MB5Z) ELAGOLIX 300 mg NORETHINDRONE (UNII: T18F433X4S) (NORETHINDRONE - UNII:T18F433X4S) NORETHINDRONE 0.5 mg ESTRADIOL (UNII: 4TI98Z838E) (ESTRADIOL - UNII:4TI98Z838E) ESTRADIOL 1 mg Inactive Ingredients Ingredient Name Strength SODIUM CARBONATE (UNII: 45P3261C7T) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) CROSPOVIDONE, UNSPECIFIED (UNII: 2S7830E561) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) WATER (UNII: 059QF0KO0R) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) STARCH, CORN (UNII: O8232NY3SJ) COPOVIDONE K25-31 (UNII: D9C330MD8B) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) TRIACETIN (UNII: XHX3C3X673) FD&C RED NO. 40 (UNII: WZB9127XOA) FD&C YELLOW NO. 5 (UNII: I753WB2F1M) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) SHELLAC (UNII: 46N107B71O) ALCOHOL (UNII: 3K9958V90M) ISOPROPYL ALCOHOL (UNII: ND2M416302) BUTYL ALCOHOL (UNII: 8PJ61P6TS3) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) AMMONIA (UNII: 5138Q19F1X) FERROSOFERRIC OXIDE (UNII: XM0M87F357) POTASSIUM HYDROXIDE (UNII: WZH3C48M4T) Product Characteristics Color white (WHITE) , yellow (YELLOW) Score no score Shape CAPSULE (CAPSULE) Size 16mm Flavor Imprint Code EL;300;AM Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 7 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA213388 05/29/2020 Part 2 of 2 ORIAHNN
elagolix capsuleProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ELAGOLIX SODIUM (UNII: 5948VUI423) (ELAGOLIX - UNII:5B2546MB5Z) ELAGOLIX 300 mg Inactive Ingredients Ingredient Name Strength SODIUM CARBONATE (UNII: 45P3261C7T) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) CROSPOVIDONE, UNSPECIFIED (UNII: 2S7830E561) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) WATER (UNII: 059QF0KO0R) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) SHELLAC (UNII: 46N107B71O) ALCOHOL (UNII: 3K9958V90M) ISOPROPYL ALCOHOL (UNII: ND2M416302) BUTYL ALCOHOL (UNII: 8PJ61P6TS3) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) AMMONIA (UNII: 5138Q19F1X) FERROSOFERRIC OXIDE (UNII: XM0M87F357) POTASSIUM HYDROXIDE (UNII: WZH3C48M4T) Product Characteristics Color white (White) , blue (Light Blue) Score no score Shape CAPSULE Size 16mm Flavor Imprint Code EL;300;PM Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 7 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA213388 05/29/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA213388 05/29/2020 Labeler - AbbVie Inc. (078458370) Registrant - AbbVie Inc. (078458370)
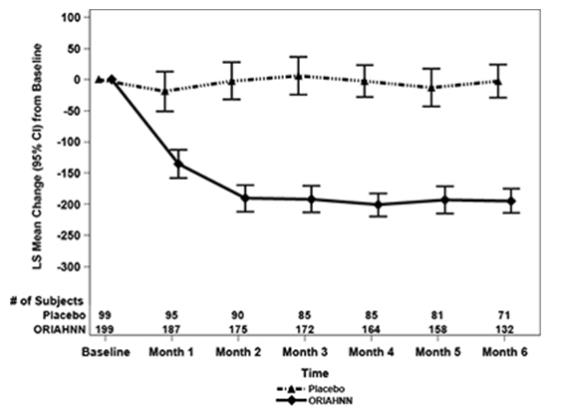
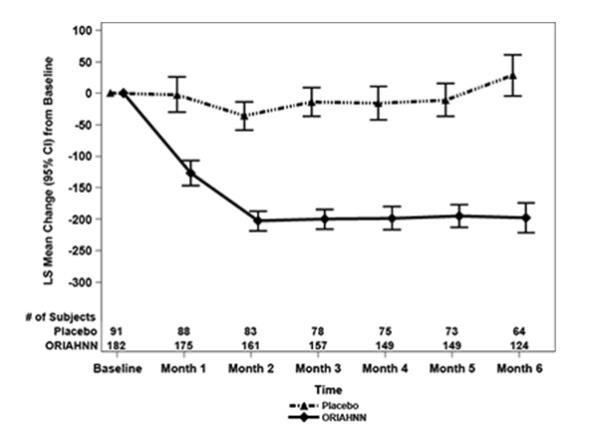
Trademark Results [Oriahnn]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ORIAHNN 88661880 not registered Live/Pending |
AbbVie Inc. 2019-10-21 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.